
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of coordinating heteroatoms on molecular structure, thermodynamic stability and redox behavior of uranyl(VI) complexes with pentadentate Schiff-base ligands†
Tomoyuki Takeyama * and
Koichiro Takao*
* and
Koichiro Takao*
Laboratory for Zero-Carbon Energy, Institute of Innovative Research, Tokyo Institute of Technology, 2-12-1 N1-32, O-Okayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: takeyama.t.ab@m.titech.ac.jp; takao.k.ac@m.titech.ac.jp
First published on 26th August 2022
Abstract
Uranyl(VI) complexes with pentadentate N3O2-, N2O3- and N2O2S1-donating Schiff base ligands, tBu,MeO–saldien–X2− (X = NH, O and S), were synthesized and thoroughly characterized by 1H NMR, IR, elemental analysis, and single crystal X-ray diffraction. The crystal structures of UO2(tBu,MeO–saldien–X) showed that the U–X bond strength follows U–O ≈ U–NH > U–S. Conditional stability constants (βX) of UO2(tBu,MeO–saldien–X) in ethanol were investigated to understand the effect of X on thermodynamic stability. The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) βX decrease in the order of UO2(tBu,MeO–saldien–NH) (logβNH = 10) > UO2(tBu,MeO–saldien–O) (logβO = 7.24) > UO2(tBu,MeO–saldien–S) (logβS = 5.2). This trend cannot be explained only by Pearson's Hard and Soft Acids and Bases (HSAB) principle, but rather follows the order of basicity of X. Theoretical calculations of UO2(tBu,MeO–saldien–X) suggested that the ionic character of U–X bonds decreases in the order of U–NH > U–O > U–S, while the covalency increases in the order U–O < U–NH < U–S. Redox potentials of all UO2(tBu,MeO–saldien–X) in DMSO were similar to each other regardless of the difference in X. Spectroelectrochemical measurements and DFT calculations revealed that the center U6+ of each UO2(tBu,MeO–saldien–X) undergoes one-electron reduction to afford the corresponding uranyl(V) complex. Consequently, the difference in X of UO2(tBu,MeO–saldien–X) affects the coordination of tBu,MeO–saldien–X2− with UO22+. However, the HSAB principle is not always prominent, but the Lewis basicity and balance between ionic and covalent characters of the U–X interactions are more relevant to determine the bond strengths.
βX decrease in the order of UO2(tBu,MeO–saldien–NH) (logβNH = 10) > UO2(tBu,MeO–saldien–O) (logβO = 7.24) > UO2(tBu,MeO–saldien–S) (logβS = 5.2). This trend cannot be explained only by Pearson's Hard and Soft Acids and Bases (HSAB) principle, but rather follows the order of basicity of X. Theoretical calculations of UO2(tBu,MeO–saldien–X) suggested that the ionic character of U–X bonds decreases in the order of U–NH > U–O > U–S, while the covalency increases in the order U–O < U–NH < U–S. Redox potentials of all UO2(tBu,MeO–saldien–X) in DMSO were similar to each other regardless of the difference in X. Spectroelectrochemical measurements and DFT calculations revealed that the center U6+ of each UO2(tBu,MeO–saldien–X) undergoes one-electron reduction to afford the corresponding uranyl(V) complex. Consequently, the difference in X of UO2(tBu,MeO–saldien–X) affects the coordination of tBu,MeO–saldien–X2− with UO22+. However, the HSAB principle is not always prominent, but the Lewis basicity and balance between ionic and covalent characters of the U–X interactions are more relevant to determine the bond strengths.
