
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
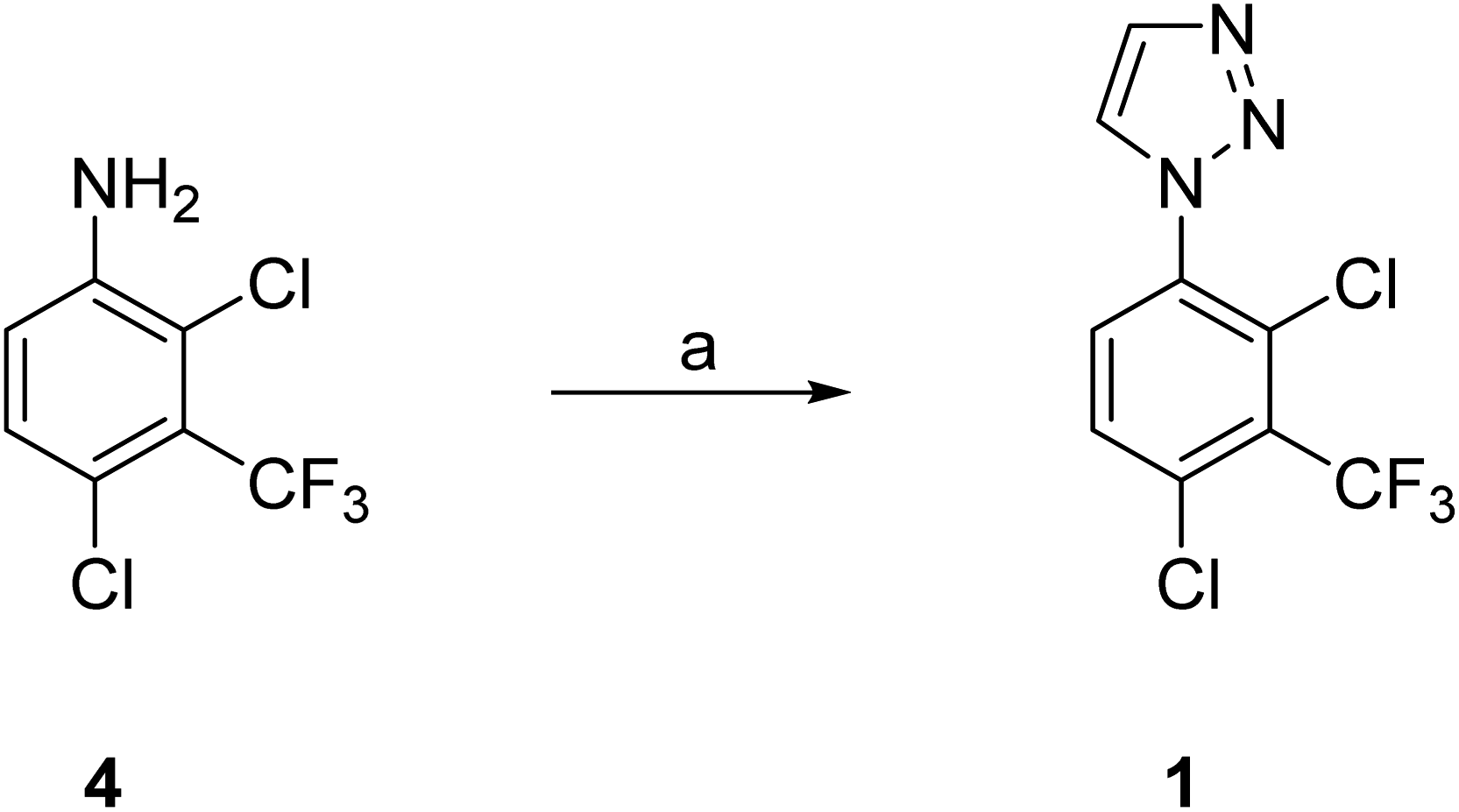
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-scale synthesis of Notum inhibitor 1-(2,4-dichloro-3-(trifluoromethyl)-phenyl)-1H-1,2,3-triazole (ARUK3001185) employing a modified Sakai reaction as the key step†
Benjamin N. Atkinson a,
Nicky J. Willisa,
Jennifer Smithb,
Rebecca Gillb,
Jody Alib,
Zhou Xuc,
Ping-Shan Laic and
Paul V. Fish*a
a,
Nicky J. Willisa,
Jennifer Smithb,
Rebecca Gillb,
Jody Alib,
Zhou Xuc,
Ping-Shan Laic and
Paul V. Fish*a
aAlzheimer's Research UK UCL Drug Discovery Institute, University College London, Cruciform Building, Gower Street, London, WC1E 6BT, UK. E-mail: p.fish@ucl.ac.uk; Tel: +44 (0)20 7679 6971
bKey Organics Ltd, Highfield Road Industrial Estate, Camelford, Cornwall, PL32 9RA, UK
cWuXi AppTec (Tianjin) Co., Ltd., 168 Nanhai Road, 10th Avenue, Tianjin Economic-Technological Development Area (TEDA), Tianjin 300457, P.R. China
First published on 16th September 2022
Abstract
1-Phenyl-1H-1,2,3-triazole 1 (ARUK3001185) was prepared on large scale from aniline 4 by application of both (1) a copper catalyzed azide–alkyne cycloaddition (CuAAC) with (trimethylsilyl)acetylene, and (2) a Clark modification of the Sakai reaction. The one-pot Sakai–Clark method with (MeO)2CHCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NNHTos (2b) proved to be superior as it was operationally simple, metal-free, and avoided the use of aryl azide 7. The Sakai–Clark method has been reliably performed on large scale to produce >100 g of 1 in good efficiency and high purity.
NNHTos (2b) proved to be superior as it was operationally simple, metal-free, and avoided the use of aryl azide 7. The Sakai–Clark method has been reliably performed on large scale to produce >100 g of 1 in good efficiency and high purity.
