DOI:
10.1039/D2RA04835C
(Paper)
RSC Adv., 2022,
12, 24830-24838
The straightforward synthesis of N-coordinated ruthenium 4-aryl-1,2,3-triazolato complexes by [3 + 2] cycloaddition reactions of a ruthenium azido complex with terminal phenylacetylenes and non-covalent aromatic interactions in structures†
Received
3rd August 2022
, Accepted 22nd August 2022
First published on 31st August 2022
Abstract
The straightforward preparation of N-coordinated ruthenium triazolato complexes by [3 + 2] cycloaddition reactions of a ruthenium azido complex [Ru]–N3 (1, [Ru] = (η5-C5H5)(dppe)Ru, dppe = Ph2PCH2CH2PPh2) with a series of terminal phenylacetylenes is reported. The reaction products, N(2)-bound ruthenium 4-aryl-1,2,3-triazolato complexes such as [Ru]N3C2H(4-C6H4CN) (2), [Ru]N3C2H(4-C6H4CHO) (3), [Ru]N3C2H(4-C6H4F) (4), [Ru]N3C2H(Ph) (5) and [Ru]N3C2H(4-C6H4CH3) (6) were produced from 4-ethynylbenzonitrile, 4-ethynylbenzaldehyde, 1-ethynyl-4-fluorobenzene, phenylacetylene and 4-ethynyltoluene, respectively, at 80 °C or above under an atmosphere of air. To the best of our knowledge, this is the first example of the preparation of N-coordinated ruthenium aryl-substituted 1,2,3-triazolato complexes by the [3 + 2] cycloaddition of a metal-coordinated azido ligand and a terminal aryl acetylene, less electron-deficient terminal aryl alkynes. All of the compounds have been fully characterized and the structures of complexes 2, 3, 5 and 6 were confirmed by single-crystal X-ray diffraction analysis. Each compound participates in non-covalent aromatic interactions in the solid-state structure which can be favorable in the binding of DNA/biomolecular targets and has shown great potential in the development of biologically active anticancer drugs.
Introduction
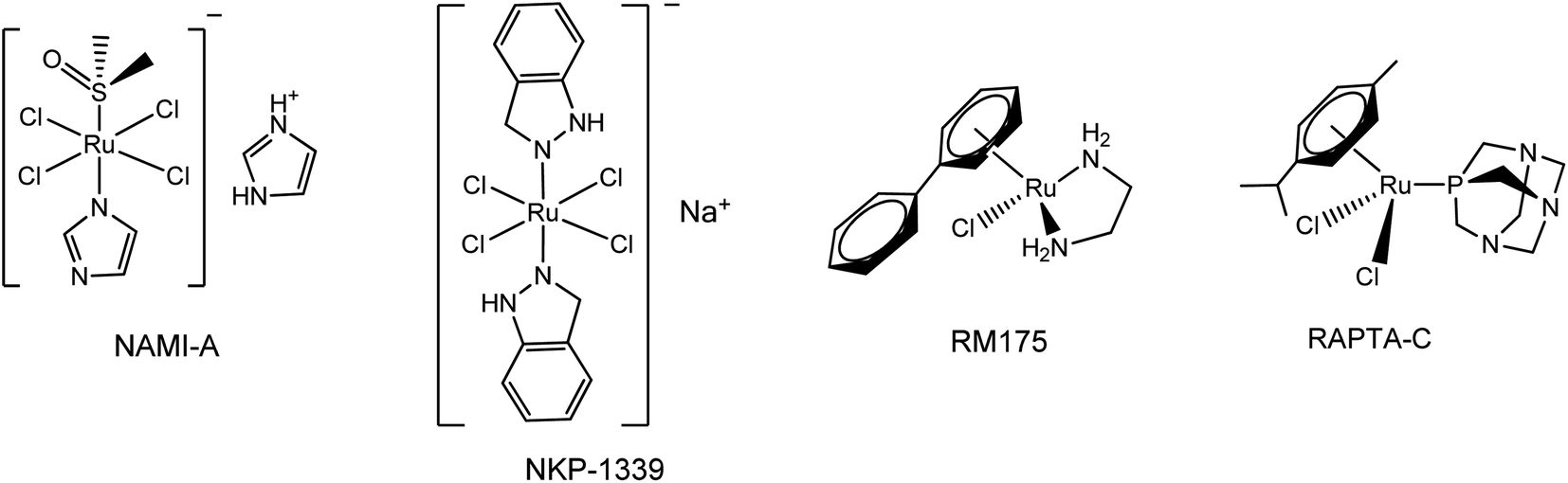
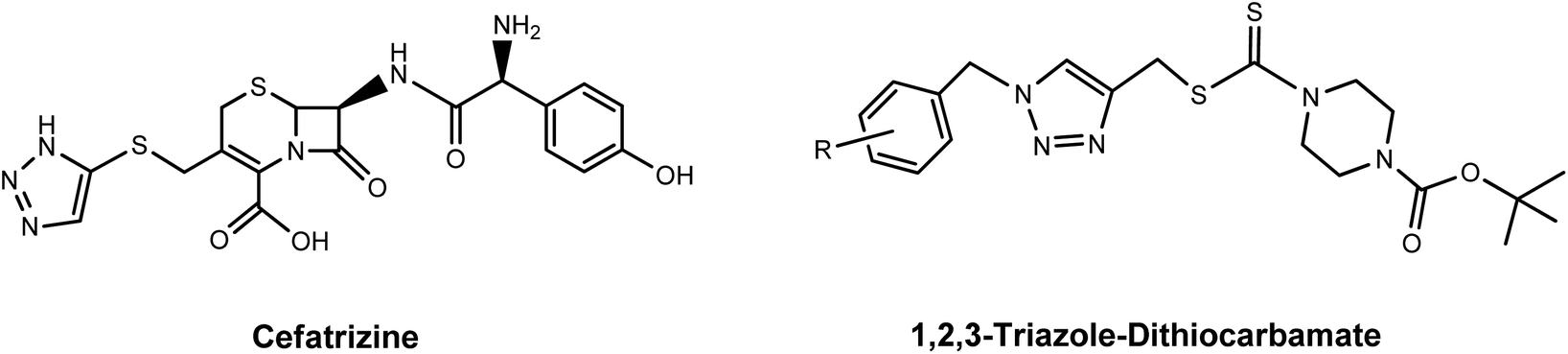
Since the successful introduction of cisplatin1 as an anticancer drug in the 1980s, interest in the field of bioorganometallic chemistry has rapidly increased and new metal agents in addition to platinum have been investigated as antitumor agents.2 Among the various transition metal complexes that have been investigated, ruthenium complexes have emerged as promising alternatives to platinum-based drugs, with three ruthenium(III) compounds, NAMI-A, NKP-1339, and TLD-1433 having proceeded to phase I clinical trials and two piano-stool Ru(II) arene complexes RM175 and RAPTA-C currently in preclinical studies.3–5 Investigations into their mechanism of action revealed that the anticancer properties of these Ru(III) and Ru(II) complexes are largely the result of metal–DNA/biomolecule non-covalent interactions such as aromatic π-stacking and C–H⋯π interactions involving the aromatic moieties on these drugs and biomolecules.6 Since the aromatic substituents on the ruthenium complexes are crucial for their biological activity, a number of efforts have been made to synthesize various ruthenium arene complexes with potential anticancer activity.7 Research on novel anticancer drugs has recently focused on ruthenium complexes bearing nitrogen-containing heterocyclic ligands, especially 1,2,3-triazole derivatives.7a,8 The 1,2,3-triazoles, one of the most important classes of nitrogen-containing heterocycles, have diverse biological activities that include antiviral, antibacterial, antifungal and antitubercular activities and excellent hydrogen donating and accepting ability, and have been applied in medicine and in pharmaceuticals during the past decades.9 Moreover, 1,2,3-triazole-containing molecules with anticancer activity are being widely synthesized and extensively tested. Some of these, such as Cefatrizine and 1,2,3-triazole–dithiocarbamate have demonstrated excellent anticancer activity and have been used in clinics for the treatment of cancer.10 Accordingly, in an attempt to synergistically combine the key structural and biological features of ruthenium, aromatic rings and 1,2,3-triazole compounds, the synthesis of novel biologically active ruthenium complexes bearing 1,2,3-triazole and aromatic rings with potential anticancer activity has attracted a broad spectrum of interest from researchers in related fields (Fig. 1 and 2).11
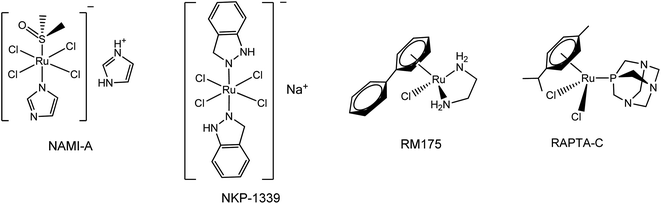 |
| | Fig. 1 Examples of ruthenium anti-cancer complexes that are currently in clinical or preclinical trials. | |
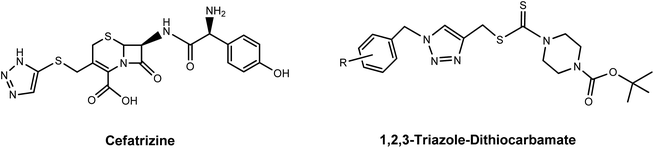 |
| | Fig. 2 The 1,2,3-triazole derivatives used in clinics for cancer treatment. | |
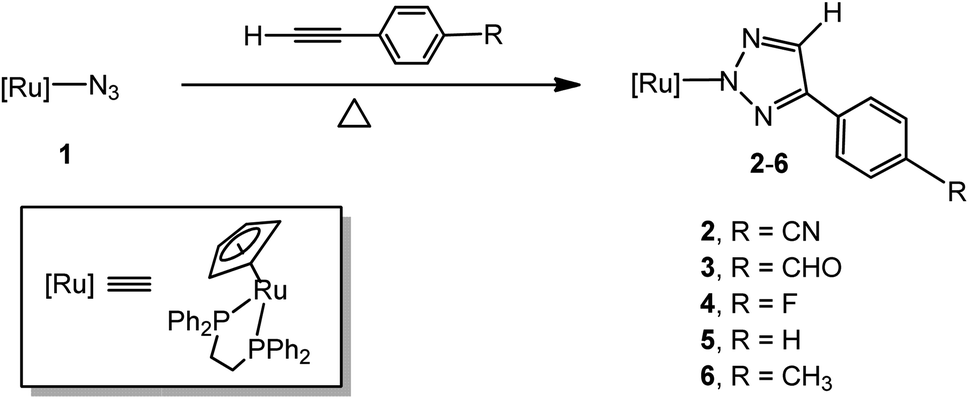
A general strategy for the synthesis of metal-coordinated triazolato complexes is the [3 + 2] cycloaddition between a metal-coordinated azide group and an activated alkyne.12 In most cases, the activated alkynes that are used as dipolarophiles are limited to highly electron-poor internal alkynes (R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R′, where R, R′ = electron withdrawing groups such as CO2Me, CO2Et, and CF3). Aryl groups can be electron withdrawing through inductive effects and resonance but their electron withdrawing ability is highly dependent on the substituent on the phenyl ring.13 Currently, the metal-coordinated aryl-substituted triazolates used for biological or medicinal evaluation are generally synthesized by ligand-exchange reactions of organometallic or inorganic complexes14,15 with organic triazoles prepared via copper(I) catalysed alkyne–azide cycloaddition (CuAAC)/click chemistry.16 These methods have resulted in significant improvements and advances in the synthesis of N-coordinated metal aryl-substituted triazolates, but most of these approaches have some limitations, which include issues such as tedious workup procedures for preparing a variety of functionalized starting materials such as organic functionalized alkynes and azides, inorganic and organometallic metal compounds and the use of strongly basic reagents such as lithium tert-butoxide or lithium methoxide. Therefore, the development of a facile and straightforward methodology for the synthesis of ruthenium aryl-substituted 1,2,3-triazoles would be of great interest. The design of aryl-functionalized ruthenium heterocyclic complexes to generate potential biologically active ruthenium heterocyclic compounds is a subject of our interest.17 In a continuation of our previous studies on ruthenium 1,3-dipolar cycloaddition18 and in view of our interest in the development of facile and convenient methods for the synthesis of biologically active ruthenium aryl-substituted triazolates, we report herein on a simple one-step synthesis of ruthenium 4-aryl-1,2,3-triazole complexes by the [3 + 2] cycloaddition of a ruthenium azido complex with a series of terminal phenylacetylenes. We now report on the results of detailed synthetic and structural investigations into this reaction, a preliminary account of the steric and electronic effects for the 1,3-dipolarophiles is given and the results of observations of the non-covalent aromatic interactions in the solid state of the thus formed products is reported as well.
