
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew dibenzocyclooctadiene lignans from Kadsura induta with their anti-inflammatory activity†
Bui Huu Taiab,
Pham Hai Yena,
Nguyen Huy Hoanga,
Phan Thi Thanh Huonga,
Nguyen Viet Dunga,
Bui Van Thanhc,
Nguyen The Cuongc,
Ngo Anh Banga,
Nguyen Xuan Nhiemab and
Phan Van Kiem *ab
*ab
aInstitute of Marine Biochemistry, Vietnam Academy of Science and Technology (VAST), 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. E-mail: phankiem@yahoo.com
bGraduate University of Science and Technology, VAST, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
cInstitute of Ecology and Biological Resources VAST, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
First published on 7th September 2022
Abstract
Five new dibenzocyclooctadiene lignans, named kadsuindutains A–E (1–5), and three known ones schizanrin F (6), schizanrin O (7), and schisantherin J (8) were isolated from the stems of Kadsura induta. Their structures were determined by analyses of HR-ESI-MS, NMR, and ECD spectra. Compounds 1–5 contain a 2′,4′-dioxygenated-2′,3′-dimethylbutyryl moiety which is rarely reported for dibenzocyclooctadiene lignans. Molecular docking predicted that compounds 1–8 displayed good binding affinity to the active site of iNOS and TNF-α proteins but unstable binding to the active site of COX-2 protein. Additionally, in vitro experiments showed that compounds 1–8 inhibited NO production in LPS-activated RAW264.7 cells with IC50 values from 10.7 μM to 34.0 μM, compared to the positive control L-NMMA (IC50 = 31.2 μM).
Introduction
The genus Kadsura (Schisandraceae family) comprises 16 species which are widely distributed in Asian countries.1 The stems, roots, and leaves of Kadsura species have been used in traditional medicines to treat arthritis and gastritis, promote the circulatory system, and relieve pain.2 Recently, a lot of phytochemical studies have been focused on Kadsura species due to being a rich source of highly oxygenated dibenzocyclooctadiene lignans and triterpenoids.2,3 Not only interesting in chemical structure, lignans and triterpenoids from Kadsura species also exerted various beneficial biological activities such as anti-inflammatory and cytotoxic activities, as well as being potent antiviral agents.4–9 Dibenzocyclooctadiene lignans have been suggested to be important sources for development of potent anti-hepatitis drugs.9 With the aim to find bioactive dibenzocyclooctadiene lignans, K. induta was selected for investigation and lead to isolation of five new and three known dibenzocyclooctadiene lignans. We describe, herein, the isolation and structural elucidation of those compounds. In addition, the anti-inflammatory activities of the isolated compounds were studied by molecular docking on iNOS, TNF-α, COX-2 proteins, and in vitro evaluated on the NO production in LPS-activated RAW264.7 cells.Results and discussion
