
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFeII/III and MnII complexes based on 2,4,6-tris(2-pyridyl)-triazine: synthesis, structures, magnetic and biological properties†
Mariana Dariia,
Annabel Mikoschb,
Jan van Leusen b,
Victor Ch. Kravtsova,
Elena G. Dvorninac,
Steliana T. Clapcoc,
Alexandra Cilocic,
Paul Kögerler*b and
Svetlana G. Baca*a
b,
Victor Ch. Kravtsova,
Elena G. Dvorninac,
Steliana T. Clapcoc,
Alexandra Cilocic,
Paul Kögerler*b and
Svetlana G. Baca*a
aInstitute of Applied Physics, Academiei 5, MD2028 Chisinau, Moldova. E-mail: svetlana.baca@ifa.md
bInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de
cInstitute of Microbiology and Biotechnology, Academiei 1, MD2028 Chisinau, Moldova
First published on 11th October 2022
Abstract
Reaction of the polypyridyl ligand 2,4,6-tris(2-pyridyl)-s-triazine (tpt) with mono- and polynuclear Fe(III) or Mn(II) precursors, specifically tri- or hexanuclear Fe(III) pivalates (piv−), [Mn6O2(piv)10(Hpiv)4] and Mn(II) isobutyrate (ib−), in various solvents and under various reaction conditions showcase the ligand’s surprising coordination characteristics. The reactions result in mononuclear [FeIII(tpt)(tptH)][FeIIICl4]4·2(thf)·0.23(H2O) (1), [FeIII(piv)(tpt)Cl2] (2), [FeII(tpt)Cl2]·2(H2O) (3a), dinuclear [FeIII2O(tpt)2Cl4] (3), and heptanuclear [FeIII7O4(piv)12(tpt-O)]·A [A = MeCN (4a) or 4(dioxane) (4b)] and [FeIII7O4(piv)11(tpt-O)(i-PrO)(i-PrOH)]·0.75(i-PrOH) (5), as well as the mononuclear compounds [MnII(tpt)(NO3)(H2O)2](NO3) (6) and [MnII(tpt)(ib)(Cl)(MeOH)]·MeOH (7). Single-crystal X-ray diffraction analyses identify tpt as a tridentate NNN donor ligand that forms two five-membered metallocycles in 1–3, 6 and 7, whereas in 4 and 5 five tpt N atoms form coordinative bonds accompanied by an unusual metal-induced oxidation of one of the carbon atoms of the central triazine core. Magnetic properties of Fe(III)–tpt (2–5), Fe(II)–tpt (3a), and Mn(II)–tpt (7) compounds show dominant antiferromagnetic coupling for polynuclear coordination cluster compounds. The Mn(II)–tpt complexes 6 and 7 exhibit efficient catalytic properties in the production of enzymes by microorganisms, which concerns the synthesis of exocellular proteases in Fusarium gibbosum CNMN FD 12 or Trichoderma koningii Oudemans CNMN FD 15 fungi strains. Thus, compounds 6 and 7 can be used for producing proteolytic enzymes with wide applications including in the food, detergents and pharmaceutical industries.
Introduction
Coordination compounds of polypyridyl derivatives have attracted great interest and enormous efforts since the discovery that such clusters can be used as potential model systems for studying and identifying fundamental biochemical processes1 as well as individual objects for realizing nano-devices in molecular electronics, spintronics, and sensor technologies.2–4 Among polypyridyl ligands, 2,4,6-tris(2-pyridyl)-s-triazine (tpt) has been widely utilized for the spectrophotometric determination of V(IV) and V(V),5 Fe(II),6 Ru(III),7 Co(II),8 Al(III)9 and Ag(I)10 ions or as a solvent extraction reagent.11 Moreover, tpt shows various pharmaceutical properties such as antiviral, antitumor, analgesic, antioxidant, antidepressant, antibacterial, and antimicrobial activities.12 This widely used ligand has a rich coordination chemistry and formed a variety of mono-, di-, tri-, tetra-, penta-, hexa-, and decanuclear metal complexes and coordination polymers. The reader is referred to one of the excellent reviews by B. Therrien13 on the coordination properties of tpt. In coordination compounds this versatile ligand may adopt different coordination modes (Scheme 1 and Table S1†). A detailed analysis of coordination modes of tpt in previously published examples of its metal complexes retrieved from the Cambridge Structural Database (CSD 2020.2)14 reveals ten different coordination types: tridentate terpyridine-like (A), bidentate bipyridine-like (B), mixed tridentate terpyridine-like and bidentate bipyridine-like (C), mixed terpyridine-like and monodentate pyridine-like (D), bis-bipyridine-like (E and F), mixed bidentate bipyridine-like and monodentate pyridine-like (G), tris-bipyridine-like (H and K), and mixed bis-bipyridine-like and monodentate pyridine-like (J). Out of the 324 hits for tpt metal complexes in the latest version of the CSD database, 279 belong to the terpyridine-like coordination mode (A). Other dominant modes of the coordination of tpt are bidentate bipyridine-like (B, 14 hits), mixed tridentate terpyridine-like and bidentate bipyridine-like (C, 10 hits) and bis-bipyridine-like (E and F, 9 hits). Type D is limited to 5 cases, G to one case. The simultaneous coordination of the tpt ligand to three metal centers is rare: 3 cases for H, 2 cases for J and one case for K (for more details and refs. see Table S1 in the ESI†).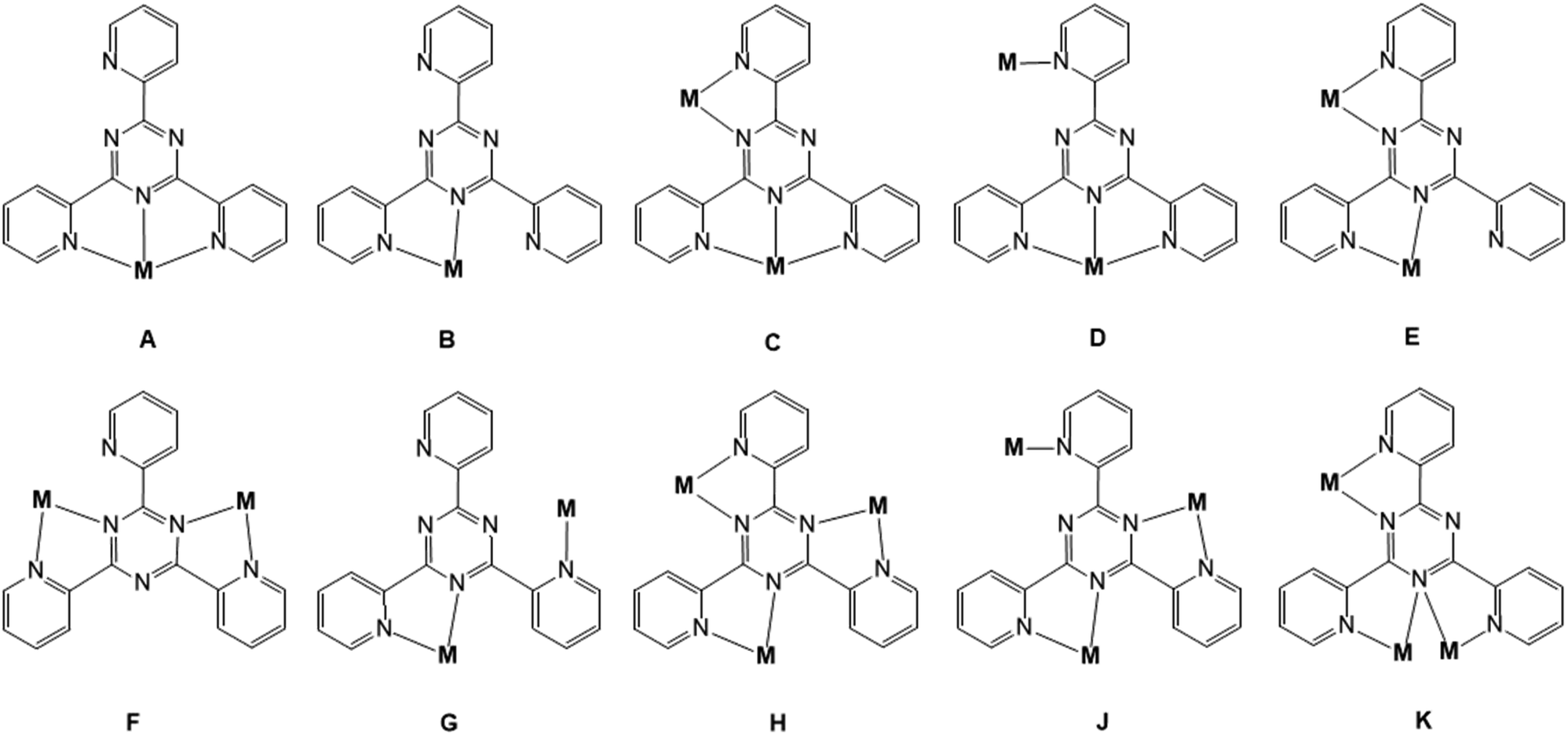 | ||
| Scheme 1 Coordination modes exhibited by the tpt ligand in metal complexes. Data retrieved from the Cambridge Structural Database (version 5.41, 12/2020). | ||
Interestingly, although tpt is usually stable toward hydrolysis and has been widely used as analytical reagent for the detection of various metal ions,5–10 it was found that tpt can undergo hydrolysis under certain conditions when metal ions such as Ru(II)15 and Cu(II)16,17 are present. Additionally, P. Paul and co-workers observed the hydrolysis of tpt to the bis(2-pyridylcarbonyl)amide anion and 2-picolinamide in Rh(III) complexes18 and metal-induced hydroxylation at one carbon atom of the triazine ring in dinuclear Ru(II) and Os(II) complexes.19 Partial hydrolysis can also be observed in reactions between ruthenium polypyridine tpt complexes with Pd(dmso)Cl2 and Pt(dmso)Cl2.20 The metal-induced methoxylation at one of the ring carbon atoms of the central triazine core of the bridging tpt ligand was detected in Re(I) complexes by J. A. Zubieta et al.21 V. Chandrasekhar and co-workers reported a ring hydroxylation at the triazine carbon atom upon metalation in an Ir(III) dimer.22
We systematically explored the reactivity of tpt towards both mono- and polynuclear Fe(III) and Mn(II) carboxylate precursors and identified a series of mono- and polynuclear Fe compounds formulated as [FeIII(tpt)(tptH)][FeIIICl4]4·2(thf)·0.23(H2O) (1), [FeIII(piv)(tpt)Cl2] (2), [FeIII2O(tpt)2Cl4] (3), [FeII(tpt)Cl2]·2(H2O) (3a), [FeIII7O4(piv)12(tpt-O)]·MeCN (4a), [FeIII7O4(piv)12(tpt-O)]·4(dioxane) (4b) [FeIII7O4(piv)11(tpt-O)(i-PrO)(i-PrOH)]·0.75(i-PrOH) (5), as well as the two mononuclear Mn compounds [MnII(tpt)(NO3)(H2O)2](NO3) (6) and [MnII(tpt)(ib)(Cl)(MeOH)]·MeOH (7). The products were characterized by elemental analysis, IR spectroscopy, ESI mass-spectrometry, and X-ray diffraction studies. In particular, single-crystal X-ray analysis revealed an unusual metal-induced oxidation at the carbon atom of the triazine ring of the bridging tpt ligand and subsequent coordination of the thus-formed O donor in the heptanuclear Fe(III) pivalate complexes. A selected range of Fe(III)/Fe(II)–tpt and Mn(II)–tpt complexes have also been characterized via magnetometry.