C–R′, where R, R′ = electron withdrawing groups such as CO2Me, CO2Et, and CF3). Aryl groups can be electron withdrawing through inductive effects and resonance but their electron withdrawing ability is highly dependent on the substituent on the phenyl ring.13 Currently, the metal-coordinated aryl-substituted triazolates used for biological or medicinal evaluation are generally synthesized by ligand-exchange reactions of organometallic or inorganic complexes14,15 with organic triazoles prepared via copper(I) catalysed alkyne–azide cycloaddition (CuAAC)/click chemistry.16 These methods have resulted in significant improvements and advances in the synthesis of N-coordinated metal aryl-substituted triazolates, but most of these approaches have some limitations, which include issues such as tedious workup procedures for preparing a variety of functionalized starting materials such as organic functionalized alkynes and azides, inorganic and organometallic metal compounds and the use of strongly basic reagents such as lithium tert-butoxide or lithium methoxide. Therefore, the development of a facile and straightforward methodology for the synthesis of ruthenium aryl-substituted 1,2,3-triazoles would be of great interest. The design of aryl-functionalized ruthenium heterocyclic complexes to generate potential biologically active ruthenium heterocyclic compounds is a subject of our interest.17 In a continuation of our previous studies on ruthenium 1,3-dipolar cycloaddition18 and in view of our interest in the development of facile and convenient methods for the synthesis of biologically active ruthenium aryl-substituted triazolates, we report herein on a simple one-step synthesis of ruthenium 4-aryl-1,2,3-triazole complexes by the [3 + 2] cycloaddition of a ruthenium azido complex with a series of terminal phenylacetylenes. We now report on the results of detailed synthetic and structural investigations into this reaction, a preliminary account of the steric and electronic effects for the 1,3-dipolarophiles is given and the results of observations of the non-covalent aromatic interactions in the solid state of the thus formed products is reported as well.
Results and discussion
Reactions of ruthenium–azido complex 1 with terminal phenylacetylenes
The treatment of [Ru]-N3 (1, [Ru] = (η5-C5H5)(dppe)Ru, dppe = Ph2PCH2CH2PPh2) with a 3-fold excess of 4-ethynylbenzonitrile in benzene at 80 °C in a silicone oil bath under an atmosphere of air for 48 hours afforded the [3 + 2] cycloaddition product, the N(2)-bound 4-(4-cyanophenyl)-1,2,3-triazolate complex [Ru]N3C2H(4-C6H4CN) (2), in an isolated yield of 89% (Scheme 1). Because no obvious color change was observed, the reaction was monitored by 31P NMR spectroscopy. When 1 was treated with 4-ethynylbenzonitrile in benzene at 80 °C in a silicone oil bath for 24 hours, the resonance at δ 81.5 for 1 became smaller and a new peak at δ 87.3, attributed to 2, appeared and became larger and sharper in the 31P NMR spectrum with time. The reaction reached completion in ca. 48 hours. The FAB mass spectrum of 2 displayed a parent peak at m/z 734.1 (M+). [Ru]N3C2H(4-C6H4CHO) (3), [Ru]N3C2H(4-C6H4F) (4), [Ru]N3C2H(Ph) (5) and [Ru]N3C2H(4-C6H4CH3) (6) were prepared in good to high yields by a procedure similar to that of 2 at higher temperature (for 3, 100 °C, 48 h; for 4, 100 °C, 72 h; for 5, 140 °C, 72 h; for 6, 140 °C, 96 h). The structures of complexes 2, 3, 5 and 6 were confirmed by single-crystal X-ray diffraction analysis and were structurally characterized as being N(2)-bound. It is notable that the time-elapsed 31P NMR study of the formation of 2–6 did not provide any evidence for the formation of the N(1)-bound isomer during the reaction. Each synthesis step involved here was straightforward and provided the desired N(2)-bound product in pure form. The evidence obtained to date indicates that either the two N(1)- and N(2)-bonded isomers are formed simultaneously or that the N(2)-bound isomer is the exclusive product.12 Since the azide ligand is N(1)-coordinated in 1, it appears that a 1,2-shift occurs during or after the cycloaddition reactions.
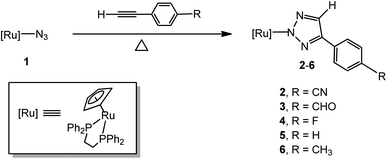 |
| | Scheme 1 Synthesis of ruthenium 4-aryl-1,2,3-triazolato complexes 2–6. | |
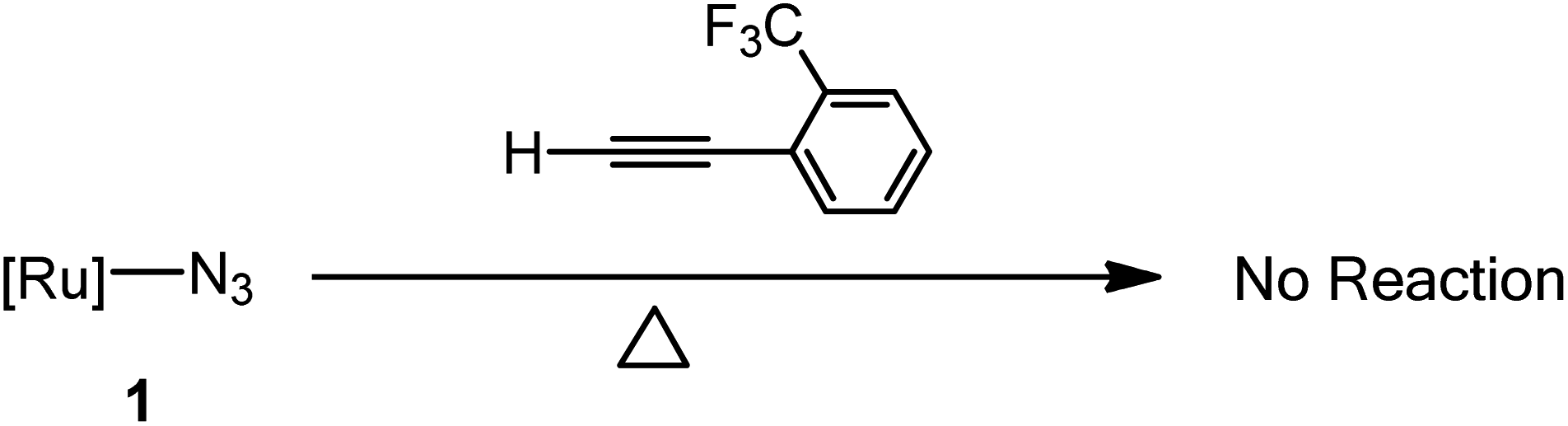
In this study, initial attempts were carried out at ambient temperature for more than one week but this resulting in the complete recovery of the starting materials. The reactions of these phenylacetylenes toward 1 were then examined at a higher temperature and the time for reaction to reach completion was noticeably longer than that of the corresponding reactions with highly electron-poor alkynes.17,18 Furthermore, the reaction of 1 with phenylacetylene and 4-ethynyltoluene proceeded noticeably slower than the corresponding reactions with 4-ethynylbenzonitrile, 4-ethynylbenzaldehyde and 1-ethynyl-4-fluorobenzene, phenyl acetylenes with an electron-withdrawing group at the 4-position of the phenyl ring. In addition, we examined on the steric effect of a substituent on the phenyl group of the acetylene derivatives by treating 2-(trifluoromethyl)phenylacetylene with 1 and resulting in the complete recovery of the starting materials when the reaction was run at 140 °C for more than 5 days (Scheme 2).
