
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon nanotube–titanium dioxide nanocomposite support for improved activity and stability of an iridium catalyst toward the oxygen evolution reaction†
Eom Ji Kim,
Ki hyun Kim,
Junu Bak,
KwangHo Lee and
EunAe Cho *
*
Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: eacho@kaist.ac.kr
First published on 15th December 2022
Abstract
In order to improve the electrocatalytic activity and stability of an iridium (Ir) nanoparticle catalyst toward the oxygen evolution reaction (OER) in acidic electrolyte, carbon nanotube and titanium dioxide nanocomposites (CNT@TiO2) are presented as a high-performance support. TiO2 was synthesized on CNTs by using a novel layer-by-layer solution coating method that mimics atomic layer deposition (ALD) but is cost-effective and scalable. In the nanocomposites, CNTs serve as the electron pathways and the surface TiO2 layers protect CNTs from corrosion under the harsh OER conditions. Thus, CNT@TiO2 demonstrates excellent corrosion resistance as well as a high electrical conductivity (1.6 ± 0.2 S cm−1) comparable to that of Vulcan carbon (1.4 S cm−1). The interaction between Ir and TiO2 promotes the formation of Ir(III) species, thereby enhancing the OER activity and stability of the Ir nanoparticle catalyst. Compared to commercial carbon-supported Ir (Ir/C) and Ir black catalysts, CNT@TiO2-supported Ir exhibits superior OER activity and stability.
1. Introduction
Renewable energy is indispensable for dealing with upcoming climate change issues. Accordingly, energy storage technologies have become crucial topics in the energy industry to address the intermittency of renewable electricity. Water electrolysis that converts electricity into hydrogen is considered as a promising candidate to store surplus renewable electricity. In particular, polymer electrolyte water electrolysis (PEMWE) thanks to their wide load range and fast response to be directly connected to renewable electricity.1,2 One of the main huddles of commercialization of the PEMWE is the usage of large amount of precious metal catalysts, particularly iridium (Ir), to overcome the sluggish oxygen evolution reaction (OER) at the anode.3 To reduce the amount of Ir catalyst usage, Ir has been alloyed or shape-controlled, exhibiting enhanced OER activity.4,5 However, stability of those catalysts is an obstacle to applications. As an alternative, support can be introduced since support not only distributes but also stabilizes the small catalyst nanoparticles, resulting in improved catalyst performance. In polymer electrolyte membrane fuel cell (PEMFC), carbon black has been widely used as a support for platinum nanoparticle catalyst.6 However, at the high anode potentials of PEMWE, carbon black is easily corroded and cannot be utilized as a support. Thus, electrically-conductive and corrosion-resistant support materials should be developed for Ir nanoparticle catalyst.7Firstly, carbon materials with higher crystallinity have been proposed as support since they have better corrosion resistance than carbon black.8 For instance, multiwall-carbon nanotube (MWCNT)-supported IrO2 has presented high OER activity in acidic electrolytes.9–11 Although MWCNT has better corrosion resistance than carbon black,8,12,13 it is also corroded at the high anode potentials and durability of MWCNT-supported IrO2 needs to be improved. Secondly, metal oxides such as titanium dioxide (TiO2) and tin dioxide (SnO2) have been explored with an advantage of excellent corrosion resistance under acidic and high potential conditions.14–20 However, they have an obvious drawback of extremely low electrical conductivity,21–23 that obstructs electron transfer during the OER and significantly increases the overpotential.17 To overcome the problem, metal oxides have been doped with various dopants such as niobium and vanadium.24,25 However, dopants-related degradation of the oxide supports has been reported particularly for doped SnO2.26
In this study, we present CNT and TiO2 nanocomposites (CNT@TiO2) that combine the electrical conductivity of CNT with corrosion resistance of TiO2 as a support for Ir nanoparticles. TiO2 was synthesized on CNTs using a novel layer-by-layer solution coating method, that mimics atomic layer deposition (ALD)27 but is cost-effective and scalable. Ir nanoparticles supported on CNT@TiO2 nanocomposite (Ir/CNT@TiO2) demonstrate superior OER activity and stability to commercial Ir black and carbon-supported Ir and catalysts. As a descriptor of OER activity, oxidation states of Ir were examined by using X-ray photoelectron spectroscopy (XPS).