Introduction
Notum is a carboxylesterase enzyme that suppresses Wnt signaling1 through deacylation of an essential palmitoleate group on Wnt proteins.2,3 There is a growing understanding of the role Notum plays in human disease such as osteoporosis4 and colorectal cancer5 supporting the need to discover improved inhibitors.6–9 Recently, Willis et al. reported ARUK3001185 (1) as a potent, selective and brain penetrant inhibitor of Notum suitable for oral delivery in rodent models of disease.10 We are investigating the role of Notum in modulating Wnt signaling in Alzheimer's disease11 and required multigram quantities of Notum inhibitor 1 for use in target validation and preclinical toxicology studies.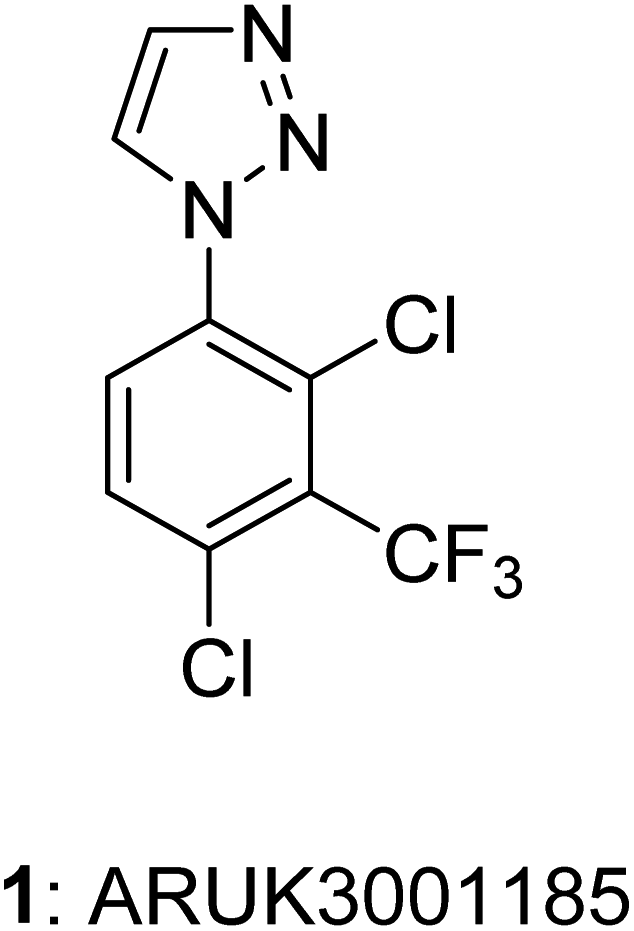
1-Aryl-1H-1,2,3-triazoles are an important structural motif in bioactive molecules and drugs11 that has led to a number of synthetic methods for their construction.12 These methods include the direct reaction of 1,2,3-triazole with a suitable aryl halide (ArBr, ArI) promoted by metal catalyzed cross-coupling. However, these tend to give mixtures of 1- and 2-aryl isomers through poor selectivity that limits its utility.13,14 Methods are preferred where the specific creation of the 1-aryl triazole of can be controlled by the appropriate choice of reagents/synthons with defined and predictable reactivity. Copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) promotes a [3 + 2] cyclisation where the three contiguous N atoms are provided by an aryl azide synthon (Ar–N1–N2–N3). Aryl azides are readily available from anilines, via the diazonium salt, and so this approach has become popular, especially when combined with a suitably protected alkyne (e.g. TMS–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, C4–C5 synthon) that can be conveniently removed when necessary.
CH, C4–C5 synthon) that can be conveniently removed when necessary.
In 1986 Sakai et al. described a new method to prepare 1,2,3-triazoles from primary amines and α,α-dichloro tosylhydrazones 2a under mild conditions as a metal-free alternative to azide–alkyne cycloaddition.15 The Sakai reaction was probably under-utilized until 2012 when Westermann et al. explored the scope and limitations of the reaction.16 The outcome and regioselectivity of the reaction is reliable in that the product triazole of the [1 + 4] cyclisation is created from the aniline (Ar–N1) and tosylhydrazone (C5–C4–N3–N2).
Recently, Clark has reported a convenient and operationally simple modification of the Sakai reaction that produces 1-aryl triazoles with a range of anilines under mildly acidic conditions.17,18 The one-pot reaction of 2,2-dimethoxyacetaldehyde ((MeO)2CHCHO) with p-toluene sulfonylhydrazide (TsNHNH2) in MeOH at RT for 2 h provides the α,α-dimethoxy tosylhydrazone 2b in situ, and then addition of the aniline and AcOH with warming to 75 °C for 16 h produces the 1-aryl triazole in good yield.18 Further modifications of the Sakai reaction were presented by Bi using a base-promoted annulation of α,α-difluoro tosylhydrazone 2c with a wide scope of amines under mild conditions.19 Proposed reaction mechanisms for these transformations have been reported.18,19
Here, we disclose the application of both a CuAAC method and a modified Sakai reagent for the large-scale synthesis of aryl triazole 1 from aniline 4 (Fig. 1).