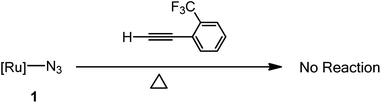 |
| | Scheme 2 The 2-(trifluoromethyl)phenylacetylene tested for its ability to undergo cycloaddition with [Ru]–N3 (1). No reaction observed in C6D6 at 140 °C in a silicone oil bath for 5 days. | |
In this study, the cycloaddition reactivity is highly related to the electron-withdrawing ability of the substituent on the phenyl group of the acetylene. Aryl groups can be electron withdrawing or electron donating through inductive effects and resonance.13 Strong electron-withdrawing groups such as –CN, –CHO and –F at the 4-position of the phenyl ring greatly activated the acetylenes and electron-donating groups such as –CH3 on the phenyl ring slightly deactivated the acetylene. The reactivity of 1 with H–CC–C6H4–R, as shown in Table 1, was comparable with the trend for electron-withdrawing ability (–CN > –CHO > –F > –H > –CH3) of the substituent on the phenyl ring. We also examined the steric effect of the substituent on the phenyl group of the acetylene by treating 2-(trifluoromethyl)phenylacetylene, which contains a strong electron-withdrawing group on 2-position of phenyl ring, in terms of its reactivity towards 1, and this resulted in the complete recovery of the starting materials. A –CF3 group at the 2-position on the phenyl ring has a mild electron-withdrawing ability, but its larger steric hindrance prevents the formation of cycloaddition products.
Table 1 Experimental conditions for cycloaddition reactions of [Ru]–N3 (1) and phenyl acetylenes. Each experiment was carried out in a sealed screw tube in a temperature-controlled silicone oil bath under an atmosphere of air
| Phenyl acetylene |
Solvent |
Conditions |
Product |
Yield (%) |
| H–CC-(4-C6H4CN) |
Benzene |
80 °C, 48 h |
2 |
89 |
| H–CC-(4-C6H4CHO) |
Benzene |
100 °C, 48 h |
3 |
88 |
| H–CC-(4-C6H4F) |
Benzene |
100 °C, 72 h |
4 |
73 |
| H–CC-(4-C6H5) |
Toluene |
140 °C, 72 h |
5 |
86 |
| H–CC-(4-C6H4CH3) |
Toluene |
140 °C, 96 h |
6 |
87 |
The formation of metal aryl-substituted triazolato complexes by the [3 + 2] cycloaddition of a metal-coordinated azide with an aryl alkyne is rare. Although T. G. Gary and N. Metzler-Nolte separately reported on the [3 + 2] cycloaddition reactions of gold azido complexes R3PAuN3 with aryl acetylenes affording a series of carbon-coordinated triazolato complexes,19 the proposed mechanism for such reactions involved the reaction proceeding via a initial ligand–exchange reaction of a gold-azide with an aryl acetylene and the subsequent cyclization of thus formed acetylide and organic azide. The cycloaddition products 2–6 prepared in this study, to the best of our knowledge, is the first example of nitrogen-coordinated metal aryl-substituted 1,2,3-triazolate products being formed by the straightforward [3 + 2] cycloaddition of a metal azide with aryl acetylenes. The ruthenium 4-aryl-1,2,3-triazolate complexes 2–6 are yellow solids, soluble in chloroform and dichloromethane, moderately soluble in acetone, methanol, benzene, toluene and ether, slightly soluble in n-hexane and n-pentane, stable in air and in moisture with a high heat tolerance. The treatment of 2–6 in CDCl3 with a trace of acid (HCl/H2O or CF3COOH) at room temperature immediately afforded a stable mixture of N(1) and N(3)-protonated 1,2,3-triazolato complexes and no Ru–N bond breaking was detected. Collectively these experiments suggest that the ruthenium–nitrogen bond is essentially stable, which bodes favorably for materials and medical applications.
Crystal structures and molecular non-covalent interactions
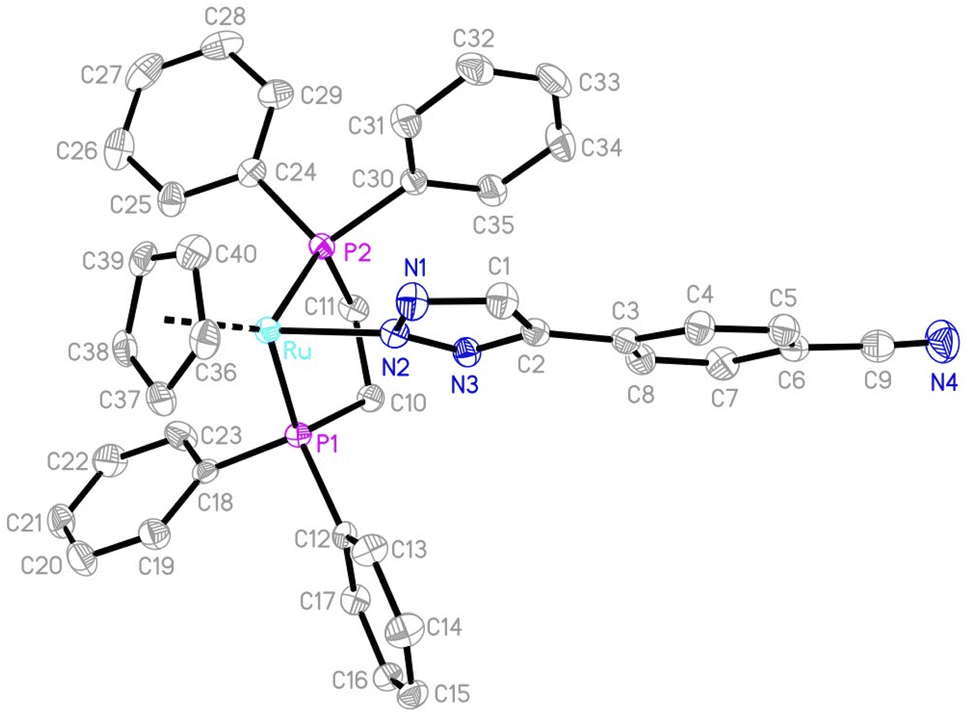
The structures of complexes 2, 3, 5 and 6 were confirmed by single-crystal X-ray diffraction analysis and structurally characterized. ORTEP diagrams are shown in Fig. 3 and S1–S3 in the ESI,† respectively, and the selected bond distances and angles are listed in Table 2. The molecular structures of complexes 2, 3, 5 and 6 can all be described as having a three-legged piano-stool geometry, with the central Ru atom being surrounded by a η5-cyclopentadiene, a N(2)-bound triazole and two phosphine ligands. As a representative example, in 2, the coordination with ruthenium is a piano-stool geometry, with the cyclopentadienyl ring occupies the apex of the stool (Ru–C = 2.205(2)–2.240(2) Å; average 2.2252 Å); the other three positions are occupied by two phosphine ligands (Ru–P1 = 2.2678(6) Å; Ru–P2 = 2.2831(5) Å) and the triazole group (Ru–N2 =2.0850(18) Å), which are comparable to those in other ruthenium triazolato complexes.17,18 The inter-atomic distances within the five-membered triazole ring (N1–N2 (1.347(2) Å), N2–N3 (1.333(2) Å), N1–C1 (1.337(3) Å), N3–C2 (1.351(3) Å) and C1–C2 (1.386(3) Å)), all display a partial double-bond character, suggest a delocalized π bonding mode and that the triazole ring has an essentially planar structure. The five-membered triazole ring exhibits an irregular pentagonal structure with bond angles of N3–N2–N1 (112.59(17)°), N2–N3–C2 (105.85(17)°), N3–C2–C1 (107.37(19)°), N1–C1–C2 (109.2(2)°) and C1–N1–N2 (104.98(18)°) all in the range of C(sp2) and N(sp2) hybridization of a five-membered heterocycle ring. The inter-atomic distances within the five-membered triazole rings are similar to that of 2 and the angles in 3, 5 and 6, as listed in Table 2, are slightly different from that of 2, but are still consistent with the delocalization of the electrons within the triazole rings. These minor differences are possibly due to the different non-covalent interactions between the molecular structures.