2. Experimental
2.1. Synthesis of CNT@TiO2 nanocomposites
To prepare CNT and TiO2 nanocomposites (CNT@TiO2), multiwall carbon nanotubes purchased from Sigma Aldrich were immersed in a solution of nitric acid and sulfuric acid at room temperature for 12 hours. TiO2 was synthesized on the acid-treated CNTs using a solution-based layer-by-layer coating method presented in Fig. S1.† 2 mL of titanium tetra isopropoxide (TTIP) were dissolved in 40 mL of isopropyl alcohol (IPA). Then, 40 mg of the acid-treated CNTs were added to the solution and sonicated for 30 min. The prepared mixture was kept at 80 °C for 2 hours for the adsorption of TTIP on the acid-treated CNTs and filtered. The collected powder was washed with ethanol. Then, DI water was dropped onto the powder for hydrolysis of TTIP adsorbed on CNT. The products were collected and dried in a convection oven at 60 °C for 3 hours. The adsorption and hydrolysis processes were alternately repeated 2, 4, 6 and 8 times to obtain corresponding number of TiO2 layers on CNT. Then, the products were heated at 350 °C in air for 2 hours for calcination.2.2. Synthesis of Ir/CNT and Ir/CNT@TiO2
Ir nanoparticles were synthesized on the acid-treated CNT and the prepared CNT@TiO2 and using a polyol reduction method.28 To load 40 wt% of Ir, 80 mg of support and 134 mg of potassium hexachloroiridiate(IV) (K2IrCl6) were mixed with 50 mL ethylene glycol and sonicated for 60 min. The solution pH was adjusted to 10 using 1 M potassium hydroxide solution. The prepared mixture was kept at 170 °C for 3 h under argon bubbling and cooled to room temperature. The precipitates were filtered, washed with DI water, and dried in a convection oven at 60 °C overnight.2.3. Materials characterization
The morphology of the samples was observed using a Hitachi SU8230 scanning electron microscope (SEM). Crystal structure was identified by X-ray diffraction (XRD) using a Rigaku Dmax-2500 with Cu Kα radiation. Transmission electron microscopy (TEM) images were obtained from a Tecnai G2 F20 S-Twin. High-angle annular dark-field (HAADF) images and energy dispersive spectroscopy (EDS) mapping images were taken using a Titan cubed G2 under 200 kV. The chemical composition of the prepared samples was obtained by inductively coupled plasma mass spectroscopy (ICP-MS). For ICP-MS, the prepared Ir/CNT@TiO2 samples were pre-treated in a mixed solution of 70% HNO3 and 35% HCl (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in vol%) at 200 °C for 30 minutes in a microwave. Electrical conductivity was measured using a home-made apparatus.29 For the measurement, powder samples were pressed to form cylindrical pellets and conductivity was obtained with applying a compressive load to the pellets.
:3 in vol%) at 200 °C for 30 minutes in a microwave. Electrical conductivity was measured using a home-made apparatus.29 For the measurement, powder samples were pressed to form cylindrical pellets and conductivity was obtained with applying a compressive load to the pellets.
X-ray photoelectron spectroscopy (XPS) was conducted using a ThermoFisher Scientific MXP10. For the measurement, catalyst-coated-carbon papers were fabricated. The catalyst inks were prepared by mixing 3 mg of catalyst powder and 20 μL of a Nafion solution (5 wt%, 1100 EW, Dupont) were dispersed in D.I. water and IPA and then sonicated for 30 min. The prepared catalyst ink was coated on a carbon paper and dried at room temperature for 30 min. The prepared catalyst-coated-carbon papers were activated in 0.05 M H2SO4 solution by conducting potential cycling between 1.0 and 1.8 VRHE for 3 cycles at a scan rate of 5 mV s−1 before XPS measurement. Binding energy values were calibrated with respect to the C 1s position (C sp2: 284.2 eV).
2.4. Electrochemical measurements
