
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel chiral spiro-β-lactams from nitrile oxides and 6-(Z)-(benzoylmethylene)penicillanate: batch, microwave-induced and continuous flow methodologies†
Américo J. S. Alves ,
João A. D. Silvestre and
Teresa M. V. D. Pinho e Melo*
,
João A. D. Silvestre and
Teresa M. V. D. Pinho e Melo*
University of Coimbra, Coimbra Chemistry Centre-Institute of Molecular Sciences and Department of Chemistry, 3004-535 Coimbra, Portugal. E-mail: tmelo@ci.uc.pt
First published on 28th October 2022
Abstract
The first examples of the diastereoselective 1,3-dipolar cycloaddition reaction of nitrile oxides and 6-alkylidene penicillanates leading to chiral spiroisoxazoline-penicillanates are reported. The synthesis of this new type of penicillanate involved the selective generation of two consecutive stereogenic centers, including a quaternary chiral center. Furthermore, the present work also describes the outcomes of these 1,3-dipolar cycloaddition reactions under three distinct reaction conditions (conventional heating, microwave irradiation and continuous flow). The successful use of the continuous flow technique as well as the proper selection of the reaction media allowed the development of a sustainable route to chiral spiroisoxazoline-penicillanates.
Introduction
Since the discovery of penicillin, the β-lactam ring has been considered a very important moiety in synthetic and medicinal chemistry due to its highly synthetic versality and biological properties.1 Spiro-β-lactams are a subclass of β-lactams characterized by having an additional ring with the fusion of the two rings in one shared sp3 carbon, the quaternary spiro carbon. Due to the tetrahedral nature of the spiro carbon, these molecules are endowed with a well-defined three-dimensional disposition, which allows an efficient interaction with a given molecular target. Therefore, these molecules have a wide spectrum of biological activities.2The above-mentioned properties of β-lactams and spiro-β-lactams have attracted the interest of organic and medicinal chemists. Thus, many researchers have turned their focus to the development of alternative synthetic approaches aiming at the synthesis of novel spiro-β-lactam derivatives.3 The synthesis of chiral spirocyclic β-lactams derived from 6-aminopenicillanic acid (6-APA) has been explored by our research group, leading to a library of chiral spiro-β-lactams with significant structural diversity. Among the synthesized spiro-β-lactams, several were identified with potent activity against both HIV and Plasmodium, the causative agents of AIDS and malaria, respectively (Fig. 1).4,5 Infections by HIV and Plasmodium remain serious public health problems, preventing economic and social progress in developing countries. The identification of spiro-β-lactams with potent activity against both infectious agents is particularly relevant due to considerable geographic overlap between HIV and Plasmodium, mainly in sub-Saharan Africa where co-infection is common, and the concerns regarding possible drug–drug interactions in patients receiving malaria and HIV treatment concomitantly.
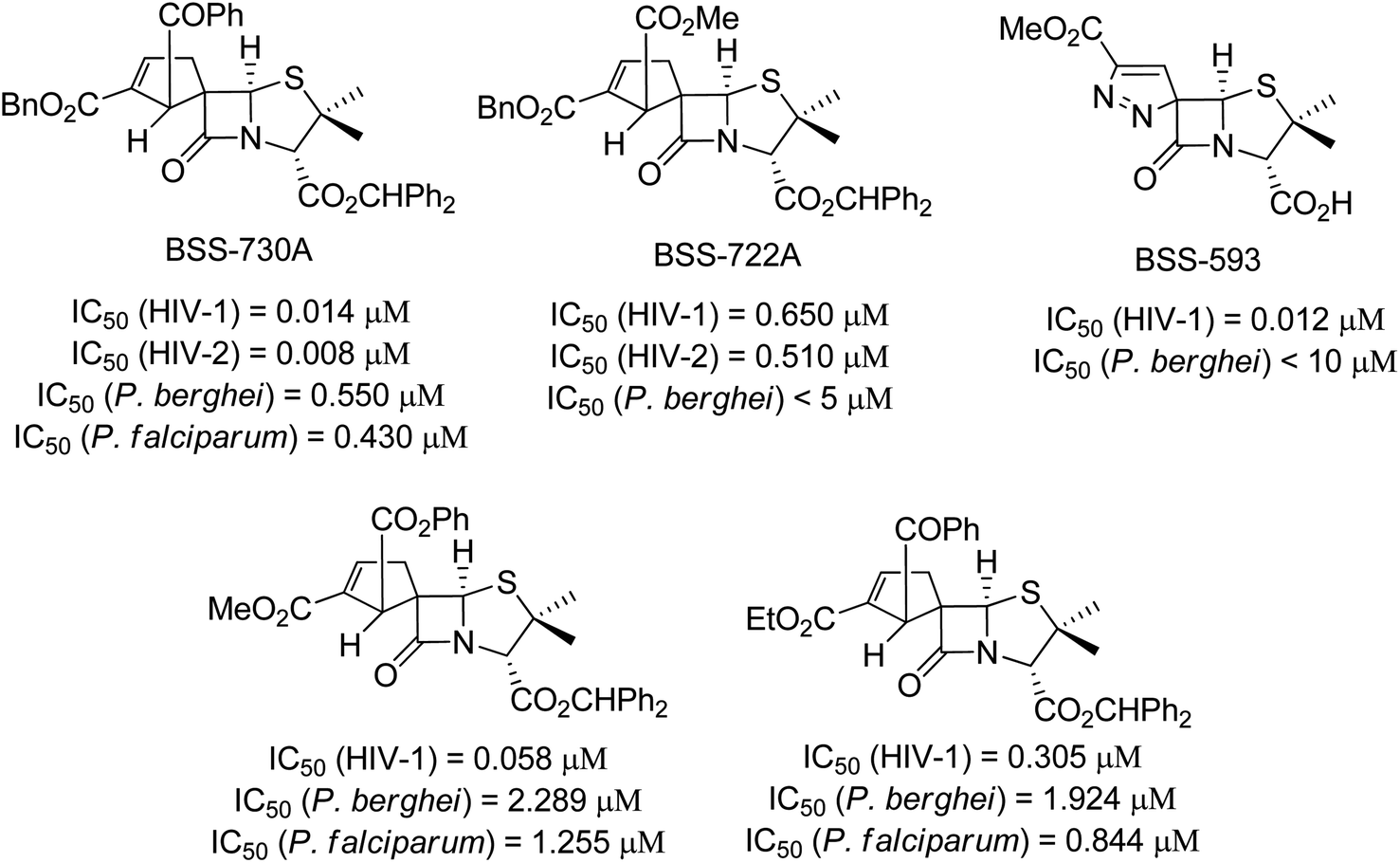 | ||
| Fig. 1 Spiro-β-lactams with potent antimicrobial activity. | ||
The spiro-β-lactams synthesized by our research group were obtained by exploring 1,3-dipolar cycloaddition and [3 + 2] annulation reactions of 6-diazopenicillanates and 6-alkylidene penicillanates allowing the synthesis of novel chiral spiropenicillanates containing carbo- or heterocyclic rings, spiro-fused to the penicillin core.4,5 Using 6-alkylidenepenicillanates as 2π-component, the phosphane-catalyzed [3 + 2] annulation with allenoates or 2-butynoates, led to chiral spirocyclopentene-β-lactams,4b,5b,c whereas 1,3-dipolar cycloaddition reactions with diazo compounds and nitrones afforded spiropyrazoline-penicillanates and spiroisoxazolidine-penicillanates, respectively (Scheme 1).5d,e
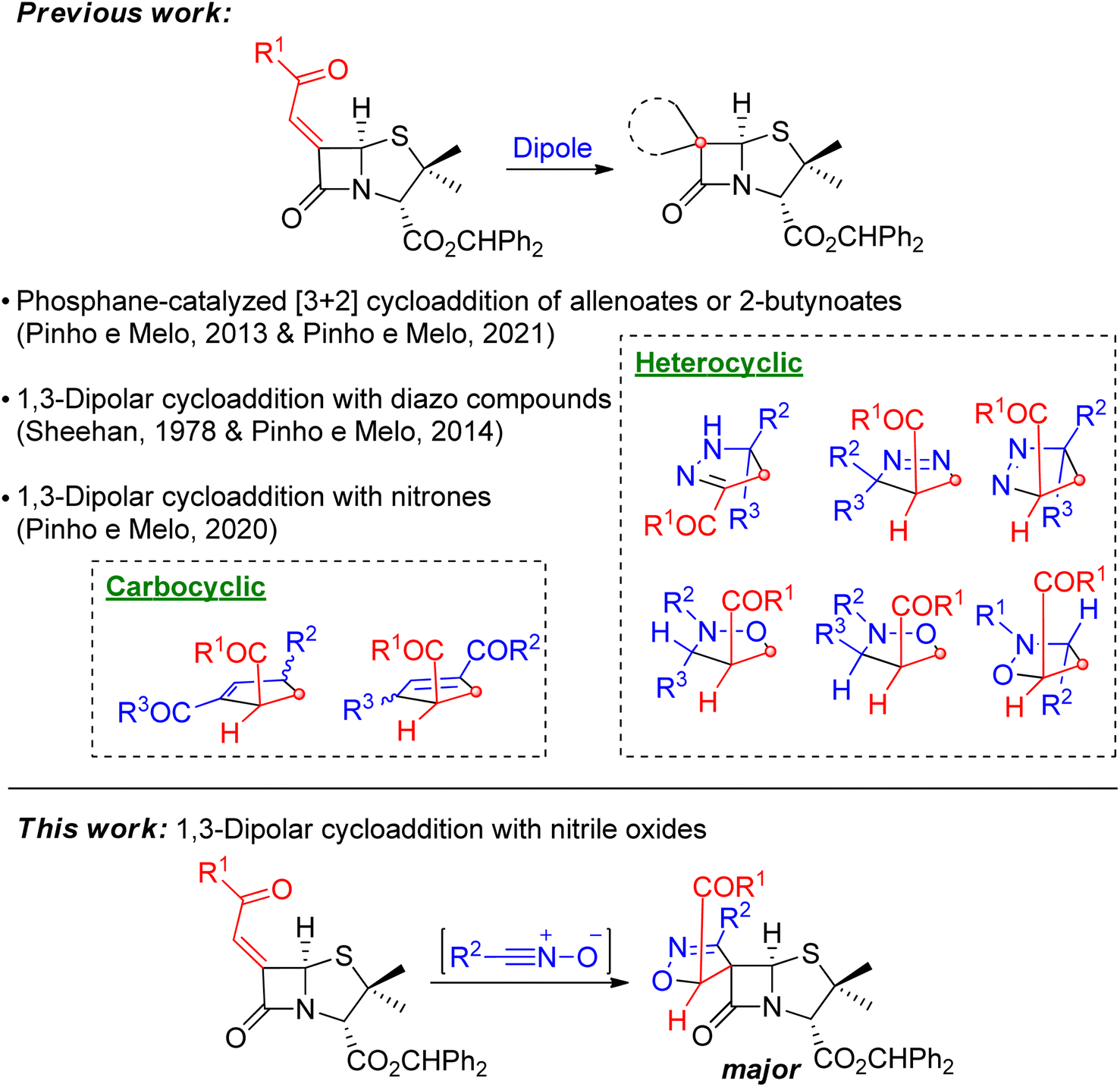 | ||
| Scheme 1 Synthesis of different classes of chiral spiro-β-lactams from 6-alkylidene penicillanates. | ||
The [3 + 2] nitrile oxide-alkene cycloaddition is an important and versatile synthetic route for the construction of isoxazoline rings.6 An important subclass of isoxazolines are the spiroisoxazolines, whose structural motif is present in natural occurring7 and synthetic compounds with biological activity, such as anticancer,7a,c,8 antibacterial,7b antimalarial,9 or antiviral activity.10
Despite the known reports on the construction of the spiroisoxazoline core, to the best of our knowledge, few of them were dedicated to alkylidene-β-lactams. In late 80 s, Corbett and co-workers reported the synthesis of spirocarbapenems, from the 1,3-dipolar cycloaddition reaction of the corresponding alkylidene-carbapenem with a nitrile oxide.11 Later, Otto and Liebscher's teams have explored the synthesis of spiro-β-lactams via 1,3-dipolar cycloaddition of nitrile oxides to α-alkylidene-β-monolactams.12 Nevertheless, none of these studies were focused on the reactivity of alkylidenepenicillanates.