The methanol extract from the stems of K. induta was separated between dichloromethane and water. The organic layer was subjected to flash column chromatography on silica gel or ODS reversed phase, and further purified by semipreparative HPLC to afford eight C18-dibenzocyclooctadiene lignans (1–8).Compound 1 was obtained as a yellow amorphous powder. Its molecular formula, C31H38O12, was determined by sodium adductive ion [M + Na]+ at m/z 625.2267 (calcd for [C31H38O12Na]+, 625.2255) and ammonium adductive ion [M + NH4]+ at m/z 620.2709 (calcd for [C31H38O12NH4]+, 620.2702) in the HR-ESI-MS. The 1H-NMR and HSQC spectra of 1 showed signals corresponding to two olefinic protons [δH 6.84 and 6.50 (each, 1H, s)], two oxygenated methine protons [δH 5.78 and 5.53 (each 1H, s)], one dioxygenated methylene group [δH 6.04 and 5.94 (each 1H, d, J = 1.5 Hz)], one oxygenate methylene protons [δH 4.16 and 3.62 (each 1H, dd, J = 4.5, 12.5 Hz)], three methoxy groups [δH 3.93, 3.84, and 3.45 (each 3H, s)], and five methyl groups [δH 1.35 (3H, s), 1.30 (3H, d, J = 7.0 Hz), 1.24 (3H, s), 0.97 (3H, d, J = 7.0 Hz), 0.84 (3H, t, J = 7.5 Hz)]. The 13C-NMR and HSQC spectra of 1 revealed 31 carbons including two carbonyl (δC 172.4 and 172.2), twelve aromatic (δC 102.6 to 152.3), one dioxymethylene (δC 101.5), five oxygen bearing sp3 hybridized (δC 72.5 to 86.6), three methoxy (δC 60.7, 60.6, 56.2), and eight aliphatic carbons (δC 8.4 to 44.1). Abovementioned NMR data as well as literature survey on the chemical components of Kasuda species2 indicated 1 to be a C18-dibenzocyclooctadiene lignan containing one dioxymethylene, three methoxy, and other two ester functional groups. Of these, the COSY cross peaks of H3-3′′ (δH 0.84)/H2-2′′ (δH 1.81 and 1.58), the HMBC correlation between H3-3′′ (δH 0.84) and C-1′′ (172.2) indicated the presence of propionyl group. On the other hand, the COSY cross peaks of H3-5′ (δH 0.97)/H-3′ (δH 1.92)/H2-4′ (δH 4.16 and, 3.62), HMBC correlation between H3-5′ (δH 0.97) and C-4′ (δC 72.5)/C-3′ (δC 42.6)/C-2′ (δC 76.7), HMBC correlation between H3-6′ (δH 1.24) and C-3′ (δC 42.6)/C-2′ (δC 76.7)/C-1′ (δC 172.4) demonstrated for the 2′,4′-dioxygenated-2′,3′-dimethylbutyryl moiety. The appearance of 2′,4′-dioxygenated-2′,3′-dimethylbutyryl moiety indicating 1 shared structural fragment of gomisin D (1a), a fantastic dibenzocyclooctadiene framework in the nature (Fig. 1).10 Locations of substituted groups were then interpreted by HMBC analysis (Fig. 2). The HMBC correlations between dioxymethylene protons (δH 6.04 and 5.94)/H-11 (δH 6.50) and C-12 (δC 148.6)/C-13 (δC 137.6), H-11 and C-9 (δC 83.9), H-9 (δH 5.53) and C-1′′ (δC 172.2) indicated dioxymethylene group at C-12/C-13 and O-propionyl group (O–Pr group) at C-9. The HMBC correlations between H-4 (δH 6.84) and C-6 (δC 86.6), H-6 (δH 5.78) and C-1′ (δC 172.4), H-4′ (δH 4.16 and 3.62) and C-14 (δC 139.1) established connection between 2′,4′-dioxygenated-2′,3′-dimethylbutyryl moiety and dibenzocyclooctadiene by an ester linkage (C-6 to C-1′) and ether linkage (C-14 to C-4′). Chemical shift of C-7 (δC 73.8), HMBC correlations between H3-17 (δH 1.35) and C-6 (δC 86.6)/C-7 (δC 73.8)/C-8 (δC 44.1) suggested the presence of hydroxy group at C-7. Three methoxy groups were assigned at C-1, C-2, and C-3 which were proved by HMBC correlation between methoxy protons and each carbon [1-OCH3 (δH 3.45)/C-1 (δC 151.5), 2-OCH3 (δH 3.84)/C-2 (δC 141.9), and 3-OCH3 (δH 3.93)/C-3 (δC 152.3)]. Finally, the stereochemistry of 1 was elucidated by combination of ECD and NOESY experiment. The ECD spectrum of 1 showed positive Cotton effect at 228 nm (+17.9 mdeg) and negative Cotton effect at 245 nm (−5.6 mdeg) which was generally assigned for S-configuration of the biphenyl system.10–12 Continuously, NOESY correlation between H-11 (δH 6.50) and H-8 (δH 1.98) revealed conformation of cyclooctadiene ring to be twist-boat-chair form and H-8 at axial-beta orientation (Fig. 3). The NOESY correlations between H-9 (δH 5.53) and H-8 (δH 1.98)/H-11 (δH 6.50), H-8 and H3-17 (δH 1.35) indicated equatorial-beta orientation of both H-9 and C-17. The NOESY correlation between H-4 (δH 6.84) and H-6 (δH 5.78) indicated for equatorial-alpha orientation of H-6. The close proximity of the two methyl groups (C-5′ and C-6′, assuming alpha orientation) was confirmed by obvious ROSEY correlation between H3-5′ (δH 0.97) and H3-6′ (δH 1.24). Consequently, the structure of 1 was established (Fig. 1) and named as kadsuindutain A.
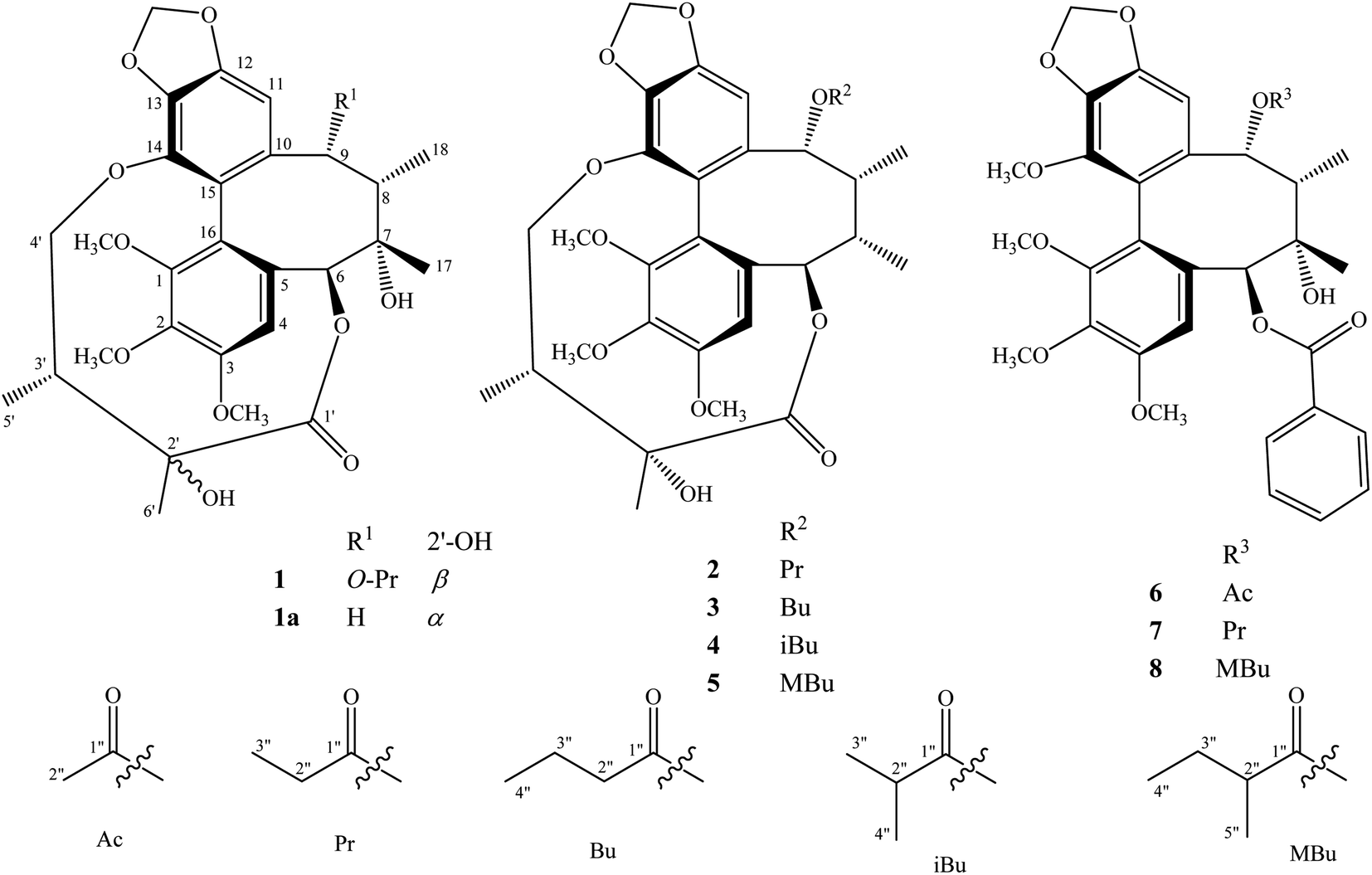 | ||
| Fig. 1 Structures of compounds 1–8 isolated from the stems of K. induta. | ||
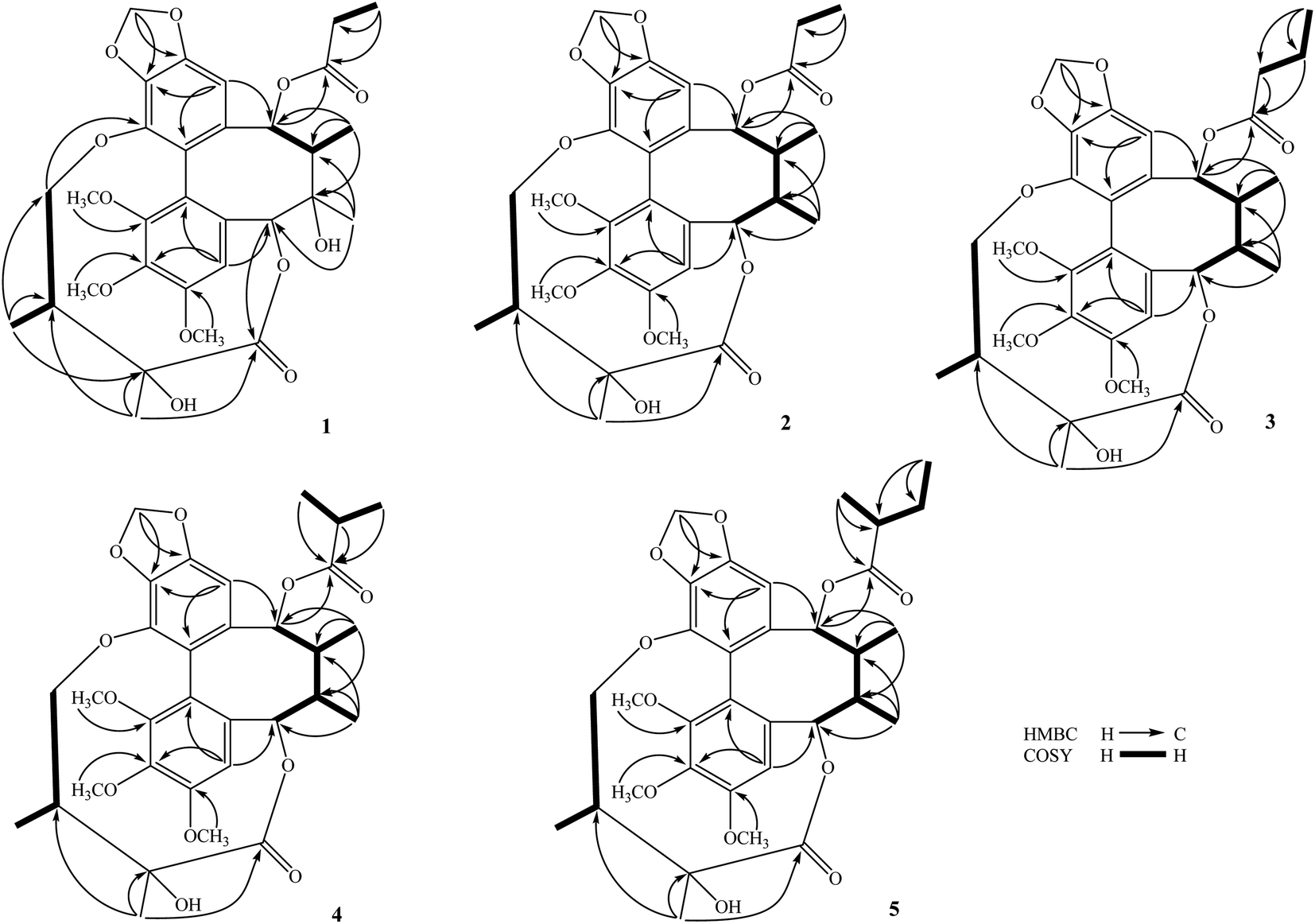 | ||
| Fig. 2 Key HMBC and COSY correlations of compounds 1–5. | ||
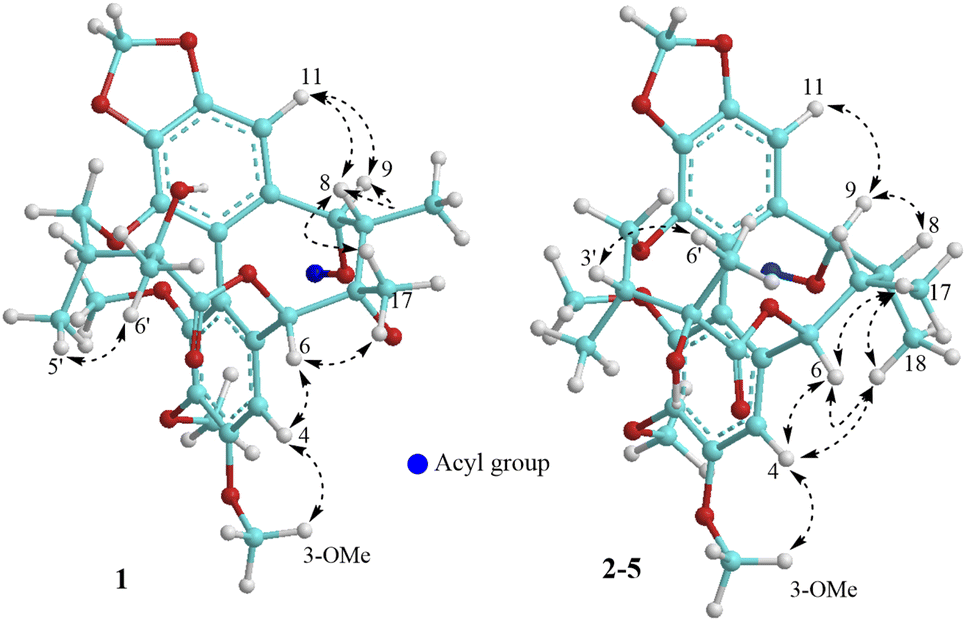 | ||
| Fig. 3 Important NOESY correlations of compounds 1–5. | ||