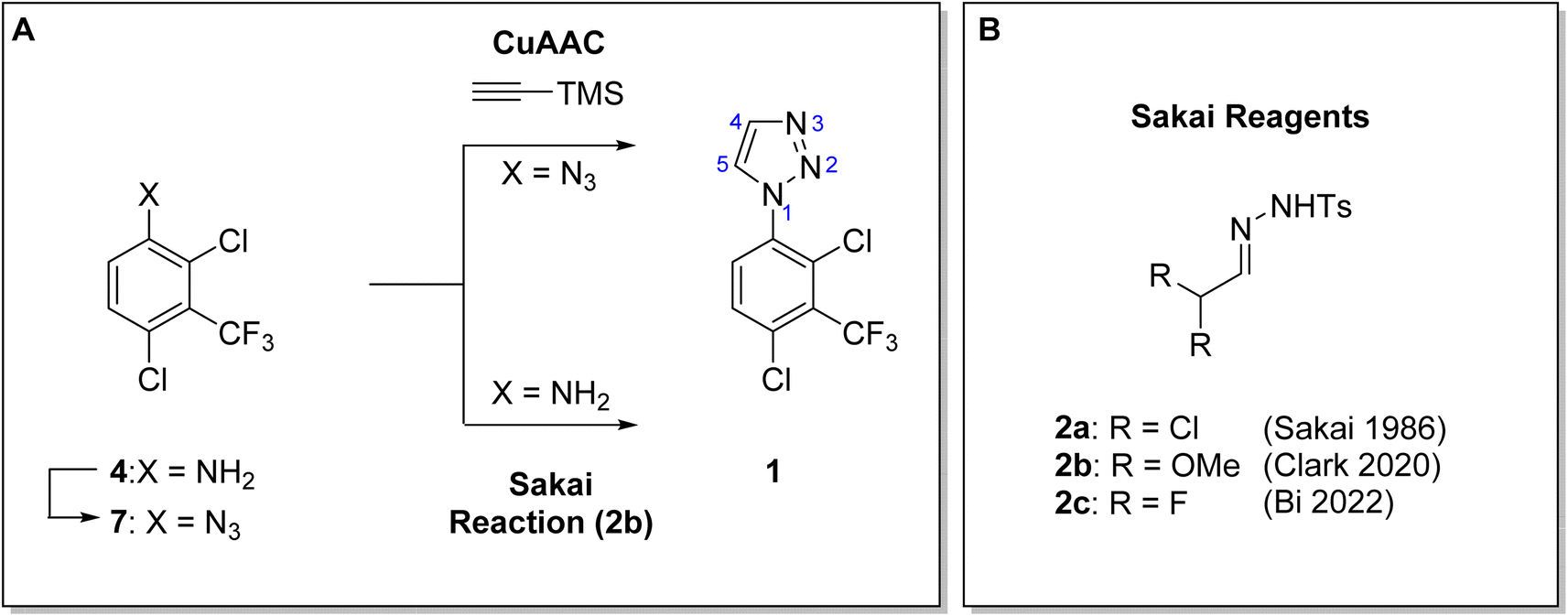 | ||
| Fig. 1 (A). Synthesis of 1-aryl-1H-1,2,3-trizole 1 using both a CuAAC method and a modified Sakai reagent from aniline 4. (B) α,α-disubstituted tosylhydrazones, Sakai reagents (2). | ||
Results and discussion
These two complementary methods to prepare 1 both required 2,4-dichloro-3-(trifluoromethyl)aniline (4) as the starting material which provided an element of efficiency and economy to this approach. Aniline 4 was either purchased from commercial suppliers or prepared by standard chlorination of 2-chloro-3-(trifluoromethyl)aniline (3) with N-chlorosuccinimide (NCS) as described below (Scheme 1).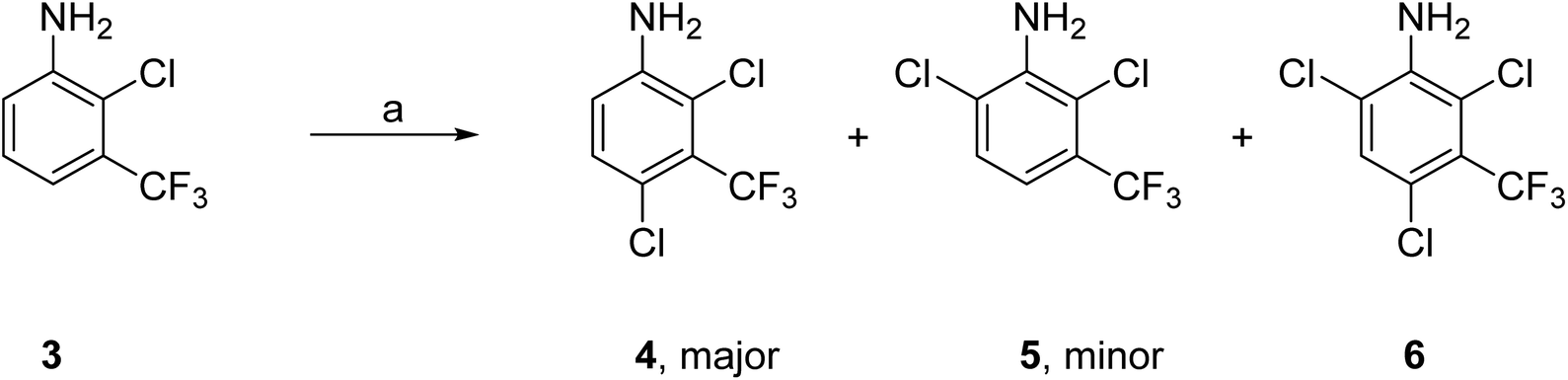 | ||
Scheme 1 Synthesis of 2,4-dichloro-3-(trifluoromethyl)aniline (4). Reagents and conditions: (a) NCS (1.1 equiv.), MeCN, 60 °C, 16 h; ratio of products 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:6, 72:19:6; 4 isolated yield 56%. :5:6, 72:19:6; 4 isolated yield 56%. | ||
Pilot reactions with 3 and NCS (1.1 equiv.) in a variety of solvents showed that the choice of solvent had a significant effect on both the conversion to 4 and the ratio of products (4–6) (Table S1†). NCS (1.1 equiv.) in MeCN gave the highest conversion to 4 with the lowest formation of 5 and 6. Treatment of 3 (50 g scale) with NCS (1.1 equiv.) in MeCN at 60 °C for 16 h gave the required 4-chlorinated aniline 4 as the major product in 56% isolated yield. Analysis of the crude product mixture showed that the 6-chloro regio isomer 5 was also produced as a minor product along with small amounts of 6 through double chlorination at both the 4- and 6-positions (3:4:5:6, 1:72:19:6). The reaction was sequentially scaled up to 3 × 200 g of 3 without issue; the distribution of products was maintained as were the isolated yields of 4 (44%). Aniline 4 was purified by chromatography to give high purity material for use in the next step. As the need for quantities of 4 increased, it became economically more viable to prepare 4 from 3 (rather than purchase 4), as 3 is significantly less expensive than 4 per unit cost.
Our efforts then focused on the development of a reliable synthesis of 50 g of 1-aryl-1H-1,2,3-triazole 1. This multigram synthesis of 1 using the CuAAC method essentially followed the discovery route but with additional precautions to minimize the risk of azide 7 on larger scale (Scheme 2).10 In brief, reaction of aniline 4 with NaNO2 in TFA gave the corresponding diazonium salt and then reaction with NaN3 gave the required azide 7. Copper catalyzed azide–alkyne cycloaddition of 7 with TMS-acetylene afforded a mixture of mostly 4-TMS triazole 8, along with 1 (8:1, 5:2), which was treated with K2CO3 in MeOH to complete the proto-desilylation to give 1.