Electrocatalytic activity and stability were evaluated using a potentiostat (Pine Instruments) with a standard three-electrode electrochemical cell. Pt mesh and a saturated calomel electrode (SCE) were used as counter electrode and reference electrode, respectively. All potentials were converted to that of a reversible hydrogen electrode (RHE). The catalyst ink prepared to fabricated the catalyst-coated-carbon papers. The prepared catalyst ink was coated on the surface of a glassy carbon electrode (GCE) and dried at room temperature for 5 min in a vacuum cell. Ir loading amount on GCE was 10 μgIr cm−2. OER polarization curves were measured in an Ar-saturated 0.05 M H2SO4 solution at a scan rate of 5 mV s−1 with a rotating speed of 2500 rpm. To estimate the stability of the catalysts, chronopotentiometry was conducted at 10 mA cm−2 with 2500 rpm. Electrochemical impedance spectroscopy was conducted at 1.51 VRHE from 10 mHz to 100 kHz.3. Results & discussion
To prepare CNT and TiO2 nanocomposites, multiwall CNTs were acid treated and then coated with TiO2 through three steps; adsorption of Ti precursor (titanium tetra isopropoxide, TTIP) on CNTs, hydrolysis of TTIP to Ti(OH)x, and calcination of Ti(OH)x to TiO2 (Fig. S1†). As-purchased and acid-treated CNTs (Fig. S1†) exhibit approximately the same one-dimensional structure with diameter of 50–60 nm, revealing that the acid treatment has negligible effects on the morphology of CNTs. However, without the acid treatment, TiO2 was not formed on CNTs, implying that the acid treatment modifies surface properties of CNTs for TTIP adsorption.30 The acid-treated multiwall CNTs are simply denoted as CNTs hereafter. Aiming at forming 2, 4, 6 and 8 layers of TiO2 on CNTs (denoted as CNT@TiO2-n L, n = 2, 4, 6 and 8), adsorption and hydrolysis steps were alternately repeated 2, 4, 6, and 8 times before calcination. To examine the effects of TiO2 coating on the pore structure, N2-adsorption–desorption isotherms were measured for CNT and CNT@TiO2-6 L (Fig. S2†). Brunauer–Emmet–Teller (BET) surface areas of CNT and CNT@TiO2-6 L were calculated and found to be 166.1 and 151.9 m2 g−1, respectively. Pore size distributions show that the average pore sizes of CNT and CNT@TiO2-6 L are 17.8 and 10.9 nm, respectively. The decreases in BET surface area and average pore size can be related to the coating heavier TiO2 layer to CNT.Fig. 1a demonstrates XRD patterns of CNT and CNT@TiO2-n L nanocomposites. CNT, CNT@TiO2-2 L, and CNT@TiO2-4 L exhibit XRD peaks from CNT whereas CNT@TiO2-6 L and CNT@TiO2-8 L demonstrate XRD peaks corresponding to anatase TiO2 in addition to the CNT peaks. The absence of the TiO2-related peaks from the XRD patterns of CNT@TiO2-2 L and CNT@TiO2-4 L could be attributed to the low contents of TiO2. CNT@TiO2-n L nanocomposites (Fig. 1b and S4†) show one-dimensional structure similar to CNTs (Fig. S3†) without precipitates or segregated particles. While the acid-treated CNTs have average wall thickness of approximately 13 nm, CNT@TiO2-2 L and CNT@TiO2-4 L demonstrate thicker walls with average thickness of approximately 14 and 16 nm, respectively (Fig. S5†). Ti compositions in CNT@TiO2-2 L and CNT@TiO2-4 L are 1.1 and 3.4 at%, respectively (Table S1†). These results demonstrate that TiO2 was mostly coated on CNTs rather than formed as a separate phase in those samples and with repeating the adsorption and hydrolysis steps, thicker TiO2 layers were formed. On the other hand, CNT@TiO2-6 L (Fig. 2) and CNT@TiO2-8 L (Fig. S5d†) have similar wall thickness to that of CNT@TiO2-4 L (approximately 16 nm) and Ti compositions of 5.3 and 5.5 at%, respectively. In particular, they demonstrate a large number of TiO2 nanoparticles well-dispersed on the TiO2-coated CNTs. Thus, it can be inferred that, using the layer-by-layer solution method proposed in this study, TiO2 was coated on CNTs up to a certain thickness (about 3 nm in this study) and excess TiO2 was formed as nanoparticles. TEM and elemental mapping images (Fig. 2 and S5d†) support formation of crystalline TiO2 nanoparticles on CNT@TiO2-6 L and CNT@TiO2-8 L, observed in the XRD patterns (Fig. 1a).