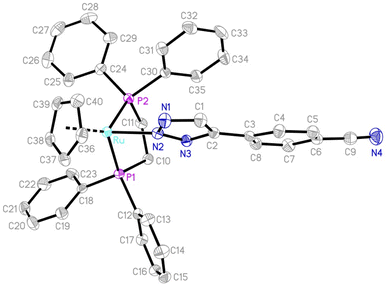 |
| | Fig. 3 An ORTEP drawing of 2 with thermal ellipsoids shown at a 50% probability level. Hydrogen atoms have been omitted for clarity. | |
Table 2 Selected bond distances (Å) and angles (deg) for 2, 3, 5 and 6
| |
2 |
3 |
5 |
6 |
| Ru–P1 |
2.2678(6) |
2.2640(5) |
2.2702(8) |
2.2837(4) |
| Ru–P2 |
2.2831(5) |
2.2803(5) |
2.2726(8) |
2.2800(4) |
| Ru–N2 |
2.0850(18) |
2.0827(16) |
2.094(3) |
2.1067(14) |
| N1–N2 |
1.347(2) |
1.347(2) |
1.332(4) |
1.333(2) |
| N2–N3 |
1.333(2) |
1.331(2) |
1.335(4) |
1.3483(19) |
| N3–C2 |
1.351(3) |
1.353(2) |
1.349(4) |
1.352(2) |
| C1–C2 |
1.386(3) |
1.385(3) |
1.382(5) |
1.383(2) |
| N1–C1 |
1.337(3) |
1.342(3) |
1.344(4) |
1.347(2) |
| P1–Ru–P2 |
83.940(19) |
84.028(18) |
84.51(3) |
83.008(14) |
| N2–Ru–P1 |
91.90(5) |
91.98(4) |
89.70(7) |
90.74(4) |
| N2–Ru–P2 |
86.49(5) |
86.64(5) |
85.03(8) |
92.48(4) |
| N1–N2–Ru |
119.57(14) |
119.18(12) |
124.0(2) |
126.91(11) |
| N3–N2–Ru |
127.21(13) |
127.55(12) |
123.2(2) |
120.98(11) |
| N2–N3–C2 |
105.85(17) |
105.94(16) |
106.1(3) |
106.25(13) |
| N1–N2–N3 |
112.59(17) |
112.65(15) |
112.7(2) |
111.75(13) |
| N2–N1–C1 |
104.98(18) |
104.95(17) |
104.8(3) |
105.86(14) |
| N1–C1–C2 |
109.2(2) |
109.09(18) |
109.5(3) |
108.91(15) |
| N3–C2–C1 |
107.37(19) |
107.36(17) |
106.8(3) |
107.23(15) |
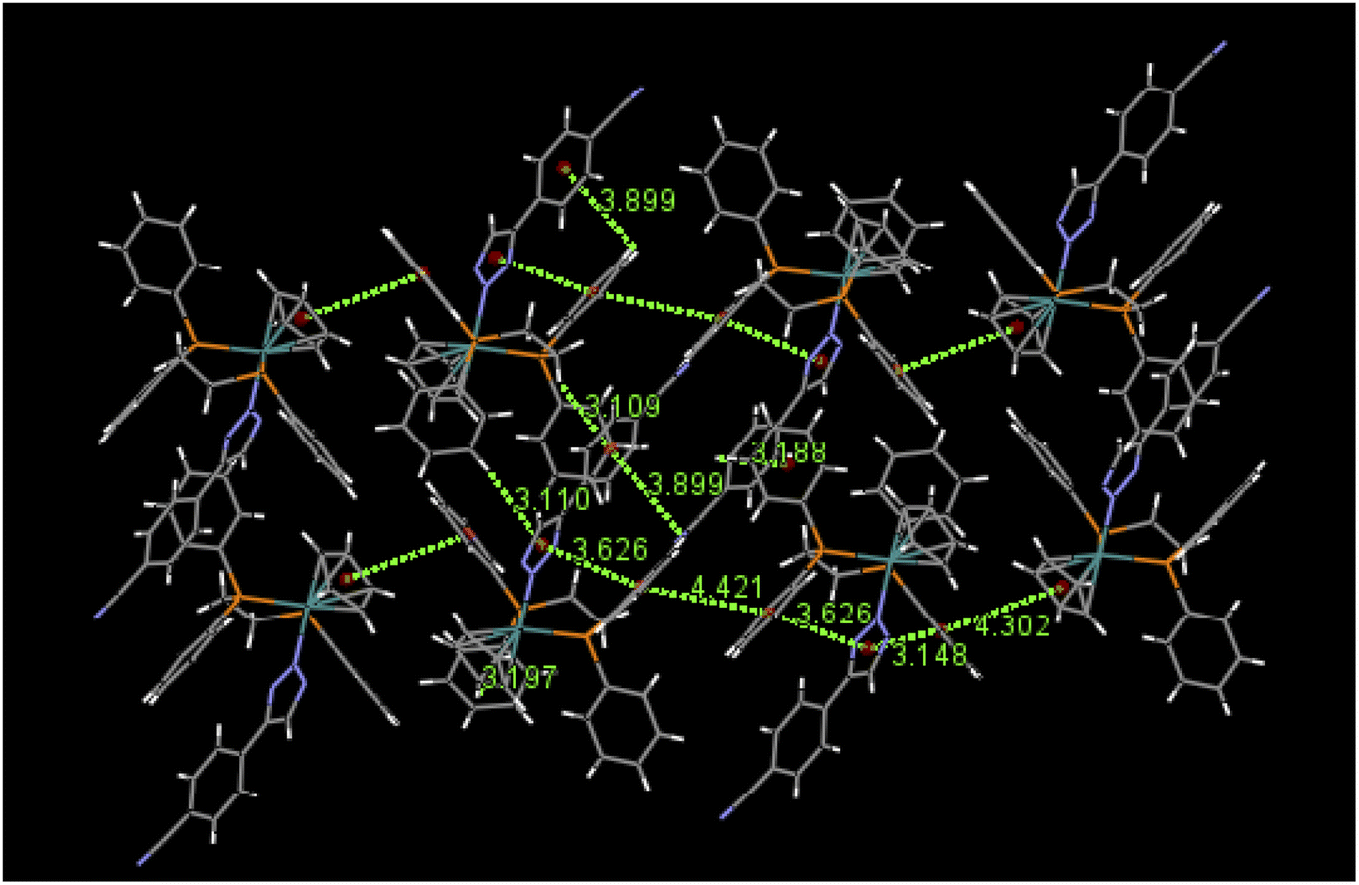
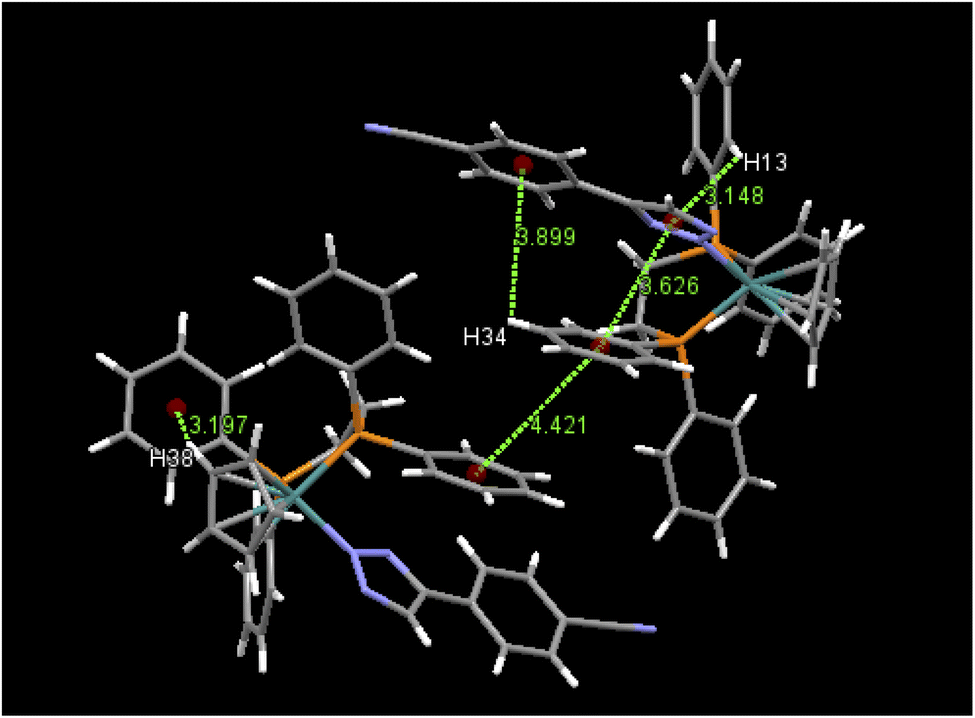
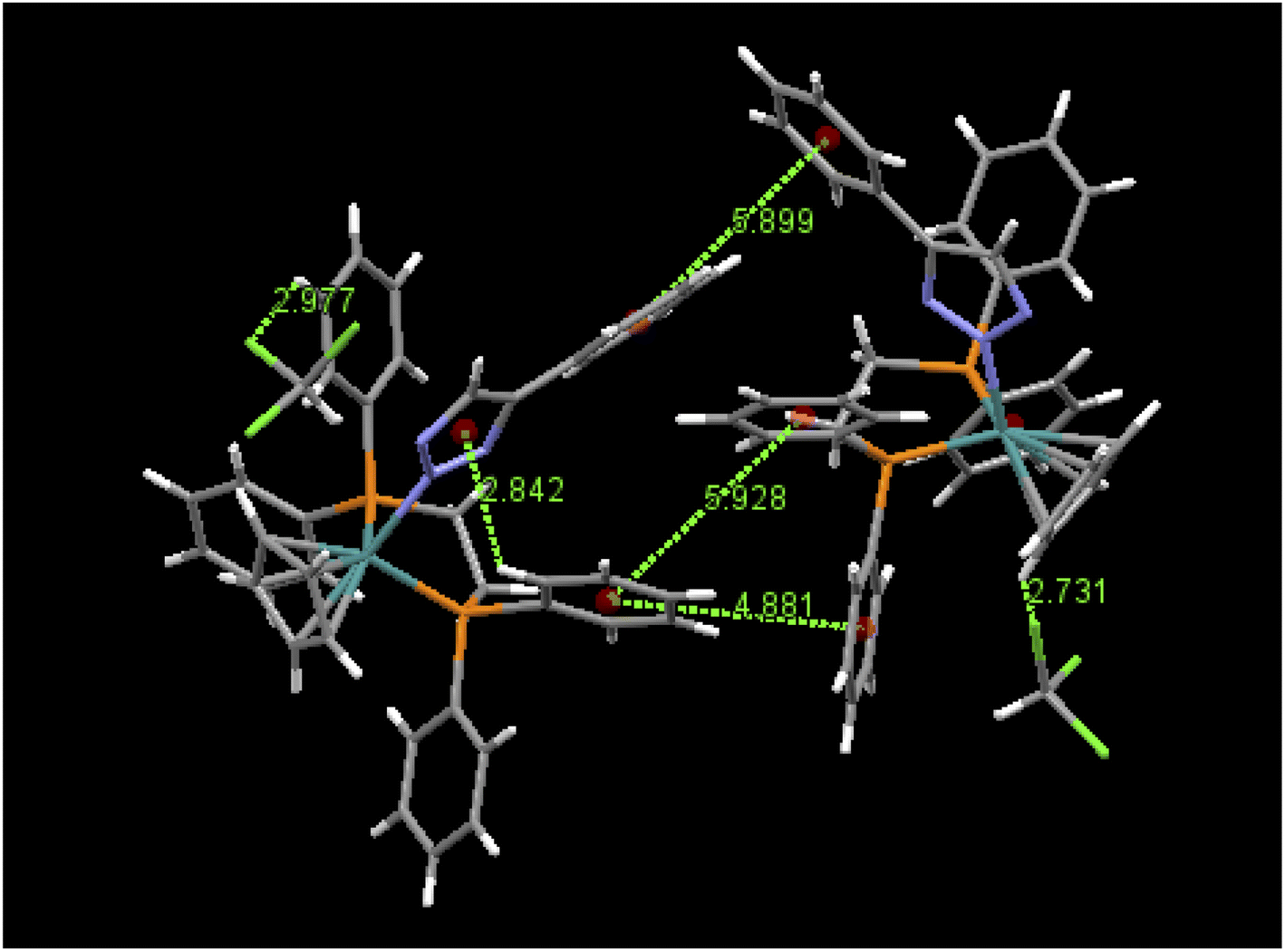
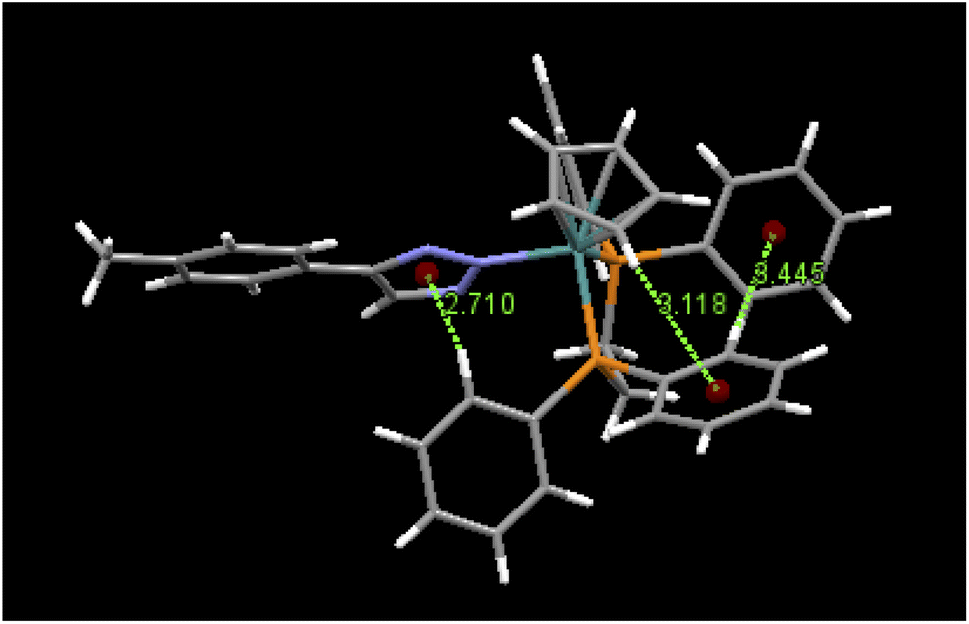
An examination of packing diagrams reveals that each of these molecules participates in significant intermolecular and intramolecular non-covalent interactions other than van der Waals contacts in the molecular packing of compounds 2, 3, 5 and 6. For example, in 2, there are various π-stacking and aromatic C–H⋯π interactions involving the aromatic moieties which include the cyclopentadienyl group, the triazole ring and phenyl groups, as shown in green dashed lines in Fig. 4. A close view of 2-dimer, as shown in Fig. 5, reveals that there is an intramolecular and an intermolecular offset face-to-face or parallel-displaced π stacking mode involving the triazole ring and neighboring phenyl rings at centroid-to-centroid distances of 3.626 and 4.421 Å, respectively, with strong electrostatic attractions between the aromatic species. Additionally, there are three intramolecular aromatic C–H⋯π interactions involving aromatic rings and hydrogen atoms on neighboring aromatic species at controid-to-H distances of 3.148, 3.197 and 3.889 Å, respectively. The crystal structure of 3, which exerts an electrostatic effect on the triazole-tethered phenyl ring similar to that of 2, exhibits a similar π stacking mode to that of 2 and shows a variety of π⋯π and C–H⋯π interactions as well, as shown in Fig. S4 in the ESI.† Interestingly, the molecular structure of 5 does not exhibit the same face-to-face π stacking as 2 and 3, but, rather, contains an intramolecular aromatic C–H⋯π interaction at a distance of 2.842 Å and different intermolecular π-stacking modes, as shown in Fig. 6. In 5, there are two intermolecular edge-to-face or T-shaped π-stacking interactions and a weak parallel-displaced π stacking and an intramolecular aromatic C–H⋯π interaction. The centroid-to-centroid distances of 4.881, 5.899 and 5.928 Å, respectively, are greater than that in 2 but remain within the range of typical π stacking interactions.20 The chloroform solvate molecules in 5 participate in intermolecular interactions by aromatic C–H⋯Cl hydrogen bonds. The structure of 6 exhibits only intramolecular aromatic C–H⋯π interactions involving the aromatic rings and hydrogen atoms on neighboring aromatic rings, as shown in Fig. 7. No significant intermolecular interaction was observed in the structure of 6.