Coordination compounds of tpt are known for their bioactivity, for example, an inhibition effect against the filarial parasite Setaria cervi that is specific to Ru-tpt compounds.23 A series of Co(II), Ni(II), Cu(II), Mn(II) and Rh(III) complexes with tpt exhibits antibacterial activity against both Gram-positive (Bacillus subtilis, Staphylococcus aureus) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacteria.24 Polynuclear Pt(II)–tpt complexes show potent cytotoxic activity in various cancer cell lines and can been used as potential chemotherapeutic agents.25 Beside the possible use of metal–tpt based complexes as antimicrobial, anticancer and antiparasitic agents, the employment of such compounds to increase the enzyme biosynthesis has not been reported. This is the first study where the prepared Mn(II)–tpt compounds have been tested as biostimulators for protease synthesis by Fusarium gibbosum CNMN FD 12 and Trichoderma koningii Oudemans CNMN FD 15 fungi strains. Note that manganese is one of the most studied metal ions among other transition metals due to its significant role in the various biological processes,26 as well as antibacterial and anticancer activities exhibited by its complexes with various organic ligands.27
Results and discussion
Synthetic aspects and preliminary characterization
Among the multiple synthetic pathways toward novel polynuclear clusters, the most common approach relies on “serendipitous” self-assembly28 where “ligands are used that display several different coordination modes, combined with metal centers that can vary their coordination geometries” in one-pot reactions. The source of metal ions can range from simple salts to preformed polynuclear metallic precursors, for example well-known trinuclear μ3-oxo-bridged metal carboxylates that can lose their terminal ligands to create vacant coordination sites and aggregate to higher nuclear clusters under various reaction conditions. The numerous Fe(III)/Fe(II)–tpt and Mn(II)–tpt clusters introduced herein are synthesized via this route. The tri- or hexanuclear Fe(III) and Mn(II,III) carboxylate precursors are soluble in various organic solvents; moreover, their terminal and their carboxylate ligands frequently are sufficiently labile, allowing the assembly into higher-nuclearity clusters. These properties made it possible to realize a very substantial range of different reaction conditions, utilizing different synthetic procedures such as stirring at room temperature in different solvents or mixtures of solvents, refluxing, sonochemical irradiation, and solvothermal reactions under high temperature and pressure. In this way, the coordination of Fe(II,III) and Mn(II,III) ions with tpt could be systematically studied, resulting in six Fe(III), one Fe(II), and two Mn(II) clusters were prepared. The reaction pathways resulting in new Fe(III)/Fe(II) coordination compounds are summarized in Scheme 2.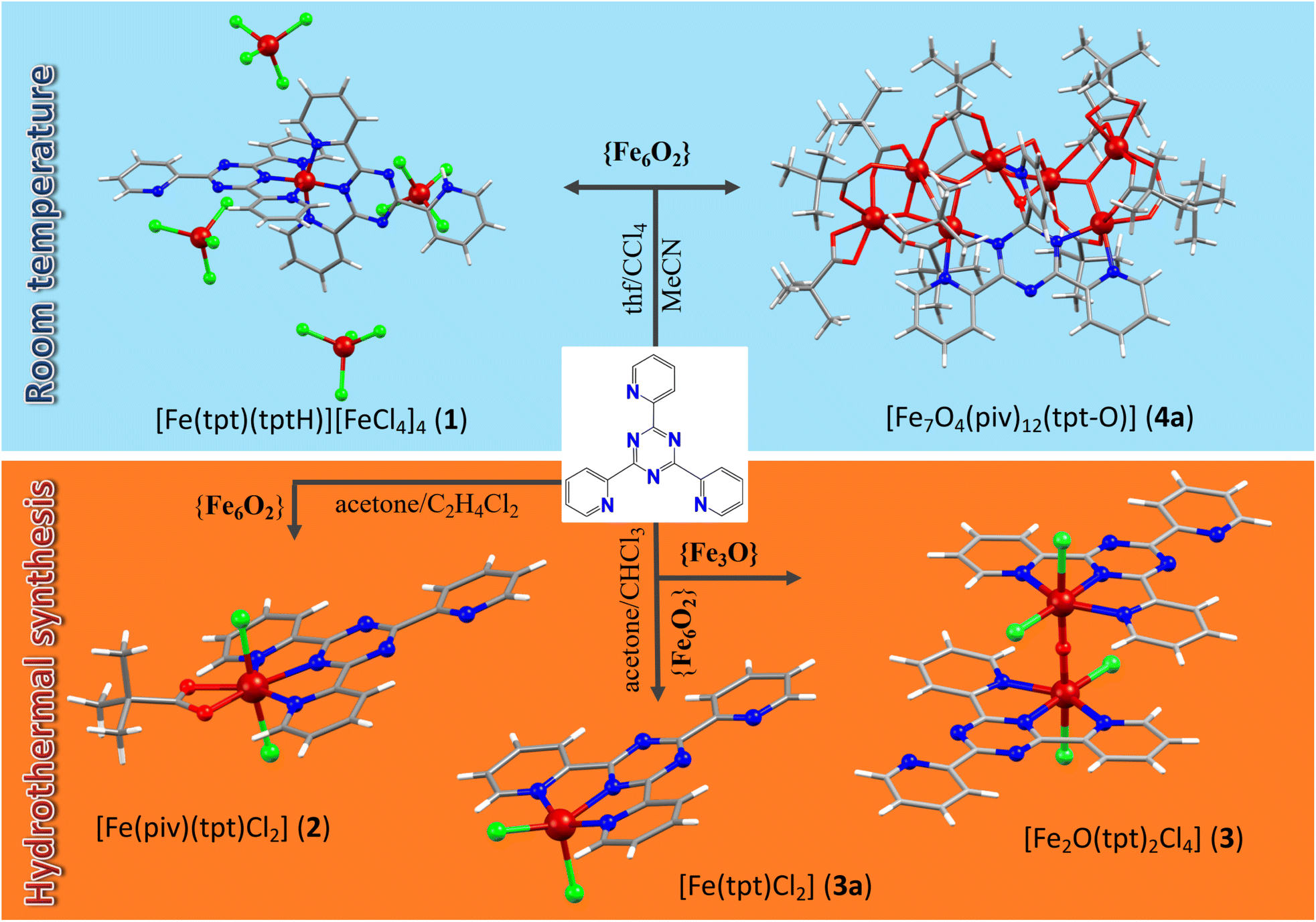 | ||
| Scheme 2 The reaction pathways resulting in Fe tpt coordination compounds 1–4. | ||
Large red-brown block-shaped crystals of compound 1, [Fe(tpt)(tptH)][FeCl4]4·2(thf)·0.23(H2O), in addition to a blue microcrystalline powder, were obtained after several weeks of crystallization from a blue-green mixture of the hexanuclear Fe(III) precursor [Fe6O2(OH)2(piv)12] and tpt in a thf/CCl4 mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) stirred at room temperature (Scheme 2). Further investigation of this synthesis revealed that visible light is critical for the formation of 1. If the reaction is carried out in the dark, only small, irregular shaped red crystals are formed from a red solution. The structure of these red crystals has not been resolved yet. The impact of the ratio of the reactants has also been examined. Doubling the amount of the tpt ligand led to an over-saturated solution and undissolved ligand that was filtered off. After several weeks only a blue precipitate was found. Another less concentrated synthesis with the same ratio of starting materials was repeated, which led to a yellow solution with a colorless precipitate. Hence, it was found that the solution needs to be highly concentrated and the 1:1 ratio of the reactants leads to more satisfying results. The blue and red crystals are well soluble in thf, acetone and chloroform, but are insoluble in CCl4, preventing the separation of two different kinds of crystals by re-crystallization. The crystals were separated manually under the microscope.
:1, v/v) stirred at room temperature (Scheme 2). Further investigation of this synthesis revealed that visible light is critical for the formation of 1. If the reaction is carried out in the dark, only small, irregular shaped red crystals are formed from a red solution. The structure of these red crystals has not been resolved yet. The impact of the ratio of the reactants has also been examined. Doubling the amount of the tpt ligand led to an over-saturated solution and undissolved ligand that was filtered off. After several weeks only a blue precipitate was found. Another less concentrated synthesis with the same ratio of starting materials was repeated, which led to a yellow solution with a colorless precipitate. Hence, it was found that the solution needs to be highly concentrated and the 1:1 ratio of the reactants leads to more satisfying results. The blue and red crystals are well soluble in thf, acetone and chloroform, but are insoluble in CCl4, preventing the separation of two different kinds of crystals by re-crystallization. The crystals were separated manually under the microscope.
Solvothermal heating of the same hexanuclear Fe(III) pivalate precursors with tpt in acetone and 1,2-dichlorethane lead to another mononuclear complex, [Fe(piv)(tpt)(Cl)2] (2) (Scheme 2). The synthesis was repeated with the ratio of the reactants according to the formula but it resulted in a worse yield. The source of Cl− anions in 1 and 2 was CCl4 and 1,2-dichloroethane, respectively, which undergo dehalogenation and introduce chloride ions into the reaction solution.
Dark-blue rod-shaped crystals of a dinuclear Fe(III)–tpt cluster, [Fe2O(tpt)2Cl4] (3), have also been received from solvothermal syntheses when using μ-oxo trinuclear Fe(III) pivalate precursors with tpt in acetone and chloroform (Scheme 2). The yield of 3 was a very low, and we noted that the reactant ratio corresponding to the composition of the product (1:3) does not improve the yield, although less concentrated solutions reveal better shaped crystals. Using hexanuclear Fe(III) pivalate precursors in similar conditions, a mononuclear Fe(II)–tpt cluster, [Fe(tpt)Cl2]·2(H2O) (3a), has been prepared. In 3 and 3a, the dehalogenation of CHCl3 under solvothermal heating yields Cl− anions, similar to 1 and 2.
All heptanuclear Fe(III)–tpt clusters, 4a/4b and 5, were obtained from reactions of preformed Fe3/Fe6 pivalate precursors with tpt under comparably mild conditions, i.e. at room temperature. Moreover, for the first time in all heptanuclear Fe(III)–tpt clusters it was found that Fe(III) ions promote the hydrolysis of tpt and coordination of the formed O atom to metal ion and formation of similar [Fe7O4(tpt-O)]12+ units. Compound [Fe7O4(piv)12(tpt-O)]·MeCN (4a) can be prepared from a 1:1 mixture of μ-oxo trinuclear Fe(III) pivalate precursors and tpt in acetonitrile (method A), or using hexanuclear [Fe6O2(OH)2(piv)12] precursors in the reaction with tpt from a 1:1 mixture of n-hexane and MeCN (method B), or from a 1:1 mixture of acetone and MeCN (method C). The complex is well soluble in acetone, thf, ethanol and methanol and is slightly soluble in n-hexane but insoluble in acetonitrile. We also run the same reactions with the ratio of starting reactants corresponding to the composition of the final product and found no improvement but a degradation of the yield. Stirring μ-oxo trinuclear [Fe3O(piv)6(H2O)3](piv)·2(Hpiv) precursors with tpt in 1,4-dioxane gave a structurally similar [Fe7O4(piv)12(tpt-O)]·4(dioxane) (4b) cluster but with solvent dioxane molecules instead of MeCN as in 4a. The yield of 16% was low, but repeating the synthesis with the starting materials in the ratio corresponding to the composition of the product gave no crystals. Finally, a further heptanuclear compound, [Fe7O4(piv)11(tpt-O)(i-PrO)(i-PrOH)]·0.75(i-PrOH) (5), has been prepared from the reaction of trinuclear Fe(III) pivalates with tpt in a mixture of iso-propanol and n-hexane (1:1) with a moderate yield of 45%. We also explored the same reaction but using only iso-propanol and found that this led to compound 5, too.
For the preparation of Mn(II)–tpt compounds, [Mn6O2(piv)10(Hpiv)4] and Mn(ib)2 have been examined as precursors. The reaction of hexanuclear [Mn6O2(piv)10(Hpiv)4] pivalate clusters with tpt and Dy(NO3)3·6H2O in EtOH solution under refluxing resulted in [Mn(NO3)(tpt)(H2O)2](NO3) (6) in a high yield (76%). Ultrasonic treatment of Mn(II) isobutyrate with tpt and 2-[bis(2-hydroxyethyl)amino]acetonitrile hydrochloride in acetonitrile solution results in [Mn(ib)(Cl)(tpt)(MeOH)]·MeOH (7) in 55% yield. Although 2-[bis(2-hydroxyethyl)amino]acetonitrile does not coordinate to Mn(II), its presence is critical for the growth of diffraction-quality crystals of 7. All bulk Fe(III)/Mn(II)–tpt samples were further characterized by IR spectroscopy and TGA to confirm their identities.