We envisaged that extending the library of spiropenicillanates by adding an isoxazoline ring, spiro-fused to the penicillin core's lactam ring, could lead to interesting new scaffolds with potential biological activity. In this context, the present work focused on the 1,3-dipolar cycloaddition reaction of a 6-alkylidenepenicillanate with a diversity of nitrile oxides as an approach to the first reported examples of chiral spiroisoxazoline-penicillanates, using batch, microwave irradiation and continuous flow techniques.
Results and discussion
1,3-Dipolar cycloaddition reactions via batch and microwave-induced methodologies
To explore the 1,3-dipolar cycloaddition of 6-(Z)-(benzoylmethylene)penicillanate to nitrile oxides, a library of nitrile oxide precursors, hydroximoyl chlorides 3, was synthesized (Scheme 2). Initially, the reaction of hydroxylamine hydrochloride with the corresponding aromatic aldehydes 1a–g was carried out in a mixture of ethanol/water with Na2SO4 as dehydrating agent under ultrasound irradiation. The condensation reaction led to aldoxime derivatives 2a–g isolated with a simple workup procedure and used without further purification. Next, treatment of aldoximes 2 with N-chlorosuccinimide (NCS) in the presence of a catalytic amount of pyridine at room temperature afforded aromatic hydroximoyl chlorides 3a–g in good to excellent yields, ranging from 52% to 98%. This synthetic approach also allowed the synthesis of aliphatic hydroximoyl chloride 3h which was obtained in moderate yield (48%).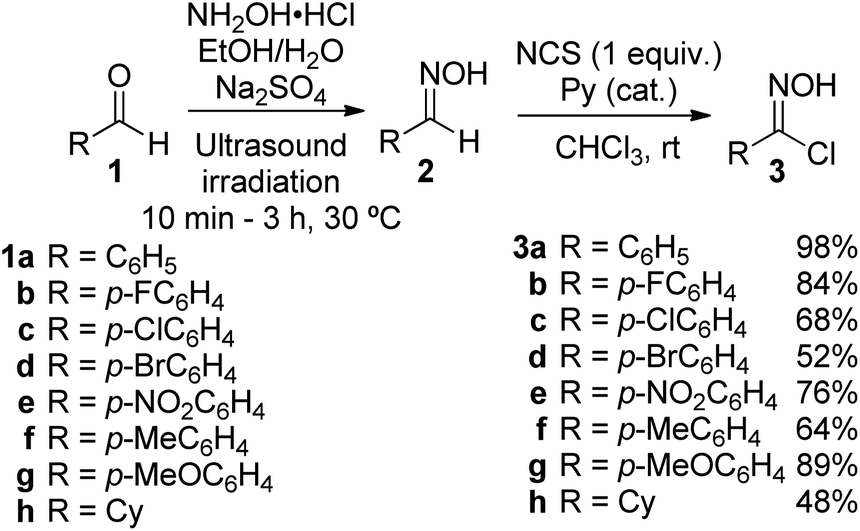 | ||
| Scheme 2 Synthesis of hydroximoyl chlorides from aldehydes. | ||
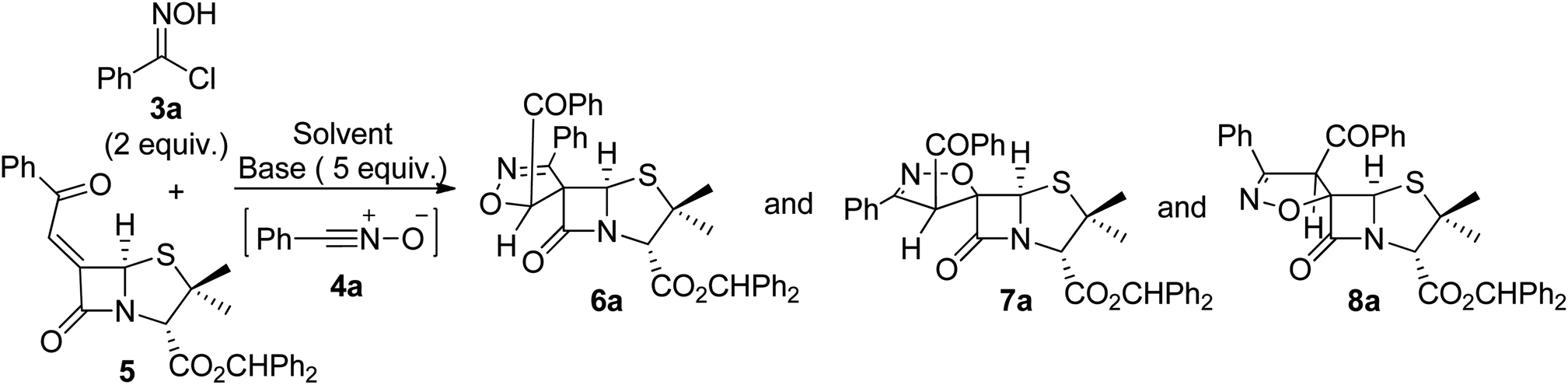
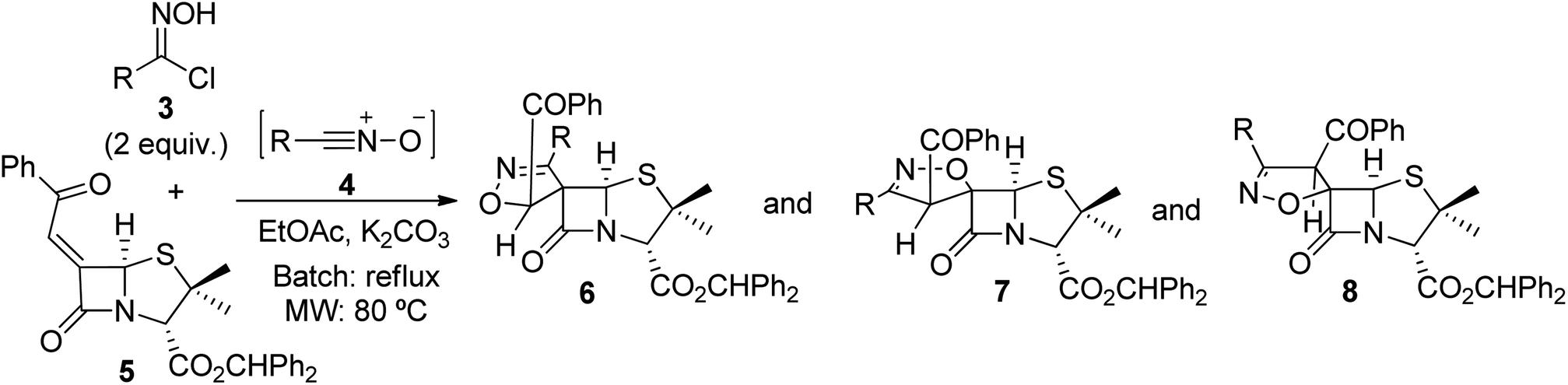
The 1,3-dipolar cycloaddition reaction of nitrile oxide 4a, generated in situ from benzaldehyde hydroximoyl chloride 3a by the action of potassium carbonate, with 6-(Z)-(benzoylmethylene)penicillanate 5 was studied for the optimization of the batch and microwave-induced reaction conditions (Table 1). The use of an inorganic base ensures a slow dehydrohalogenation of hydroximoyl chlorides 3, allowing a controlled formation of the corresponding nitrile oxide 4 which readily reacts with the desired dipolarophile. Thus, the reaction of the transient nitrile oxide 4a with 6-(Z)-(benzoylmethylidene)penicillanate 5 was carried out in ethyl acetate at room temperature for 7.5 h affording compound 6a as major product in 60% yield. From this reaction an inseparable mixture of compounds 7a and 8a was also isolated in 32% yield (66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :34 ratio) (entry 1). The increase of the reaction time to 24 h was counterproductive regarding the overall yield but led to compound 6a as single product isolated in 49% yield (entry 2). However, better results were obtained by increasing the temperature. Carrying out the reaction of 4a with 5 under refluxing ethyl acetate the desired products were obtained in excellent overall yield (97%) with a good selectivity for compound 6a which was isolated in 59% yield (entry 3). Curiously, the same overall yield was obtained by performing the reaction in toluene at 80 °C (entry 4) with a slight increase in the yield of 6a (entry 3: 59% vs. entry 4: 65%) and without the formation of 8a. Additionally, an alternative base for the dehydrohalogenation of hydroximoyl chloride 3a was also explored. Performing the reaction in the presence of triethylamine, the target spiro-β-lactams could also be obtained by carrying out the reaction either in ethyl acetate (in excellent overall yield, entry 5) or in toluene (in moderate overall yield, entry 6).