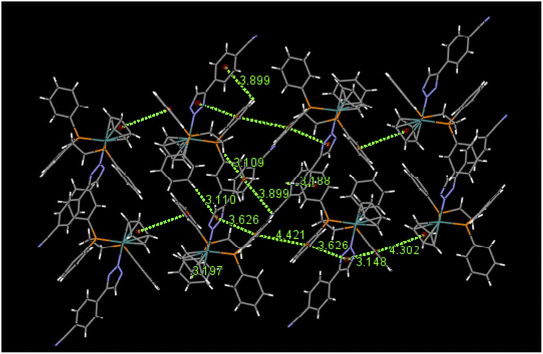 |
| | Fig. 4 Selected π⋯π and aromatic C–H⋯π interactions in the packing of 2. | |
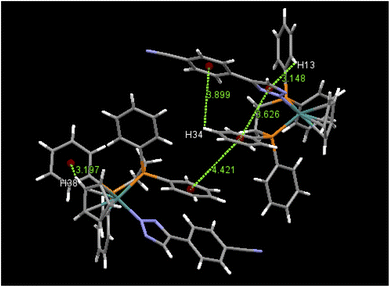 |
| | Fig. 5 Selected intramolecular and intermolecular π⋯π and aromatic C–H⋯π interactions in 2-dimer and their distances (Å). | |
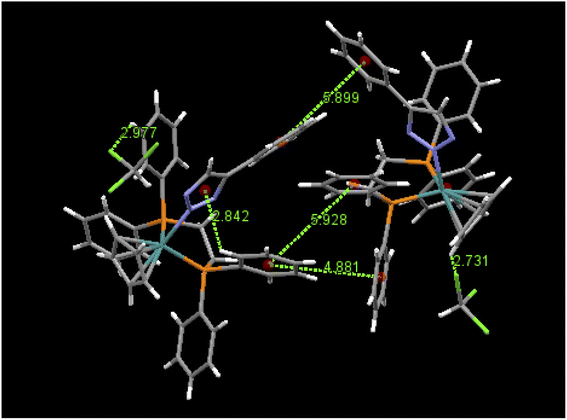 |
| | Fig. 6 Intermolecular π⋯π interactions, aromatic C–H⋯Cl interactions and an intramolecular C–H⋯π interaction in 5·CHCl3-dimer and their distances (Å). | |
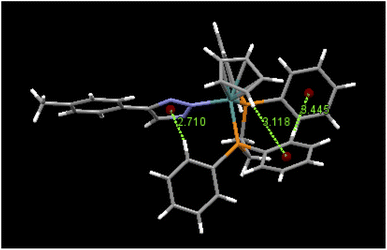 |
| | Fig. 7 The intramolecular aromatic C–H⋯π interactions in 6 and their distances (Å). | |
In summary, in comparison with 2, the fewer π⋯π and C–H⋯π interactions exhibited in 5 indicate that 5 has a weaker π-stacking ability than that of 2 and compound 6 possess the weakest π-stacking ability. These examples indicate that π-stacking interactions can vary greatly in geometrical orientation even in the case of compounds that are structurally quite similar. In 2 and 3, the molecular interactions increase as the triazole-tethered phenyl ring becomes more electron-deficient resulting from electron-withdrawing group substituents, such as –CN and –CHO in 2 and 3, respectively, and a decrease resulting from the addition of the electron-donating group such as –CH3 on the triazole-tethered phenyl ring in 6 when compared to that of 5. Such substituent effect on intermolecular π⋯π interactions has already been confirmed by Sun and co-workers and the result in this work compares well to that in Sun's study.21
Non-covalent interactions such as π⋯π, C–H⋯π interaction and hydrogen bond play a critical role in scaffolding in large biomolecular binding, such as DNA, RNA, proteins and nucleic acids. The non-covalent interactions that take place within such biomolecules contribute to their structural integrity and thereby influence their functions.22 The cytotoxic activity of Ru(II) arene complexes have been confirmed to result from their aromatic interactions with DNA, proteins and with other biological targets, thus influencing different cellular mechanisms6 and the anticancer activity of a series of Ru(II) arene complexes was shown to be highly dependent on the nature and position of the substituent on the aromatic moiety. A variety of arene-based triazole complexes containing substituents such as cyano, halogen and ethyl groups at the para position showed excellent activity against a proliferative cancer cell line.9b,23 It is also noteworthy that C–H⋯π interactions function not only in non-polar solvents but also in polar, protic solvents such as chloroform and water. This is of importance in regards to the biochemical effects of these derivatives.24 In this study, our ruthenium 4-aryl-1,2,3-triazole products 2–6 have been confirmed to participated in various intramolecular and intermolecular aromatic interactions in the solid-state structures which can be favorable for the binding of biomolecular targets and have shown great potential in the development of biologically active anticancer drugs.