The IR spectra of carboxylate free compounds (1, 3, and 6) show the characteristic bands of coordinated tpt ligand (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and νCN stretching vibrations of the pyridine and triazine rings) in the region of 1637–1441 cm−1, which are shifted compare to a free tpt ligand with a strong band appeared at 1522 cm−1, and weak bands at 1469 and 1433 cm−1.29,30 In the spectra of the carboxylate-containing compounds 2, 4 and 7 these bands overlap with strong vibrations associated with methyl and carboxylate groups of pivalates and isobutyrates. The moderately intense bands in the region 3066–2872 cm−1 belong to the C–H asymmetric and symmetric stretching vibrations of the tpt ligand and methyl groups of the pivalates (2 and 4) or isobutyrates (7). In carboxylate containing compounds 4a and 7 these are accompanied by a strong single band at 1485 cm−1 (4a) and 1474 cm−1 (7) and a typical doublet at 1378 and 1362 (4a) and 1387 and 1377 cm−1 (7) arising from the asymmetric and symmetric bending vibrations of methyl groups. The strong and broad bands arising from asymmetric and symmetric stretching vibrations of the coordinated carboxylate groups are found in 4a at 1543 and 1424 cm−1, in 7 in the 1544–1532 cm−1 region and at 1427 cm−1. The strong bands at 1388 and 1377 cm−1 attributed to the uncoordinated and coordinated nitrate groups in 6. The broad bands between 3731 and 3068 cm−1 correspond to O–H [water molecules (6) and methanol (7)] stretching vibrations.
C and νCN stretching vibrations of the pyridine and triazine rings) in the region of 1637–1441 cm−1, which are shifted compare to a free tpt ligand with a strong band appeared at 1522 cm−1, and weak bands at 1469 and 1433 cm−1.29,30 In the spectra of the carboxylate-containing compounds 2, 4 and 7 these bands overlap with strong vibrations associated with methyl and carboxylate groups of pivalates and isobutyrates. The moderately intense bands in the region 3066–2872 cm−1 belong to the C–H asymmetric and symmetric stretching vibrations of the tpt ligand and methyl groups of the pivalates (2 and 4) or isobutyrates (7). In carboxylate containing compounds 4a and 7 these are accompanied by a strong single band at 1485 cm−1 (4a) and 1474 cm−1 (7) and a typical doublet at 1378 and 1362 (4a) and 1387 and 1377 cm−1 (7) arising from the asymmetric and symmetric bending vibrations of methyl groups. The strong and broad bands arising from asymmetric and symmetric stretching vibrations of the coordinated carboxylate groups are found in 4a at 1543 and 1424 cm−1, in 7 in the 1544–1532 cm−1 region and at 1427 cm−1. The strong bands at 1388 and 1377 cm−1 attributed to the uncoordinated and coordinated nitrate groups in 6. The broad bands between 3731 and 3068 cm−1 correspond to O–H [water molecules (6) and methanol (7)] stretching vibrations.
The solution behavior of 2–5 and 7 has been characterized by electrospray ionization mass spectrometry (Table S2†). The positive-ion ESI-MS spectrum of 2 shows the most intense peak appear at m/z 434, corresponding to the cationic fragment [FeIII(tpt)(Cl)(MeO)]+ (100%). A multiplet at m/z 430 is assigned to the cation [FeIII(tpt)(MeO)2]+. Similar to 2, the positive ESI-MS spectrum of 3 also shows multiplets centered at m/z 430 and 434 with relative intensity of 75% and 71%, respectively. A multiplet at m/z 856 belongs to the cationic fragment [FeIII2O(tpt)2Cl3]+. In the ESI+ pattern of the mononuclear Fe(II) compound 3a, the most intense peak (100%) corresponds to the cationic fragment [FeII(tpt)(Cl)(thf)2(MeOH)2(H2O)2]+ that appears at m/z 647. Another multiplet with relative intensity of 89% corresponds to the cation [FeII(tpt)2]2+ centered at m/z 340.
The ESI+ mass spectrometry pattern of a heptanuclear Fe(III) cluster 4a shows a multiplet with a full intensity at m/z 1913 that belongs to the cationic fragment [Fe7O4(piv)11(tpt-O)(H2O)]+. Other important fragments are [Fe7O4(piv)11(tpt-O)(thf)(H2O)(MeCO2H)]+ (m/z 2045), [Fe7O4(piv)11(tpt-O)(H2O)(MeCO2H)]+ (m/z 1973), and [Fe7O4(piv)9(tpt-O)(MeCO2)(OH)]+ (m/z 1770). Additionally, two most intense peaks appear in the negative ESI-MS pattern of 4a at m/z 1614 (100%) and 1926 (20%), which correspond to the anionic [FeIII7O5(piv)11(MeO)]− and [FeIII7O5(piv)9(tpt-O)(MeO)3(MeCN)3]− fragments, respectively. The ESI+ patterns of the heptanuclear Fe(III) cluster compounds 4b and 5 reveal several series of peaks (Table S2†). A difference between neighboring peaks in each series of Δm/z = 70 can be identified as the gradual replacement of one negatively charged piv− moiety by one MeO− group in the cluster core of 4b and 5. In 4b, the peak of 100% relative intensity at m/z 1597 is attributed to a {[FeIII7O4(piv)5(tpt-O)(MeO)7(H2O)5] + H}+ fragment ion and additional peaks result from replacement of piv− by MeO− in the fragment with general formula of {[FeIII7O4(piv)12−x(tpt-O)(MeO)x(H2O)5] + H}+ (x = 6, 8; m/z 1667 and 1527). The next series of peaks assignable to the cationic [FeIII7O4(piv)11−x(tpt)(MeO)x]+ (x = 0–5, m/z 1910, 1840, 1770, 1700, and 1630) fragments. Furthermore, the ESI+ pattern of 5 in addition to mentioned above series of peaks {[FeIII7O4(piv)12−x(tpt-O)(MeO)x(H2O)5] + H}+ (x = 6, 8) and [FeIII7O4(piv)11−x(tpt)(MeO)x]+ (x = 0–3) show signals at m/z 1975 and 2045, assigned as [FeIII7O5(piv)8(tpt-O)(MeO)(i-PrOH)(thf)3]+ and [FeIII7O5(piv)9(tpt-O)(i-PrOH)(thf)3]+ fragments, respectively (see Table S2† for more data). Their ESI-MS patterns in negative-ion mode reveal the following fragments: in 4b, a series of peaks at m/z 1487, 1417, 1347, and 1277 are assignable to [FeIII7O5(piv)8−x(tpt-O)(OH)(MeO)x(MeOH)4]− (x = 3–6). The pattern also displays signals for the heptanuclear core plus a dioxane molecule at m/z 1699, 1629, and 1559, which arise from replacement of piv− by MeO− in {[FeIII7O5(piv)7−x(tpt-O)(OH)5(MeO)x(dioxane)(H2O)]+H}− (x = 0–2) anionic fragments. The negative ESI pattern of 5 also exhibit a series of peaks at m/z 1799, 1729, 1659, 1589, and 1519, which corresponds to sequential replacement of piv− by MeO− in [FeIII7O5(piv)10−x(tpt-O)(i-PrO)(MeO)x]− (x = 1–5) fragments. The second most intense peaks could be assigned to [FeIII7O4(piv)14−x(tpt)(MeO)x]− (x = 2–4; m/z 2042, 1972, and 1902) units. For Mn-tpt compound, the most intense fragment [Mn(ib)(tpt)]+ in the positive ESI mass spectrum of 7 were detected centered at m/z 454.
Structural analysis
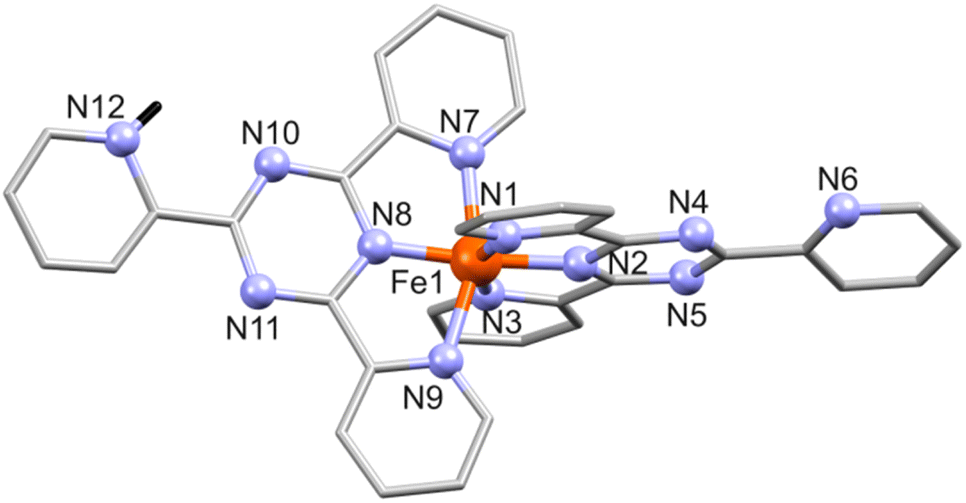 | ||
| Fig. 1 Structure of [Fe(tpt)(Htpt)]4+ in 1 with numbering of N sites. All hydrogen atoms except the protonated NH (H: black stick) in tpt are omitted for clarity. | ||
In 2, the Fe(III) center (BVS: 2.94, Table S6†) is hepta-coordinated and adopts a distorted pentagonal bipyramidal N3O2Cl2 geometry defined by the three N atoms of the fully planar tpt ligand (Fe–N: 2.170(3)–2.293(3) Å), two piv O atoms (Fe–O: 2.110(2) and 2.151(2) Å), and two chloride groups (Fe–Cl: 2.300(1)–2.342(1) Å), see Fig. 2. The Fe–N and Fe–Cl bond distances are identical to those in the similar complex [Fe(tpt)Cl3].31
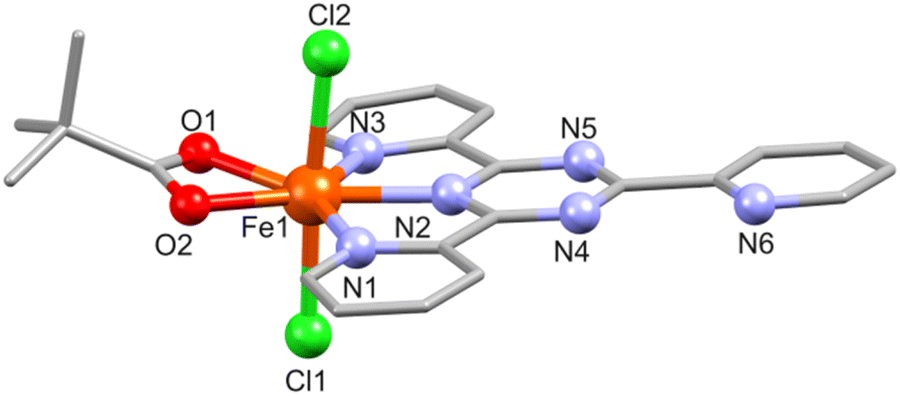 | ||
| Fig. 2 Molecular structure of 2 with a partial atom numbering. All hydrogen atoms are omitted for clarity. | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit of 3 containing one Fe(III) atom (BVS: 2.95), one tpt ligand, two Cl− and one μ-O2− center (see Fig. 3 for the molecular structure of 3 and Table S4† for selected bond distances). The N3Cl2O coordination environment here is formed by three tpt N atoms (mode A; Fe–N: 2.136(9)–2.218(8) Å), two Cl (Fe–Cl: 2.332(3) and 2.398(4) Å, axial) and one μ-oxo atom at the inversion center (Fe–O: 1.805(2) Å). The uncoordinated pyridyl group (containing N6) is rotated by 6.34° and one coordinated pyridyl ring (N5) by 5.02° with respect to the equatorial plane. The linear Fe1–O1–Fe1 (−x, 1 − y, 2 − z) bridge is slightly longer (Fe⋯Fe: 3.610(2) Å) compared to the 3.536(2) and 3.526(3) Å found in comparable μ-O-bridged dinuclear compounds [Fe2O(tpt)2Cl4]·2H2O32 and {Fe2O(tpt)2[N(CN)2]2(NO3)2}.33 In the crystal lattice the complexes stack along the c axis, and adjacent dimers are connected by π⋯π interactions between the uncoordinated pyridyl ring of one dimer and one coordinated pyridyl ring of another dimer (Fig. S3†). The distances between the centroids of the rings range from 3.708 to 4.066 Å. Within the dimeric structure, the π⋯π contact between the coordinated pyridyl rings equals 4.060 Å.