:34 ratio) (entry 1). The increase of the reaction time to 24 h was counterproductive regarding the overall yield but led to compound 6a as single product isolated in 49% yield (entry 2). However, better results were obtained by increasing the temperature. Carrying out the reaction of 4a with 5 under refluxing ethyl acetate the desired products were obtained in excellent overall yield (97%) with a good selectivity for compound 6a which was isolated in 59% yield (entry 3). Curiously, the same overall yield was obtained by performing the reaction in toluene at 80 °C (entry 4) with a slight increase in the yield of 6a (entry 3: 59% vs. entry 4: 65%) and without the formation of 8a. Additionally, an alternative base for the dehydrohalogenation of hydroximoyl chloride 3a was also explored. Performing the reaction in the presence of triethylamine, the target spiro-β-lactams could also be obtained by carrying out the reaction either in ethyl acetate (in excellent overall yield, entry 5) or in toluene (in moderate overall yield, entry 6).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Reaction conditions | Isolated yields (ratio)a | Overall yield | ||
| a Ratio determined by 1H NMR.b Trace amount of 6a. | |||||||
| Batch | 1 | K2CO3 | AcOEt | r.t., 7.5 h | 6a, 60% | 7a/8a, 32% (66:34) |
92% |
| 2 | K2CO3 | AcOEt | r.t., 24 h | 6a, 49% | — | 49% | |
| 3 | K2CO3 | AcOEt | Reflux, 5 h | 6a, 59% | 7a/8a, 38% (84:16) |
97% | |
| 4 | K2CO3 | Toluene | 80 °C, 4 h | 6a, 65% | 7a, 32% | 97% | |
| 5 | NEt3 | AcOEt | r.t., 24 h | 6a, 47% | 7a/8a, 51% (63:37) |
98% | |
| 6 | NEt3 | Toluene | 80 °C, 4 h | 6a, 11% | 7a/8a, 37% (38:62) |
48% | |
| MW | 7 | K2CO3 | AcOEt | 80 °C, 30 min | 6a, 32% | 7a, 28% | 60% |
| 8 | K2CO3 | AcOEt | 80 °C, 1 h | 6a, 35% | 7a, 34% | 69% | |
| 9 | K2CO3 | Toluene | 80 °C, 30 min | 6a, 42% | 7a, 32% | 74% | |
| 10 | K2CO3 | Toluene | 120 °C, 10 min | — | — | —b | |
| 11 | NEt3 | Toluene | 80 °C, 30 min | Complex mixture | — | ||
The reaction was also carried out under microwave irradiation (entries 7–11). It was observed that carrying out the reaction in ethyl acetate under microwave irradiation for 30 minutes at 80 °C afforded compounds 6a and 7a in 32% and 28% yield, respectively (entry 7). The yields were slightly improved using the same conditions for 1 hour, giving products 6a and 7a in 35% and 34% yield, respectively (entry 8). Changing the reaction solvent to toluene allowed the synthesis of 6a and 7a in 74% overall yield after 30 minutes of microwave irradiation at 80 °C, with compound 6a being obtained as major product in 42% yield (entry 9). On the other hand, by increasing the temperature to 120 °C for 10 min, the consumption of the reagents was observed, but only a trace amount of 6a was detected (entry 10). Finally, carrying out the microwave-induced reaction in toluene at 80 °C for 30 min using triethylamine as the dehydrohalogenating agent, a complex mixture was obtained. It should be noted that the cycloaddition reaction between alkylidenepenicillanate 5 and nitrile oxide 4a under microwave irradiation proved to be more selective, without the formation of spiro-β-lactam 8a.
The structural assignment of compounds 6a and 7a was supported by one and two-dimensional NMR spectra (1H NMR, 13C NMR and HSQC; see ESI†). As expected, the 1H NMR spectrum of derivatives 6a and 7a show signals corresponding to two methyl groups from the penicillanate core, protons corresponding to aromatic protons and to the benzhydryl proton (CHPh2, 6a: 6.98 ppm; 7a: 6.93 ppm). The 1H NMR and HSQC spectra allowed us to assign the signals corresponding to protons H-3, H-5 of the penicillanate core and the proton of the isoxazoline ring of both compounds (Fig. 2). For compound 6a the following chemical shifts were observed: H-3 at 4.65 ppm, H-5 at 5.86 ppm and isoxazoline proton at 6.17 ppm. Major difference was observed for the proton of the isoxazoline ring in the case of compound 7a with a chemical shift of 5.67 ppm. This difference can be explained by the presence of an oxygen atom in a vicinal position to the proton in compound 6a which promotes a downfield shift. Additionally, from the HSQC spectrum it was possible to confirm the assignment of the spirocyclic carbon of both spiro-β-lactams (6a: 76.6 ppm; 7a: 102.0 ppm). The difference in the chemical shift of the spirocyclic carbons of these two regioisomers can be explained considering that only in the case of compound 7a is the oxygen atom attached to the spirocyclic carbon. This NMR data is in agreement with the proposed structures of spiro-β-lactams 6a and 7a resulting from opposite regioselectivity, with 6a and 7a being a spiro[isoxazoline-4′,6-penicillanate] (major) and a spiro[isoxazoline-5′,6-penicillanate], respectively. It is known that due to the bicyclic β-lactam-thiazolidine ring system of the penicillin core which exists in a butterfly-like structure, the approach of a given reactant usually occurs by the convex face (α-side) of the penicillin derivative.3–5 This is in agreement with the observed stereoselectivity, with both compounds 6a and 7a resulting from the addition of the dipole to the less sterically hindered α-side of the penicillanates. The stereoselectivity observed in the formation of compound 8a can be rationalized by an approach of the dipole through the β-side of the penicillanate, with the regioselective formation of the cycloadduct where the dipole's oxygen adds to the forthcoming spirocyclic carbon. The 13C chemical shift of the spirocyclic carbons are similar for compounds 7a and 8a (7a: 102.2 ppm; 8a: 100.1 ppm), as expected for diastereoisomers with the same regiochemistry.
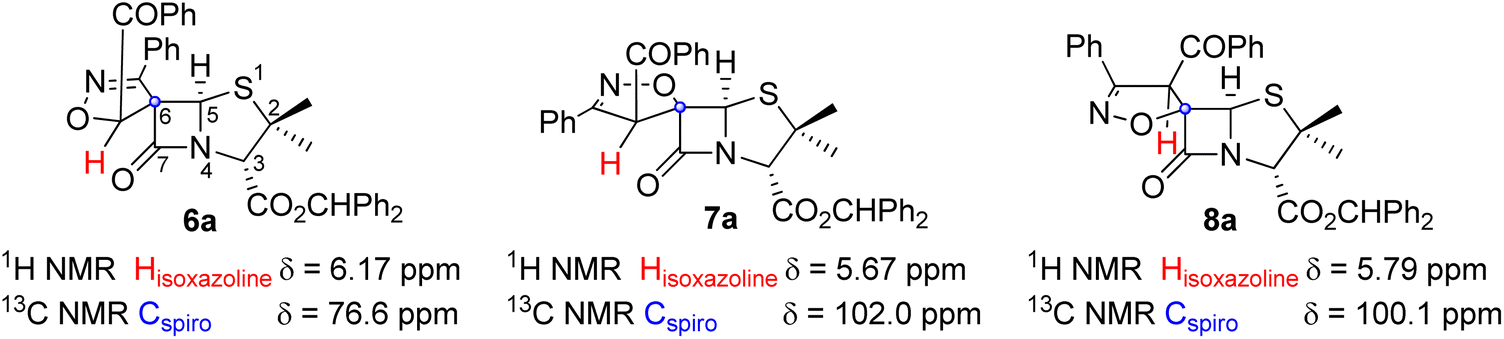 | ||
| Fig. 2 Structures of selected spiro-β-lactams highlighting the most relevant 1H and 13C chemical shifts for the stereochemistry assignment. | ||
The same stereo- and regioselectivity was previously described by Corbett, while exploring the 1,3-dipolar cycloaddition reaction of nitrile oxides with an alkylidene-carbapenem, aiming at the synthesis of spiroisoxazoline-carbapenem.11 In this work, it was observed that three products were obtained from this cycloaddition reaction. Two of them were the expected cycloadducts from the addition of the dipole to the α-side of the carbapenem: one results from the attack of the dipole's oxygen to the terminal carbon of the exocyclic double bond of the alkylidene (major product) and the other from the addition of the dipole's oxygen to the forthcoming spirocyclic carbon. The formation of a third cycloadduct was also observed, a diastereoisomer formed by the addition of the nitrile oxide to the β-side. It should be noted that the carbapenem bicyclic system is similar to the penicillanic core in terms of preferred conformation, leading to a more favorable addition of incoming reactants by the α-face.
Although a small difference was observed in the isolated yields of products of the 1,3-dipolar cycloaddition of 6-(Z)-(benzoylmethylene)penicillanate and phenyl-nitrile carrying out the reactions in ethyl acetate or in toluene, ethyl acetate was selected for further studies since it is a “greener” solvent than toluene.13 On the other hand, potassium carbonate was selected over triethylamine also aiming at developing a more sustainable synthetic methodology.
Next, we extended the study of the reactivity of 6-(Z)-(benzoylmethylidene)penicillanate (5) to other nitrile oxides under batch and microwave irradiation optimized reaction conditions. However, reactions carried out under conventional heating were monitored by TLC and the reaction time determined by the total consumption of alkylidenepenicillanate 5, ranging from 15 minutes to 8 hours.
The 1,3-dipolar cycloaddition reactions with nitrile oxides bearing electron-withdrawing and electron-donating substituents at para-position of the phenyl ring were explored (Table 2). To our delight, both types of dipole activation led to the expected cycloadducts, with pure chiral spiroisoxazoline-β-lactams 6b–f being obtained as major products, in yields ranging from 49–72% and 31–47%, under conventional heating and microwave irradiation, respectively (entries 1–12).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 4 | R | Methodology | Reaction time (h) | Isolated yields (ratio)a | Overall yield | |
| a Ratio determined by 1H NMR. | |||||||
| 1 | 4b | p-FC6H4 | Batch | 6 | 6b, 72% | 7b, trace amounts | 72% |
| 2 | 4b | p-FC6H4 | MW | 1 | 6b, 39% | 7b/8b, 32% (84:16) |
71% |
| 3 | 4c | p-ClC6H4 | Batch | 8 | 6c, 49% | 7c/8c, 38% (71:29) |
87% |
| 4 | 4c | p-ClC6H4 | MW | 1 | 6c, 41% | 7c/8c, 29% (90:10) |
70% |
| 5 | 4d | p-BrC6H4 | Batch | 2 | 6d, 49% | 7d/8d, 43% (65:35) |
92% |
| 6 | 4d | p-BrC6H4 | MW | 1 | 6d, 44% | 7d/8d, 33% (58:42) |
77% |
| 7 | 4e | p-NO2C6H4 | Batch | 2 | 6e, 54% | 7e/8e, 35% (34:66) |
98% |
| 8 | 4e | p-NO2C6H4 | MW | 1 | 6e, 47% | 7e/8e, 32% (50:50) |
79% |
| 9 | 4f | p-MeC6H4 | Batch | 5 | 6f, 50% | 7f/8f, 38% (68:32) |
88% |
| 10 | 4f | p-MeC6H4 | MW | 1 | 6f, 31% | 7f/8f, 20% (85:15) |
51% |
| 11 | 4g | p-MeOC6H4 | Batch | 0.5 | 6g/8g, 52% (69:31) |
52% | |
| 12 | 4g | p-MeOC6H4 | MW | 0.25 | 6g/8g, 36% (69:31) |
7g, 21% | 57% |
| 13 | 4h | Cy | Batch | 6 | 6h, 79% | 7h/8h, 15% (17:83) |
94% |
| 14 | 4h | Cy | MW | 1 | 6h, 60% | 7h/8h, 10% (10:90) |
70% |
The reaction of alkylidenepenicillanate 5 with nitrile oxide 4b (R = p-FC6H4) under conventional heating was highly selective, leading to the exclusive formation of a single product (6b) in 72% yield (entry 1). On the other hand, performing the same reaction under microwave irradiation (entry 2) led to the synthesis of the same spiro-β-lactam 6b (39% yield) together with spiro-β-lactams 7b/8b (32% yield). Carrying out the cycloaddition reaction with p-chloro or p-bromo substituted nitrile oxides (4c and 4d, respectively) the corresponding spiroisoxazoline-β-lactams 6 (6c and 6d) were obtained as major products under both methodologies in yields ranging from 41% to 49% (entries 3–6). From these reactions, mixtures of isomeric compounds 7c/8c and 7d/8d were also obtained leading to overall yields up to 92% (entries 3–6). The cycloaddition reaction of nitrile oxide 4e, bearing a nitro group at the para position of the aromatic ring, with alkylidenepenicillanate 5 was also explored under the optimized conditions (entries 7 and 8). This reaction allowed the synthesis of 6e as major product in 54% yield under conventional heating (entry 7) as well as under microwave conditions, with compound 6e being obtained in 47% yield (entry 8). In both reactions, mixture of compounds 7e/8e were also obtained in 32–35% yield (entries 7 and 8). Notably, concerning the overall yields, the 1,3-dipolar cycloaddition reaction between 4e and 5 was the most efficient under both conventional heating and microwave irradiation (overall yields: 98% and 79%, respectively).