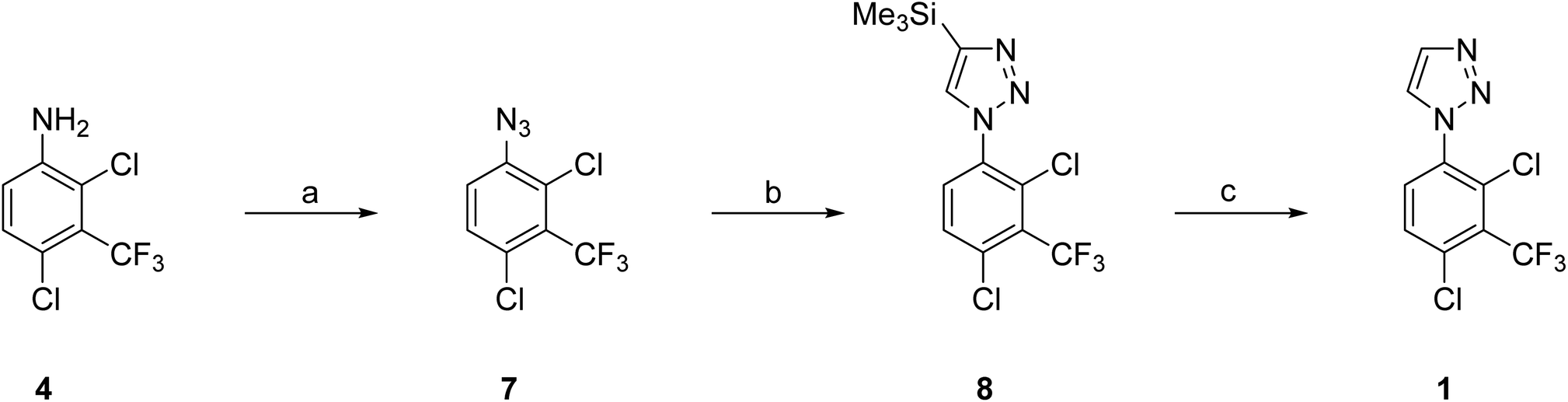 | ||
| Scheme 2 First generation multigram synthesis of 1 using CuAAC method. Reagents and conditions: (a) (i) NaNO2 (4.8 equiv.), CF3CO2H, 0 °C, 0.5 h → RT, 1.5 h; (ii) NaN3 (4.8 equiv.), H2O, 0 °C → RT, 16 h; (iii) NaOH, 0 °C; (b) HCCSiMe3 (2.0 equiv.), sodium L-ascorbate (0.95 equiv.), CuSO4·5H2O (0.5 equiv.), MeOH-tBuOH-H2O, RT, 16 h; (c) K2CO3 (2 equiv.), MeOH, RT, 3 d; 3-step overall total yield 65 g, 53%. | ||
Azides are potentially explosive reagents and should be treated with caution (see Experimental section for details). Differential scanning calorimetry (DSC) analysis of azide 7 established 42 °C as the maximum safe handling temperature for its preparation and use.10 In addition, azide 7 was maintained in solution throughout with solvent swapping as necessary. Reactions were performed batchwise (5 × 20 g of 4) to limit the quantity of sodium azide used per reaction, and were carried through the 3-step sequence without purification until the end. Purification by chromatography gave 1 (13.5–15.8 g, 55–64%). These five batches were then pooled for a final purification to give a homogenous, crystalline product 1 (65 g, 53%) of very high purity.
Although successful in meeting its objective of producing >50 g of high purity 1, the CuAAC method was inefficient through repetition of the 3-step procedure and would be scale limited without a more detailed understanding of the energetics of the azide 7 and the overall process. An improved procedure was required for scale-up to 100 g of 1 and larger quantities.
The Sakai reaction was an attractive alternative because of its simplicity through N1-aryl specificity of the construction of the 1,2,3-triazole, readily available reagents, precedent for a toleration of wide variety of substituents on the aryl ring, and because it has been used to make representative bioactive molecules on gram scale.17–19 A pilot reaction with 4 (334 mg) using α,α-dimethoxy tosylhydrazone 2b in MeOH at 75 °C for 16 h following the published conditions18 gave the required triazole 1 in 65% yield demonstrating the feasibility of this approach. Switching the solvent to THF improved the yield to 76%, whereas the use of DMF as solvent only gave recovered aniline. A few exploratory reactions of α,α-difluoro tosylhydrazone 2c with 4 by the two reported methods (NaH/EtOAc or EtNiPr2/MeOH) failed to produce 1 despite the reported success with similar anilines.19 Hence 2b in THF was selected as the preferred reagent and solvent respectively for the larger scale procedures.
Three large scale reactions were then performed in parallel (4:25 g and 2 × 140 g) and the crude products combined for purification. A mixture of TsNHNH2 and (MeO)2CHCHO in THF was stirred at 20 °C for 2 h to create the α,α-dimethoxy tosylhydrazone 2b in situ and then addition of aniline 4 and AcOH in THF with warming to 75 °C for 5 h produced crude 1-aryl triazole 1 (Scheme 3). The reaction mixture was cooled to RT, washed with brine and concentrated. The combined products from these three reactions was purified by column chromatography followed by preparative HPLC. The appropriate fractions were combined and partially concentrated, which prompted crystallisation. This approach proved to be very effective in producing 1 (113 g, 30%) of very high purity (99.8% pure by HPLC), although the overall yield was somewhat lower than the pilot reactions.20 This chemistry, and these yields, have not been optimized and a more systematic approach to this process would be justified as the next step.