The HR-ESI-MS of 2 showed protonated molecule [M + H]+ at m/z 587.2484 (calcd for [C31H39O11]+, 587.2487) and ammonium adductive ion [M + NH4]+ at m/z 604.2755 (calcd for [C31H38O11NH4]+, 604.2752) which indicated molecular formula of 2, C31H38O11, less than one oxygen atom (16 atomic mass unit) in compared to 1. The 1H and 13C-NMR spectral data of 2 were close similarity with those of 1 except the appearance of methine group (δC-7 40.3 and δH-7 1.76) instead of oxygenated tertiary carbon (δC-7 73.8). The change in HR-ESI-MS and NMR spectral data between 2 and 1 suggested the gross structure of 2 were different to 1 by absence of hydroxy group at C-7. The ECD spectrum of 2 showed positive Cotton effect at 227 nm (+20.0 mdeg) and negative Cotton effect at 244 nm (−8.9 mdeg) indicated for S-configuration of the biphenyl system as previously described.10–12 Interestingly, NOESY correlation between H3-18 (δH 0.74) and H-4 (δH 6.76) revealed conformation of cyclooctadiene ring in 2 to be twist-boat form and C-18 at axial-alpha orientation which are different to the twist-boat-chair form and C-18 at equatorial-alpha orientation in 1 (Fig. 3). The NOESY correlation between H3-18 (δH 0.74) and H3-17 (δH 1.06), H3-17/H3-18/H-4 (δH 6.76) and H-6 δH (5.07) indicated equatorial-alpha orientation of C-17 and H-6. On the other hand, the obvious NOESY correlation between H3-6′ (δH 1.27) and H-3′ (δH 2.11) indicated relative alpha and beta configurations of C-5′ and C-6′, respectively. Consequently, the structure of 2 was established (Fig. 1) and named as kadsuindutain B.
The molecular formulas of 3–5 were determined as C32H40O11 (3 and 4) and C33H42O11 (5) by their corresponding protonated molecule [M + H]+ at m/z 601.2653, m/z 601.2660, and m/z 615.2822 in the HR-ESI-MS (calcd for [C32H41O11]+, 601.2643; and calcd for [C33H43O11]+, 615.2800), respectively. The 1D, 2D-NMR, and ECD spectral data of compounds 3–5 were recognized identical with those of 2. The slight difference between them was due to an acyl group at C-9. In compound 3, the COSY cross peaks of H3-4′′ (δH 0.80)/H2-3′′ (δH 1.40)/H2-2′′ (δH 1.90 and 1.81), HMBC correlations between H2-3′′ (δH 1.40)/H2-2′′ (δH 1.90 and 1.81)/H-9 (δH 5.91) and C-1′′ (δC 173.0) indicated the acyl group at C-9 to be butyryl group (Bu group). Meanwhile, signals of isopropyl coupled protons in the 1H-NMR spectrum of 4 [δH 0.85 (3H, d, J = 7.0 Hz), 0.93 (3H, d, J = 7.0 Hz), 2.08 (1H, m)], the HMBC correlations between H3-3′′ (δH 0.93)/H3-4′′ (δH 0.85)/H-9 (δH 5.92) and C-1′′ (δC 176.5) demonstrated for the presence of isobutyryl group (iBu group) at C-9. The acyl group at C-9 of compound 5 was determined to be α-methylbutyryl group (MBu group) which was confirmed by COSY cross peaks of H3-4′′ (δH 0.73)/H2-3′′ (δH 1.37 and 1.17)/H-2′′ (δH 1.98)/H3-5′′ (δH 0.91), HMBC correlations between H3-5′′ (δH 0.91)/H2-3′′ (δH 1.37 and 1.17)/H-9 (δH 5.91) and C-1′′ (δC 176.2). Thus, the structures of 3–5, kadsuindutains C–E, were established as shown in the Fig. 1.