The cycloaddition reaction of spiro-β-lactam 5 with para-methyl derivative 4f also afforded the expected major spiroisoxazoline-β-lactam 6f in 50% yield under conventional heating (entry 9), together with spiro-β-lactams 7f/8f (38% yield). However, under microwave irradiation (entry 10) the overall yield of this reaction was modest, leading to compounds 6f and 7f/8f in 31% and 20% yield, respectively.
The 1,3-dipolar cycloaddition using as 1,3-dipole nitrile oxide 4g, bearing a stronger electron-donating group (R = p-MeOC6H4), was extremely fast under conventional heating (15 minutes) leading to an inseparable mixture of 6g/8g in 52% yield (entry 11). Under microwave irradiation for 30 minutes the same mixture (6g/8g) was isolated in 36% yield, together with pure spiro-β-lactam 7g obtained in 20% yield (entry 12).
The synthesis of spiroisoxazoline-β-lactams derived from an alkyl-substituted nitrile oxide were also explored (entries 13 and 14). Under conventional heating, the 1,3-dipolar cycloaddition reaction of dipole 4h, bearing a cyclohexyl moiety, with alkylidenepenicillanate 5 led to pure spiroisoxazoline-β-lactam 6h in an exceptional yield of 79%. The formation of 7h/8h was also observed in 15% yield. Conducting the same reaction under microwave irradiation allowed the synthesis of compound 6h in 60% yield, together with the mixture of 7h/8h isolated in low yield (10%). It should be noted that this cycloaddition was highly selective for the synthesis of spiro-β-lactam 6h and the yields were comparable with the ones observed in the cycloaddition of spiro-β-lactam 5 with aryl-substituted nitrile oxides.
Briefly, both methodologies led to pure chiral spiroisoxazoline-β-lactams 6a–h (with exception of 6g which is obtained in a mixture with 8g). To our surprise, the reaction under microwave irradiation took longer than expected, and in most cases, there were no substantial differences regarding reaction times using conventional heating or microwave irradiation. In addition, we should highlight the synthesis of spiro-β-lactams 6 in higher yields under conventional heating (conventional heating: 49–79% vs. microwave irradiation: 31–60%) as well as higher overall yields (conventional heating: 52–98% vs. microwave irradiation: 51–79%).
Continuous flow 1,3-dipolar cycloaddition reactions
Continuous flow chemistry is a term used to describe chemical reactions performed in a continuous manner.14 This approach has been widely used in the past years due to its advantages, such as faster reactions, improved safety, high reproducibility, scalability, facile automation, and a precise control of reaction conditions. Furthermore, the continuous flow technology also allows to carry out reactions in a safer, greener, and sustainable way, making flow systems extraordinary useful.15 Continuous flow apparatus enables the construction of different systems by coupling several and distinct reactors (e.g. coil reactor, microchips and packed-bed column) in any sequence. This characteristic extends the versality of the methodology allowing to carry out multistep transformations.16 As consequence, continuous flow chemistry has improved the synthesis of complex molecules such as pharmaceuticals17 and natural products.18Thus, we have extended our efforts to the development of a continuous flow approach to chiral spiroisoxazoline-penicillanates via 1,3-dipolar cycloaddition reaction of alkylidenepenicillanate 5 to nitrile oxides 4 (Table 3). The approach for the dehydrohalogenation of hydroximoyl chlorides 3 leading to the in situ generation of the corresponding nitrile oxides 4, relied on the use of a packed-bed reactor filled with fresh potassium carbonate.
|
|||||
|---|---|---|---|---|---|
| Entry | 4 | R | Isolated yields (ratio)a | Overall yields | |
| a Ratio determined by 1H NMR.b 50 μL min−1 flow rate, tR = 24 min.c Reaction carried out at 80 °C.d 4 equiv. of the nitrile oxide. | |||||
| 1 | 4a | C6H5 | 6a, 54% | 7a/8a, 35% (33:67) |
89% |
| 2 | 4a b | C6H5 | 6a, 50% | 7a/8a, 45% (45:55) |
95% |
| 3 | 4a c | C6H5 | 6a, 10% | — | 10% |
| 4 | 4a c,d | C6H5 | 6a, 22% | — | 22% |
| 5 | 4b | p-FC6H4 | 6b, 49% | 7b/8b, 38% (53:47) |
87% |
| 6 | 4c | p-ClC6H4 | 6c, 45% | 7c/8c, 29% (65:35) |
74% |
| 7 | 4d | p-BrC6H4 | 6d, 57% | 7d/8d, 35% (75:25) |
92% |
| 8 | 4f | p-MeC6H4 | 6f, 51% | 7f/8f, 23% (39:61) |
74% |
| 9 | 4g | p-MeOC6H4 | 6g/8g, 54% (51:49) |
54% | |
| 10 | 4h | Cy | 6h, 40% | 7h/8h, 3% (39:61) |
43% |

The set-up was composed of two inlets, one containing a solution of 6-(Z)-(benzoylmethylidene)penicillanate 5 and another one containing a solution of the appropriate hydroximoyl chloride 3, with both solutions being mixed right before entering the packed-bed reactor. Carrying out the reaction of alkylidene 5 with 1,3-dipole 4a at room temperature with a flow rate of 100 μL min−1 of each solution led to the expected spirocyclic compounds, in 89% overall yield, with the major compound 6a being obtained in 54% yield (Table 3, entry 1). The reaction conditions were then optimized. The influence of the residence time, tR, was investigated by tuning the flow rates of reagents (entry 2). Decreasing the flow rate of each solution to 50 μL min−1, an improvement on the overall yield was observed (95%), however, with a slight decrease in the yield of the major compound 6a (50%). Unlike what was observed for reactions carried out in batch or under microwave irradiation, the increase of the temperature to 80 °C under flow conditions did not lead to better results even with the simultaneous increase of nitrile oxide equivalents (entries 3 and 4). Considering the above-presented results, we selected a flow rate of 100 μL min−1, together with room temperature and 2 equiv. of the nitrile oxide as the best conditions to continue our studies.
Next, continuous flow 1,3-dipolar cycloaddition of 6-(Z)-(benzoylmethylidene)penicillanate 5 to nitrile oxides 4b–h was explored (Table 3, entries 5–10). Using p-halophenyl-nitrile oxides (R = F, Cl, Br) the expected spiro-β-lactams were obtained in excellent overall yields, ranging from 74% to 92% (entries 5–7). Chiral spiro-β-lactam 6b, 6c and 6d were obtained as major products in 49%, 45% and 57%, respectively. The continuous flow also proved to be a viable approach for the synthesis of spiroisoxazoline-penicillanates using nitrile oxides containing electron-donating groups at the phenyl para-position, giving the expected cycloadducts in yields as good as under conventional heating (entries 8 and 9). Unfortunately, among the set of aryl-substituted nitrile oxides used in the present work it was not possible to study the behavior of nitrile oxide 4e (R = p-NO2C6H4) under flow conditions due to its partial insolubility in a wide range of solvents.
Finally, the 1,3-dipolar cycloaddition of alkyl-substituted dipole 4h was studied. In this case, the major spirocyclic adduct 6h was obtained in moderate yield (40%) together with a mixture of 7h/8h (39:61 ratio) in 3% yield. Thus, the synthesis of chiral spiroisoxazoline-penicillanates 6h/7h/8h under continuous flow conditions proved to be less efficient than under conventional heating or microwave irradiation.
These results have shown that continuous flow is an interesting alternative approach to the other methodologies discussed in the present work, to carry out 1,3-dipolar cycloaddition reactions leading to spiro isoxazoline penicillanates. It is noteworthy that the continuous flow technique allowed the synthesis of the target compounds with a very short reaction time (tR = 12 minutes) having the great advantage of enabling easy scale-up processes. In fact, as outlined in Fig. 3, continuous flow proved to be the best methodology for the synthesis of the major spiro-β-lactams 6 with production rates ranging from 200 to 400 mg h−1, values considerably higher than the ones observed for batch and microwave-induced methodologies.