 | ||
| Scheme 3 Second generation multigram synthesis of 1 using Sakai–Clark method. Reagents and conditions: (a) (i) TsNHNH2 (2.0 equiv.), (MeO)2CHCHO (3.3 equiv.), THF, RT, 2 h; (ii) 4 (1.0 equiv.), AcOH (3.0 equiv.), THF, 75 °C, 5 h, 30%. | ||
Conclusion
A one-pot synthesis of Notum inhibitor ARUK3001185 (1) has been developed enabling the delivery of multi-gram quantities of 1 for use in target validation and preclinical toxicology studies. 1-Phenyl-1H-1,2,3-triazole 1 was prepared from aniline 4 by a modification of the Sakai reagent. The one-pot Sakai–Clark method with α,α-dimethoxy tosylhydrazone 2b proved to be superior to the classical CuAAC method as it was operationally simple, metal-free, and avoided the use of aryl azide 7. This method has been reliably performed on large scale to produce >100 g of 1 in good efficiency and high purity.Experimental section
General information
Unless preparative details are provided, all reagents and solvents were purchased from commercial suppliers and used without further purification. Petroleum ether refers to fraction bp 60-90 °C. Thin-layer chromatography (TLC) was carried out on aluminum-backed Silica gel 60 F254 plates. Organic solvent layers were routinely dried with anhydrous Na2SO4 or MgSO4 and concentrated under reduced pressure using a Büchi rotary evaporator. Compound purification by column chromatography was performed using either a Biotage Isolera using prepacked Biotage Sfär silica cartridges, or silica gel packed columns. 1H, 13C and 19F NMR spectra were recorded in deuterated (≥99.5%) solvents on a Bruker Avance III 400 with ultrashield magnet and B-ACS-60 autosampler. Chemical shifts (δ) are reported as parts per million (ppm), coupling constants (J) are reported in Hz, and signal multiplicities are reported as singlet (s), doublet (d), doublet of doublets (dd), quartet (q), or multiplet (m).Liquid chromatography-mass spectrometry (LCMS) analysis was performed on an Agilent 1260 infinity HPLC with Agilent 6130 single quadrupole MS in electrospray mode. Method 1: column, Phenomenex Kinetex XB-C18, 50 × 4.6 mm, 2.6 μm; column temperature, 40 °C; flow rate, 2 mL min−1. Method 2: column, Luna C18, 50 × 2.0 mm, 5 μm; column temperature, 40 °C; flow rate, 1 mL min−1. Solvent system and elution profiles, see Table 1.
| Time (minutes) | % Aqueous (A) (0.1% formic acid in water) | % Organic (B) (100% acetonitrile) |
|---|---|---|
| Method 1 | ||
| 0 | 95 | 5 |
| 1.37 | 2 | 98 |
| 1.83 | 95 | 5 |
| 2.25 end | ||
|
||
| Method 2 | ||
| 0 | 95 | 5 |
| 3.00 | 5 | 95 |
| 4.00 | 95 | 5 |
| 4.50 end | ||
Analytical high performance liquid chromatography (HPLC) was performed on either an Agilent 1260 (method 1) or Shimadzu LC-20AB (method 2): column, XBridge C18, 50 × 2.1 mm, 5 μm; column temperature, 40 °C; flow rate, 0.8–1.2 mL min; detection at λ 220 and 254 nm; solvent system and elution profile, see Table 2.
| Time (minutes) | % Aqueous (A) (10 mM NH4HCO3 in water) | % Organic (B) (100% acetonitrile) | Flow rate (mL min−1) |
|---|---|---|---|
| Method 1 | |||
| 0.00 | 90 | 10 | 0.8 |
| 4.00 | 20 | 80 | 0.8 |
| 4.90 | 90 | 10 | 1.2 |
| 6.00 end | |||
|
|||
| Method 2 | |||
| 0.00 | 90 | 10 | 0.8 |
| 4.50 | 20 | 80 | 0.8 |
| 5.40 | 90 | 10 | 1.2 |
| 6.00 end | |||
Preparative high performance liquid chromatography (Prep-HPLC) was performed on Shimadzu LC-8A: column, Phenomenex luna c18 250 × 100 mm, 10 μm; column temperature, 40 °C; flow rate, 250 mL min; detection at λ 220 and 254 nm; solvent system and elution profile, see Table 3.
| Time (minutes) | % Aqueous (A) (0.09% TFA in water) | % Organic (B) (100% acetonitrile) |
|---|---|---|
| 0 | 60 | 40 |
| 20 | 30 | 70 |
| 30 | 2 | 98 |
| 35 end |
Chemical reactions were frequently performed in parallel batches and then combined for purification for expediency. Chemical yields refer to isolated and purified products unless otherwise stated. Purity of compounds was evaluated by NMR spectroscopy, HPLC and/or LCMS analysis. All compounds had purity ≥95%.
Full spectroscopic and analytical data for 1 have been reported.10 Additional spectroscopic and analytical data for 1 from this work are presented in the ESI† (Fig. S1–S6†).
3 × 200 g) and then combined for purification.N-Chlorosuccinimide (150.0 g, 1.12 mol, 1.1 equiv.) was added to a stirred solution of 2-chloro-3-(trifluoromethyl)aniline (3) (200.0 g, 1.02 mol) in MeCN (2.4 L) at 20 °C, and the mixture was then heated at 60 °C for 16 h. The mixture was cooled to RT, concentrated, and the residue triturated with EtOAc (1.0 L).
The combined EtOAc filtrates (ca 3 L) from these three reactions was concentrated, and the crude product purified by column chromatography on silica gel (1–10% EtOAc in petroleum ether) to afford 4 as brown oil (310 g, 1.35 mol, 44%) containing some residual EtOAc (ca. 0.15 equiv.) by 1H NMR.
1H NMR (400 MHz, CDCl3) 7.19 (d, J = 8.8 Hz, 1H), 6.84 (d, J = 8.9 Hz, 1H), 4.28 (br s, 2H);19F NMR (376 MHz, CDCl3)-55.71; HPLC (method 1): Rt 3.84 minutes, 99.6%.
Azide 7 was maintained in solution throughout its preparation and use, with solvent swapping as necessary. Solutions of 7 should not be evaporated to dryness. Differential scanning calorimetry (DSC) analysis of 7 established 42 °C as the maximum safe handling temperature.10
Reactions were performed batchwise (5 × 20 g of 4) to limit the quantity of sodium azide used per reaction, and carried through the 3-step sequence without purification until the end. A representative experiment is presented.
Step 1. 2,4-Dichloro-3-(trifluoromethyl)aniline (4) (20.0 g, 87.0 mmol, 1.0 equiv.) was dissolved in 2,2,2-trifluoroacetic acid (400 mL) under nitrogen and the mixture was cooled to 0 °C with efficient stirring. Sodium nitrite (7.30 mol L−1, 56.5 mL, 412 mmol, 4.8 equiv.) in water was added dropwise over 90 min, whilst maintaining the reaction mixture at ca. 0 °C throughout. The mixture was stirred at 0 °C for an additional 30 min., the ice bath was removed, and mixture allowed to warm slowly for 1 h (final temp. 5–10 °C). The solution of the diazonium salt was re-cooled to 0 °C, and sodium azide (3.85 mol L−1, 107 mL, 412 mmol, 4.8 equiv.) in water was added dropwise over ca. 90 min., keeping the reaction temperature below 0 °C throughout. The reaction mixture was left in the ice bath and allowed to warm slowly to RT overnight. The solution of azide 7 in TFA-water was cooled to 0 °C and basified with the dropwise addition of aqueous sodium hydroxide (5.00 mol L−1, 1045 mL, 5.23 mol) over ca. 5 h whilst maintaining the temperature at 0 °C. The mixture was warmed to RT, diluted with brine (500 mL) and extracted into Et2O (3 × 700 mL). The combined organic extracts were dried (MgSO4) and partially concentrated in vacuo (water bath set to 24 °C) to a volume of ca. 250 mL. tert-BuOH (200 mL) was then added, and the mixture evaporated until final traces of Et2O have been removed to give 1-azido-2,4-dichloro-3-(trifluoromethyl)benzene (7) as solution in tert-BuOH (119 g total weight), which was used directly in the next step.