Compounds 6–8 were previously described C18-dibenzocyclooctadiene lignans schizanrin F,13 schizanrin O,14 and schisantherin J,15 respectively. Their NMR spectral data was well consisted with those reported in the literature.13–15
Literature survey showed that C18-dibenzocyclooctadiene lignans are very valuable ingredients from two genera Kadsura and Schisandra in Schisandraceae family.2,9 Their structures are derived by combination between absolute configuration of biaryl axis and oxygen substituted groups in the dibenzocyclooctadiene skeleton.16 However, dibenzocyclooctadiene lignans containing 2′,4′- dioxygenated-2′,3′-dimethylbutyryl moiety as in compounds 1–5 are rarely found in nature. To the best of our knowledge, two compounds gomisins D and E are typically isolated and only reported either in Schisandra or Kadsura genus.10,17,18 Numerous studies reported dibenzocyclooctadiene lignans showing potent anti-inflammation, anti-oxidant, anti-cancer, and anti-virus.8,19 The stems of K. induta are commonly used in folk medicines for treatment of rheumatic arthritis.14,20 Therefore, anti-inflammatory activity of dibenzocyclooctadienes 1–8 were attempted to study by in silico and in vitro experiments. Firstly, molecular docking approaches were performed on important anti-inflammatory targeted proteins including iNOS, TNF-α, and COX-2. Docking results (Table 2) of compounds 1–8 at active site of iNOS had binding energy ranging from −6.43 kcal mol−1 to −8.00 kcal mol−1 which are close with that of positive drug S71 (binding energy −7.71 kcal mol−1).21 In the interaction with TNF-α protein, compounds 1–8 exhibited a bit weak binding affinity to active site (−6.15 kcal mol−1 to −7.81 kcal mol−1) in comparison with positive drug 307 (binding energy −8.62 kcal mol−1).22 However, compounds 1–8 exhibited unstable binding to active site of COX-2 (positive binding energy, from +16.60 kcal mol−1 to +90.84 kcal mol−1) as expected to positive drug celecoxib (binding energy −11.12 kcal mol−1).23 Docking results and chemical structures of compounds 1–8 indicated that the presence of 2′,4′-dioxygenated-2′,3′-dimethylbutyryl moiety (compounds 1–5, binding energy −6.43 kcal mol−1 to −7.17 kcal mol−1) reduced the binding affinity with active site of iNOS protein (compounds 6–8, binding energy −6.82 kcal mol−1 to −8.00 kcal mol−1). In contrast, 2′,4′-dioxygenated-2′,3′-dimethylbutyryl moiety increased binding affinity of C18-dibenzocyclooctadiene lignans to active site of TNF-α. Additionally, compounds 1–8 expected to be poor COX-2 inhibitors. The positive binding energy of compounds 1–8 to active site of COX-2 could be due to the large in volume of their molecules which are unable fitting with active pocket of COX-2 (ESI material†). Based on docking results, compounds 1–8 were evaluated for their inhibitory effects on the NO production in LPS activated RAW264.7 cells. At concentration of 100 μM, all compounds 1–8 did not show significant cytotoxic effect to the RAW264.7 cells (data was not shown). Therefore, NO inhibitory activity of the compounds was assayed at diluted concentrations from 0.4 μM to 100 μM. As shown in the Table 3, compounds 1–8 potentially inhibited NO production in LPS activated RAW264.7 cells with the IC50 values in range of 10.7 μM to 34.0 μM in compared to positive control L-NMMA (IC50 of 31.2 μM). Our results indicated compounds 1–8 could be active anti-inflammatory activity constituents of K. induta by inhibiting NO and TNF-α secretion.
| No. | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| 1 | 151.5 | — | 151.6 | — | 151.6 | — | 151.7 | — | 151.6 | — |
| 2 | 141.9 | — | 142.2 | — | 142.3 | — | 142.3 | — | 142.3 | — |
| 3 | 152.3 | — | 152.3 | — | 152.3 | — | 152.4 | — | 152.3 | — |
| 4 | 111.2 | 6.84 (s) | 112.3 | 6.76 (s) | 112.3 | 6.76 (s) | 112.4 | 6.77 (s) | 112.3 | 6.76 (s) |
| 5 | 130.6 | — | 130.1 | — | 130.1 | — | 130.0 | — | 129.9 | — |
| 6 | 86.6 | 5.78 (s) | 86.8 | 5.07 (d, 7.5) | 86.8 | 5.07 (d, 7.5) | 86.8 | 5.07 (d, 7.0) | 86.8 | 5.07 (d, 7.5) |
| 7 | 73.8 | — | 40.3 | 1.76 (m) | 40.3 | 1.76 (m) | 40.3 | 1.78 (m) | 40.3 | 1.77 (m) |
| 8 | 44.1 | 1.98 (m) | 41.7 | 2.25 (m) | 41.7 | 2.25 (m) | 41.8 | 2.26 (m) | 41.8 | 2.24 (m) |
| 9 | 83.9 | 5.53 (s) | 76.9 | 5.90 (d, 9.5) | 76.8 | 5.91 (d, 9.5) | 76.8 | 5.92 (d, 9.5) | 77.0 | 5.91 (d, 9.5) |