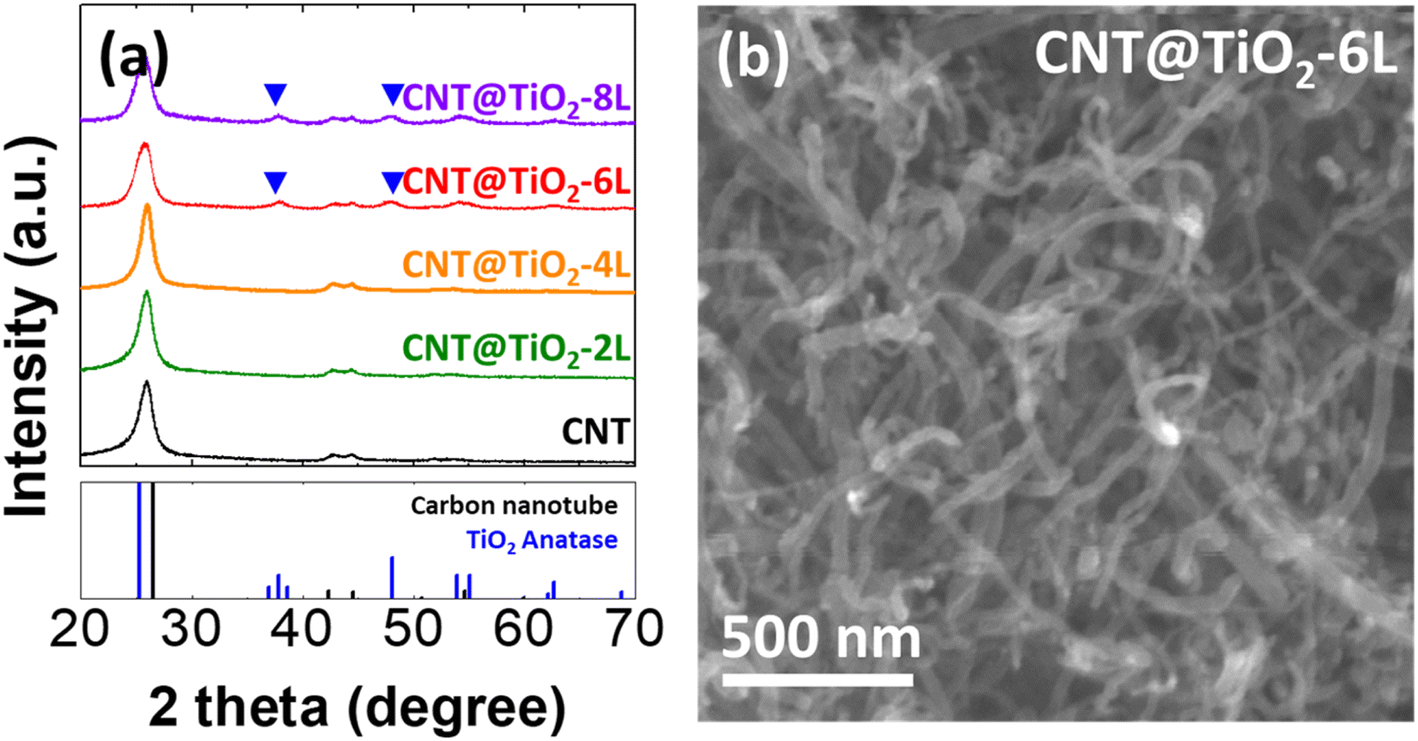 | ||
| Fig. 1 (a) XRD patterns of acid-treated CNT and CNT@TiO2-n L nanocomposites (n = 2, 4, 6 and 8). (b) SEM image of CNT@TiO2-6 L nanocomposites. | ||
 | ||
| Fig. 2 (a) and (b) TEM images, (c) HAADF STEM image and EDS elemental mapping for (d) carbon, (e) oxygen and (f) titanium of CNT@TiO2-6 L nanocomposites. | ||
To evaluate performance of the prepared supports, Ir nanoparticles were synthesized on CNT (Ir/CNT) and CNT@TiO2-n L with Ir contents of about 40 wt% (Ir/CNT@TiO2-n L, Table S2†). The OER activity of the prepared Ir/CNT and Ir/CNT@TiO2-n L catalysts was evaluated by measuring polarization curves in an Ar-saturated 0.05 M H2SO4 solution at a scan rate of 5 mV s−1, in comparison to commercial Ir black and Ir/C (Fig. 4a and S5†). The measured polarization curves were iR-corrected. The loading amounts of Ir on the rotating disk electrode (RDE) were fixed at 10 μgIr cm−2. Exceptively, Ir black was coated on the RDE with loading amount of 37.5 μgIr cm−2 to maintain the catalyst film quality. Among the prepared Ir/CNT@TiO2-n L samples, Ir/CNT@TiO2-6 L exhibited slightly higher OER activity than the others (Fig. S6†). Accordingly, CNT@TiO2-6 L was investigated as a representative support and denoted simply as CNT@TiO2 hereafter. Considering the Ir loading amounts, the OER polarization curves were normalized by Ir mass in Fig. 4a. The mass activity at 1.55 VRHE for the Ir black, Ir/C, Ir/CNT, and Ir/CNT@TiO2 were found to be 247, 616, 927, and 1055 A gIr−1, respectively. Compared to Ir black, the supported catalysts exhibit higher mass activity, that can be associated with the well distribution of Ir nanoparticles and efficient utilization of Ir catalyst. Among the supported catalysts, Ir/CNT@TiO2 presents the highest mass activity. Compared to the previously-reported oxide-supported Ir-based catalysts, Ir/CNT@TiO2 demonstrates excellent OER activity, as summarized in Table S3.†
Tafel slopes for Ir black, Ir/C, Ir/CNT, and Ir/CNT@TiO2 were found to be 48, 50, 49, and 50 mV dec−1, respectively (Fig. S7a†), reflecting similar OER mechanisms on all of the prepared sample. Charge transfer resistances of the supported catalysts estimated from Nyquist plots (Fig. S7b†) were 500, 370 and 240 ohm for Ir/C, Ir/CNT, and Ir/CNT@TiO2, respectively, in accordance with the mass activity. Due to the higher Ir loading for Ir black, it is not suitable to compare the charge transfer resistance of Ir black to those of supported catalysts.
Stability of the prepared catalysts was evaluated by conducting chronopotentiometry at a current density of 10 mA cm−2, which was selected as an accelerated stability test (AST) condition based on the chronopotentiometry outcomes collected for Ir black at 1, 5, and 10 mA cm−2 (Fig. S8†). As shown in Fig. 3b, potentials of Ir black, Ir/C and Ir/CNT increase gradually and abruptly increase to 2.0 VRHE at 140, 50 and 235 min, respectively. On the other hand, Ir/CNT@TiO2 demonstrates potentials lower than 1.75 V during 360 min (the saw-toothed potentials is associated with the generation and removal of oxygen gas bubbles). Ir/C shows inferior stability to Ir black despite a better OER performance. These outcomes reflect that carbon black easily corroded under the OER condition and the carbon corrosion has crucial effects on the stability of the supported catalyst.7 The better stability of Ir/CNT than Ir black and Ir/C reveals that CNT has an improved corrosion resistance than black carbon. Furthermore, Ir/CNT@TiO2 shows the best durability superior to Ir/CNT.