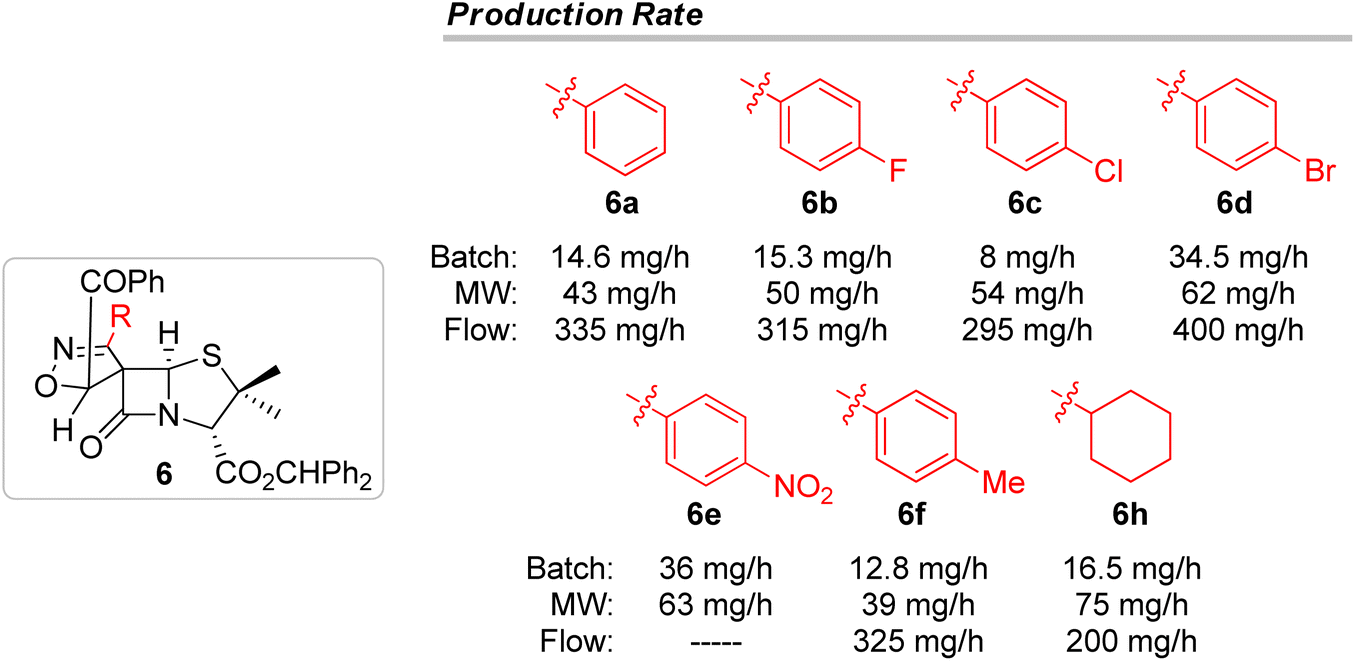 | ||
| Fig. 3 Production rates in mg h−1 for the synthesis of the major products, spiro-β-lactams 6. | ||
Conclusion
Herein, a 1,3-dipolar cycloaddition-based approach to a new type of chiral spiropenicillanates has been disclosed. 6-(Z)-(Benzoylmethylidene)penicillanate reacted with in situ generated nitrile oxides to afford novel spiro isoxazoline penicillanates with the diastereoselective creation of two consecutive stereogenic centers, including a quaternary chiral center. Spiro[isoxazoline-4′,6-penicillanates] were obtained as the major stereo- and regioisomers resulting from the addition of the dipole to the less sterically hindered α-side of the 6-alkylidene penicillanate and the attack of the dipole's oxygen to the terminal carbon of the exocyclic double bond of the alkylidene.It was demonstrated that these spirocyclic adducts could be obtained using three different methodologies: conventional heating, microwave irradiation and continuous flow. Both conventional heating and continuous flow proved to be better approaches than the microwave-induced methodology leading to the major product in higher yields (conventional heating: 49–79%; continuous flow: 40–57%), and in high overall yields (conventional heating: 52–98%; continuous flow: 43–92%). Nevertheless, the continuous flow 1,3-dipolar cycloaddition reaction stands out for allowing very short reaction times, and by its inherent characteristics that ensure easy scale-up processes. In this study it was possible to observe that continuous flow conditions led to higher production rates than batch or microwave-induced methodologies, with the major products being obtained in up to 400 mg h−1.
Experimental section
Thin-layer chromatography (TLC) analyses were performed using precoated silica gel plates. Flash column chromatography was performed with silica gel 60 as the stationary phase. 1H Nuclear magnetic resonance (NMR) spectra (400 MHz) and 13C NMR spectra (100 MHz) were recorded using CDCl3 as solvent. Chemical shifts are expressed in parts per million (ppm) relatively to internal tetramethylsilane (TMS) and coupling constants (J) are expressed in Hertz (Hz). Infrared spectra (IR) were recorded in a Fourier transform spectrometer coupled with a diamond Attenuated Total Reflectance (ATR) sampling accessory. High-resolution mass spectra (HRMS) were obtained on a Orbitrap q-Exactive Focus spectrometer coupled to a Vanquish HPLC with ESI. Melting points (mp) were determined in open glass capillaries. Optical rotations were measured on an Optical Activity AA-5 electrical polarimeter. 6-(Z)-(benzoylmethylidene)penicillanate 5 (ref. 5b) was prepared as described in the literature.General procedure for the synthesis of hydroximoyl chlorides 3
Hydroximoyl chlorides 3 were prepared based on previously developed procedures.19 The appropriate aldehyde (1 equiv.) was dissolved in ethanol (15 mL). A solution of hydroxylamine hydrochloride (1.25 equiv.) in water (3 mL) and anhydrous sodium sulfate (1 equiv.) were added. The reaction mixture was irradiated in the ultrasonic bath at 30 °C for the time indicated in each case. The mixture was filtered, and the solvent was evaporated under reduced pressure. The residue was dissolved in CH2Cl2, washed with water, and extracted with CH2Cl2. The combined organic layers were dried over anhydrous NaSO4, filtered, and the solvents were evaporated affording oximes 2 which were used without further purification. Oximes 2 were dissolved in chloroform (11 mL) and a drop of pyridine was added. After 5 min, N-chlorosuccinimide (1 equiv.) was added stepwise to the stirring reaction mixture. The reaction mixture was stirred for the time indicated in each case, monitored by TLC until completion. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography.:1) afforded compound 3a as a yellow solid (917 mg, 5.89 mmol, 98%).3a: mp 45.1–47.1 °C (lit.20 47–48 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.40–7.46 (m, 3H), 7.85 (dd, J = 8.1 and 1.5 Hz, 2H), 8.25 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.21
:1) afforded compound 3b as a white solid (438 mg, 2.52 mmol, 84%).3b: mp 75.1–77.1 °C (lit.22 74.5–75 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.10 (t, J = 8.7 Hz, 2H), 7.84 (dd, J = 9.0 and 5.3 Hz, 2H), 7.91 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.23
:1) afforded compound 3c as a white solid (389 mg, 2.05 mmol, 68%).3c: mp 86.4–88.4 °C (lit.20 88.5–89.5 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.39 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 8.8 Hz, 2H), 27.96 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.24
:1) afforded compound 3d as a white solid (363 mg, 1.55 mmol, 52%).3d: mp 78.3–80.3 °C (lit.25 76–78 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.55 (d, J = 8.7 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.84 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.23
:1) afforded compound 3e as a yellow solid (460 mg, 2.29 mmol, 76%).3e: mp 121.4–123.4 °C (lit.20 124–125 °C); 1H NMR (CDCl3, 400 MHz) δ = 8.05 (d, J = 9.0 Hz, 2H), 8.27 (d, J = 9.0 Hz, 2H), 8.32 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.23
:1) afforded compound 3f as a white solid (323 mg, 1.91 mmol, 64%).3f: mp 67.4–69.4 °C (lit.26 71–72 °C); 1H NMR (CDCl3, 400 MHz) δ = 2.38 (s, 3H), 7.21 (d, J = 8.1 Hz, 2H), 7.65 (s, 1H), 7.73 (d, J = 8.3 Hz, 2H). The 1H NMR spectral data are in good agreement with the literature data.24
:1) afforded compound 3g as a white solid (498 mg, 2.68 mmol, 89%).3g: mp 85.7–87.7 °C (lit.20 87.5–89 °C); 1H NMR (CDCl3, 400 MHz) δ = 3.85 (s, 3H), 6.91 (d, J = 9.0 Hz, 2H), 7.65 (s, 1H), 7.79 (d, J = 9.0 Hz, 2H). The 1H NMR spectral data are in good agreement with the literature data.27
:1) afforded compound 3h as a colorless oil (235 mg, 1.45 mmol, 48%).3h: 1H NMR (CDCl3, 400 MHz) δ = 1.23–1.35 (m, 1H), 1.39–1.50 (m, 2H), 1.66–1.70 (m, 1H), 1.78–1.82 (m, 2H), 1.92–1.96 (m, 2H), 2.46 (tt, J = 11.5 and 3.4 Hz, 1H), 8.10 (s, 1H). The 1H NMR spectral data are in good agreement with the literature data.28
General procedure for the 1,3-dipolar cycloaddition of nitrile oxides with 6-(Z)-(benzoylmethylidene)penicillanate
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-phenyl-isoxaxoline)-4′,6-penicillanate] (6a) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-phenyl-isoxaxoline)-5′,6-penicillanate] (7a) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-phenyl-isoxaxoline)-5′,6-penicillanate] (8a). Obtained from hydroximoyl chloride 3a (64 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). Method A, gave, in order of elution, 6a as a white solid (73 mg, 0.121 mmol, 59%) and a mixture of 7a/8a (84:16) as a yellow oil (47 mg, 0.078 mmol, 38%). Method B, gave, in order of elution, 6a as a white solid (43 mg, 0.071 mmol, 35%) and 7a as a yellow oil (42 mg, 0.070 mmol, 34%). Method C, gave, in order of elution, 6a as a white solid (67 mg, 0.111 mmol, 54%) and a mixture of 7a/8a (33:67) as a yellow oil (43 mg, 0.071 mmol, 35%).6a: mp 74.0–76.0 °C; [α]25D = +360 (c 0.5 in CH2Cl2); IR (ATR): ν = 881, 1083, 1156, 1449, 1496, 1586, 1678, 1740 and 1780 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.15 (s, 3H), 1.57 (s, 3H), 4.65 (s, 1H), 5.86 (s, 1H), 6.17 (s, 1H), 6.98 (s, 1H), 7.27–7.35 (m, 12H), 7.43 (t, J = 7.5 Hz, 1H), 7.52–7.56 (m, 2H), 7.66 (t, J = 7.4 Hz, 1H), 7.81–7.83 (m, 2H), 8.04–8.06 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ = 26.2, 32.1, 63.7, 68.0, 69.9, 76.6, 78.8, 81.8, 126.7, 127.3, 127.4, 127.5, 127.7, 128.5, 128.6, 128.6, 128.8, 128.9, 129.1, 129.2, 129.4, 131.0, 134.5, 135.0, 139.1, 139.2, 155.6, 166.5, 170.7, 191.6; HRMS (ESI) m/z: calcd for C36H31N2O5S [M + H]+ 603.1948; found 666.1941.
7a: mp low melting solid; [α]25D = +280 (c 0.25 in CH2Cl2); IR (ATR): ν = 910, 1080, 1176, 1448, 1497, 1595, 1676, 1741 and 1777 cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.12 (s, 3H), 1.46 (s, 3H), 4.60 (s, 1H), 5.67 (s, 1H), 5.68 (s, 1H), 6.93 (s, 1H), 7.22–7.24 (m, 2H), 7.30–7.35 (m, 11H), 7.48–7.55 (m, 4H), 7.63 (t, J = 7.4 Hz, 1H), 7.95–7.97 (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 25.5, 33.4, 57.9, 63.7, 68.9, 72.9, 77.5, 78.7, 102.2, 127.2, 127.3, 127.5, 127.6, 128.4, 128.6, 128.6, 128.7, 128.8, 128.8, 128.9, 129.0, 129.3, 131.0, 134.5, 137.2, 139.1, 139.1, 156.7, 166.2, 170.3, 194.6; HRMS (ESI) m/z: calcd for C36H31N2O5S [M + H]+ 603.1948; found 666.1945.
Recorded as a mixture of 7a/8a (33:67): 8a: 1H NMR (CDCl3, 400 MHz) δ = 1.30 (s, 3H), 1.33 (s, 3H), 4.46 (s, 1H), 5.56 (s, 1H), 5.79 (s, 1H), 6.93 (s, 1H), 7.22–7.24 (m, 2H), 7.30–7.35 (m, 11H), 7.48–7.55 (m, 4H), 7.63 (t, J = 7.4 Hz, 1H), 8.11 (d, J = 7.2 Hz, 2H).