| 10 | 133.0 | — | 133.5 | — | 133.5 | — | 133.5 | — | 133.6 | — |
| 11 | 102.6 | 6.50 (s) | 103.5 | 6.49 (s) | 103.6 | 6.48 (s) | 103.7 | 6.48 (s) | 103.6 | 6.48 (s) |
| 12 | 148.6 | — | 148.3 | — | 148.3 | — | 148.3 | — | 148.3 | — |
| 13 | 137.6 | — | 138.3 | — | 138.3 | — | 138.3 | — | 138.1 | — |
| 14 | 139.1 | — | 139.0 | — | 139.0 | — | 139.0 | — | 139.0 | — |
| 15 | 121.3 | — | 122.6 | — | 122.6 | — | 122.7 | — | 122.7 | — |
| 16 | 121.7 | — | 124.0 | — | 124.0 | — | 124.0 | — | 124.0 | — |
| 17 | 28.4 | 1.35 (s) | 20.6 | 1.06 (d, 7.0) | 20.6 | 1.06 (d, 7.0) | 20.7 | 1.06 (d, 7.0) | 20.6 | 1.06 (d, 7.0) |
| 18 | 17.9 | 1.30 (d, 7.0) | 9.4 | 0.74 (d, 7.5) | 9.5 | 0.74 (d, 7.5) | 9.4 | 0.75 (d, 7.5) | 9.7 | 0.76 (d, 7.5) |
| –OCH2O– | 101.5 | 5.94 (d, 1.5) | 101.5 | 5.96 (d, 1.5) | 101.5 | 5.96 (d, 1.5) | 101.5 | 5.96 (d, 1.5) | 101.5 | 5.96 (d, 1.5) |
| 6.04 (d, 1.5) | 6.06 (d, 1.5) | 6.06 (d, 1.5) | 6.06 (d, 1.5) | 6.06 (d, 1.5) | ||||||
| 3-OCH3 | 56.2 | 3.93 (s) | 56.2 | 3.92 (s) | 56.2 | 3.92 (s) | 56.2 | 3.92 (s) | 56.1 | 3.92 (s) |
| 2-OCH3 | 60.6 | 3.84 (s) | 60.5 | 3.91 (s) | 60.5 | 3.91 (s) | 60.4 | 3.90 (s) | 60.5 | 3.91 (s) |
| 1-OCH3 | 60.7 | 3.45 (s) | 60.6 | 3.72 (s) | 60.6 | 3.72 (s) | 60.6 | 3.74 (s) | 60.6 | 3.74 (s) |
| 1′ | 172.4 | — | 177.0 | — | 177.0 | — | 177.0 | — | 177.0 | — |
| 2′ | 76.7 | — | 74.6 | — | 74.7 | — | 74.8 | — | 74.7 | — |
| 3′ | 42.6 | 1.92 (m) | 38.2 | 2.11 (m) | 38.4 | 2.10 (m) | 38.2 | 2.11 (m) | 38.3 | 2.11 (m) |
| 4′ | 72.5 | 3.62 (dd, 4.5, 12.5) | 72.4 | 3.57 (m) | 72.4 | 3.58 (m) | 72.4 | 3.58 (m) | 72.4 | 3.59 (m) |
| 4.16 (dd, 4.5, 12.5) | 3.88 (m) | 3.88 (m) | 3.86 (m) | 3.88 (m) | ||||||
| 5′ | 12.8 | 0.97 (d, 7.0) | 11.3 | 1.06 (d, 7.0) | 11.3 | 1.06 (d, 7.0) | 11.3 | 1.06 (d, 7.0) | 11.3 | 1.06 (d, 7.0) |
| 6′ | 21.5 | 1.24 (s) | 25.3 | 1.27 (s) | 25.3 | 1.27 (s) | 25.3 | 1.27 (s) | 25.4 | 1.27 (s) |
| 1′′ | 172.2 | — | 173.8 | — | 173.0 | — | 176.5 | — | 176.2 | — |
| 2′′ | 26.6 | 1.58 (m)/1.81 (m) | 27.2 | 1.90 (m) | 35.8 | 1.81 (m)/1.90 (m) | 33.6 | 2.08 (m) | 40.3 | 1.98 (m) |
| 3′′ | 8.4 | 0.84 (t, 7.5) | 8.9 | 0.90 (t, 7.5) | 18.1 | 1.40 (m) | 18.1 | 0.93 (d, 7.0) | 26.6 | 1.17 (m)/1.37 (m) |
| 4′′ | 13.6 | 0.80 (t, 7.0) | 19.5 | 0.85 (d, 7.0) | 11.2 | 0.73 (t, 7.5) | ||||
| 5′′ | 15.4 | 0.91 (d, 6.5) | ||||||||
| Comp | Binding energy (kcal mol−1) | ||
|---|---|---|---|
| iNOS | TNF-α | COX-2 | |
| a Original inhibitors S71, 307, and celecoxib were used to be positive drugs and locate active site of iNOS (4CX7), TNF-α (2AZ5), and COX-2 (3LN1) proteins.21–23 | |||
| 1 | −6.75 | −6.49 | +90.84 |
| 2 | −6.90 | −7.55 | +56.32 |
| 3 | −7.17 | −7.64 | +58.30 |
| 4 | −6.43 | −7.81 | +55.54 |
| 5 | −6.57 | −7.51 | +59.33 |
| 6 | −6.82 | −6.33 | +19.94 |
| 7 | −7.41 | −6.35 | +16.60 |
| 8 | −8.00 | −6.15 | +20.04 |
| S71a | −7.71 | — | — |
| 307a | — | −8.62 | — |
| Celecoxiba | — | — | −11.12 |
Experimental
General experimental procedures
Optical rotation was measured on a Jasco P2000 polarimeter. ECD spectrum was measured on a Chirascan spectrometer. NMR spectra were obtained on a Bruker AvanceIII 500 MHz FT-NMR spectrometer. HR-ESI-MS was analysed on an Agilent 6530 Accurate-Mass QTOF. Semi-preparative HPLC was performed on an Agilent 1260 infinity II system including binary pump, autosampler, DAD detector, and YMC J'sphere ODS-H80 (20 × 250 mm, 4 μm) HPLC column. Mobile phase was an isocratic system of methanol/water or acetonitrile/water at flow rate of 3 mL min−1. Thin layer chromatography was performed on pre-coated plates with silica gel or reversed phase C18 (RP-18). Column chromatography was performed using silica gel (40–63 μm) or RP-18 (150 μm) as adsorbents.Plant material
Plant samples were collected in February 2022 at Sapa, Lao Cai province, Vietnam and taxonomically identified to be Kadsura induta A. C.·Sm. by Dr Bui Van Thanh and Dr Nguyen The Cuong, Institute of Ecology and Biological Resources, VAST. Voucher specimen (IMBC-NCXS-02) was kept at the Institute of Marine Biochemistry, VAST.Extraction and isolation