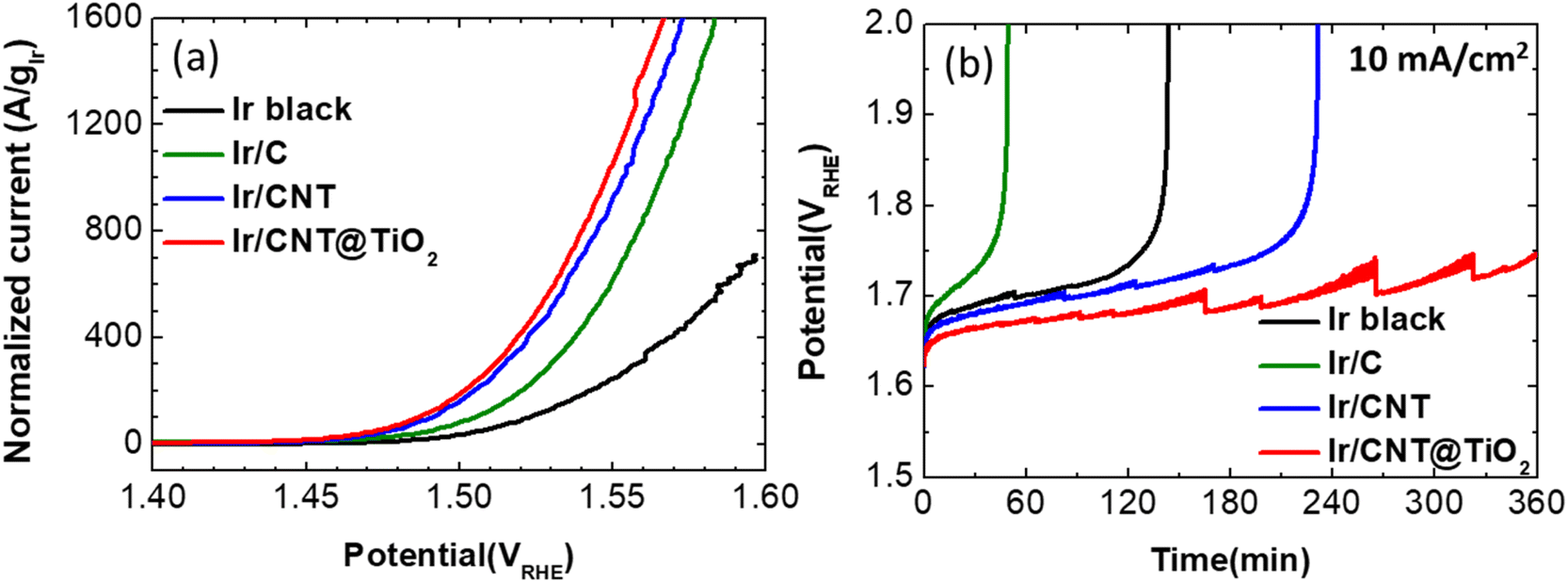 | ||
| Fig. 3 (a) OER polarization curves normalized by Ir loading amounts on RDE and (b) durability measurement at 10 mA cm−2 in Ar-saturated 0.05 M H2SO4 electrolyte for Ir black, Ir/C, Ir/CNT and Ir/CNT@TiO2. | ||
As a support for Ir catalyst, TiO2 has been known to demonstrate the poor performance due to the extremely low electrical conductivity.17 However, Ir/CNT@TiO2 exhibits excellent OER performance and stability compared to Ir black, Ir/C and Ir/CNT. In that respect, electrical conductivity of CNTs and CNT@TiO2 was measured and found to be 3.2 and 1.6 ± 0.2 S cm−1, respectively, using the previously-reported method.29 Even though lower than that of CNT, electrical conductivity of CNT@TiO2 is significantly higher than that of TiO2 nanoparticles (∼10−6 S cm−1) and even comparable with that of Vulcan carbon (1.4 S cm−1), one of the most widely-used support materials.29 These results demonstrate that CNT in CNT@TiO2 nanocomposite effectively contributes to the formation of the electron-conduction paths, enabling CNT@TiO2 to serve as a support.
Fig. 4a and c illustrate that Ir/CNT and Ir/CNT@TiO2 have similar sizes and distributions of Ir nanoparticles (Fig. S9–S11†), with average particle sizes of 1.61 and 1.64 nm, respectively. However, Ir/CNT@TiO2 display higher OER activity than Ir/CNT despite lower electrical conductivity. These outcomes imply that the CNT@TiO2 nanocomposites support, particularly TiO2, affects electrocatalytic activity of Ir. The electronic structure of Ir is considered a crucial factor determining the catalytic performance of the OER. In particular, iridium hydroxo surface species (Ir–OH) has higher intrinsic activity toward the OER than iridium oxide surface species.31 Since the Ir(III) species is known to be related to Ir–OH, composition of Ir(III) has been proposed as an activity descriptor for the OER.32 Furthermore, it has been reported that the electronic structure can be modulated through interaction between Ir and oxide support.7,29 Effects of TiO2 on the electronic structure of Ir were investigated. XPS spectra in the Ir 4f region of Ir/CNT and Ir/CNT@TiO2 (Fig. 5) display presence of the Ir(0), Ir(III), and Ir(IV) species.33 In Ir/CNT, Ir species is composed of approximately 14% Ir(0), 22% Ir(III), and 64% Ir(IV) (Fig. S12†). Ir/CNT@TiO2 presents corresponding compositions of 15, 36, and 49%. The higher composition of Ir(III) species in Ir/CNT@TiO2 demonstrates that TiO2 promotes the formation of Ir(III) species,14,29 thereby enhancing the OER activity.