Concluding remarks
In recent years, new classes of piano-stool Ru(II) arene compounds have been found to have promising anticancer activity and nitrogen-coordinated ruthenium aryl triazolato complexes have become the major targets in the drug design of biologically active medicines. In this study, we successfully synthesized a series of piano-stool ruthenium 4-aryl-1,2,3-triazole complexes by thermal [3 + 2] cycloaddition reactions of a ruthenium azido complex [Ru]–N3 (1, [Ru] = (η5-C5H5)(dppe)Ru, dppe = Ph2PCH2CH2PPh2) with terminal phenylacetylenes, the less electron-deficient alkynes, under an atmosphere of air. The single crystal X-ray structures of 2, 3, 5 and 6 were obtained and verified that all of the thus formed 1,2,3-triazole coordinate the metal center through the N(2) atom. To the best of our knowledge, this is the first example of the straightforward preparation of N-coordinated ruthenium aryl-substituted 1,2,3-triazole complexes by [3 + 2] cycloaddition reactions of a metal-azide with terminal phenylacetylenes. Inspections on the solid-state structures of these complexes indicate that there are abundant intramolecular and intermolecular non-covalent interactions such as the π⋯π and C–H⋯π interactions that involve aromatic moieties including cyclopentadienyl groups, triazole rings and phenyl rings in 2, 3 and 5, which can be favorable for the binding of DNA/biomolecular targets and has shown great potential in the development of biologically active anticancer drugs. The focus of this study was on the facile synthesis of a series of ruthenium aryl-substituted 1,2,3-triazole derivatives because of their excellent biological activities resulting from their great ability to participate in aromatic interactions. Such a preparation would be expected to find applications in the areas of medicine, pharmaceuticals and material chemistry. We are currently in the process of exploring the synthesis and the reactivity of new ruthenium aryl triazolato complexes and new triazole derivatives further. Studies of related reactions and application of these complexes are currently underway.
Experimental
General procedures
The reagents used in this study were obtained from commercial suppliers. All solvents and reagents were of reagent grade and were used without further purification. Elemental analyses were performed on a PerkinElmer 2400 CHN elemental analyzer. HR & LR-FAB mass spectra were conducted on a JMS-700 double focusing mass spectrometer (JEOL, Tokyo, Japan) with a resolution of 8000 (3000) (5% valley definition). IR spectra were obtained on a PerkinElmer Paragon 1000 FT-IR spectrometer in the range of 4000–400 cm−1 using KBr pellets. NMR spectra were recorded on Bruker AVA-300 NMR spectrometers at room temperature and are reported in units of δ with residual protons in the solvents as an internal standard (CDCl3, δ 7.24; C6D6, δ 7.26). Complexes [Ru]–N3 (1, [Ru] = (η5-C5H5)(dppe)Ru, dppe = Ph2PCH2CH2PPh2) was prepared following methods reported in the literature.25 Elemental analyses and X-ray diffraction studies were carried out at the Instrumentation Center located at the National Taiwan University.
Preparation of N(2)-bound ruthenium aryl triazolato complexes 2–6
To a screw tapped tube charged with [Ru]–N3 (1) (110.2 mg, 0.182 mmol), 4-ethynylbenzonitrile (71.6 mg, 97% purity, 0.546 mmol) and benzene (10 mL) were added and the tube was then sealed with a screw cap. The mixture was allowed to stand at 80 °C in a silicone oil bath under an atmosphere of air for 48 hours and the volume of the solvent was then reduced to 2 mL by a rotary evaporator. To the residue, 25 mL of n-pentane was added to give a yellow precipitate. After isolating the precipitate on a filter, it was washed with 2 × 5 mL of n-pentane and dried under a vacuum to give [Ru]N3C2H(4-C6H4CN) (2, 119 mg, 0.162 mmol, 89.3%). The spectroscopic data for 2 are as follows: IR (KBr, cm−1): 3435 (w), 3237 (w), 3056 (w), 2908 (w), ν(CN) 2219 (s), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1605 (vs), ν(NN) 1434 (s), 1171 (s), 1100 (s), 998 (w), 973 (m), ν(triazolato ring) 844 (s), 794 (m), 748 (m), 695 (vs), 528 (vs). 1H NMR (CDCl3): δ 7.60–6.91 (m, 24H, Ph), 5.26 (s, 1H, N3CH), 4.61 (s, 5H, Cp), 3.21, 2.66 (2m, PCH2CH2P). 31P NMR (CDCl3): δ 87.3. 13C NMR (CDCl3): δ 143.6 (N3CH), 137.9 (NC(Ph)), 143.0–124.6, 107.4 (Ph), 118.2 (CN), 82.0 (Cp), 29.07 (t, PCH2CH2P, JC–P = 22.6 Hz). MS (m/z, Ru102): 734.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C40H34N4P2Ru: C, 65.47; H, 4.67; N, 7.64 found: C, 65.62; H, 4.69; N, 7.54. [Ru]N3C2H(4-C6H4CHO) (3, 214 mg, 0.29 mmol, 88.1% yield from 200.1 mg, 0.330 mmol of 1 at 100 °C for 48 h), [Ru]N3C2H(4-C6H4F) (4, 91.9 mg, 0.127 mmol, 73% yield from 105.7 mg, 0.174 mmol of 1 at 100 °C for 72 h), [Ru]N3C2H(Ph) (5, 514.5 mg, 0.727 mmol, 85.6% yield from 514.6 mg, 0.849 mmol of 1 at 140 °C for 72 h) and [Ru]N3C2H(4-C6H4CH3) (6, 591 mg, 0.819 mmol, 87% yield from 570.5 mg, 0.941 mmol of 1 at 140 °C for 96 h) were prepared using a procedure similar to that used for preparing 2. Spectroscopic data for 3 are as follows: IR (KBr, cm−1): 3647 (w), 3277 (w), 3052 (w), 2802 (w), 2732 (w), 2053 (w), 1979 (w), ν(CO) 1692 (s), 1600 (vs), ν(NN) 1483 (w), 1434 (s), 1303 (m), 1211 (m), 1163 (m), 1100 (m), 999 (w), 972 (w), ν(triazolato ring) 834 (vs), 744 (m), 695 (s), 529 (s). 1H NMR (CDCl3): δ 9.78 (s, 1H, CHO), 7.82–6.95 (m, 25H, Ph and N3CH), 4.62 (Cp), 3.24, 2.66 (2m, PCH2CH2P). 31P NMR (CDCl3): δ 87.3. 13C NMR (CDCl3): 191.7 (CHO), 144.0 (N3CH), 139.6 (N3C(Ph)), 142.8–124.5 (Ph), 81.9 (Cp), 28.9 (t, PCH2CH2P, JC–P = 22.6 Hz). MS (m/z, Ru102): 737.1 (M+), 565.1 (M+–triazole ring). Anal. calcd. for C40H35N3OP2Ru: C, 63.82; H, 4.69; N, 5.58 found: C, 63.98; H, 4.71; N, 5.49. Spectroscopic data for 4 are as follows: IR (KBr, cm−1): 3052 (m), 2916 (w), 2078 (m), 1965 (w), 1888 (w), 1812 (w), 1540 (m), 1478 (s), ν(NN) 1434 (vs), 1213 (m), 1097 (s), 974 (m), ν(triazolato ring) 837 (s), 810 (s), 744 (s), 695 (vs), 609 (m), 530 (vs), 499 (m). 1H NMR (CDCl3): δ 7.90–6.69 (m, 25H, Ph and N3CH), 4.61 (Cp), 3.22, 2.65 (2m, PCH2CH2P) 31P NMR (CDCl3): δ 87.4. 13C NMR (CDCl3): δ 144.5 (N3CH), 143.2–126.0, 115.7–114.2 (N3C(Ph) and Ph), 81.9 (Cp), 29.0 (t, PCH2, JC–P = 22.6 Hz). MS (m/z, Ru102): 727.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C39H34N3FP2Ru: C, 64.46; H, 4.71; N, 5.78 found: C, 64.68; H, 4.74; N, 5.62. Spectroscopic data for 5 are as follows: IR (KBr, cm−1): 3052 (m), 2920 (m), 2853 (w), 2075 (w), 1980 (w), 1605 (m), ν(NN) 1483 (w), 1404 (s), 1363 (w), 1308 (w), 1175 (m), 1098 (m), 999 (w), 973 (m), ν(triazolato ring) 833 (w), 798 (m), 744 (m), 700 (vs), 531 (s), 498 (m). 1H NMR (CDCl3): δ 7.54–6.87 (m, 26H, Ph and N3CH), 4.60 (Cp), 3.25, 2.63 (2m, PCH2) 31P NMR (CDCl3): δ 87.6. 13C NMR (CDCl3): δ 145.4 (N3CH), 143.4–124.8 (N3C(Ph) and Ph), 81.9 (Cp), 29.1 (t, PCH2CH2P, JC–P = 22.6 Hz). MS (m/z, Ru102): 709.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C39H35N3P2Ru: C, 66.09; H, 4.98; N, 5.93 found: C, 66.15; H, 5.02; N, 5.85. Spectroscopic data for 6 are as follows: IR (KBr, cm−1): 3051 (m), 2922 (w), 2038 (w), 1981 (w), 1710 (m), 1604 (w), ν(NN) 1479 (m), 1434 (s), 1361 (w), 1307 (w), 1273 (w), 1180 (m), 1097 (m), 999 (w), 970 (m), ν(triazolato ring) 824 (w), 795 (m), 740 (m), 700 (vs), 530 (s), 499 (m). 1H NMR (CDCl3): δ 7.90–6.75 (m, 25H, Ph and N3CH), 4.60 (Cp), 3.25, 2.63 (2m, PCH2), 2.21 (s, 3H, CH3). 31P NMR (CDCl3): δ 87.6. 13C NMR (CDCl3): δ 145.3 (N3CH), 143.4–127.5, 124.8 (N3C(Ph) and Ph), 81.9 (Cp), 29.0 (t, PCH2CH2P, JC–P = 22.6 Hz), 21.0 (CH3). MS (m/z, Ru102): 723.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C40H37N3P2Ru: C, 66.47; H, 5.16; N, 5.81 found: C, 66.64; H, 5.18; N, 5.74.