. The asymmetric unit of 3 containing one Fe(III) atom (BVS: 2.95), one tpt ligand, two Cl− and one μ-O2− center (see Fig. 3 for the molecular structure of 3 and Table S4† for selected bond distances). The N3Cl2O coordination environment here is formed by three tpt N atoms (mode A; Fe–N: 2.136(9)–2.218(8) Å), two Cl (Fe–Cl: 2.332(3) and 2.398(4) Å, axial) and one μ-oxo atom at the inversion center (Fe–O: 1.805(2) Å). The uncoordinated pyridyl group (containing N6) is rotated by 6.34° and one coordinated pyridyl ring (N5) by 5.02° with respect to the equatorial plane. The linear Fe1–O1–Fe1 (−x, 1 − y, 2 − z) bridge is slightly longer (Fe⋯Fe: 3.610(2) Å) compared to the 3.536(2) and 3.526(3) Å found in comparable μ-O-bridged dinuclear compounds [Fe2O(tpt)2Cl4]·2H2O32 and {Fe2O(tpt)2[N(CN)2]2(NO3)2}.33 In the crystal lattice the complexes stack along the c axis, and adjacent dimers are connected by π⋯π interactions between the uncoordinated pyridyl ring of one dimer and one coordinated pyridyl ring of another dimer (Fig. S3†). The distances between the centroids of the rings range from 3.708 to 4.066 Å. Within the dimeric structure, the π⋯π contact between the coordinated pyridyl rings equals 4.060 Å.
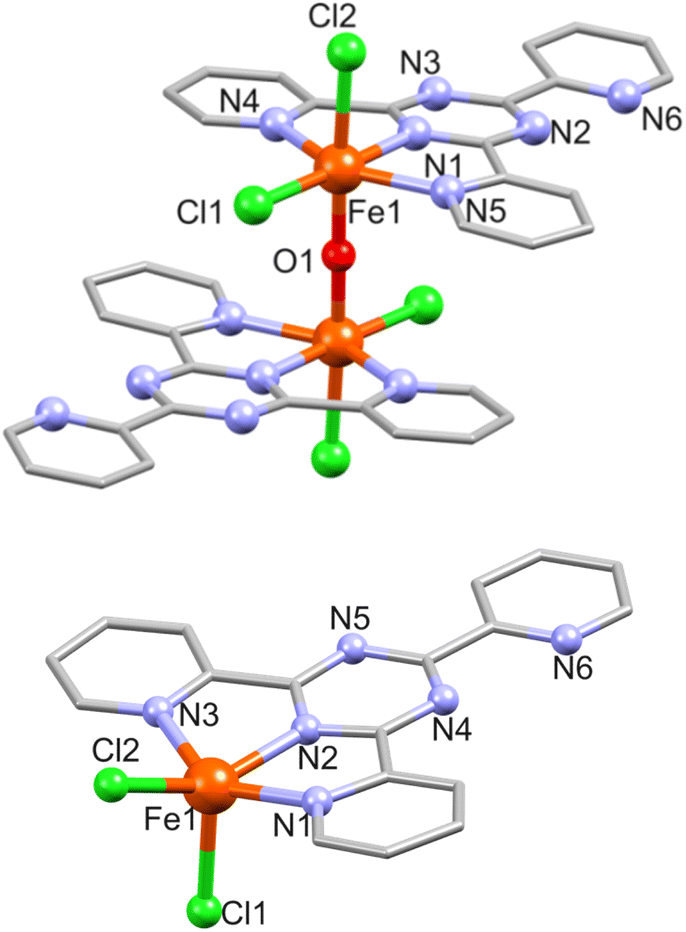 | ||
| Fig. 3 Structure of 3 (top) and 3a (bottom) with a partial atom numbering. Hydrogen atoms are omitted for clarity. Solvent water molecules are omitted for clarity in 3a. | ||
3a consists of a charge–neutral complex [Fe(tpt)Cl2] (Fig. 3) and two solvate water molecules. The coordination environment around the Fe(II) ion (BVS: 2.02) is approx. square pyramidal and comprises three tpt N atoms and two Cl− ions (Fe–N: 2.102(4)–2.220(4), Fe–Cl: 2.2930(2)/2.3278(14) Å). The two coordinated pyridine rings are almost coplanar with the central triazine and the largest deviation is 0.369 and 0.466 Å for the rings containing N1 and N3, respectively, while the uncoordinated pyridine ring is twisted by 5.1° with respect to the triazine ring.
, respectively. The structure of [Fe7O4(piv)12(tpt-O)] in 4a and 4b is shown in Fig. 4, S4 and S5.†4a and 4b comprise a very similar [FeIII7O4(piv)12(tpt-O)] core and differ in their crystal solvent. Thus, we here only discuss in detail compound 4a. Its molecular structure consists of seven Fe(III) atoms (BVS: 2.99–3.13) interlinked by three μ3-O and one μ2-O site, forming a [Fe7(μ3-O)3(μ2-O)] core with ten bridging pivalates as shown in Fig. 4. The core shows two subunits, a tetranuclear {Fe4(μ3-O)2} unit defined by Fe1, Fe2, Fe3 and Fe4 aggregated with a trinuclear {Fe3(μ3-O)} unit (Fe5, Fe6 and Fe7) via a μ2-O bridge (O4) and a bridging pivalate. Additionally, Fe atoms are bridged by one tpt ligand. Here, the tpt ligand acts as bidentate and monodentate ligand coordinating to Fe2 and Fe7, and Fe4, respectively. A μ-O bridge (O5) connecting the tpt triazine ring with Fe4 and Fe5 (Fig. 4, S4 and S5†) causes a distorted-tetragonal geometry around the carbon atom of the triazine ring with bond angles showing slight deviation (94.8(2)–115.8(4)°). This unusual behavior of tpt has already been observed when the triazine ring of tpt was hydroxylated15–19 or methoxylated21 as mentioned above, but for the first time, this O atom is coordinated to metal atoms.
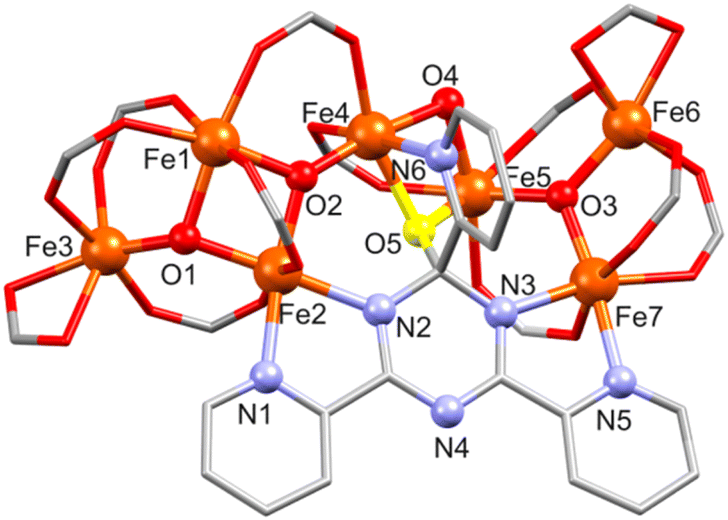 | ||
| Fig. 4 Structure of [Fe7O4(piv)12(tpt-O)] in 4a and 4b with a partial atom numbering. The oxygen atom (O5) of the triazine ring of tpt is shown in yellow. CMe3 groups and H atoms are omitted for clarity. | ||
Within the tpt structure, the saturated carbon atom is not in plane with the other five atoms of the ring (0.195 Å above the plane of the other five atoms). The plane of the N6-pyridyl ring substituted to this carbon atom is almost perpendicular to the plane of the triazine ring (83.83°), whereas the other two pyridyl rings are rotated against the triazine plane by 4.23° (N1) and 9.76° (N5). Two pivalates act as chelating ligands and complete the coordination sphere around Fe3 and Fe6.
All Fe atoms reside in distorted octahedral environments: O6 for Fe1, Fe3, Fe5 and Fe6, N2O4 for Fe2 and Fe7, and NO5 for Fe4 (Table S5†). For Fe1, the FeO6 unit contains two μ3-O2− ions and four O sites of different bridging carboxylates; for Fe3/Fe6 one μ3-O2−, three O atoms from three bridging pivalates and two O sites from a chelating pivalate; finally, for Fe5 one μ3-O2−, two μ-O atoms (one of them originating from the unprecedented oxidation of the C atom of the central triazine core), and three O atoms from bridging carboxylates. For Fe2, the FeN2O4 environment consists of two μ3-O2− ions, two O atoms from different bridging carboxylates, and two tpt N centers; for Fe7: one μ3-O2−, three O atoms from separate bridging carboxylates, and two tpt N atoms. The NO5 environment of Fe4 consists of a μ3-O2−, two μ-O atoms (one of them from tpt), two O atoms from bridging carboxylates, and one tpt N site (Fe–O: 1.843(6)–2.167(6) Å, Fe–N: 2.106(7)–2.175(7) Å).
and its asymmetric unit contains two crystallographically independent heptanuclear [FeIII7O4(piv)11(tpt-O)(i-PrO)(i-PrOH)] clusters and iso-propanol as crystal solvent (Fig. S6†). The {Fe7} core is similar to that of 4a/4b including a trinuclear {Fe3O} and a tetranuclear {Fe4O2} unit bridged by μ-oxo atom, tpt-O and pivalate ligands as shown in Fig. 5. The main difference between 5 and 4a/4b concerns the coordination of two iso-propanol ligands. In 5, an iso-propanolate group bridges two Fe centers of the {Fe3O} unit, and an i-PrOH coordinates to the outer Fe atom of the {Fe4O2} unit substituting one C–O bond between a pivalate and an Fe atom and forming a short intramolecular O–H⋯O hydrogen bond (2.53(1) and 2.56(1) Å) with a mono-coordinated pivalate ligand (Fig. 5). Similar to 4, the Fe coordination environment are distorted octahedral (O6 for Fe1, Fe2, Fe5, Fe6, Fe8, Fe10, Fe12, Fe13; N2O4 for Fe3, Fe7 and Fe9, Fe14; NO5 for Fe4 and Fe11); Fe–O: 1.819(5)–2.127(5) Å, Fe–N: 2.108(6)–2.195(7) Å.
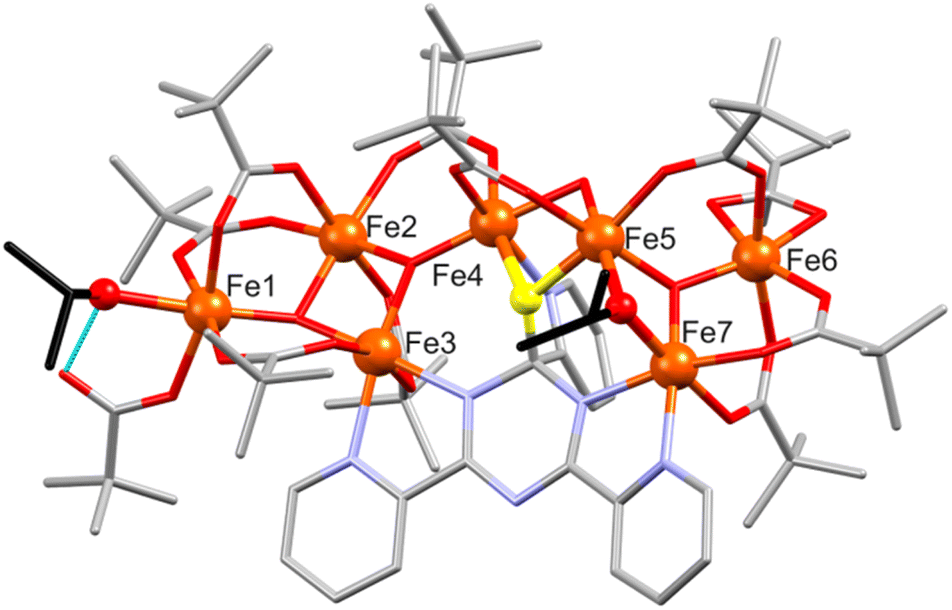 | ||
| Fig. 5 Structure of [Fe7O4(piv)11(tpt-O)(i-PrO)(i-PrOH)] in 5. Color scheme: Fe, orange spheres, O, red, N, blue, C, grey sticks. In i-PrO ligands C atoms are shown in black color and O atoms are shown as red balls; O atom in tpt is highlighted. Hydrogen atoms and crystal solvent molecules are omitted for clarity. | ||
contains a neutral [Mn(ib)(Cl)(tpt)(MeOH)] unit and a solvate MeOH molecule (Fig. 6, bottom). In both complexes tpt serves as a tridentate NNN ligand that forms two five-membered metallacycles (Mn–N: 2.232(2)–2.378(2) Å in 6; 2.295(2)–2.412(2) Å in 7; Table S4†). The Mn–N bond distances are in line with other tpt-containing manganese(II) complexes.34–45 In both 6 and 7, the tpt ligand is slightly twisted, with dihedral angles between the central triazine ring and the attached pyridine rings of 5.93 (N1), 3.35 (N3), and 0.69° (N6) in 6, whereas in 7 these values are 1.99 (N1), 9.14 (N3), and 3.28° (N6). Additionally, Mn coordinates to one NO3− anion and two water molecules in 6, and to isobutyrate and Cl− anions and a methanol group in 7, resulting in pentagonal bipyramidal Mn environments (Table S5†). The NO3− and ib− anions coordinate in a bidentate mode (Mn–O: 2.266(2) and 2.411(2) Å in 6, 2.226(2) and 2.307(2) Å in 7). The Mn–O bond distances to the apical H2O ligands equal 2.140(2) and 2.191(2) Å in 6. In 7 the Mn–Cl bond amounts to 2.486(1), the Mn–O(MeOH) bond to 2.294(2) Å. In the crystal lattice of 6 the cationic complexes are linked through bifurcated hydrogen bonds (2.881(3) and 2.810(3) Å) between the apical aqua ligand (O1W) and a tpt N atom (N6; symmetry code −x + 1/2, y + 1/2, −z + 1/2) and an oxygen atom (O1; symmetry code −x + 1/2, y − 1/2, −z + 1/2) from the coordinated nitrate anion of two adjacent cationic fragments forming chains parallel to the b axis.