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-(4-fluorophenyl)-isoxaxoline)-4′,6-penicillanate] (6b) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-(4-fluorophenyl)-isoxaxoline)-5′,6-penicillanate] (7b) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-(4-fluorophenyl)-isoxaxoline)-5′,6-penicillanate] (8b). Obtained from hydroximoyl chloride 3b (71 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). Method A gave 6b as a white solid (92 mg, 0.148 mmol, 72%). Method B, gave, in order of elution, 6b as a white solid (50 mg, 0.081 mmol, 39%) and 7b/8b (84:16) as a yellow oil (41 mg, 0.066 mmol, 32%). Method C, gave, in order of elution, 6b as a white solid (63 mg, 0.101 mmol, 49%) and a mixture of 7b/8b (53:47) as a yellow oil (49 mg, 0.079 mmol, 38%).6b: mp 167.1–169.1 °C; [α]25D = +380 (c 0.5 in CH2Cl2); IR (ATR): ν = 838, 1081, 1156, 1449, 1509, 1598, 1676, 1736 and 1778 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.17 (s, 3H), 1.57 (s, 3H), 4.65 (s, 1H), 5.84 (s, 1H), 6.17 (s, 1H), 6.90 (t, J = 8.7 Hz, 2H), 6.98 (s, 1H), 7.30–7.36 (m, 10H), 7.53–7.56 (m, 2H), 7.66 (t, J = 7.4 Hz, 1H), 7.79–7.82 (m, 2H), 8.04–8.06 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ = 26.1, 32.3, 63.8, 68.0, 69.9, 76.6, 78.9, 81.8, 116.5 (d, J = 22 Hz), 123.4 (d, J = 3 Hz), 127.2, 127.7, 128.5, 128.7, 128.8, 128.8, 129.0, 129.2, 129.4, 129.4, 129.5, 134.5, 134.9, 139.0 (d, J = 2 Hz), 154.6, 164.4 (d, J = 252 Hz), 166.5, 170.4, 191.6; 19F NMR (CDCl3, 376 MHz) δ = −108.5 (s, 1F); HRMS (ESI) m/z: calcd for C36H29N2O5FNaS [M + Na]+ 643.1673; found 643.1671.
Recorded as a mixture of 7b/8b (53:47): 7b: 1H NMR (CDCl3, 400 MHz) δ = 1.12 (s, 3H), 1.46 (s, 3H), 4.60 (s, 1H), 5.63 (s, 1H), 5.66 (s, 1H), 6.91–6.96 (3H), 7.30–7.35 (m, 10H), 7.47–7.56 (m, 4H), 7.62–7.66 (m, 1H), 7.93–7.95 (m, 2H); 8b: 1H NMR (CDCl3, 400 MHz) δ = 1.30 (s, 3H), 1.34 (s, 3H), 4.46 (s, 1H), 5.56 (s, 1H), 5.74 (s, 1H), 6.91–6.96 (3H), 7.30–7.35 (m, 10H), 7.47–7.56 (m, 4H), 7.62–7.66 (m, 1H), 8.08–8.10 (m, 2H).
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-(4-chlorophenyl)-isoxaxoline)-4′,6-penicillanate] (6c) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-(4-chlorophenyl)-isoxaxoline)-5′,6-penicillanate] (7c) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-(4-chlorophenyl)-isoxaxoline)-5′,6-penicillanate] (8c). Obtained from hydroximoyl chloride 3c (78 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). Method A, gave, in order of elution, 6c as a white solid (64 mg, 0.100 mmol, 49%) and a mixture of 7c/8c (71:29) as a yellow oil (50 mg, 0.078 mmol, 38%). Method B, gave, in order of elution, 6c as a white solid (54 mg, 0.084 mmol, 41%) and 7c/8c (90:10) as a yellow oil (38 mg, 0.21 mmol, 29%). Method C, gave, in order of elution, 6c as a white solid (59 mg, 0.093 mmol, 45%) and a mixture of 7c/8c (65:35) as a yellow oil (38 mg, 0.060 mmol, 29%).6c: mp 198.1–199.4 °C; [α]25D = +450 (c 0.5 in CH2Cl2); IR (ATR): ν = 975, 1086, 1156, 1203, 1450, 1492, 1595, 1667, 1736 and 1779 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.17 (s, 3H), 1.57 (s, 3H), 4.65 (s, 1H), 5.85 (s, 1H), 6.18 (s, 1H), 6.99 (s, 1H), 7.16 (d, J = 8.7 Hz, 2H), 7.31–7.36 (m, 10H), 7.53–7.56 (m, 2H), 7.66 (t, J = 7.4 Hz, 1H), 7.73–7.77 (m, 2H), 8.03–8.05 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ = 26.1, 32.8, 63.8, 68.0, 69.9, 76.5, 79.0, 81.9, 125.7, 127.2, 127.7, 128.6, 128.8, 129.0, 129.2, 129.4, 129.6, 134.6, 134.9, 137.2, 139.0, 154.6, 166.4, 170.4, 191.5; HRMS (ESI) m/z: calcd for C36H29N2O5Cl2S [M + Cl]+ 671.1180; found 671.1183.
Recorded as a mixture of 7c/8c (90:10): 7c: 1H NMR (CDCl3, 400 MHz) δ = 1.12 (s, 3H), 1.46 (s, 3H), 4.60 (s, 1H), 5.64 (s, 1H), 5.67 (s, 1H), 6.93 (s, 1H), 7.21–7.23 (m, 2H), 7.30–7.36 (m, 10H), 7.46–7.53 (m, 4H), 7.62–7.66 (m, 1H), 7.93–7.95 (m, 2H); 8c: 1H NMR (CDCl3, 400 MHz) δ = 1.30 (s, 3H), 1.34 (s, 3H), 4.46 (s, 1H), 5.56 (s, 1H), 5.74 (s, 1H), 6.93 (s, 1H), 7.21–7.23 (m, 2H), 7.30–7.36 (m, 10H), 7.46–7.53 (m, 4H), 7.62–7.66 (m, 1H), 8.07–8.09 (m, 2H).
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-(4-bromophenyl)-isoxaxoline)-4′,6-penicillanate] (6d) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-(4-bromophenyl)-isoxaxoline)-5′,6-penicillanate] (7d) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-(4-bromophenyl)-isoxaxoline)-5′,6-penicillanate] (8d). Obtained from hydroximoyl chloride 3d (97 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). Method A, gave, in order of elution, 6d as a white solid (69 mg, 0.101 mmol, 49%) and a mixture of 7d/8d (65:35) as a yellow oil (60 mg, 0.088 mmol, 43%). Method B, gave, in order of elution, 6d as a white solid (62 mg, 0.091 mmol, 44%) and 7d/8d (58:42) as a yellow oil (47 mg, 0.069 mmol, 33%). Method C, gave, in order of elution, 6d as a white solid (80 mg, 0.117 mmol, 57%) and a mixture of 7d/8d (75:25) as a yellow oil (49 mg, 0.072 mmol, 35%).6d: mp 196.7–198.4 °C; [α]25D = +410 (c 0.5 in CH2Cl2); IR (ATR): ν = 866, 1086, 1155, 1203, 1450, 1492, 1594, 1667, 1736 and 1779 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.18 (s, 3H), 1.57 (s, 3H), 4.65 (s, 1H), 5.86 (s, 1H), 6.18 (s, 1H), 6.90 (t, J = 8.7 Hz, 2H), 6.96 (s, 1H), 7.30–7.37 (m, 10H), 7.53–7.56 (m, 2H), 7.65–7.70 (m, 3H), 8.03–8.06 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ = 26.1, 32.4, 63.8, 68.0, 69.9, 76.5, 79.0, 81.9, 125.7, 126.2, 127.2, 127.7, 128.6, 128.7, 128.8, 129.0, 129.2, 129.4, 132.5, 134.6, 134.9, 139.0, 154.6, 166.4, 170.4, 191.5; HRMS (ESI) m/z: calcd for C36H30N2O5BrS [M + H]+ 681.1053; found 681.1049.
Recorded as a mixture of 7d/8d (58:42): 7d: 1H NMR (CDCl3, 400 MHz) δ = 1.12 (s, 3H), 1.46 (s, 3H), 4.60 (s, 1H), 5.64 (s, 1H), 5.66 (s, 1H), 6.93 (s, 1H), 7.31–7.35 (m, 10H), 7.38–7.40 (m, 3H), 7.47–7.53 (m, 3H), 7.63–7.66 (m, 1H), 7.93–7.95 (m, 2H); 8d: 1H NMR (CDCl3, 400 MHz) δ = 1.30 (s, 3H), 1.34 (s, 3H), 4.46 (s, 1H), 5.56 (s, 1H), 5.73 (s, 1H), 6.94 (s, 1H), 7.31–7.35 (m, 10H), 7.38–7.40 (m, 3H), 7.47–7.53 (m, 3H), 7.63–7.66 (m, 1H), 8.07–8.09 (m, 2H).
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-(4-nitrophenyl)-isoxaxoline)-4′,6-penicillanate] (6e) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-(4-nitrophenyl)-isoxaxoline)-5′,6-penicillanate] (7e) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-(4-nitrophenyl)-isoxaxoline)-5′,6-penicillanate] (8e). Obtained from hydroximoyl chloride 3e (83 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). Method A, gave, in order of elution, 6e as a white solid (72 mg, 0.111 mmol, 54%) and a mixture of 7e/8e (34:66) as a yellow oil (47 mg, 0.072 mmol, 35%). Method B, gave, in order of elution, 6e as a white solid (63 mg, 0.097 mmol, 47%) and 7e/8e as a yellow oil (50:50) (43 mg, 0.066 mmol, 32%).6e: mp 171.7.-173.7 °C; [α]25D = +370 (c 0.5 in CH2Cl2); IR (ATR): ν = 884, 1082, 1155, 1344, 1449, 1497, 1518, 1597, 1669, 1744 and 1777 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.20 (s, 3H), 1.58 (s, 3H), 4.68 (s, 1H), 5.88 (s, 1H), 6.25 (s, 1H), 7.00 (s, 1H), 7.33–7.38 (m, 10H), 7.54–7.58 (m, 2H), 7.69 (t, J = 7.4 Hz, 1H), 7.98–8.06 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ = 26.1, 32.5, 64.0, 67.9, 69.9, 76.4, 79.2, 82.3, 124.4, 127.1, 127.8, 128.2, 128.6, 128.8, 128.8, 128.9, 128.9, 129.0, 129.3, 129.4, 133.4, 134.8, 138.8, 149.1, 153.8, 166.3, 169.9, 191.4; HRMS (ESI) m/z: calcd for C36H30N3O7S [M + H]+ 648.1799; found 648.1792.