O) 1605 (vs), ν(NN) 1434 (s), 1171 (s), 1100 (s), 998 (w), 973 (m), ν(triazolato ring) 844 (s), 794 (m), 748 (m), 695 (vs), 528 (vs). 1H NMR (CDCl3): δ 7.60–6.91 (m, 24H, Ph), 5.26 (s, 1H, N3CH), 4.61 (s, 5H, Cp), 3.21, 2.66 (2m, PCH2CH2P). 31P NMR (CDCl3): δ 87.3. 13C NMR (CDCl3): δ 143.6 (N3CH), 137.9 (NC(Ph)), 143.0–124.6, 107.4 (Ph), 118.2 (CN), 82.0 (Cp), 29.07 (t, PCH2CH2P, JC–P = 22.6 Hz). MS (m/z, Ru102): 734.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C40H34N4P2Ru: C, 65.47; H, 4.67; N, 7.64 found: C, 65.62; H, 4.69; N, 7.54. [Ru]N3C2H(4-C6H4CHO) (3, 214 mg, 0.29 mmol, 88.1% yield from 200.1 mg, 0.330 mmol of 1 at 100 °C for 48 h), [Ru]N3C2H(4-C6H4F) (4, 91.9 mg, 0.127 mmol, 73% yield from 105.7 mg, 0.174 mmol of 1 at 100 °C for 72 h), [Ru]N3C2H(Ph) (5, 514.5 mg, 0.727 mmol, 85.6% yield from 514.6 mg, 0.849 mmol of 1 at 140 °C for 72 h) and [Ru]N3C2H(4-C6H4CH3) (6, 591 mg, 0.819 mmol, 87% yield from 570.5 mg, 0.941 mmol of 1 at 140 °C for 96 h) were prepared using a procedure similar to that used for preparing 2. Spectroscopic data for 3 are as follows: IR (KBr, cm−1): 3647 (w), 3277 (w), 3052 (w), 2802 (w), 2732 (w), 2053 (w), 1979 (w), ν(CO) 1692 (s), 1600 (vs), ν(NN) 1483 (w), 1434 (s), 1303 (m), 1211 (m), 1163 (m), 1100 (m), 999 (w), 972 (w), ν(triazolato ring) 834 (vs), 744 (m), 695 (s), 529 (s). 1H NMR (CDCl3): δ 9.78 (s, 1H, CHO), 7.82–6.95 (m, 25H, Ph and N3CH), 4.62 (Cp), 3.24, 2.66 (2m, PCH2CH2P). 31P NMR (CDCl3): δ 87.3. 13C NMR (CDCl3): 191.7 (CHO), 144.0 (N3CH), 139.6 (N3C(Ph)), 142.8–124.5 (Ph), 81.9 (Cp), 28.9 (t, PCH2CH2P, JC–P = 22.6 Hz). MS (m/z, Ru102): 737.1 (M+), 565.1 (M+–triazole ring). Anal. calcd. for C40H35N3OP2Ru: C, 63.82; H, 4.69; N, 5.58 found: C, 63.98; H, 4.71; N, 5.49. Spectroscopic data for 4 are as follows: IR (KBr, cm−1): 3052 (m), 2916 (w), 2078 (m), 1965 (w), 1888 (w), 1812 (w), 1540 (m), 1478 (s), ν(NN) 1434 (vs), 1213 (m), 1097 (s), 974 (m), ν(triazolato ring) 837 (s), 810 (s), 744 (s), 695 (vs), 609 (m), 530 (vs), 499 (m). 1H NMR (CDCl3): δ 7.90–6.69 (m, 25H, Ph and N3CH), 4.61 (Cp), 3.22, 2.65 (2m, PCH2CH2P) 31P NMR (CDCl3): δ 87.4. 13C NMR (CDCl3): δ 144.5 (N3CH), 143.2–126.0, 115.7–114.2 (N3C(Ph) and Ph), 81.9 (Cp), 29.0 (t, PCH2, JC–P = 22.6 Hz). MS (m/z, Ru102): 727.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C39H34N3FP2Ru: C, 64.46; H, 4.71; N, 5.78 found: C, 64.68; H, 4.74; N, 5.62. Spectroscopic data for 5 are as follows: IR (KBr, cm−1): 3052 (m), 2920 (m), 2853 (w), 2075 (w), 1980 (w), 1605 (m), ν(NN) 1483 (w), 1404 (s), 1363 (w), 1308 (w), 1175 (m), 1098 (m), 999 (w), 973 (m), ν(triazolato ring) 833 (w), 798 (m), 744 (m), 700 (vs), 531 (s), 498 (m). 1H NMR (CDCl3): δ 7.54–6.87 (m, 26H, Ph and N3CH), 4.60 (Cp), 3.25, 2.63 (2m, PCH2) 31P NMR (CDCl3): δ 87.6. 13C NMR (CDCl3): δ 145.4 (N3CH), 143.4–124.8 (N3C(Ph) and Ph), 81.9 (Cp), 29.1 (t, PCH2CH2P, JC–P = 22.6 Hz). MS (m/z, Ru102): 709.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C39H35N3P2Ru: C, 66.09; H, 4.98; N, 5.93 found: C, 66.15; H, 5.02; N, 5.85. Spectroscopic data for 6 are as follows: IR (KBr, cm−1): 3051 (m), 2922 (w), 2038 (w), 1981 (w), 1710 (m), 1604 (w), ν(NN) 1479 (m), 1434 (s), 1361 (w), 1307 (w), 1273 (w), 1180 (m), 1097 (m), 999 (w), 970 (m), ν(triazolato ring) 824 (w), 795 (m), 740 (m), 700 (vs), 530 (s), 499 (m). 1H NMR (CDCl3): δ 7.90–6.75 (m, 25H, Ph and N3CH), 4.60 (Cp), 3.25, 2.63 (2m, PCH2), 2.21 (s, 3H, CH3). 31P NMR (CDCl3): δ 87.6. 13C NMR (CDCl3): δ 145.3 (N3CH), 143.4–127.5, 124.8 (N3C(Ph) and Ph), 81.9 (Cp), 29.0 (t, PCH2CH2P, JC–P = 22.6 Hz), 21.0 (CH3). MS (m/z, Ru102): 723.1 (M+), 565.1 (M+–triazolato ring). Anal. calcd. for C40H37N3P2Ru: C, 66.47; H, 5.16; N, 5.81 found: C, 66.64; H, 5.18; N, 5.74.
X-ray analysis of 2, 3, 5 and 6
Single crystals of complex 2, 3, 5 and 6 were grown by allowing n-hexane to diffuse into a dichloromethane or chloroform solution of the complexes. A single crystal of 2 with dimensions 0.173 × 0.165 × 0.033 mm3 was mounted on a Bruker SMART APEX diffractometer equipped with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å). Initial lattice parameters were determined from 25 reflections with 10.0° < 2θ < 25°. Data were collected using the θ/2θ scan method. Data collection was executed using the SMART program; cell refinement and data reduction were performed with the SAINT program. The structure was determined by the SHELXTL/PC26 program and refined by the full-matrix least-squares methods on F2. Hydrogen atoms were placed geometrically using the riding model with thermal parameters set to 1.2 times that for the atoms to which the hydrogen is attached and 1.5 times that for the methyl hydrogens. The merging of equivalent and duplicate reflections gave a total of 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 728 unique measured data points from which 7638 were considered to be observed (I > 2σ(I)). Final refinement using the full-matrix, least-squares converged smoothly to the value of Rf = 0.0297 and Rw = 0.0630. Details of the crystal data, data collections and structure refinements are summarized in Table 3. The procedures for the structure determination of 3 (0.290 × 0.164 × 0.079 mm3), 5 (0.141 × 0.131 × 0.094 mm3) and 6 (0.266 × 0.215 × 0.206 mm3) were similar. Other relevant crystal data for all crystals are also given in Table 3.
728 unique measured data points from which 7638 were considered to be observed (I > 2σ(I)). Final refinement using the full-matrix, least-squares converged smoothly to the value of Rf = 0.0297 and Rw = 0.0630. Details of the crystal data, data collections and structure refinements are summarized in Table 3. The procedures for the structure determination of 3 (0.290 × 0.164 × 0.079 mm3), 5 (0.141 × 0.131 × 0.094 mm3) and 6 (0.266 × 0.215 × 0.206 mm3) were similar. Other relevant crystal data for all crystals are also given in Table 3.
Table 3 Crystal and intensity collection data for complexes 2, 3, 5 and 6
| |
2 |
3 |
5·CHCl3a |
6 |
| Crystals grown from a chloroform–n-hexane mixture are found to incorporate a chloroform molecule. Rf = ∑(Fo − Fc)/∑(Fo); Rw = [∑(w(Fo − Fc)2)/∑(wFo2)]1/2. |
| Formula |
C40H34N4P2Ru |
C40H35N3OP2Ru |
C40H36Cl3N3P2Ru |
C40H37N3P2Ru |
| M |
733.72 |
736.72 |
828.08 |
722.73 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| T/K |
150(2) |
150(2) |
150(2) |
150(2) |
| Space group |
C2/c |
C2/c |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å |
39.0862(13) |
39.1531(12) |
9.0659(2) |
11.5899(3) |
| b/Å |
9.1717(3) |
9.1339(2) |
37.8048(10) |
11.8945(3) |
| c/Å |
18.6636(6) |
18.7253(5) |
10.7826(3) |
12.2685(7) |
| α/deg |
90 |
90 |
90 |
87.7701(8) |
| β/deg |
93.3599(11) |
94.2890(9) |
97.1681(7) |
77.0758(6) |
| γ/deg |
90 |
90 |
90 |
84.2939(7) |
| V/Å3 |
6679.2(4) |
6677.8(3) |
3666.69(16) |
1640.03(7) |
| Z |
8 |
8 |
4 |
2 |
| μ/mm−1 |
0.601 |
0.603 |
0.767 |
0.610 |
| Unique reflections collected |
23728 |
26171 |
28929 |
18965 |
| Observed data [I > 2σ(I)] |
7638 |
9736 |
8400 |
9569 |
| Data/parameters |
7638/424 |
9736/449 |
8400/452 |
9569/416 |
| Final Rf, Rw indices [I > 2σ(I)] |
0.0297, 0.0630 |
0.0332, 0.0685 |
0.0518, 0.0938 |
0.0290, 0.0613 (Rf, Rw)b |
CCDC 2191749, 2191750, 2191751 and 2191752 contain the supplementary crystallographic data for this paper.†
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors wish to thank Prof. Shie-Ming Peng, the head of the Instrumentation Center located in National Taiwan University, for technical assistance from the Instrumentation Center and the authors also wish to thank the Ministry of Science and Technology of Taiwan for financial support.