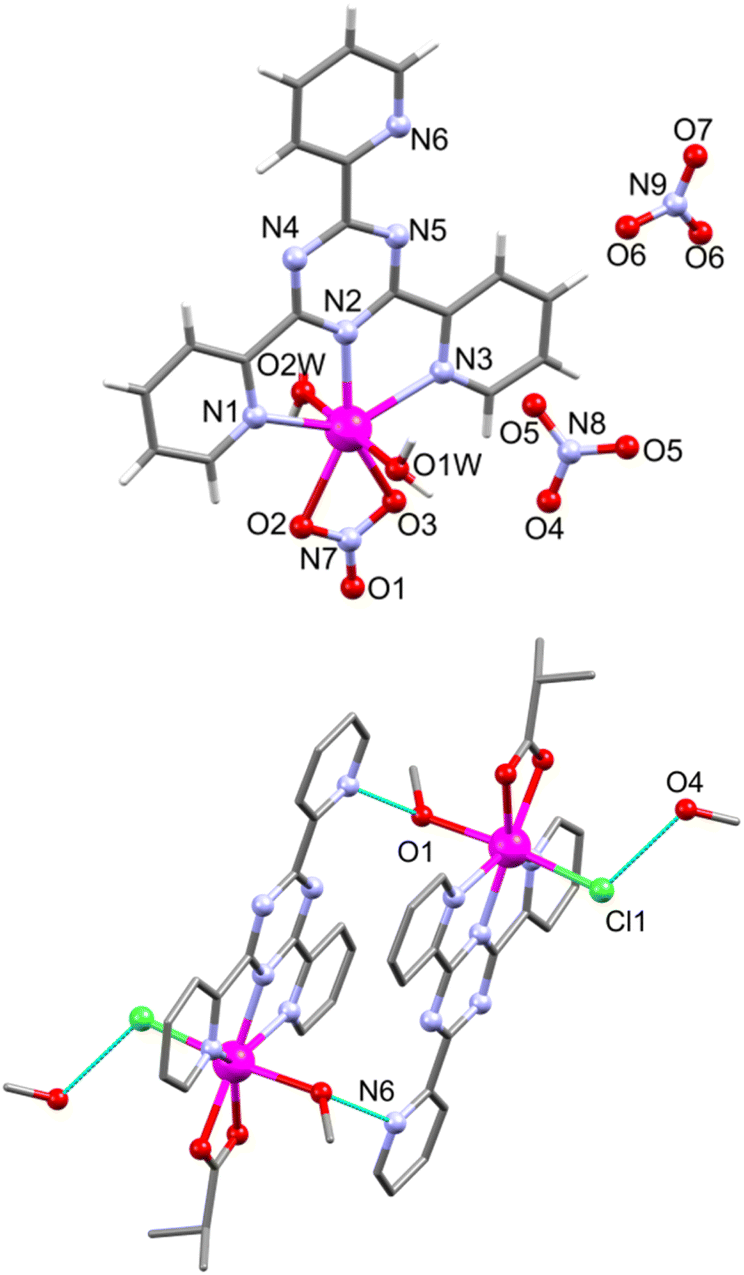 | ||
| Fig. 6 Structure of [Mn(NO3)(tpt)(H2O)2](NO3) (6, top) and a hydrogen-bonded (cyan dotted lines) dimer in [Mn(ib)(Cl)(tpt)(MeOH)] (7, bottom) with a partial atom numbering. | ||
These chains are connected via bifurcated hydrogen O–H⋯O bonds (2.79(5) and 2.951(4) Å) between the second apical aqua ligand (O2W) and two solvate nitrate anions (O5 and O6) resulting in a supramolecular 2D network (Fig. S7†). In 7, the coordinated MeOH molecules form intermolecular O1–H1⋯N6 (−x + 2, −y + 2, −z + 1) hydrogen bonds (2.848(3) Å) with a nitrogen atom of the pyridyl ring of tpt from an adjacent complex, resulting in a hydrogen-bonded dimer (Fig. 6, bottom). This supramolecular dimer exhibits additional weak bonds (3.161(5) Å) between chloride ligands and O4 of solvate MeOH molecules.
Magnetic studies
Magnetic data were evaluated for the title compounds that exhibited unexpected features or comprised interesting spin connectivities.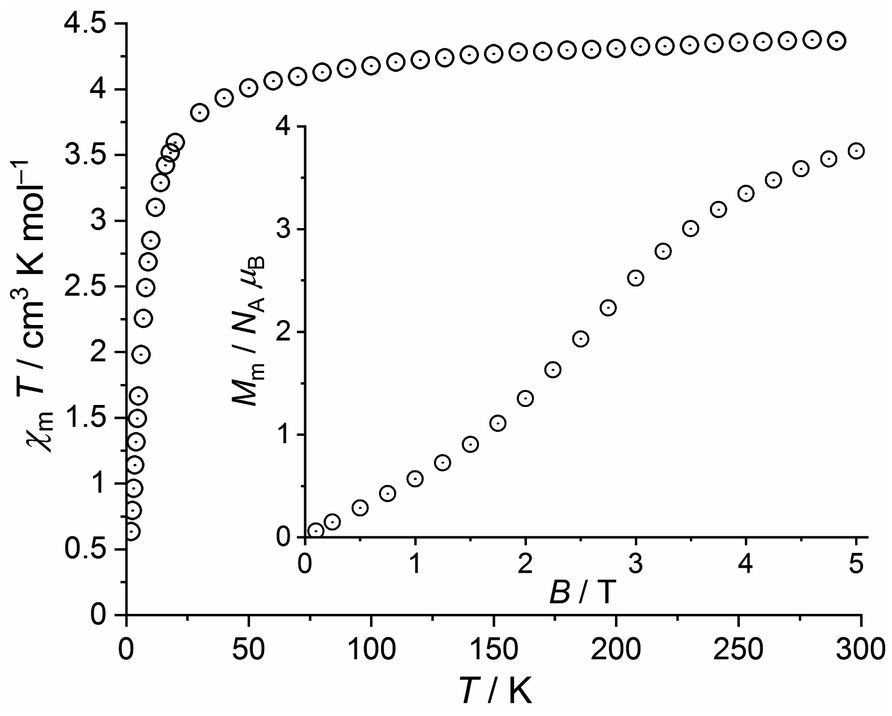 | ||
| Fig. 7 Temperature dependence of χmT at 1.0 T of 2; inset: field dependence of the molar magnetization Mm at 2.0 K. | ||
At 290 K and 1.0 T, the χmT value of 4.37 cm3 K mol−1 is well within the expected46 range 4.1–4.5 cm3 K mol−1 of an isolated high-spin Fe(III) center (μeff = 5.91 μB). Upon cooling, χmT slightly decreases to 4.01 cm3 K mol−1 at 50 K, and subsequently drops off reaching 0.64 cm3 K mol−1 at 2.0 K. At this temperature, the field dependent magnetization curve shows a nearly sigmoidal shape with an inflection point at 2.5 T. At 5.0 T, the curve shows a distinct slope and a value of Mm = 3.8 NA μB, both indicating that the saturation of Mm,sat = 5.0 NA μB is not reached. The observed data are remarkable, since the ground state of the free Fe(III) ion is (almost exclusively) 6S5/2. Thus, the high-spin Fe(III) centers are isotropic and spin-like. Since the Fe(III) centers are widely separated in 2 (closest intermolecular Fe⋯Fe distance: ca. 8.2 Å) and in the absence of alternative exchange pathways such as π⋯π stacking, the χmT vs. T plot should be almost constant above 10 K considering the magnitude of Zeeman effect at 1.0 T. In addition, the magnetization Mm at 2.0 K should be a continuously growing linear or concave function of B, and approach the saturation value Mm,sat at 5.0 T. However, the data below 50 K are in conflict with this simple situation, which has yet to be resolved.
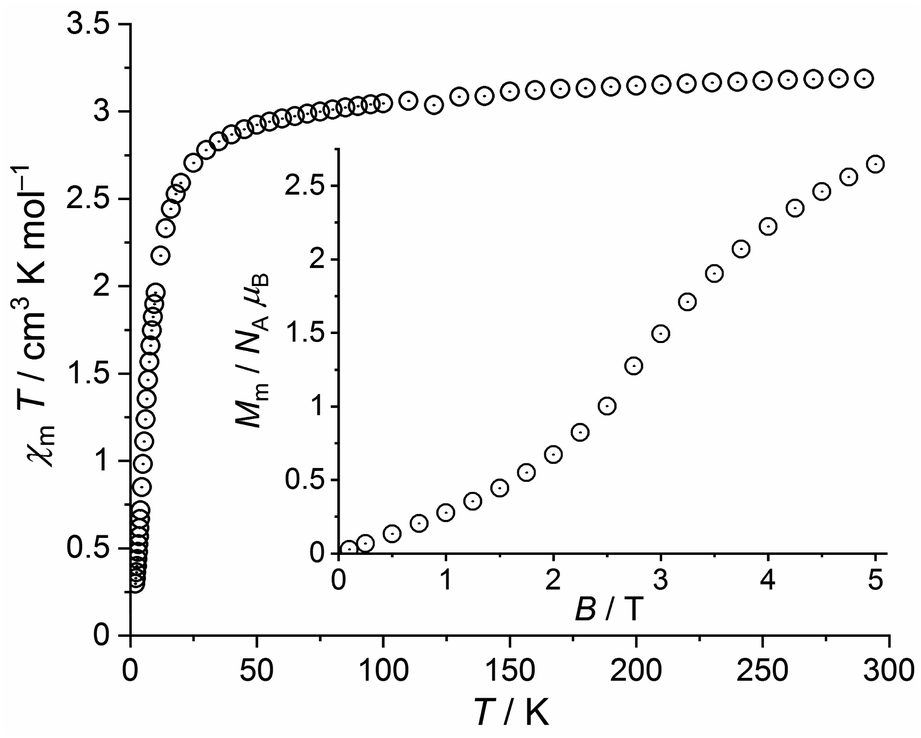 | ||
| Fig. 8 Temperature dependence of χmT at 0.1 T of 3a; inset: field dependence of the molar magnetization Mm at 2.0 K. | ||
This value is slightly below the lower bound of the expected42 range 3.2–4.1 cm3 K mol−1 for an isolated high-spin Fe(II) center. χmT continuously decrease upon cooling the compound and reaches a value of 2.92 cm3 K mol−1 at 50 K. Subsequently, χmT drops to 0.30 cm3 K mol−1 at 2.0 K. The molar magnetization as a function of the applied field is characterized by a sigmoidal shape with an inflection point at 2.9 T and a value of 2.6 NA μB at 5.0 T and 2.0 K. The magnetic properties of 2 and 3a thus show a few generic similarities but differ in detail. Besides the natural differences in the magnitudes of χmT and Mm due to their different 3dn configurations, the χmT values of 3a change from 290 K to 50 K by a larger amount (in relation to 2) due to relevant contributions of the orbital momentum. In addition, the inflection point of the Mm vs. B curve is observed at a slightly larger field (ca. 5% larger).