1H NMR (400 MHz, CDCl3) δ 7.45 (dd, J = 8.8, 0.8 Hz, 1H), 7.26 (dd, J = 8.7, 0.7 Hz, 1H); LCMS (method 1): Rt 1.97 minutes, 100%.
Step 2. Azide 7 as solution in tert-BuOH was dissolved in MeOH (200 mL) under nitrogen at RT. Copper sulfate pentahydrate (0.400 mol L−1, 102 mL, 40.9 mmol, 0.47 equiv.) in water, sodium-L-ascorbate (1.01 mol L−1, 81.8 mL, 82.6 mmol, 0.95 equiv.) in water and trimethylsilylacetylene (17.1 g, 174 mmol, 2.0 equiv.) were sequentially added, and the mixture stirred at RT overnight. TLC (20% EtOAc in hexane) showed no azide remaining. The mixture was extracted with CH2Cl2 (3 × 200 mL), dried (MgSO4) and evaporated to give mostly 1-(2,4-dichloro-3-(trifluoromethyl)phenyl)-4-trimethylsilyl-1H-1,2,3-triazole (8) along with triazole 1 (33 g, 8
:1, 5:2), which was used directly in the next step to complete the desilylation.1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.66–7.59 (m, 2H), 0.39 (s, 9H); LCMS (method 1): 1 Rt 1.70 minutes, 26% and 8 Rt 1.98 minutes, 68%.
Step 3. Crude 4-TMS triazole 8 was dissolved in MeOH (550 mL), potassium carbonate (24.0 g, 174 mmol, 2.0 equiv.) was added, and the mixture stirred at RT under nitrogen for 3 d. The reaction mixture was carefully concentrated under reduced pressure to remove the MeOH, and the residue suspended in CH2Cl2 (300 mL). This solution was filtered through a pad of silica gel and the silica pad was then washed with 20% MeOH in CH2Cl2 to ensure all product 1 was collected. The filtrate was evaporated, and the crude product (ca. 32 g) (range 28–35 g) purified by (1) column chromatography (0–100% CH2Cl2 in hexane), (2) column chromatography (0–20% EtOAc in hexane) and (3) triturated in hexane to give 1 as a white solid (14.7 g, 59%) (range 13.5–15.8 g, 55–64%).
Five batches of 1 were combined (73.4 g) and purified by column chromatography (15–20% EtOAc in hexane) to give 1 (65.3 g, 53%) as a crystalline, white solid of high purity.
Mp 107.2–108.6 °C; 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 1.1 Hz, 1H), 7.90 (d, J = 1.1 Hz, 1H), 7.70–7.61 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 136.37, 135.77, 133.93, 131.54, 131.01, 130.83, 127.99 (q, J = 30.95 Hz), 126.04, 122.04 (q, J = 276.76 Hz); LCMS (method 1): Rt 1.69 minutes, 100%, MS+ m/z 282.0 (100%).
Small scale pilot reactions. THF (3.0 mL) was added to 4-methylbenzenesulfonohydrazide (250 mg, 1.34 mmol, 1.0 equiv.) in a thick-walled reaction vial with stir bar and the vial was sealed with a crimped septum cap. An aqueous solution of dimethoxyacetaldehyde (0.22 mL, 60% in H2O, 1.48 mmol, 1.1 equiv.) was added via syringe and the mixture stirred at RT for 2 h. Separately, 2,4-dichloro-3-(trifluoromethyl)aniline (4) (334 mg, 1.48 mmol, 1.1 equiv.) and AcOH (80 μL, 1.34 mmol, 1.0 equiv.) was dissolved in THF (0.5 mL), and this solution was added to via syringe. The mixture was then heated at 75 °C for 18 h in a DrySyn block. TLC (30% EtOAc in cyclohexane) showed no aniline remaining. The reaction mixture was cooled to RT, concentrated and the residue partitioned between CH2Cl2 (30 mL) and water (20 mL) with vigorous mixing for 5 minutes. The 2-phase mixture was passed through a phase separator and the organic phase concentrated. The crude product was purified by chromatography on a 25 g Biotage SNAP KP Si cartridge (2-45% EtOAc in cyclohexane) to give 1 (286 mg, 1.01 mmol, 76%) as an off-white solid.
Three large scale reactions were then performed in parallel (4:25 g and 2 × 140 g) and combined for purification.
25 g scale. A mixture of 4-methylbenzenesulfonohydrazide (40.5 g, 0.217 mol, 2.0 equiv.) and 2,2-dimethoxyacetaldehyde (62.2 g, 60 wt% in H2O, 0.359 mol, 3.3 equiv.) in THF (500 mL) was stirred at 20 °C for 2 h. A solution of 4 (25 g, 0.109 mol) and AcOH (19.6 g, 0.326 mol, 3.0 equiv.) in THF (250 mL) was added, and the mixture heated at 75 °C for 5 h. The mixture was cooled to RT, washed with brine (2 × 355 mL), dried (Na2SO4), and concentrated. The crude product was combined with additional batches for purification.
2 × 140 g scale. A mixture of 4-methylbenzenesulfonohydrazide (226.7 g, 1.217 mol, 2.0 equiv.) and 2,2-dimethoxyacetaldehyde (348.4 g, 60 wt% in H2O, 2.01 mol, 3.3 equiv.) in THF (2.8 L) was stirred at 20 °C for 2 h. A solution of 4 (140 g, 0.609 mol) and AcOH (109.6 g, 1.825 mol, 3.0 equiv.) in THF (1.4 L) was added, and the mixture heated at 75 °C for 5 h. The mixture was cooled to RT, washed with brine (2 × 2 L), dried (Na2SO4), and concentrated.