Dried powdered K. induta stems (6 kg) was ultrasonically macerated with methanol at room temperature for three times (15 L and 2 h each time) to give methanolic residue. The obtained residue (150 g) was well suspended with 3.0 L of water and then successively partitioned with dichloromethane and ethyl acetate to give dichloromethane, ethyl acetate, and water-soluble extracts. The dichloromethane extract (52 g) was separated on a silica gel column, eluting with gradient solvent system of n-hexane/acetone (1/0–0/1, v/v) to give six fractions, KID1-KID6. Fraction KID3 (17 g) was continuously chromatographed on a silica gel column, eluting with n-hexane/acetone (7/1, v/v) to give three fractions, KID3A-KID3C. Fraction KID3A was purified by semi-preparative HPLC (SemiPre-HPLC) using acetonitrile/water (3/1, v/v) to yield compounds 5 (12.5 mg, tR 61.3 min) and 8 (14.1 mg, tR 68.1 min). Fraction KID4 (12 g) was subjected to a silica gel column and eluted with n-hexane/acetone (5/1, v/v) to give seven fractions, KID4A-KID4G. Fraction KID4B was first separated on a RP-18 column, eluting with methanol/water (3/1, v/v) and then purified by SemiPre-HPLC with 78% methanol in water to give compounds 4 (7.6 mg, tR 56.0 min) and 3 (7.3 mg, tR 58.2 min). Fraction KID4C was chromatographed on a RP-18 column, eluting with acetone/water (2/1, v/v) to give three fractions, KID4C1-KID4C3. Fractions KID4C2 and KID4C3 were separately purified by SemiPre-HPLC using acetonitrile/water (3/1, v/v) to give compounds 2 (10.3 mg, tR 39.7 min) and 7 (7.9 mg, tR 42.0 min), respectively. Fraction KID4E was purified on a RP-18 column, eluting with methanol/water (3/1, v/v) to give compound 6 (50.6 mg). Fraction KID4F was first separated on a RP-18 column, eluting with methanol/water (2/1, v/v) and then purified by SemiPre-HPLC with 50% acetonitrile in water to give compound 1 (8.4 mg, tR 67.7 min).Molecular docking
Molecular docking was carried out using AutoDock v4.2 program as previously described.21–24 The crystal structure of inducible nitric oxide synthase with inhibitor (S71), TNF-α with inhibitor (307), and COX-2 with inhibitor (celecoxib) were obtained from the RCSB Protein Database Bank (PDB ID: 4CX7, 2AZ5, and 3LN1, respectively). The 3D structures of compounds 1–8 were obtained by Spartan18 software. Conformational search was started with the MMFF molecular mechanics set and then minimized energy by PM3 semi-empirical condition. A conformer with lowest energy was used as a ligand for molecular docking. Receptor was prepared as following steps: delete water molecules, remove initial ligands, repair the missing residues, add polar hydrogen atoms, add and spread Kollman charges over all macro-molecule. The grid box dimensions were 40 × 40 × 40 Å with 0.5 Å grid spacing and center coordinates at original inhibitor to locate active site of the receptors (x: −15.0, y: −66.0, z: 17.0 for iNOS 4CX7; x: −9.3, y: 68.7, z: 20.2 for TNF-α 2AZ5, and x: 29.8, y: −21.3, z: −17.4 for COX-2 3LN1). AutoDock calculations were run by Lamarckian genetic algorithm and set at running times of 50, population sizes of 300, and default setting for other miscellaneous parameters. Docked complex with the lowest binding energy was selected to represent the most favourable interaction between the ligand and receptor. Results were re-rendered using Chimera v1.16.Conclusions
Our phytochemical investigation on the stems of K. induta resulted eight dibenzocyclooctadiene lignans comprising five new (1–5) and three known (6–8) ones. Compounds 1–5 possessed unique structure and rarely found in the nature. Molecular docking suggested that compounds 1–8 are good binding affinity with anti-inflammatory mediators including iNOS and TNF-α. Additionally, compounds 1–8 inhibited NO production in LPS activated RAW 264.7 cells with IC50 values from 10.7 μM to 34.0 μM which are almost smaller than IC50 value of positive control, L-NMMA.Author contributions
PV Kiem, BH Tai, NX Nhiem, contributed to research idea and writing; BV Thanh, NT Cuong contributed to sample collection and taxonomical identification. NH Hoang, PTT Huong, NV Dung, NA Bang contributed to isolation; PH Yen, NX Nhiem, PV Kiem, BH Tai contributed to structure elucidation; BH Tai, NX Nhiem contributed to molecular docking and NO assay.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by Vietnam Academy of Science and Technology (VAST) under grant number NCXS01.02/22-24.Notes and references
- J. S. Liu, Y. D. Qi, H. W. Lai, J. Zhang, X. G. Jia, H. T. Liu, B. G. Zhang and P. G. Xiao, Phytomedicine, 2014, 21, 1092–1097 CrossRef CAS PubMed.