We therefore use the effective spin-only option of the computational framework CONDON47 considering Heisenberg exchange interactions (using the ‘−2Jex’ notation of the Hamiltonian) and the Zeeman effect to fit the data. We generate starting values of the exchange interaction parameters Jik between the seven isotropic S = 5/2 (geff = 2.00) centers using wxJFinder.48 The least-squares fit yield the solid lines in Fig. 9. Note that we restricted the fitting range of the magnetization to data below 2 T to avoid the regions of potential saturation effects, which are not comprehensively considered in the used effective spin model. The values of the fit parameters are given in Table 1. The relevant exchange interactions between the seven Fe(III) centers are all antiferromagnetic with the exception of the coupling between Fe1 and Fe2. However, the magnetic properties of 4a are barely affected by a larger variation of the corresponding parameter value, which is also represented by its rather large error margin. According to this set of parameters, the molecular ground state is characterized by Stotal = 1/2. Magnetization Mm vs. applied magnetic field B at 2.0 K are shown in Fig. 9 for 4a. At 290 K, χmT amounts to 7.94 cm3 K mol−1, which is well below the range 28.4–31.5 cm3 K mol−1 expected42 for seven isolated high-spin Fe(III) centers. Upon cooling the compound, χmT almost linearly decreases, showing a weak dent at about 9 K, and subsequently drops down to 0.81 cm3 K mol−1 at 2.0 K.
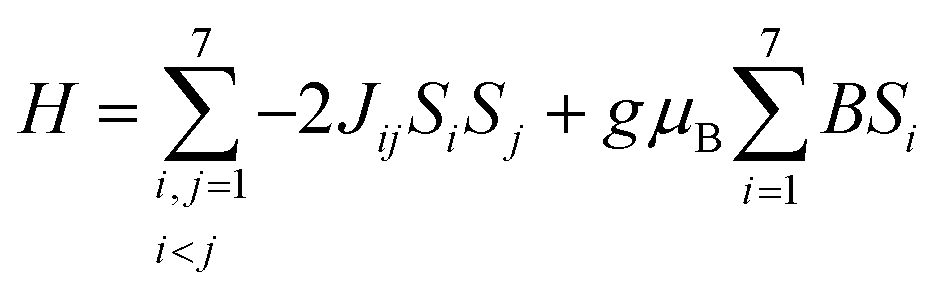
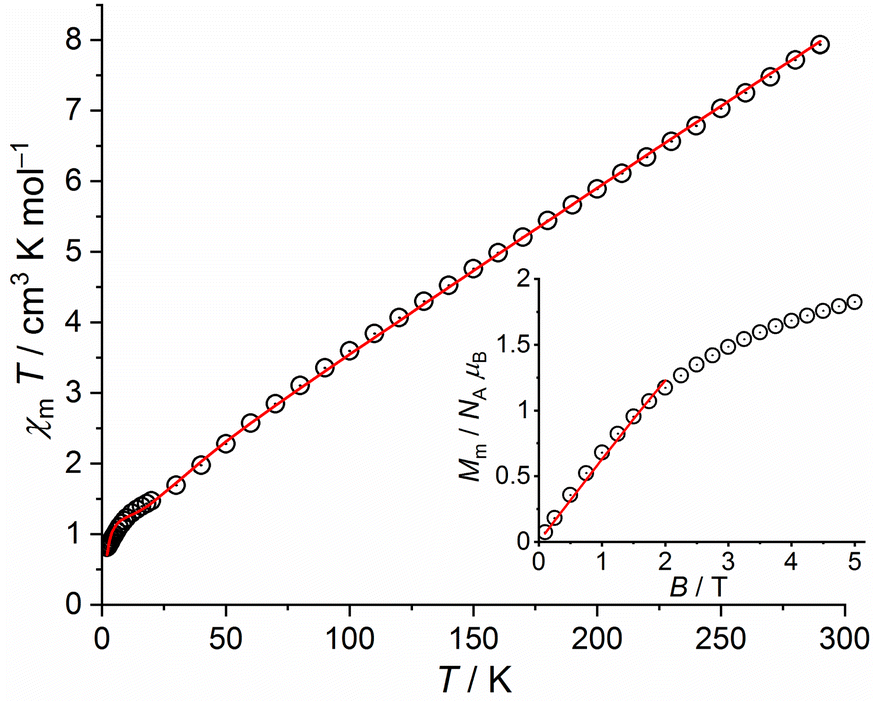 | ||
| Fig. 9 Temperature dependence of χmT at 0.1 T of 4a; inset: field dependence of the molar magnetization Mm at 2.0 K (data: symbols, least-squares fit: solid line). | ||
| 4a | 4b | 5 | ||
|---|---|---|---|---|
| J12 | +7.6 ± 7.4 | +7.9 ± 6.1 | J23 | +4.7 ± 1.3 |
| J13 | −26.6 ± 2.0 | −22.6 ± 2.4 | J12 | −24.0 ± 2.9 |
| J23 | −31.9 ± 9.0 | −30.9 ± 2.1 | J13 | −23.1 ± 2.9 |
| J14 | −30.4 ± 2.2 | −30.1 ± 4.4 | J24 | −18.9 ± 3.1 |
| J24 | −44.4 ± 1.6 | −43.2 ± 3.2 | J34 | −20.1 ± 3.1 |
| J45 | −2.8 ± 0.3 | −5.9 ± 1.3 | J45 | −2.0 ± 1.2 |
| J56 | −20.8 ± 2.3 | −18.1 ± 1.2 | J56 | −39.6 ± 6.5 |
| J57 | −16.7 ± 3.1 | −14.4 ± 1.1 | J57 | −12.5 ± 0.8 |
| J67 | −29.5 ± 2.1 | −25.0 ± 2.9 | J67 | −59.3 ± 4.0 |
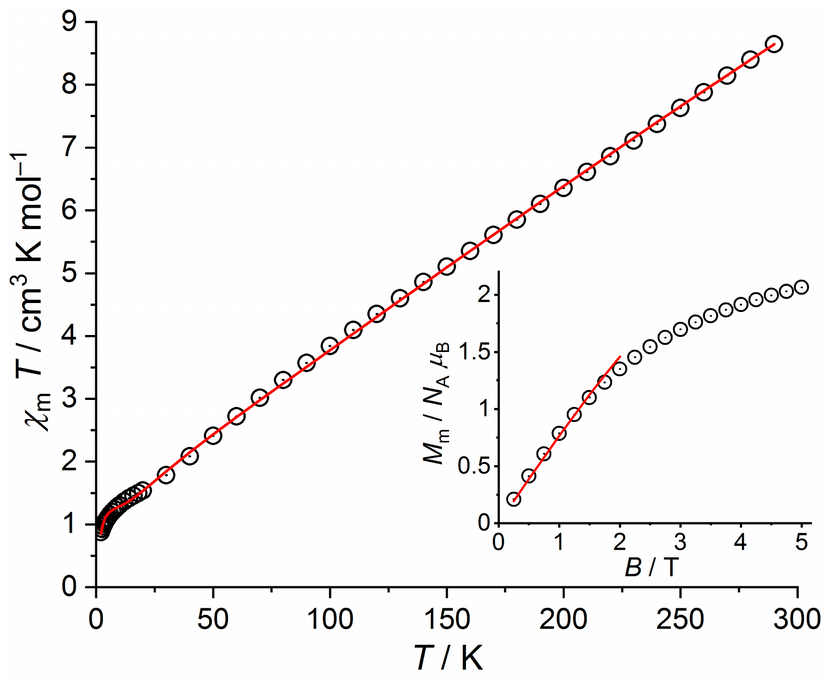 | ||
| Fig. 10 Temperature dependence of χmT at 1.0 T of 4b; inset: field dependence of the molar magnetization Mm at 2.0 K (data: symbols, least-squares fit: solid line). | ||
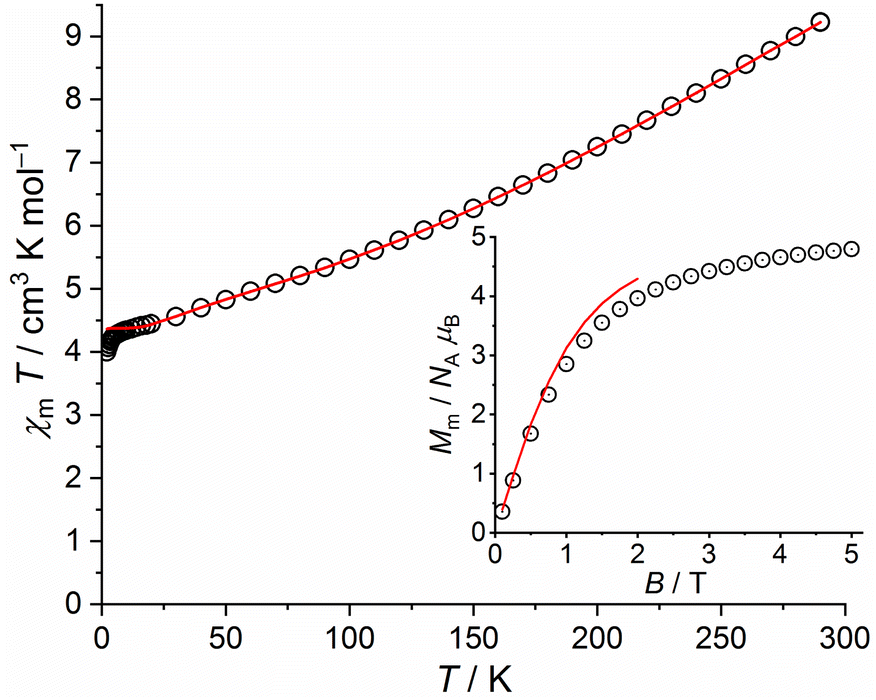 | ||
| Fig. 11 Temperature dependence of χmT at 0.1 T of 5; inset: field dependence of the molar magnetization Mm at 2.0 K (data: symbols, least-squares fit: solid line). | ||
Fitting these data, we find the exchange interaction parameters shown in Table 1, corresponding to the solid lines in Fig. 11. Note that the deviation of data and fit in the χmT vs. T plot at T < 5 K is due to the applied simple model. In comparison to 4a and 4b, the exchange interaction parameters of 5 are stronger within the triangular unit and weaker within the rhombic unit but do not change their signs. The total effective ground state spin is Stotal = 5/2.
The χmT vs. T plot at 0.1 T and the Mm vs. B plot at 2.0 K of 7 are shown in Fig. 12. At 290 K, χmT is 4.37 cm3 K mol−1, i.e. within the expected42 range 4.0–4.7 cm3 K mol−1 for an isolated high-spin Mn(II) center (μeff = 5.91 μB). This value stays almost constant down to 20 K, at which a value of 4.28 cm3 K mol−1 is observed. Upon further cooling, χmT noticeably decreases to reach 4.14 cm3 K mol−1 at 2.0 K. At this temperature, the molar magnetization continuously increases with decreasing slope without hinting at an inflection point in the experimental data. At 5.0 T, the magnetization is significantly affected by saturation effects and reaches 4.8 NA μB.