The crude product from these three reactions was purified by column chromatography on silica gel (10–40% EtOAc in petroleum ether) and then by preparative HPLC (40–98% MeCN in water containing 0.09% TFA) (Table 3). The appropriate fractions were combined and partially concentrated to remove most of MeCN, which prompted crystallisation. The solid was collected, washed with water, and dried to afford 1 as white solid (113 g, 401 mmol, 30%).
Mp 108.2–109.2 °C;1H NMR (400 MHz, CDCl3) 7.98 (d, J = 1.1 Hz, 1H), 7.91 (d, J = 1.1 Hz, 1H), 7.72–7.62 (m, 2H); 13C NMR (101 MHz, CDCl3) 136.35, 135.74, 133.94, 131.55, 131.04, 130.82, 127.93 (q, J = 31.51 Hz), 126.09, 122.03 (q, J = 278.26 Hz); 19F NMR (376 MHz, CDCl3) −55.79; HPLC (method 2): Rt 3.29 minutes, 99.8%. LCMS (method 2): Rt 2.50 minutes, 100%, MS+ m/z 282.0 (100%).
Funding
This work was supported by Alzheimer's Research UK (ARUK) and the Alzheimer's Drug Discovery Foundation (ADDF). The ARUK UCL Drug Discovery Institute is core funded by Alzheimer's Research UK (560832). The Alzheimer's Drug Discovery Foundation and Harrington Discovery Institute at University Hospitals in Cleveland granted an ADDF-Harrington Scholar Award to P.·V.·F.Author contributions
The research was performed through contributions of all authors.Conflicts of interest
B. N. A., N. J. W, and P. V. F. are co-inventors of patent application WO 2020043866, which describes inhibitors of Notum. The authors have no other relevant affiliations or financial involvement apart from those disclosed.Abbreviations
| CuAAC | copper(I)-catalyzed azide–alkyne cycloaddition |
| DMF | dimethylformamide |
| DSC | differential scanning calorimetry |
| HPLC | high performance liquid chromatography |
| LCMS | Liquid chromatography-mass spectrometry |
| NCS | N-chlorosuccinimide |
| RT | room temperature |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TLC | thin-layer chromatography |
| TMS | trimethylsilyl |
Acknowledgements
Our advisors from the Alzheimer's Drug Discovery Foundation and Harrington Discovery Institute Therapeutic Development Center are gratefully acknowledged for their expert guidance: Peter Bernstein, Jeffery Klein, Robert Lipper, Meriel Owen, Frank Richardson, Ashley Roe, and Alessio Travaglia. We thank staff at the UCL Department of Chemistry for spectroscopic and analytical services.Notes and References
- R. Nusse and H. Clevers, Wnt/β-catenin signaling, disease, and emerging therapeutic modalities, Cell, 2017, 169, 985–999 CrossRef CAS PubMed.
- S. Kakugawa, P. F. Langton, M. Zebisch, S. A. Howell, T. H. Chang, Y. Liu, T. Feizi, G. Bineva, N. O'Reilly, A. P. Snijders, E. Y. Jones and J.-P. Vincent, Notum deacylates Wnt proteins to suppress signalling activity, Nature, 2015, 519, 187–192 CrossRef CAS PubMed.
- X. Zhang, S. M. Cheong, N. G. Amado, A. H. Reis, B. T. MacDonald, M. Zebisch, E. Y. Jones, J. G. Abreu and X. He, NOTUM is required for neural and head induction via Wnt deacylation, oxidation, and inactivation, Dev. Cell, 2015, 32, 719–730 CrossRef CAS PubMed.
- R. Brommage, J. Liu, P. Vogel, F. Mseeh, A. Y. Thompson, D. G. Potter, M. K. Shadoan, G. M. Hansen, S. Jeter-Jones, J. Cui, D. Bright, J. P. Bardenhagen, D. D. Doree, S. Movérare-Skrtic, K. H. Nilsson, P. Henning, U. H. Lerner, C. Ohlsson, A. T. Sands, J. E. Tarver, D. R. Powell, B. Zambrowicz and Q. Liu, NOTUM inhibition increases endocortical bone formation and bone strength, Bone Res., 2019, 7, 2 CrossRef CAS PubMed.
- D. J. Flanagan, N. Pentinmikko, K. Luopajärvi, N. J. Willis, K. Gilroy, A. P. Raven, L. Mcgarry, J. I. Englund, A. T. Webb, S. Scharaw, N. Nasreddin, M. C. Hodder, R. A. Ridgway, E. Minnee, N. Sphyris, E. Gilchrist, A. K. Najumudeen, B. Romagnolo, C. Perret, A. C. Williams, H. Clevers, P. Nummela, M. Lähde, K. Alitalo, V. Hietakangas, A. Hedley, W. Clark, C. Nixon, K. Kirschner, E. Y. Jones, A. Ristimäki, S. J. Leedham, P. V. Fish, J.-P. Vincent, P. Katajisto and O. J. Sansom, NOTUM from Apc-mutant cells biases clonal competition to initiate cancer, Nature, 2021, 594, 430–435 CrossRef CAS PubMed.
- J. E. Tarver, P. K. Pabba, J. Barbosa, Q. Han, M. W. Gardyan, R. Brommage, A. Y. Thompson, J. M. Schmidt, A. G. E. Wilson, W. He, V. K. Lombardo and K. G. Carson, Stimulation of cortical bone formation with thienopyrimidine based inhibitors of Notum pectinacetylesterase, Bioorg. Med. Chem. Lett., 2016, 26, 1525–1528 CrossRef CAS PubMed.
- R. M. Suciu, A. B. Cognetta, Z. E. Potter and B. F. Cravatt, Selective irreversible inhibitors of the Wnt deacylating enzyme NOTUM developed by activity-based protein profiling, ACS Med. Chem. Lett., 2018, 9, 563–568 CrossRef CAS.
- E. D. Bayle, F. Svensson, B. N. Atkinson, D. Steadman, N. J. Willis, H. L. Woodward, P. Whiting, J.-P. Vincent and P. V. Fish, Carboxylesterase Notum is a druggable target to modulate Wnt signalling, J. Med. Chem., 2021, 64, 4289–4311 CrossRef CAS PubMed.