 | ||
| Fig. 4 TEM images of Ir nanoparticles of (a), (b) the Ir/CNT catalyst and (c), (d) the Ir/CNT@TiO2 catalyst (a), (c) before and (b), (d) after the durability test. | ||
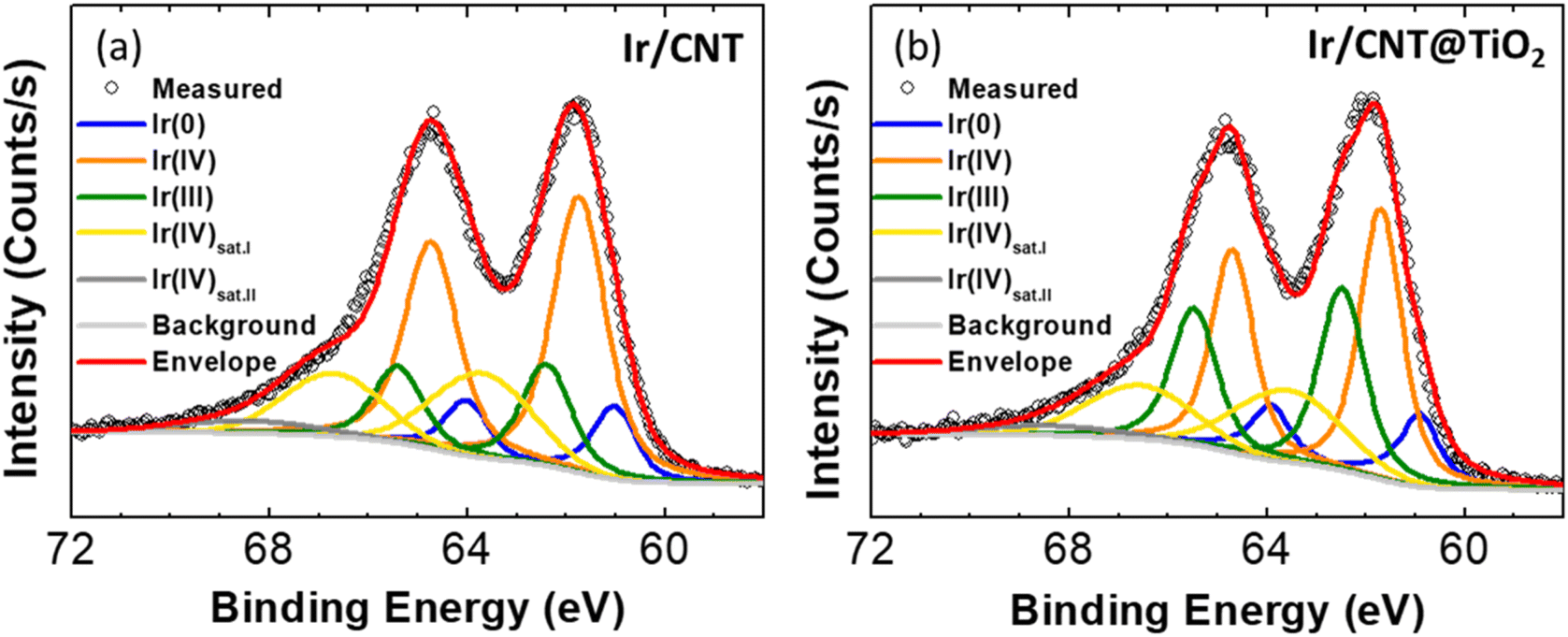 | ||
| Fig. 5 XPS Ir 4f spectra of (a) Ir/CNT and (b) Ir/CNT@TiO2. | ||
In addition to the improvement of the OER activity, the strong interaction between Ir and TiO2 could affect stability of Ir catalyst.34 To examine effects of TiO2 on the degradation of Ir nanoparticles, Ir/CNT and Ir/CNT@TiO2 were exposed to the AST condition of 10 mA cm−2 for ten hours and then observed using TEM and EDS (Fig. 4b, d, S9 and S10†). During the AST, Ir nanoparticles were substantially lost from Ir/CNT, whereas most of Ir nanoparticles were well preserved in Ir/CNT@TiO2. In addition, the average Ir particle size of Ir/CNT was reduced to 1.49 nm (Fig. S11†), whereas that of Ir/CNT@TiO2 was kept similar (1.68 nm, Fig. S11†). These results support that the strong interaction between Ir and TiO2 significantly suppressed dissolution and loss of Ir nanoparticles,14,34 thereby improving stability of Ir nanoparticle catalyst. The loss of Ir nanoparticles is known as the main origin of catalyst degradation during the OER. The enhanced stability of the Ir/CNT@TiO2 catalyst can be attributed to (1) the higher corrosion resistance of the TiO2 layers on CNT than that of the carbon support under OER conditions and (2) the strong interaction between Ir and metal-oxide support. These results demonstrate that introduction of CNT@TiO2 nanocomposites support can be an effective way to improve activity and stability of Ir nanoparticle catalyst in the PEMWE anode.