Recorded as a mixture of 7e/8e (50:50): 7e: 1H NMR (CDCl3, 400 MHz) δ = 1.13 (s, 3H), 1.46 (s, 3H), 4.61 (s, 1H), 5.69 (s, 1H), 5.72 (s, 1H), 6.94 (s, 1H), 7.31–7.36 (m, 10H), 7.48–7.56 (m, 2H), 7.76–7.78 (m, 2H), 8.08–8.12 (m, 2H), 8.18–8.20 (m, 2H), 8.33–8.36 (m, 1H); 8e: 1H NMR (CDCl3, 400 MHz) δ = 1.31 (s, 3H), 1.33 (s, 3H), 4.47 (s, 1H), 5.58 (s, 1H), 5.82 (s, 1H), 6.94 (s, 1H), 7.31–7.36 (m, 10H), 7.48–7.56 (m, 2H), 7.69–7.72 (m, 2H), 7.94–7.96 (m, 2H), 8.08–8.12 (m, 2H), 8.33–8.36 (m, 1H).
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-(4-methylphenyl)-isoxaxoline)-4′,6-penicillanate] (6f) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-(4-methylphenyl)-isoxaxoline)-5′,6-penicillanate] (7f) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-(4-methylphenyl)-isoxaxoline)-5′,6-penicillanate] (8f). Obtained from hydroximoyl chloride 3f (70 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). Method A, gave, in order of elution, 6f as a white solid (64 mg, 0.104 mmol, 50%) and a mixture of 7f/8f (68:32) as a yellow oil (48 mg, 0.078 mmol, 38%). Method B, gave, in order of elution, 6f as a white solid (39 mg, 0.063 mmol, 31%) and 7f/8f (85:15) as a yellow oil (26 mg, 0.042 mmol, 20%). Method C, gave, in order of elution, 6f as a white solid (65 mg, 0.105 mmol, 51%) and a mixture of 7f/8f (39:61) as a yellow oil (29 mg, 0.047 mmol, 23%).6f: mp 88.4–90.4 °C; [α]25D = +420 (c 0.5 in CH2Cl2); IR (ATR): ν = 876, 1156, 1449, 1497, 1596, 1676, 1742 and 1774 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.16 (s, 3H), 1.56 (s, 3H), 2.34 (s, 3H), 4.65 (s, 1H), 5.88 (s, 1H), 6.16 (s, 1H), 6.98 (s, 1H), 7.04 (d, J = 8.0 Hz, 2H), 7.26–7.36 (m, 10H), 7.52–7.55 (m, 2H), 7.65 (t, J = 7.4 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 8.04–8.06 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ = 21.6, 26.2, 32.1, 63.6, 68.0, 69.8, 76.6, 78.8, 81.6, 124.4, 127.3, 127.3, 127.6, 128.5, 128.5, 128.8, 128.9, 129.1, 129.4, 130.0, 134.4, 135.0, 139.1, 139.2, 141.3, 155.5, 166.6, 170.7, 191.6; HRMS (ESI) m/z: calcd for C37H33N2O5S [M + H]+ 617.2105; found 617.2098.
Recorded as a mixture of 7f/8f (85:15): 7f: 1H NMR (CDCl3, 400 MHz) δ = 1.12 (s, 3H), 1.46 (s, 3H), 2.27 (s, 3H), 4.59 (s, 1H), 5.66 (s, 1H), 5.66 (s, 1H), 6.93 (s, 1H), 7.04 (d, J = 8.1 Hz, 2H), 7.30–7.35 (m, 10H), 7.43 (d, J = 8.2 Hz, 2H), 7.46–7.52 (m, 2H), 7.62 (t, J = 7.4 Hz, 1H), 7.94–7.96 (m, 2H); 8f: 1H NMR (CDCl3, 400 MHz) δ = 1.30 (s, 3H), 1.34 (s, 3H), 2.32 (s, 3H), 4.46 (s, 1H), 5.56 (s, 1H), 5.75 (s, 1H), 6.93 (s, 1H), 7.13 (d, J = 8.1 Hz, 2H), 7.30–7.35 (m, 10H), 7.42–7.44 (m, 2H), 7.46–7.52 (m, 2H), 7.56–7.58 (m, 1H), 8.09 (d, J = 7.3 Hz, 2H).
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-(4-methoxyphenyl)-isoxaxoline)-4′,6-penicillanate] (6g) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-(4-methoxyphenyl)-isoxaxoline)-5′,6-penicillanate] (7g) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-(4-methoxyphenyl)-isoxaxoline)-5′,6-penicillanate] (8g). Obtained from hydroximoyl chloride 3g (76 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). The crude product was purified by flash chromatography (hexane/ethyl acetate, 3:1). Method A gave a mixture of 6g/8g (69:31) as a yellow oil (68 mg, 0.107 mmol, 52%). Method B, gave, in order of elution, a mixture of 6g/8g (69:31) as a yellow oil (47 mg, 0.074 mmol, 36%) and 7g as a white solid (28 mg, 0.044 mmol, 21%). Method C gave a mixture of 6g/8g (75:25) as a yellow oil (70 mg, 0.111 mmol, 54%).Recorded as a mixture of 6g/8g (69:31): 6g: 1H NMR (CDCl3, 400 MHz) δ = 1.16 (s, 3H), 1.57 (s, 3H), 3.78 (s, 3H), 4.65 (s, 1H), 5.89 (s, 1H), 6.15 (s, 1H), 6.76 (d, J = 8.9 Hz, 2H), 6.99 (s, 1H), 7.31–7.36 (m, 10H), 7.51–7.55 (m, 2H), 7.63–7.65 (m, 1H), 7.78–7.80 (m, 2H), 8.03–8.06 (m, 2H); 8g: 1H NMR (CDCl3, 400 MHz) δ = 1.16 (s, 3H), 1.57 (s, 3H), 3.86 (s, 3H), 4.66 (s, 1H), 5.89 (s, 1H), 6.16 (s, 1H), 6.65 (d, J = 8.9 Hz, 1H), 6.99 (s, 1H), 7.31–7.36 (m, 10H), 7.51–7.55 (m, 2H), 7.63–7.65 (m, 1H), 7.75 (dd, J = 8.9 and 2.2 Hz, 2H), 7.90 (d, J = 2.2 Hz, 1H), 8.03–8.06 (m, 2H).
7g: mp 94.3–96.3 °C; [α]25D = +260 (c 0.25 in CH2Cl2); IR (ATR): ν = 911, 1717, 1256, 1457, 1516, 1607, 1740 and 1781 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.12 (s, 3H), 1.46 (s, 3H), 3.75 (s, 3H), 4.60 (s, 1H), 5.62 (s, 1H), 5.65 (s, 1H), 6.74 (d, J = 8.9 Hz, 2H), 6.92 (s, 1H), 7.30–7.35 (m, 10H), 7.47–7.51 (m, 4H), 7.62 (t, J = 7.4 Hz, 1H), 7.94–7.96 (m, 2H);13C NMR (CDCl3, 100 MHz) δ = 25.5, 33.4, 55.4, 58.5, 63.7, 68.9, 72.9, 78.7, 101.9, 114.4, 119.7, 127.3, 127.6, 128.4, 128.6, 128.7, 128.8, 128.8, 129.2, 129.3, 134.4, 137.2, 139.1, 139.1, 156.3, 161.7, 166.2, 170.5, 194.6; HRMS (ESI) m/z: calcd for C37H33N2O6S [M + H]+ 633.2054; found 633.2048.
(4′R,5′S)-Benzhydryl spiro[(5-benzoyl-3-cyclohexyl-isoxaxoline)-4′,6-penicillanate] (6h) and (4′S,5′S)-benzhydryl spiro[(4-benzoyl-3-cyclohexyl-isoxaxoline)-5′,6-penicillanate] (7h) and (4′S,5′R)-benzhydryl spiro[(4-benzoyl-3-cyclohexyl-isoxaxoline)-5′,6-penicillanate] (8h). Obtained from hydroximoyl chloride 3h (67 mg, 0.412 mmol) and 6-(Z)-(benzoylmethylidene)penicillanate 5 (100 mg, 0.206 mmol) as described in the general procedure. The crude product was purified by flash chromatography (hexane/ethyl acetate, 3
:1). The crude product was purified by flash chromatography (hexane/ethyl acetate, 3:1). Method A, gave, in order of elution, 6h as a white solid (99 mg, 0.163 mmol, 79%) and a mixture of 7h/8h as a colorless oil (13:87) (19 mg, 0.031 mmol, 15%). Method B, gave, in order of elution, 6h as a white solid (75 mg, 0.123 mmol, 60%) and a mixture of 7h/8h as a colorless oil (10:90) (13 mg, 0.021 mmol, 10%). Method C, gave, in order of elution, 6h as a white solid (40 mg, 0.066 mmol, 35%) and a mixture of 7h/8h (39:61) as a yellow oil (9 mg, 0.015 mmol, 8%).6h: mp 182.7–185.0 °C; [α]25D = +340 (c 0.25 in CH2Cl2); IR (ATR): ν = 882, 1154, 1176, 1450, 1495, 1597, 1668, 1746, 1773, 2856 and 2390 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 1.19 (s, 3H), 1.22–1.26 (m, 3H), 1.53 (s, 3H), 1.61–1.79 (m, 5H), 1.97–2.00 (m, 2H), 2.47 (tt, J = 11.4 and 3.3 Hz, 1H), 4.59 (s, 1H), 5.84 (s, 1H), 5.94 (s, 1H), 6.97 (s, 1H), 7.32–7.38 (m, 10H), 7.49–7.53 (m, 2H), 7.63 (t, J = 7.4 Hz, 1H), 7.99–8.01 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ = 25.8, 26.0, 26.0, 26.2, 31.4, 31.9, 32.7, 36.1, 63.6, 68.0, 70.0, 78.2, 78.7, 80.0, 127.2, 127.6, 128.5, 128.7, 128.8, 128.9, 129.0, 129.4, 134.3, 135.1, 139.1, 139.3, 160.8, 166.5, 170.7, 192.0; HRMS (ESI) m/z: calcd for C36H37N2O5S [M + H]+ 609.2418; found 609.2413.
Recorded as a mixture of 7h/8h (10:90): 7h: 1H NMR (CDCl3, 400 MHz) δ = 1.10 (s, 3H), 1.38 (s, 3H), 1.40–1.88 (m, 10H), 2.03–2.10 (m, 1H), 4.40 (s, 1H, 5.40 (s, 1H), 5.46 (s, 1H), 6.91 (s, 1H), 7.29–7.36 (m, 10H), 7.53 (t, J = 7.8 Hz, 2H), 7.66 (t, J = 7.4 Hz, 1H), 7.95–7.97 (m, 2H); 8h: 1H NMR (CDCl3, 400 MHz) δ = 1.10 (s, 3H), 1.38 (s, 3H), 1.40–1.88 (m, 10H), 2.03–2.10 (m, 1H), 4.53 (s, 1H), 5.25 (s, 1H), 5.62 (s, 1H), 6.91 (s, 1H), 7.29–7.36 (m, 10H), 7.46–7.50 (m, 2H), 7.60 (t, J = 7.4 Hz, 1H), 8.02 (d, J = 7.2 Hz, 2H).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Coimbra Chemistry Centre – Institute of Molecular Sciences (CQC-IMS) is supported by the Portuguese Agency for Scientific Research, “Fundação para a Ciência e a Tecnologia” (FCT) through projects UIDB/00313/2020 and UIDP/00313/2020 (National Funds) and the IMS special complementary funds provided by FCT. Américo J. S. Alves thank FCT for fellowship SFRH/BD/128910/2017. The authors also acknowledge the UC-NMR facility for obtaining the NMR data (https://www.nmrccc.uc.pt).References
- (a) N. Arya, A. Y. Jagdale, T. A. Patil, S. S. Yeramwar, S. S. Holikatti, J. Dwivedi, C. J. Shishoo and K. S. Jain, Eur. J. Med. Chem., 2014, 74, 619–656 CrossRef CAS PubMed; (b) P. D. Mehta, N. P. S. Sengar and A. K. Pathak, Eur. J. Med. Chem., 2010, 45, 5541–5560 CrossRef CAS.