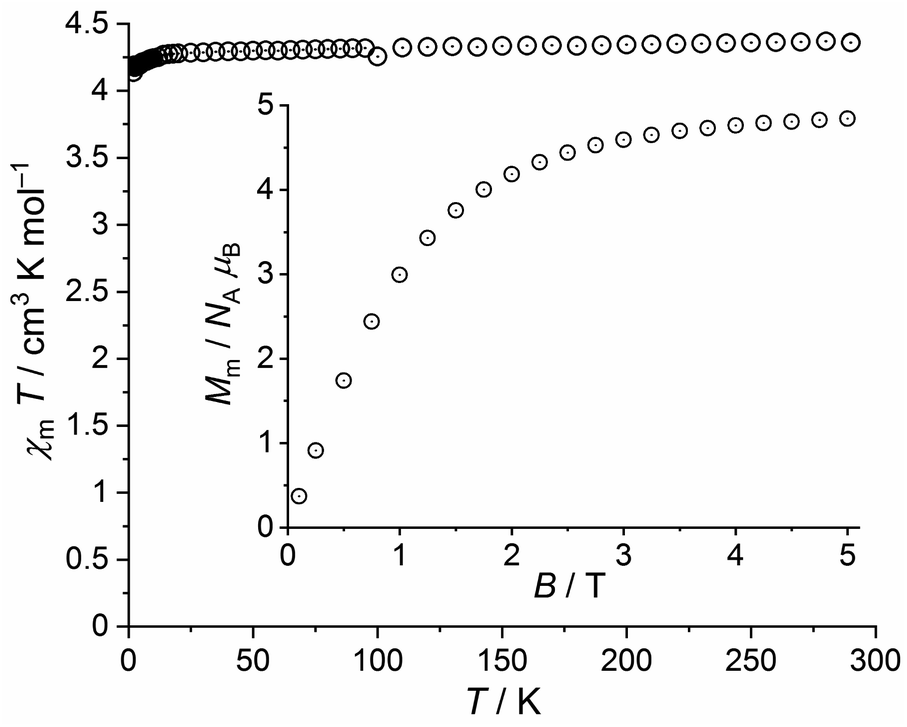 | ||
| Fig. 12 Temperature dependence of χmT at 0.1 T of 7; inset: field dependence of the molar magnetization Mm at 2.0 K. | ||
Therefore, the data for 7 are fully in line with a regular high-spin Mn(II) center at probed temperatures, in apparent contrast to the similar iron(III) tpt complex 2. Note, however, that the decrease of χmT below 20 K is only marginally due to the Zeeman effect, which generates relevant contributions not until below 5 K at 0.1 T (about 0.02 cm3 K mol−1 at 2.0 K). These observed data can be explained by a small zero field splitting due to the ligand field.
Biological studies
In view of the general bioactivity of Mn(II)–tpt compounds, we assessed the potential biostimulator functionality of compounds [Mn(NO3)(tpt)(H2O)2](NO3) (6) and [Mn(ib)(Cl)(tpt)(MeOH)]·MeOH (7). We found that the addition of 6 and 7 to a nutrient medium of fungal strain Fusarium gibbosum CNMN FD 12 in concentrations of 0.0005 to 0.0015% essentially increases fungal proteolytic activity by up to 24.8–355.4% (Table S7†).According to the obtained data, the control presented the maximal activity of neutral (33.01 μ mL−1) and alkaline (21.77 μ mL−1) proteases at the sixth day of growth. The activity of neutral proteases of samples cultivated in the presence of coordination compounds ranged from 21.05 to 63.41 μ mL−1, the highest values being revealed at the fifth and sixth day for compound 6 and 7, respectively. It has to be mentioned that in the case of compound 6, the activity of neutral proteases at the 5th day exceeds the value of maximal control activity achieved on the 6th day of cultivation by 104.4% (concentration 0.0010%) and 71.3% (0.0015%). The highest activity was observed at the complex concentration of 0.0010%. When the compound 7 was introduced into the nutrient medium, the maximal activity of neutral proteases (49.25–63.41 μ mL−1) was revealed at 6th day, similar to the control. The activity increased by 49.2–92.1%, with the maximum established, also, at the concentration of 0.0010%.
The study indicated that both 6 and 7 showed positive effect on alkaline protease activity and accelerated the biosynthesis of enzymes, reducing the cycle of cultivation by 24 h. Thus, the highest values of protease activity were found at the 5th day of growth. Activity ranged from 59.96 to 88.57 μ mL−1, being by 208.3–355.4% and 175.4–306.8% higher than the control from 5th and 6th day, respectively. Similar to neutral proteases, the most effective concentration for enzyme production was 0.0010%. The results regarding the influence of compounds 6 and 7 on proteolytic activity of micromycete Trichoderma koningii CNMN FD 15 are summarized in Table S8.† Maximum of enzymatic activity in control was observed at the 9th day of cultivation, constituting 51.03 and 23.55 μ mL−1 for neutral and alkaline proteases, respectively. A more prolonged cultivation leads to a considerable decreasing of proteolytic activity (13.92 and 8.92 μ mL−1).
A significant difference was found in the effect of coordination compounds 6 and 7: the activity of neutral proteases is preferentially higher than the control, while the activity of alkaline proteases is mostly inhibited. The highest protease production was shown by samples grown on the medium supplemented with compound 6 in concentration of 0.0005% and 7 in concentration of 0.0005 and 0.0010%, respectively. Activity of neutral proteases under these conditions was 94.39 μ mL−1 (6) and, respectively, 71.91 and 79.41 μ mL−1 (7). The stimulating effect constituted 84.9%, 40.9% and 55.6%, respectively. Increasing of coordination compound concentration leads to a decrease in the activity of neutral proteases and to a corresponding decrease of stimulating effect. According to the obtained data, the optimal concentration for both complex compounds is 0.0005%, providing a maximal increase in the activity of neutral proteases.
The use of studied compounds in the process of T. koningii cultivation negatively affects the alkaline proteases, leading to a decrease of activity by 42.4–55.3% compared to the maximum control level revealed at the 9th day of growth. At the same time, in samples cultivated in the presence of coordination compounds during ten days, the activity of alkaline proteases was increased, reaching at a concentration of 0.0015% the maximal level of control achieved at the 9th day. Thus, under these conditions the activity of samples cultivated in the media supplemented with coordination compounds were 23.91 μ mL−1 and 25.52 μ mL−1 for compounds 6 and 7, respectively.
According to the data, the both examined coordination compounds showed a stimulator effects on proteases production by fungal strains Fusarium gibbosum CNMN FD 12 and Trichoderma koningii CNMN FD 15 and can be used to improve the proteolytic activity of micromycetes. The most notable result was the increase of proteolytic activity of F. gibbosum. Thus, the use of complex compounds 6 and 7 in the optimal concentration of 0.0010% during submerged cultivation of the producer for 5 and, respectively, 6 days provide an increase in the level of neutral protease activity by 104.4% (compound 6) and 92.1% (compound 7) and of alkaline proteases by 264.8% (6) and 306.8% (7) compared to the highest value of control. Also, the possibility to reduce the duration of cultivation of the producer by 24 hours and, respectively, to obtain enzymatic preparations with high activity over shorter periods of time and with less energy expenditure has been revealed. Regarding the strain Trichoderma koningii CNMN FD 15, the most promising assay is the use of compound 7 in concentration of 0.0005%, which ensures the greatest enhancement of the biosynthesis of neutral proteases.
Experimental
Materials and methods
All reactions were conducted under aerobic conditions using commercially available chemicals and solvents as received without further purification. [Fe3O(piv)6(H2O)3]piv·2(Hpiv), [Fe6O2(OH)2(piv)12] and Mn(ib)2 has been prepared as described elsewhere.51,52 The precursor [Mn6O2(piv)10(Hpiv)4] has been prepared with a slight modification53 by the reaction of manganese(II) acetate with an excess of pivalic acid followed by adding MeCN solution to the reaction mixture. For solvothermal syntheses the reaction mixtures were placed in a sealed PTEE-lines steel autoclave. The temperature program started with heating from room temperature up to 120 °C within 1 h, temperature was held constant for 4 h and then slowly cooled down to room temperature within 48 h. For elemental analysis the analyser Elementar Vario El was used. The IR spectra were recorded on a PerkinElmer IR spectrometer using KBr discs in the region 4000–400 cm−1 (1–5) or PerkinElmer Spectrum 100 FT-IR in the region 4000–650 cm−1 (6–7). ESI mass spectra were recorded on a ThermoFinnigan LCQ Deca XP plus mass spectrometer with a thf/MeOH/acetic acid gel. The sets of PXRD data (Fig. S8–S11†) were collected at room temperature with the aid of a STOE StadiP diffractometer (Stoe&Cie, Darmstadt, Germany; area detector; Cu Kα radiation; λ = 1.54059 Å). A WiseClean ultrasonic bath operating at 40 kHz with a maximum output of 140 W was used for the ultrasonic irradiation.Synthetic procedures
By using the solvothermal procedure describe above for 3 with [Fe6O2(OH)2(piv)12] (0.161 g, 0.10 mmol) instead of [Fe3O(piv)6(H2O)3](piv)·2(Hpiv) as starting material, compound 3a was obtained. Yield: 0.049 g (17% based on Fe). Elemental analysis calculated for 3a, C18H16Cl2FeN6O2 (475.12 g mol−1): C 45.5%; H 3.39%; N 17.69%. Found: C 45.37%; H 3.3%; N 17.6%.
Method A. [Fe3O(piv)6(H2O)3](piv)·2(Hpiv) (0.2 g, 1.7 mmol) and tpt (0.033 g, 1.05 mmol) were dissolved in MeCN (10 mL) and stirred for 1.5 h at room temperature. After one month the cap of the vial was perforated to accelerate crystallization and eleven days later fine red-brown needle-shaped crystals suitable for X-ray measurements were received. They were filtered off, washed with MeCN and dried in air. Mother liquid was put for further crystallization and red-brown powder was received after two more weeks. Infrared spectra and elemental analysis confirmed the powder to be of the same composition as the crystals received earlier. Yield: 0.087 g (57% based on Fe). Elemental analysis calculated for 4a without MeCN, C78H120N6O29Fe7 (1996.73 g mol−1): C 46.92; H 6.06; N 4.21. Found: C 46.77%; H 5.74; N 4.2%. IR data (KBr pellet, cm−1): 3509 br.w, 2962 m, 2929 sh, 2870 sh, 1576 sh, 1561 s, 1543 vs., 1485 vs., 1462 m, 1424 vs., 1378 m, 1362 m, 1262 w, 1229 m, 1156 w, 1131 w, 1088 w, 1030 w, 907 w, 787 w, 683 w, 605 m, 510 w, 436 m.
Method B. A solution of [Fe6O2(OH)2(piv)12] (0.162 g, 0.1 mmol) and tpt (0.031 g, 0.1 mmol) in a 1
:1 mixture of n-hexane and MeCN (10 mL) was stirred at room temperature for 2.5 h and filtered. The filtrate was stored in a closed vial at room temperature. After three weeks, the brown crystals of 4a suitable for X-ray diffraction analysis were filtered off, washed with acetonitrile and dried in air. Yield: 0.099 g (58% based on Fe).
Method C. A solution of [Fe6O2(OH)2(piv)12] (0.162 g, 0.1 mmol) and tpt (0.0316 g, 0.1 mmol) in a 1
:1 mixture of acetone (5 mL) and MeCN (5 mL) were stirred at room temperature for 2 h and filtered. The filtrate was stored in a closed vial at room temperature. After three weeks, very thin crystals were received from the filtrate, which were recrystallized from acetone. Crystallization occurred three weeks later, crystals were filtered off, washed with acetonitrile and dried in air. Yield: 0.003 g (2% based on Fe).The identity of 4a prepared by methods B and C was established from their IR data, elemental and TG analyses, as well as by a single-crystal X-ray diffraction analysis.