- L. Zhang, Y. Z. Jia, B. Li, C. Y. Peng, Y. P. Yang, W. Wang and C. X. Liu, Chin. Herb. Med., 2021, 13, 157–166 CrossRef PubMed.
- Y. Q. Zhang, Y. Liu, Z. P. Zhang, D. D. Wu, L. X. Zhuang, A. M. Algradi, H. X. Kuang and B. Y. Yang, Fitoterapia, 2022, 161, 105230 CrossRef CAS.
- L. H. Tram, T. T. Huong, L. T. Thuy, N. V. Thong, N. T. Anh, N. H. Minh, T. T. Minh, N. T. H. Phuong, T. T. Ha, N. H. Dang, L. D. Dat, P. P. Dien and M. Lee, Phytochem. Lett., 2021, 45, 57–62 CrossRef.
- J. B. Liu, P. K. Pandey, X. J. Wang, K. Adams, X. Z. Qi, J. B. Chen, H. Sun, Q. Hou, D. Ferreira, R. J. Doerksen, M. T. Hamann and S. Li, J. Nat. Prod., 2019, 82, 2842–2851 CrossRef CAS PubMed.
- X. Z. Qi, J. B. Liu, J. B. Chen, Q. Hou and S. Li, Chin. Chem. Lett., 2020, 31, 423–426 CrossRef CAS.
- N. Shehla, B. Li, J. P. Zhao, L. Cao, Y. Q. Jian, I. A. Khan, D. F. Liao, A. U. Rahman, M. I. Choudhary and W. Wang, Nat. Prod. Res., 2022, 36, 8–17 CrossRef CAS PubMed.
- X. Y. Xu, D. Y. Wang, Y. P. Li, S. T. Deyrup and H. J. Zhang, Phytochem. Rev., 2022, 21, 239–289 CrossRef CAS.
- Y. P. Yang, Y. Q. Jian, S. W. Cheng, Y. Z. Jia, Y. B. Liu, H. H. Yu, L. Cao, B. Li, C. Y. Peng, M. Iqbal Choudhary, A. U. Rahman and W. Wang, Bioorg. Chem., 2021, 115, 105277 CrossRef CAS PubMed.
- Y. Ikeya, H. Taguchi, I. Yosioka, Y. Iitaka and H. Kobayashi, Chem. Pharm. Bull., 1979, 27, 1395 CrossRef CAS.
- J. S. Liu and L. Li, Phytochemistry, 1993, 32, 1293–1296 CrossRef CAS.
- Y. Ikeya, H. Taguchi, I. Yosioka, Y. Iitaka and H. Kobayashi, Chem. Pharm. Bull., 1979, 27, 1395–1401 CrossRef CAS PubMed.
- M. D. Wu, R. L. Huang, L. M. Y. Kuo, C. C. Hung, C. W. Ong and Y. H. Kuo, Chem. Pharm. Bull., 2003, 51, 1233–1236 CrossRef CAS PubMed.
- P. T. H. Minh, D. T. Lam, N. Q. Tien, N. N. Tuan, V. P. Nhung, N. V. Hai, P. V. Kiem, N. X. Nhiem, C. V. Minh, S. J. Park and S. H. Kim, Bull. Korean Chem. Soc., 2014, 35, 1859–1862 CrossRef CAS.
- J. S. Liu and Y. P. Pan, Acta Chim. Sin., 1991, 49, 308–312 CAS.
- J. Chang, J. Reiner and J. Xie, Chem. Rev., 2005, 105, 4581–4609 CrossRef CAS PubMed.
- Y. Ikeya, H. Taguchi, I. Yosioka and H. Kobayashi, Chem. Pharm. Bull., 1979, 27, 2695 CrossRef CAS.
- L. N. Li, H. Xue and R. Tan, Planta Med., 1985, 297–300 CAS.
- L. Opletal, H. Sovova and M. Bartlova, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 812, 357–371 CrossRef CAS.
- W. H. Ma, X. L. Ma, H. Huang, P. Zhou and D. F. Chen, Chem. Biodivers., 2007, 4, 966–972 CrossRef CAS PubMed.
- F. Arias, F. Franco-Montalban, M. Romero, M. D. Carrion and M. E. Camacho, Bioorg. Med. Chem., 2021, 44, 116294 CrossRef CAS PubMed.
- A. M. Srour, H. H. Fahmy, M. A. Khater, E. S. Zarie, S. S. Mohamed and M. F. Abdelhameed, J. Mol. Struct., 2022, 1266, 133499 CrossRef CAS.
- K. R. A. Abdellatif, E. K. A. Abdelall, P. F. Lamie, M. B. Labib, M. M. Abdelhakeem, M. M. Abdel-Fattah and E. S. El-Nahaas, J. Mol. Struct., 2022, 1266, 133529 CrossRef CAS.
- D. T. T. Hang, D. T. Trang, D. T. Dung, D. T. H. Yen, N. H. Hoang, N. A. Bang, N. T. Cuc, N. X. Nhiem, P. T. T. Huong, B. H. Tai and P. V. Kiem, Phytochemistry, 2021, 190, 112889 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: HR-ESI-MS, NMR, ECD spectra of new compounds, and molecular docking results of all isolated compounds would be found. See https://doi.org/10.1039/d2ra05052h |
| This journal is © The Royal Society of Chemistry 2022 |