- Y. Zhao, S. Jolly, S. Benvegnu, E. Y. Jones and P. V. Fish, Small molecule inhibitors of carboxylesterase Notum, Future Med. Chem., 2021, 13, 1001–1015 CrossRef CAS PubMed.
- N. J. Willis, W. Mahy, J. Sipthorp, Y. Zhao, H. Woodward, B. N. Atkinson, E. D. Bayle, F. Svensson, S. Frew, F. Jeganathan, A. Monaghan, S. Benvegnu, S. Jolly, L. Vecchia, R. R. Ruza, S. Kjaer, S. Howell, A. Snijders, M. Bictash, P. C. Salinas, J.-P. Vincent, E. Y. Jones, P. Whiting and P. V. Fish, Design of a potent, selective and brain penetrant inhibitor of Wnt-deactivating enzyme Notum by optimization of a crystallographic fragment hit, J. Med. Chem., 2022, 65, 7212–7230 CrossRef CAS PubMed.
- Recent review of bioactive 1,2,3-triazoles: S. M. L. daForezi, C. G. S. Lima, A. A. P. Amaral, P. G. Ferreira, M. C. B. V. de Souza, A. C. Cunha, F. de C. da Silva and V. F. Ferreira, Bioactive 1,2,3-triazoles: an account on their synthesis, structural diversity and biological applications, Chem. Rec., 2021, 21, 2782–2807 CrossRef PubMed.
- Reviews of general methods to prepare 1,2,3-triazoles:
(a) A. C. Tome, Product class 13: 1,2,3-triazoles, Sci. Synth., 2004, 13, 415–601 CAS;
(b) N. Z. Fantoni, A. H. El-Sagheer and T. Brown, A hitchhiker's guide to click-chemistry with nucleic acids, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed;
(c) M. M. Matin, P. Matin, M. R. Rahman, T. Ben Hadda, F. A. Almalki, S. Mahmud, M. M. Ghoneim, M. Alruwaily and S. Alshehri, Triazoles and their derivatives: chemistry, synthesis and therapeutic applications, Front. Mol. Biosci., 2022, 9, 864286 CrossRef CAS PubMed;
(d) Y. Zheng, L. Tian, V. Ramadoss, H. Zhang, L.-L. Zhu and Y. Wang, Recent developments in N2-selective functionalization of 1,2,3-triazoles, Synthesis, 2022, 54, 2548–2560 CrossRef CAS.
- Cross-coupling methods to N1-aryl 1,2,3-triazoles: (a) R.-J. Song, C.-L. Deng, Y.-X. Xie and J.-H. Li, Solvent-free copper/ion co-catalyzed N-arylation reactions of nitrogen containing heterocycles with trimethoxysilanes in air, Tetrahedron Lett., 2007, 48, 7845–7848 CrossRef CAS; (b) B.-X. Tang, S.-M. Guo, M.-B. Zhang and J.-H. Li, N-Arylations of nitrogen-containing heterocycles with aryl and heteroaryl halides using copper(I) oxide nanoparticle/1,10-phenanthroline catalytic system, Synthesis, 2008, 11, 1707–1716 Search PubMed.
- Cross-coupling methods to N2-aryl 1,2,3-triazoles: S. Ueda, M. Su and S. L. Buchwald, Highly N2-selective palladium-catalyzed arylation of 1,2,3-triazoles, Angew. Chem., Int. Ed., 2011, 50, 8944–8947 CrossRef CAS.
- K. Sakai, N. Hida and K. Kondo, Reactions of α-polyhalo ketone tosylhydrazones with sulfide ion and primary amines. Cyclization to 1,2,3-thiadiazoles and 1,2,3-triazoles, Bull. Chem. Soc. Jpn., 1986, 59, 179–183 CrossRef CAS.
- S. S. van Berkel, S. Brauch, L. Gabriel, M. Henze, S. Stark, D. Vasilev, L. A. Wessjohann, M. Abbas and B. Westermann, Traceless tosylhydrazone-based triazole formation: a metal-free alternative to strain-promoted azide-alkyne cycloaddition, Angew. Chem., Int. Ed., 2012, 51, 5343–5346 CrossRef CAS PubMed.
- P. R. Clark, G. D. Williams, J. F. Hayes and N. C. O. Tomkinson, A scalable, metal-, azide-, and halogen-free method for the preparation of triazoles, Angew. Chem., Int. Ed., 2020, 59, 6740–6744 CrossRef CAS PubMed.
- S. J. M. Patterson, P. R. Clark, G. D. Williams and N. C. O. Tomkinson, An azide and acetylene free synthesis of 1-substituted 1,2,3-triazoles, Tetrahedron Lett., 2020, 61, 152483 CrossRef CAS.
- H. Wang, Y. Ning, P. Sivaguru, G. Zanoni and X. Bi, [4+1] Annulation of in situ generated azoalkenes with amines: a powerful approach to access 1-substituted 1,2,3-triazoles, Chin. Chem. Lett., 2022, 33, 1550–1554 CrossRef CAS.
- The single impurity (0.2%) in this batch was identified as 1-(2-chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazole (9). It is likely that 9 was formed from 2-chloro-3-(trifluoromethyl)aniline (3), which was the only detected impurity (0.34%) in the starting material 4 (99.66% pure by LCMS) for the Sakai-Clark reaction. A small sample was collected for characterization and as an analytical standard. Data for 9: 1H NMR (400 MHz, CDCl3) 8.03 (d, J = 1.0 Hz, 1H), 7.95–7.88 (m, 2H), 7.81 (dd, J = 8.0, 1.3 Hz, 1H), 7.60 (dt, J = 8.0, 0.6 Hz, 1H); 19F NMR (376 MHz, CDCl3) −62.54; HPLC (method 2): Rt 3.00 minutes, 97.8%; LCMS (method 2): Rt 2.33 minutes, 99.3%, MS+ m/z 247.9 (100%).
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and analytical data for 1 (DSC, 1H NMR, 13C and DEPT NMR, 19F NMR, HPLC, LCMS), 4 (1H NMR, 19F NMR, HPLC) and 9 (1H NMR, 19F NMR, HPLC, LCMS). Table of pilot reactions for the chlorination of 2-chloro-3-(trifluoromethyl)aniline (3) with N-chlorosuccinimide in various solvents. See https://doi.org/10.1039/d2ra05132j |
| This journal is © The Royal Society of Chemistry 2022 |