4. Conclusion
In this study, we present CNT and TiO2 nanocomposite (CNT@TiO2) as a high-performance support for Ir nanoparticle catalyst toward the OER in the PEMWE anode. TiO2 was synthesized in a form of thin layers first and then nanoparticles on acid-treated CNTs. CNT@TiO2 nanocomposites show a high electrical conductivity value of 1.6 ± 0.2 S cm−1, that is lower than that of CNT but comparable to that of carbon black. Through XPS analysis, it was demonstrated that TiO2 in CNT@TiO2 promotes the formation of OER-active Ir(III) species in Ir/CNT@TiO2. The strong interaction between Ir and TiO2 prevents loss of Ir nanoparticles during OER, improving stability of Ir/CNT@TiO2. Ir/CNT@TiO2 demonstrated superior OER activity and stability to Ir black, Ir/C and Ir/CNT. Thus, introduction of CNT@TiO2 nanocomposites support can be an effective way to improve performance and durability of Ir nanoparticle catalyst in the PEMWE anode. In addition to the high-performing support material, a novel solution-based layer-by-layer coating method that mimics atomic layer deposition (ALD) is presented with advantages of easy control of TiO2 loading amount, cost-effectiveness and scalability.Conflicts of interest
There are no conflicts to declare.References
- S. Shiva Kumar and V. Himabindu, Hydrogen production by PEM water electrolysis – a review, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, A comprehensive review on PEM water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, Electrocatalysts for acidic oxygen evolution reaction: achievements and perspectives, Nano Energy, 2020, 78, 105392 CrossRef CAS.
- Y. Zhang, X. Zhu, G. Zhang, P. Shi and A.-L. Wang, Rational catalyst design for oxygen evolution under acidic conditions: strategies toward enhanced electrocatalytic performance, J. Mater. Chem. A, 2021, 9, 5890–5914 RSC.
- M.-C. Chuang and J.-a. A. Ho, Efficient electrocatalytic oxidation of water: minimization of catalyst loading by an electrostatic assembly of hydrous iridium oxide colloids, RSC Adv., 2012, 2, 4092–4096 RSC.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C.-j. Liu, A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: nanostructure, activity, mechanism and carbon support in PEM fuel cells, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- H.-S. Oh, H. N. Nong, D. Teschner, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schloegl and P. Strasser, Electrochemical catalyst-support effects and their stabilizing role for IrOx nanoparticle catalysts during the oxygen evolution reaction (OER), J. Am. Chem. Soc., 2016, 138(38), 12552–12563 CrossRef CAS PubMed.
- X. Wang, M. Waje and Y. Yan, CNT-based electrodes with high efficiency for PEMFCs, Electrochem. Solid-State Lett., 2004, 8, A42 CrossRef.
- R. Badam, M. Hara, H.-H. Huang and M. Yoshimura, Synthesis and electrochemical analysis of novel IrO2 nanoparticle catalysts supported on carbon nanotube for oxygen evolution reaction, Int. J. Hydrogen Energy, 2018, 43(39), 18095–18104 CrossRef CAS.
- X. Wen, L. Bai, M. Li and J. Guan, Ultrafine iridium oxide supported on carbon nanotubes for efficient catalysis of oxygen evolution and oxygen reduction reactions, Mater. Today Energy, 2018, 10, 153–160 CrossRef.
- T. Fujigaya, Y. Shi, J. Yang, H. Li, K. Ito and N. Nakashima, A highly efficient and durable carbon nanotube-based anode electrocatalyst for water electrolyzers, J. Mater. Chem. A, 2017, 5, 10584–10590 RSC.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Carbon nanotubes for lithium ion batteries, Energy Environ. Sci., 2009, 2, 638–654 RSC.
- X. Li, W.-X. Chen, J. Zhao, W. Xing and Z.-D. Xu, Microwave polyol synthesis of Pt/CNTs catalysts: effects of pH on particle size and electrocatalytic activity for methanol electrooxidization, Carbon, 2005, 43, 2168–2174 CrossRef CAS.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, IrO2–TiO2: A High-Surface-Area, Active, and Stable Electrocatalyst for the Oxygen Evolution Reaction, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- M. Yuan, Y. Zhu, L. Deng, R. Ming, A. Zhang, W. Li, B. Chai and Z. Ren, IrO2–TiO2 electrocatalysts for the hydrogen evolution reaction in acidic water electrolysis without activation, New J. Chem., 2017, 41(14), 6152–6159 RSC.
- H. Lv, G. Zhang, C. Hao, C. Mi, W. Zhou, D. Yang, B. Li and C. Zhang, Activity of IrO2 supported on tantalum-doped TiO2 electrocatalyst for solid polymer electrolyte water electrolyzer, RSC Adv., 2017, 7, 40427–40436 RSC.
- R. V. Genova-Koleva, F. Alcaide, G. Álvarez, P. L. Cabot, H.-J. Grande, M. V. Martínez-Huerta and O. Miguel, Supporting IrO2 and IrRuOx nanoparticles on TiO2 and Nb-doped TiO2 nanotubes as electrocatalysts for the oxygen evolution reaction, J. Energy Chem., 2019, 34, 227–239 CrossRef.