Crystallography
X-ray data sets for 1–5 were collected on a Bruker APEX CCD-diffractometer, for 3a on a STOE STADIVARI diffractometer, and for 6 and 7 on an Oxford Xcalibur diffractometer equipped with a CCD detector using graphite monochromator utilizing MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refine by full-matrix least-squares on weighted F2 values for all reflections using SHELXL suite of programs.54 All non-H atoms in the compounds were refined with anisotropic displacement parameters except in the case of water molecule in 1 and disordered solvent molecules in 5. Some of [FeCl4]− anions in 1 and some pivalate and iso-propanol groups in 4a, 4b, and 5 were found to be disordered over two positions. Therefore, SIMU, DELU, SADI, and ISOR restrains were used to deal with the disordered moieties in the structures and to obtain reasonable geometrical parameters and thermal displacement coefficients. The positions of hydrogen atoms in the structures were located on difference Fourier maps or calculated geometrically and refined isotropically in the “rigid body” model with U = 1.2 Ueq or 1.5 Ueq of corresponding O and C atoms. Some H atoms of solvent molecules (H2O in 1 and hydroxyl hydrogen of i-PrOH molecules in 5) have not been localized. Crystallographic data and structure refinements of 1–7 are summarized in Table S2.† The figures were drawn using the Mercury program. CCDC reference numbers for the structures are CCDC reference numbers for the structures are 2108859 (1), 2108860 (2), 2108861 (3), 2108862 (3a), 2108863 (4a), 2108864 (4b), 2108865 (5), 210956 (6), and 2109057 (7).†Biological studies
The biological properties of coordinative compounds were evaluated on the base of their effect on the proteolytic activity of micromycetes Fusarium gibbosum CNMN FD 12 and Trichoderma koningii CNMN FD 15 in the dynamic of submerged cultivation. The producers’ strains were grown in Erlenmeyer flasks with a capacity of 0.75 L, which contained 0.2 L nutrient medium with the following composition, %: corn flour, 2.0; soy flour, 1.0; CaCO3, 0.2; (NH4)2SO4, 0.1; compounds 6 or 7, 0.0005–0.0015; the rest was a tap water. Initial pH of the medium was 6.25. The nutrient medium was inoculated with a suspension of spores and mycelium in an amount of 5% v/v, which were obtained by washing with sterile distilled water of the culture grown on sloping surfaces of malt-agar for 12–14 days. Cultivation was performed at 28 °C under continuous stirring (180–200 rpm) for 5–6 days in the case of Fusarium gibbosum CNMN FD 12 strain and for 9–10 days for Trichoderma koningii Oudemans CNMN FD 15. Coordination compounds were introduced into a sterile nutrient medium, simultaneously with inoculum, in the concentration of 0.0005, 0.0010 and 0.0015%. A sample cultivated on nutrient medium without coordination compounds served as a control. The activity of proteolytic enzymes was essayed by the Anson method55 based on the degree of sodium caseinate hydrolysis.Conclusions
In conclusion, a series of Fe(III)/Fe(II)/Mn(II)–tpt compounds of different nuclearity ranging from mononuclear up to heptanuclear form in the reaction of Fe/Mn pivalates or isobutyrates and the tpt ligand. Solvothermal reactions lead to the precipitation of mono- or dinuclear Fe-tpt compounds (1–3), whereas stirring of precursor solutions at room temperature afforded the heptanuclear Fe-tpt compounds 4 and 5. 4 and 5 represent condensed coordination cluster compounds with unprecedented structural features, in which an unexpected metal-induced oxidation at the carbon atom of the triazine ring of the bridging tpt ligand and the coordination of the thus-resulting O atom to metal ion were observed. Previously, a complexation-induced hydrolysis of tpt was only observed in Ru, Rh, Os, Pd, Pt or Cu complexes, in which the formed O center did however not engage in coordination. Thus, our research, to some extent, provides a new path for the design and synthesis of specific Fe(III)-(tpt-O) cluster materials under soft conditions. The heptanuclear Fe(III) compounds (4a, 4b, 5), the iron-oxo core structures result in the anticipated dominant antiferromagnetic exchange coupling. Finally, the Mn-tpt compounds studied in this work exhibit promising catalytic properties in the production of enzymes by microorganisms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work in part was supported by State Programs of the National Agency for Research and Development of R. Moldova ANCD 20.80009.5007.15 and ANCD 20.80009.5007.28. S.G.B. is grateful to the Alexander von Humboldt Foundation for Georg Förster Research Award.Notes and references
- Ch. R. K. Glasson, L. F. Lindoy and G. V. Meehana, Coord. Chem. Rev., 2008, 252, 940–963 CrossRef CAS.
- M.-H. Yu, X.-T. Liu, B. Space, Z. Chang and X.-H. Bu, Coord. Chem. Rev., 2021, 427, 213518 CrossRef CAS.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- S. U. Lee, R. V. Belosludov, H. Mizuseki and Y. Kawazoe, Small, 2008, 7, 962–969 CrossRef PubMed.
- J. Wei, N. Teshima and T. Sakai, Anal. Sci., 2008, 24, 371–376 CrossRef CAS PubMed.
- (a) G. S. R. Krishnamurti and P. M. Huang, Talanta, 1990, 37, 745–748 CrossRef CAS PubMed; (b) B. G. Stephens, H. L. Felkel Jr and W. M. Spinelli, Anal. Chem., 1974, 46, 692–696 CrossRef CAS; (c) A. A. Schilt and P. J. Taylor, Anal. Chem., 1970, 42, 220–224 CrossRef CAS PubMed.
- (a) Y. Sasaki, Anal. Chim. Acta, 1978, 98, 335–342 CrossRef CAS; (b) W. A. Embry and G. H. Ayres, Anal. Chem., 1968, 40, 1499–1501 CrossRef CAS.
- M. J. Janmohamed and G. H. Ayres, Anal. Chem., 1972, 44, 2263–2268 CrossRef CAS.
- Sh. G. Azahari, M. H. Abdel-Rhman and M. M. Mostafa, Spectrochim. Acta A, 2014, 132, 165–173 CrossRef CAS PubMed.
- K. P. P. R. Mohan Reddy, P. Govind Chowdary, V. Krishna Reddy and P. Raveendra Reddy, Ann. Chim., 2007, 97, 1207–1215 CrossRef.
- C. Musikas, X. Vitart, J. Y. Pasquiou and R. Hoel, in Chemical Separations, ed. C. J. King and J. D. Navratil, Litarvan Literature, Denver, 1986, Vol. 1 Search PubMed.
- S. Singh, M. K. Mandal, A. Masih, A. Saha, S. K. Ghosh, H. R. Bhat and U. P. Singh, Arch. Pharm., 2021, 354, 2000363 CrossRef CAS PubMed.
- B. Therrien, J. Organomet. Chem., 2011, 696, 637–651 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., 2002, B58, 380–388 CAS.
- N. C. Tomas, B. L. Foley and A. L. Rheingold, Inorg. Chem., 1988, 27, 3426–3429 CrossRef.
- A. Cantarero, J. M. Amigo, J. Faus, M. Julve and T. Debaerdemaeker, J. Chem. Soc., Dalton Trans., 1988, 2033–2039 RSC.
- J. Faus, M. Julve, J. M. Amigo and T. Debaerdemaeker, J. Chem. Soc., Dalton Trans., 1989, 1681–1687 RSC.
- P. Paul, B. Tyagi, A. K. Bilakhiya, M. M. Bhadbhade, E. Suresh and G. Ramachandraiah, Inorg. Chem., 1998, 37, 5733–5742 CrossRef CAS.
- P. Paul, B. Tyagi, A. K. Bilakhiya, P. Dastidar and E. Suresh, Inorg. Chem., 2000, 39, 14–22 CrossRef CAS PubMed.
- M. Schwable, M. Karnahl, H. Görls, D. Chartrand, F. laverdiere, G. S. Hanan, S. Tschierlei, B. Dietzek, M. Schmitt, J. Popp, J. G. Vos and S. Rau, Dalton Trans., 2009, 4012–4022 Search PubMed.
- X. Chen, F. J. Femia, J. W. Babich and J. A. Zubieta, Inorg. Chem., 2001, 40, 2769–2777 CrossRef CAS PubMed.
- T. Hajra, J. K. Bera and V. Chandrasekhar, Inorg. Chim. Acta, 2011, 372, 53–61 CrossRef CAS.
- S. Sharma, M. Chandra and D. Sh. Pandey, Eur. J. Inorg. Chem., 2004, 3555–3563 CrossRef CAS.
- M. M. Najafpour, M. Hołynska, M. Amini, S. H. Kazemi, T. Lis and M. Bagherzadeh, Polyhedron, 2010, 29, 2837–2843 CrossRef CAS.
- S. Rubino, P. Portanova, A. Girasolo, G. Calvaruso, S. Orecchio and G. C. Stocco, Eur. J. Med. Chem., 2009, 44, 1041–1048 CrossRef CAS PubMed.
- E. Salomon, N. Keren, M. Kanteev and N. Adir, Manganese in Biological Systems: Transport and Function, in PATAI’s Chemistry of Functional Groups; The Chemistry of Organomanganese Compounds, ed. Z. Rappoport, John Wiley & Sons, 2011 Search PubMed.
- S. Uddin, Md. S. Hossain, Md. A. Latif, Md. R. Karim, R. K. Mohapatra and Md. Kudrat-E-Zahan, Am. J. Heterocycl. Chem., 2019, 5, 27–36 CrossRef.
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC.
- C. P. Raptopoulou, Y. Sanakis and A. K. Boudalis, Eur. J. Inorg. Chem., 2008, 5632–5641 CrossRef CAS.
- M. M. Najafpour, D. M. Boghaei and V. McKee, Polyhedron, 2010, 29, 3246–3250 CrossRef CAS.
- S. A. Cotton, V. Franckevicius and J. Fawcett, Polyhedron, 2002, 20, 2055–2061 CrossRef.
- R. Zibaseresht, W. T. Robinson and R. M. Hartshorn, Acta Crystallogr., 2006, E62, m1150–m1153 Search PubMed.
- G. P. Majumder, M. Salah El Fallah, J. Ribas and S. Mitra, Inorg. Chim. Acta, 2007, 360, 2307–2312 CrossRef.
- K. Ha, Acta Crystallogr, 2010, E66, m262 Search PubMed.
- K. Ha, Acta Crystallogr, 2011, E67, m1238–m1239 Search PubMed.
- K. Ha, Z. Kristallogr., 2011, 226, 57–58 CAS.
- K. Ha, Z. Kristallogr., 2011, 226, 483–485 CrossRef CAS.
- G.-Y. Hsu, P. Misra, S.-C. Cheng, H.-H. Wie and S. Mohanta, Polyhedron, 2006, 25, 3393–3398 CrossRef CAS.
- H. Zhao, M. Shatruk, A. V. Prosvirin and K. R. Dunbar, Chem. – Eur. J., 2007, 13, 6573–6589 CrossRef CAS PubMed.
- X.-P. Sun, W. Gu and X. Liu, Acta Crystallogr., 2007, E63, m1027–m1028 Search PubMed.
- K. M. Lo and S. W. Ng, Acta Crystallogr., 2009, E65, m591–m592 Search PubMed.
- P. Tyagi and U. P. Singh, J. Coord. Chem., 2009, 62, 1613–1622 CrossRef CAS.
- M. M. Najafpour, D. M. Boghaei and V. McKee, Polyhedron, 2010, 29, 3246–3250 CrossRef CAS.
- A. Grirrane, A. Pastor, A. Galindo, E. Alvarez, C. Mealli, A. Ienco, A. Orlandini, P. Rosa, A. Caneschi, A.-L. Barra and J. F. Sanz, Chem. – Eur. J., 2011, 17, 10600–10617 CrossRef CAS PubMed.
- M. M. Najafpour, M. Holynska, A. N. Shamkhali, M. Amini, S. H. Kazemi, S. Zaynalpoor, R. Mohamadi, M. Bagherzadeh and T. Lis, Polyhedron, 2012, 34, 202–209 CrossRef CAS.
- H. Lueken, Magnetochemie, Teubner Verlag, Stuttgart, 1999 Search PubMed.
- M. Speldrich, J. van Leusen and P. Kögerler, J. Comput. Chem., 2018, 39, 2133–2145 CrossRef CAS PubMed.
- S. G. Baca, T. Secker, A. Mikosch, M. Speldrich, J. van Leusen, A. Ellern and P. Kögerler, Inorg. Chem., 2013, 52, 4154–4156 CrossRef CAS PubMed.
- J. S. Griffith, The Theory of Transition-Metal Ions, Cambridge University Press, Cambridge, 1980 Search PubMed.
- E. König and S. Kremer, Magnetism Diagrams for Transition Metal Ions, Plenum Press, New York, 1979 Search PubMed.
- N. V. Gerbeleu, A. S. Batsanov, G. A. Timko, Iu. T. Struchkov, K. M. Indrichan and G. A. Popovich, Dokl. Akad. Nauk SSSR, 1987, 293, 364–367 CAS.
- S. G. Baca, Yu. Sevryugina, R. Clerac, Iu. Malaestean, N. Gerbeleu and M. A. Petrukhina, Inorg. Chem. Commun., 2005, 8, 474–478 CrossRef CAS.
- S. G. Baca, I. L. Malaestean, T. D. Keene, H. Adams, M. D. Ward, J. Hauser, A. Neels and S. Decurtins, Inorg. Chem., 2008, 47, 11108–11119 CrossRef CAS PubMed.
- M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- M. Gracheva, Yu. P. Grachev, M. Mosichev and E. G. Borisenko, Laboratornyi praktikum po tekhnologii fermentnykh preparatov (Laboratory Works on the Technology of Enzymatic Preparations), Legk. i pishch. promt’, Moscow, 1982, p. 240 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2108859–2108865, 210956 and 2109057. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra04868j |
| This journal is © The Royal Society of Chemistry 2022 |