- (a) G. Chaubet, T. Coursindel, X. Morelli, S. Betzi, P. Roche, Y. Guari, A. Lebrun, L. Toupet, Y. Collette, I. Parrot and J. Martinez, Org. Biomol. Chem., 2013, 11, 4719–4726 RSC; (b) E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Molecules, 2019, 24 Search PubMed; (c) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS.
- (a) A. Jarrahpour, E. Ebrahimi, E. De Clercq, V. Sinou, C. Latour, L. Djouhri Bouktab and J. M. Brunel, Tetrahedron, 2011, 67, 8699–8704 CrossRef CAS; (b) G. S. Singh, M. D’hooghe and N. De Kimpe, Tetrahedron, 2011, 67, 1989–2012 CrossRef CAS; (c) H. D. Thi and M. D'Hooghe, Arkivoc, 2018, 314–347 Search PubMed; (d) N. G. Alves, A. J. S. Alves, M. I. L. Soares and T. M. V. D. Pinho e Melo, Adv. Synth. Catal., 2021, 363, 2464–2501 CrossRef CAS.
- (a) A. J. S. Alves, N. G. Alves, C. C. Caratão, M. I. M. Esteves, D. Fontinha, I. Bártolo, M. I. L. Soares, S. M. M. Lopes, M. Prudêncio, N. Taveira and T. M. V. D. Pinho e Melo, Curr. Top. Med. Chem., 2020, 20, 140–152 CrossRef CAS; (b) N. G. Alves, I. Bártolo, A. J. S. Alves, D. Fontinha, D. Francisco, S. M. M. Lopes, M. I. L. Soares, C. J. V. Simões, M. Prudêncio, N. Taveira and T. M. V. D. Pinho e Melo, Eur. J. Med. Chem., 2021, 219, 113439 CrossRef CAS PubMed; (c) I. Bártolo, B. S. Santos, D. Fontinha, M. Machado, D. Francisco, B. Sepodes, J. Rocha, H. Mota-Filipe, R. Pinto, M. E. Figueira, H. Barroso, T. Nascimento, A. P. Alves de Matos, A. J. S. Alves, N. G. Alves, C. J. V. Simões, M. Prudêncio, T. M. V. D. Pinho e Melo and N. Taveira, ACS Infect. Dis., 2021, 7, 421–434 CrossRef.
- (a) B. S. Santos, S. C. C. Nunes, A. A. C. C. Pais and T. M. V. D. Pinho e Melo, Tetrahedron, 2012, 68, 3729–3737 CrossRef CAS; (b) B. S. Santos and T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2013, 2013, 3901–3909 CrossRef CAS; (c) A. J. S. Alves, N. G. Alves, I. Bártolo, D. Fontinha, S. Caetano, M. Prudêncio, N. Taveira and T. M. V. D. Pinho e Melo, Front. Chem., 2022, 10, 1017250 CrossRef PubMed; (d) B. S. Santos, C. S. B. Gomes and T. M. V. D. Pinho e Melo, Tetrahedron, 2014, 70, 3812–3821 CrossRef CAS; (e) A. J. S. Alves and T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2020, 2020, 6259–6269 CrossRef CAS.
- (a) F. M. Cordero, D. Giomi and L. Lascialfari, in Prog. Heterocycl. Chem., ed. G. W. Gribble and J. A. Joule, Elsevier, 2014, vol. 26, pp. 319–348 Search PubMed; (b) G. Kumar and R. Shankar, ChemMedChem, 2021, 16, 430–447 CrossRef CAS PubMed; (c) V. Kumar and K. Kaur, J. Fluorine Chem., 2015, 180, 55–97 CrossRef CAS; (d) J. Liao, L. Ouyang, Q. Jin, J. Zhang and R. Luo, Org. Biomol. Chem., 2020, 18, 4709–4716 RSC.
- (a) M. P. Badart, C. M. L. Squires, S. K. Baird and B. C. Hawkins, Tetrahedron Lett., 2016, 57, 5108–5111 CrossRef CAS; (b) S. Bardhan, D. C. Schmitt and J. A. Porco, Org. Lett., 2006, 8, 927–930 CrossRef CAS PubMed; (c) P. Das and A. T. Hamme II, Eur. J. Org. Chem., 2015, 2015, 5159–5166 CrossRef CAS.
- (a) A. Abolhasani, F. Heidari, S. Noori, S. Mousavi and H. Abolhasani, Curr. Chem. Biol., 2020, 14, 38–47 CrossRef CAS; (b) É. Frank, D. Kovács, G. Schneider, J. Wölfling, T. Bartók and I. Zupkó, Mol. Diversity, 2014, 18, 521–534 CrossRef PubMed; (c) M. Gul and A. Tutar, J. Heterocycl. Chem., 2014, 51, 327–335 CrossRef CAS; (d) D. M. Reddy, N. A. Qazi, S. D. Sawant, A. H. Bandey, J. Srinivas, M. Shankar, S. K. Singh, M. Verma, G. Chashoo, A. Saxena, D. Mondhe, A. K. Saxena, V. K. Sethi, S. C. Taneja, G. N. Qazi and H. M. Sampath Kumar, Eur. J. Med. Chem., 2011, 46, 3210–3217 CrossRef CAS PubMed; (e) C. J. A. Ribeiro, J. D. Amaral, C. M. P. Rodrigues, R. Moreira and M. M. M. Santos, Bioorg. Med. Chem., 2014, 22, 577–584 CrossRef CAS.
- S. Pratap, F. Naaz, S. Reddy, K. K. Jha, K. Sharma, D. Sahal, M. Akhter, D. Nayakanti, H. M. S. Kumar, V. K. Pandey and S. Shafi, Arch. Pharm., 2019, 352, 1800192 Search PubMed.
- (a) P. Das, S. Boone, D. Mitra, L. Turner, R. Tandon, D. Raucher and A. T. Hamme, RSC Adv., 2020, 10, 30223–30237 RSC; (b) P. Das, M. H. Hasan, D. Mitra, R. Bollavarapu, E. J. Valente, R. Tandon, D. Raucher and A. T. Hamme, J. Org. Chem., 2019, 84, 6992–7006 CrossRef CAS.
- D. F. Corbett, J. Chem. Soc., Perkin Trans. 1, 1986, 421–428 RSC.
- (a) S. Anklam and J. Liebscher, Tetrahedron, 1998, 54, 6369–6384 CrossRef CAS; (b) A. Strauss and H.-H. Otto, Helv. Chim. Acta, 1997, 80, 1823–1830 CrossRef CAS.
- (a) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustain. Chem. Process., 2016, 4, 7 CrossRef; (b) R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC; (c) D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (b) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC.
- (a) P. Brandão, M. Pineiro and T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2019, 2019, 7188–7217 CrossRef; (b) R. Gérardy, N. Emmanuel, T. Toupy, V.-E. Kassin, N. N. Tshibalonza, M. Schmitz and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2018, 2301–2351 CrossRef; (c) S. V. Ley, Chem. Rec., 2012, 12, 378–390 CAS; (d) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC; (e) L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC; (f) L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC.
- (a) P. Bana, R. Örkényi, K. Lövei, Á. Lakó, G. I. Túrós, J. Éles, F. Faigl and I. Greiner, Bioorg. Med. Chem., 2017, 25, 6180–6189 CrossRef CAS PubMed; (b) J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC; (c) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed; (d) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC; (e) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (b) D. L. Hughes, Org. Process Res. Dev., 2018, 22, 13–20 CrossRef CAS; (c) S. G. Koenig and H. F. Sneddon, Green Chem., 2017, 19, 1418–1419 RSC; (d) L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed; (e) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS; (f) A. R. Bogdan and A. W. Dombrowski, J. Med. Chem., 2019, 62, 6422–6468 CrossRef CAS PubMed.
- (a) M. Baumann, I. R. Baxendale and S. V. Ley, Mol. Diversity, 2011, 15, 613–630 CrossRef CAS PubMed; (b) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- (a) A. V. Dubrovskiy and R. C. Larock, Org. Lett., 2010, 12, 1180–1183 CrossRef CAS PubMed; (b) J.-T. Li, X.-L. Li and T.-S. Li, Ultrason. Sonochem., 2006, 13, 200–202 CrossRef CAS PubMed.
- K. J. Dignam, A. F. Hegarty and P. L. Quain, J. Chem. Soc., Perkin trans. II, 1977, 1457–1462 RSC.
- K. Livingstone, S. Bertrand, A. R. Kennedy and C. Jamieson, Chem. - Eur. J., 2020, 26, 10591–10597 CrossRef CAS PubMed.
- L. K. Gibbons, C. J. Peake, W. N. Harnish and F. M. C. Corporation, N-(Sulfonyloxy) benzimidoyl halides as bactericidal or fungicidal agents, US Pat., US3983246A, 1976.
- B. C. Sanders, F. Friscourt, P. A. Ledin, N. E. Mbua, S. Arumugam, J. Guo, T. J. Boltje, V. V. Popik and G.-J. Boons, J. Am. Chem. Soc., 2011, 133, 949–957 CrossRef CAS PubMed.
- Q. Feng, H. Huang and J. Sun, Org. Lett., 2021, 23, 2431–2436 CrossRef PubMed.
- A. Q. Hussein, M. M. El-Abadelah and W. S. Sabri, J. Heterocycl. Chem., 1983, 20, 301–304 CrossRef CAS.
- Y. Uchida and S. Kozuka, Bull. Chem. Soc. Jpn., 1984, 57, 2011–2012 CrossRef CAS.
- S. Castellano, D. Kuck, M. Viviano, J. Yoo, F. López-Vallejo, P. Conti, L. Tamborini, A. Pinto, J. L. Medina-Franco and G. Sbardella, J. Med. Chem., 2011, 54, 7663–7677 CrossRef CAS PubMed.
- S. Stotani, V. Gatta, F. Medda, M. Padmanaban, A. Karawajczyk, P. Tammela, F. Giordanetto, D. Tzalis and S. Collina, Molecules, 2018, 23, 2545 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra04848e |
| This journal is © The Royal Society of Chemistry 2022 |