- S. Schlicht, P. Büttner and J. Bachmann, Highly Active Ir/TiO2 Electrodes for the Oxygen Evolution Reaction Using Atomic Layer Deposition on Ordered Porous Substrates, ACS Appl. Energy Mater., 2019, 2, 2344–2349 CrossRef CAS.
- C. V. Pham, M. Bühler, J. Knöppel, M. Bierling, D. Seeberger, D. Escalera-López, K. J. J. Mayrhofer, S. Cherevko and S. Thiele, IrO2 coated TiO2 core–shell microparticles advance performance of low loading proton exchange membrane water electrolyzers, Appl. Catal., B, 2020, 269, 118762 CrossRef CAS.
- X. Wang, X. Wan, X. Qin, C. Chen, X. Qian, Y. Guo, Q. Xu, W.-B. Cai, H. Yang and K. Jiang, Electronic Structure Modulation of RuO2 by TiO2 Enriched with Oxygen Vacancies to Boost Acidic O2 Evolution, ACS Catal., 2022, 12, 9437–9445 CrossRef CAS.
- E. Oakton, D. Lebedev, A. Fedorov, F. Krumeich, J. Tillier, O. Sereda, T. J. Schmidt and C. Copéret, A simple one-pot Adams method route to conductive high surface area IrO2–TiO2 materials, New J. Chem., 2016, 40, 1834–1838 RSC.
- P. Mazúr, J. Polonský, M. Paidar and K. Bouzek, Non-conductive TiO2 as the anode catalyst support for PEM water electrolysis, Int. J. Hydrogen Energy, 2012, 37, 12081–12088 CrossRef.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef.
- C. Hao, H. Lv, Q. Zhao, B. Li, C. Zhang, C. Mi, Y. Song and J. Ma, Investigation of V-doped TiO2 as an anodic catalyst support for SPE water electrolysis, Int. J. Hydrogen Energy, 2017, 42, 9384–9395 CrossRef CAS.
- C. Hao, H. Lv, C. Mi, Y. Song and J. Ma, Investigation of Mesoporous Niobium-Doped TiO2 as an Oxygen Evolution Catalyst Support in an SPE Water Electrolyzer, ACS Sustainable Chem. Eng., 2016, 4, 746–756 CrossRef CAS.
- G. C. da Silva, S. I. Venturini, S. Zhang, M. Löffler, C. Scheu, K. J. J. Mayrhofer, E. A. Ticianelli and S. Cherevko, Oxygen evolution reaction on tin oxides supported iridium catalysts: do we need dopants?, ChemElectroChem, 2020, 7, 2330–2339 CrossRef CAS.
- C. Glynn and C. O'Dwyer, Solution Processable Metal Oxide Thin Film Deposition and Material Growth for Electronic and Photonic Devices, Adv. Mater. Interfaces, 2017, 4, 1600610 CrossRef.
- F. Bonet, V. Delmas, S. Grugeon, R. Herrera Urbina, P. Y. Silvert and K. Tekaia-Elhsissen, Synthesis of monodisperse Au, Pt, Pd, Ru and Ir nanoparticles in ethylene glycol, Nanostruct. Mater., 1999, 11, 1277–1284 CrossRef CAS.
- E.-J. Kim, J. Shin, J. Bak, S. J. Lee, K. h. Kim, D. Song, J. Roh, Y. Lee, H. Kim, K.-S. Lee and E. Cho, Stabilizing role of Mo in TiO2-MoOx supported Ir catalyst toward oxygen evolution reaction, Appl. Catal., B, 2021, 280, 119433 CrossRef CAS.
- S.-T. Kang, J.-Y. Seo and S.-H. Park, The Characteristics of CNT/Cement Composites with Acid-Treated MWCNTs, Adv. Mater. Sci. Eng., 2015,(2015), 308725 Search PubMed.
- D. F. Abbott, D. Lebedev, K. Waltar, M. Povia, M. Nachtegaal, E. Fabbri, C. Copéret and T. J. Schmidt, Iridium Oxide for the Oxygen Evolution Reaction: Correlation between Particle Size, Morphology, and the Surface Hydroxo Layer from Operando XAS, Chem. Mater., 2016, 28, 6591–6604 CrossRef CAS.
- C. Spöri, P. Briois, H. N. Nong, T. Reier, A. Billard, S. Kühl, D. Teschner and P. Strasser, Experimental Activity Descriptors for Iridium-Based Catalysts for the Electrochemical Oxygen Evolution Reaction (OER), ACS Catal., 2019, 6653–6663 CrossRef.
- V. Pfeifer, T. E. Jones, J. J. Velasco Velez, C. Massue, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Havecker, A. Knop-Gericke and R. Schlögl, The Electronic Structure of Iridium Oxide Electrodes Active in Water Splitting, Phys. Chem. Chem. Phys., 2016, 18, 2292 RSC.
- F. Claudel, L. Dubau, G. Berthomé, L. Sola-Hernandez, C. Beauger, L. Piccolo and F. Maillard, Degradation Mechanisms of Oxygen Evolution Reaction Electrocatalysts: A Combined Identical-Location Transmission Electron Microscopy and X-ray Photoelectron Spectroscopy Study, ACS Catal., 2019, 9, 4688–4698 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05027g |
| This journal is © The Royal Society of Chemistry 2022 |