References
-
(a) P. B. Tchounwou, S. Dasari, F. K. Noubissi, P. Ray and S. Kumar, J. Exp. Pharmacol., 2021, 13, 303 CrossRef PubMed;
(b) R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728 CrossRef CAS;
(c) S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364 CrossRef CAS PubMed.
-
(a) G. Jaouen, A. Vessi_eres and S. Top, Chem. Soc. Rev., 2015, 44, 8802 RSC;
(b) A. R. Rubio, R. González, N. Busto, M. Vaquero, A. L. Iglesias, F. A. Jalón, G. Espino, A. M. Rodríguez, B. García and B. R. Manzano, Pharmaceutics, 2021, 13(10), 1540 CrossRef CAS PubMed;
(c) P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5 CrossRef CAS.
-
(a) C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140 CrossRef CAS PubMed;
(b) R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925 RSC.
- Q. Chen, V. Ramu, Y. Aydar, A. Groenewoud, X. Q. Zhou, M. J. Jager, H. Cole, C. G. Cameron, S. A. McFarland, S. Bonnet and B. E. Snaar-Jagalska, Cancers, 2020, 12(3), 587 CrossRef CAS PubMed.
-
(a) S. Y. Lee, C. Y. Kim and T. G. Nam, Drug Des., Dev. Ther., 2020, 14, 5375 CrossRef CAS PubMed;
(b) L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771 RSC.
-
(a) C. Riccardi, D. Musumeci, M. Trifuoggi, C. Irace, L. Paduano and D. Montesarchio, Pharmaceuticals, 2019, 12(4), 146 CrossRef CAS PubMed;
(b) E. Alessio and L. Messori, Molecules, 2019, 24, 1995 CrossRef CAS PubMed;
(c) M. Piccolo, M. G. Ferraro, F. Raucci, C. Riccardi, A. Saviano, I. R. Krauss, M. Trifuoggi, M. Caraglia, L. Paduano, D. Montesarchio, F. Maione, G. Misso, R. Santamaria and C. Irace, Cancers, 2021, 13, 5164 CrossRef CAS PubMed;
(d) J. P. C. Coverdale, T. Laroiya-McCarron and I. Romero-Canelon, Inorganics, 2019, 7, 31 CrossRef CAS.
-
(a) T. R. Steel, F. Walsh, A. Wieczorek-, M. Hanif and C. G. Hartinger, Coord. Chem. Rev., 2021, 439, 213890 CrossRef CAS;
(b) Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 38, 4764 RSC;
(c) G. Süss-Fink, Dalton Trans., 2010, 39, 1673 RSC;
(d) L. Wang, Y. He, G. Xiang and X. Shang, Appl. Organomet. Chem., 2018, 32, e4311 CrossRef.
-
(a) K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Dalton
Trans., 2014, 43, 1443 RSC;
(b) M. Djukić, M. S. Jeremić, R. Jelić, O. Klisurić, V. Kojić, D. Jakimov, P. Djurdjević and Z. D. Matović, Inorg. Chim. Acta, 2018, 483, 359 CrossRef;
(c) M. G. Mendoza-Ferri, C. G. Hartinger, A. A. Nazarov, R. E. Eichinger, M. A. Jakupec, K. Severin and B. K. Keppler, Organometallics, 2009, 28(21), 6260 CrossRef CAS;
(d) C. Sonkar, N. Malviya, N. Sinha, A. Mukherjee, S. Pakhira and S. Mukhopadhyay, BioMetals, 2021, 34, 795 CrossRef CAS PubMed.
-
(a) D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30 CrossRef CAS PubMed;
(b) A. Rani, G. Singh, A. Singh, U. Maqbool, G. Kaur and J. Singh, RSC Adv., 2020, 10, 5610 RSC;
(c) S. Kantheti, R. Narayan and K. V. S. N. Raju, RSC Adv., 2015, 5, 3687 RSC.
-
(a) M. Hrimla, A. Oubella, Y. Laamari, L. Bahsis, A. Ghaleb, M. Y. A. Itto, A. Auhmani, H. Morjani, M. Julve and S.-E. Stiriba, Biointerface Res. Appl. Chem., 2022, 12(6), 7633 CAS;
(b) Z. Xu, S.-J. Zhao and Y. Liu, Eur. J. Med. Chem., 2019, 183, 111700 CrossRef CAS PubMed;
(c) K. I. Slavova, L. T. Todorov, N. P. Belskaya, M. A. Palafox and I. P. Kostova, Anticancer Agents Med. Chem., 2020, 15(2), 92 CAS.
-
(a) O. A. Lenis-Rojas, R. Cabral, B. Carvalho, S. Friães, C. Roma-Rodrigues, J. A. A. Fernández, S. F. Vila, L. Sanchez, C. S. B. Gomes, A. R. Fernandes and B. Royo, Inorg. Chem., 2021, 60(11), 8011 CrossRef CAS PubMed;
(b) C. A. Riedl, M. Hejl, M. H. M. Klose, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2018, 47, 4625 RSC;
(c) C. K. Rono, W. K. Chu, J. Darkwa, D. Meyer and B. C. E. Makhubela, Organometallics, 2019, 38, 3197 CrossRef CAS;
(d) A. H. Alkhzem, T. J. Woodman and I. S. Blagbrough, RSC Adv., 2022, 12, 19470 RSC.
-
(a) W. P. Fehlhammer and W. Z. Beck, Z. Anorg. Allg. Chem., 2015, 641, 1599 CrossRef CAS;
(b) H. W. Fruhauf, Chem. Rev., 1997, 97, 523 CrossRef PubMed;
(c) P. Schmid, M. Maier, H. Pfeiffer, A. Belz, L. Henrt, A. Freidrich, F. Schonfeld, K. Edkins and U. Schatzschneider, Dalton Trans., 2017, 46, 13386 RSC;
(d) A. D. Mironova, M. A. Mikhailov, K. A. Brylev, A. L. Gushchin, T. S. Sukhikh and M. N. Sokolov, New J. Chem., 2020, 44, 20620 RSC.
- D. E. Lewis, Advanced organic chemistry, Oxford University Press, New York, 2016 Search PubMed.
-
(a) T. J. Robilotto, D. S. Alt, H. A. von Recum and T. G. Gray, Dalton Trans., 2011, 40, 8083–8085 RSC;
(b) J. E. Heckler, N. Deligonul, A. L. Rheingold and T. G. Gray, Chem. Commun., 2013, 49, 5990 RSC;
(c) J. E. Heckler, B. L. Anderson and T. G. Gray, J. Organomet. Chem., 2016, 818, 68 CrossRef CAS.
-
(a) D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 554 CrossRef CAS;
(b) T. Hosseinnejad, F. Ebrahimpour-Malmir and B. Fattahi, RSC Adv., 2018, 8, 12232 RSC.
-
(a) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed;
(b) A. Rani, G. Singh, A. Singh, U. Maqbool, G. Kaur and J. Singh, RSC Adv., 2020, 10, 5610 RSC;
(c) C. Wang, Q. Li, S. Wang, G. Zhu and A. Zhu, RCS Adv., 2021, 11, 38108 CAS;
(d) N. V. Sokolova and V. G. Nenajdenko, RSC Adv., 2013, 3, 16212 RSC.
-
(a) C. W. Chang and G. H. Lee, Dalton Trans., 2019, 48, 2028 RSC;
(b) C. W. Chang, M. C. Cheng, G. H. Lee and S. M. Peng, Dalton Trans., 2019, 48, 11732 RSC.
-
(a) C. W. Chang and G. H. Lee, Inorg. Chim. Acta, 2019, 494, 232 CrossRef CAS;
(b) C. W. Chang and G. H. Lee, J. Organomet. Chem., 2019, 896, 146 CrossRef CAS.
-
(a) D. V. Partyka, J. B. Updegraff III, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2007, 26, 183 CrossRef CAS;
(b) S. D. Köster, H. Alborzinia, S. Can, I. Kitanovic, S. Wölfl, R. Rubbiani, I. Ott, P. Riesterer, A. Prokop, K. Merz and N. Metzler-Nolte, Chem. Sci., 2012, 3, 2062 RSC.
- R. Anjana, M. K. Vaishnavi, D. Sherlin, S. P. Kumar, K. Naveen, P. S. Kanth and K. Sekar, Bioinformation, 2012, 8(24), 1220 CrossRef PubMed.
- J. D. Mottishaw and H. Sun, J. Phys. Chem. A, 2013, 117, 7970 CrossRef CAS PubMed.
-
(a) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC;
(b) B. K. Mishra, S. Karthikeyan and V. Ramanathan, J. Chem. Theory Comput., 2012, 8(6), 1935 CrossRef CAS PubMed;
(c) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed. Engl., 2003, 42, 1210 CrossRef CAS PubMed.
-
(a) R. Huang, G. Langille, R. K. Gill, C. M. J. Li, Y. Mikata, M. Q. Wong, D. T. Yapp and T. Storr, J. Biol. Inorg Chem., 2013, 18, 831 CrossRef CAS PubMed;
(b) H. C. Bertrand, S. Clède, R. Guillot, F. Lambert and C. Policar, Inorg. Chem., 2014, 53, 6204 CrossRef CAS PubMed;
(c) F. Naaz, M. C. Preeti Pallavi, S. Shafi, N. Mulakayala, M. S. Yar and H. M. S. Kumar, Bioorg. Chem., 2018, 81, 1 CrossRef CAS PubMed;
(d) M. U. Rahman, Y. Mohammad, K. M. Fazili, K. A. Bhat and T. Ara, Steroids, 2017, 118, 1 CrossRef PubMed.
- F. Trudu, F. Amato, P. Vaňhara, T. Pivetta, E. M. Peña-Méndez and J. Havel, J. Appl. Biomed., 2015, 13, 79 CrossRef.
- C. W. Chang and G. H. Lee, Organometallics, 2003, 22, 3107 CrossRef CAS.
- SHELXTL: Structure Analysis Program, version 5.04, Siemens Industrial Automation Inc., Madison, WI, 1995 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Chi-Rung Leeb,
Gene-Hsiang Leec and
Kuang-Lieh Lu*d
*a,
Chi-Rung Leeb,
Gene-Hsiang Leec and
Kuang-Lieh Lu*d