Introduction
Uranium is the most important element in nuclear engineering. The chemistry of uranium plays important roles in nuclear fuel fabrication and spent fuel reprocessing. Under ambient conditions, uranium is most commonly present as a hexavalent uranyl(VI) ion, UO22+, with a typical linear [O![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) UVIO]2+ structure. The chemical separation of UO22+ from various aqueous systems such as feed solutions of spent nuclear fuels and even seawater is one of the important research topics in nuclear chemistry. In the usual sense, coordination chemistry provides very powerful tools for chemical separation. Hence, the complexation between UO22+ and organic ligands has been widely studied.1–13
UVIO]2+ structure. The chemical separation of UO22+ from various aqueous systems such as feed solutions of spent nuclear fuels and even seawater is one of the important research topics in nuclear chemistry. In the usual sense, coordination chemistry provides very powerful tools for chemical separation. Hence, the complexation between UO22+ and organic ligands has been widely studied.1–13
Pearson's Hard and Soft Acids and Bases (HSAB) principle is quite useful to describe preferential interactions between specific metal ions and coordinating atoms of ligands and to design organic molecules selectively coordinating with a target metal ion,14,15 although this principle is rather empirical. In the HSAB principle, UO22+ is classified as a hard acid,14,15 and therefore, generally tends to more strongly interact with hard bases like N, O and F, compared with softer ones such as heavier congeners like P, S, and Cl.14,15 Indeed, thermodynamic stability of a UO22+-halido complex in DMF follows the order of hardness of halide ligands, Cl− > Br− > I−.16 In contrast, such a trend in complexation between UO22+ and heteroatoms like N, O and S, seems not to be well understood systematically, although it would provide essential information to understand the fundamental nature of UO22+ in more depth and to design molecular structures of ligands exclusively interacting with UO22+. Indeed, several extracting reagents have been successfully developed for separation of Am(III) and Cm(III) from Ln(III) on the basis of difference in coordinating affinities of these metal ions with soft-donor atoms incorporated in the designed ligands.17–20
In this study, we discuss strengths of U–N, U–O and U–S interactions formed in UO22+ complexes having analogous coordination geometries. For this purpose, it is first necessary to choose a suitable ligand system. Previously, we reported UO22+ complexes with N3O2-pentadentate Schiff base ligands, UO2(R1,R2,–Rsaldien), shown in Fig. 1(a).21,22 Its NR moiety can be substituted with O or S to provide the similar UO22+ complexes, UO2(tBu,MeO–saldien–X) (UO2(LX), X = NH, O, S, Fig. 1(b)), where the U–O or U–S bond will be formed instead of U–NR. Here, we report synthesis and characterization of UO2(LX) (X = NH, O, S) to discuss effects of X to the U–X bond strength and thermodynamic stability as well as redox chemistry of this class of UO22+ complexes.
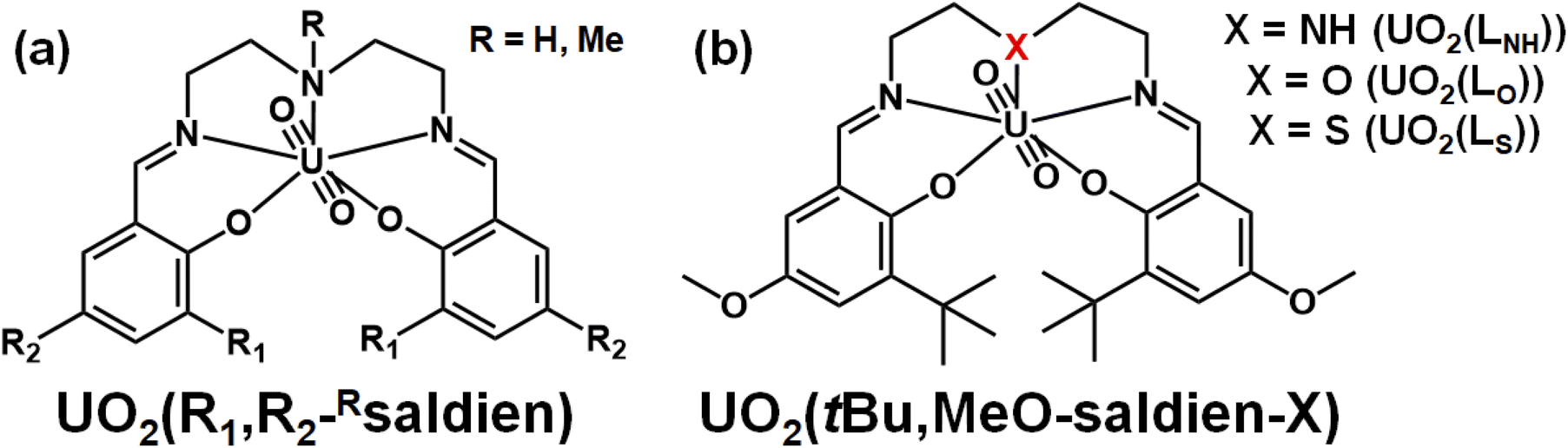 | ||
| Fig. 1 Schematic structures of UO2(R1,R2,–Rsaldien)21,22 (a) and UO2(tBu,MeO–saldien–X) complexes (UO2(LX)); (X = NH, O, S) (b). | ||
Experimental section
Materials and syntheses
All reagents used were of reagent grade and used as received, if not specified. 3-tert-Butyl-5-methoxysalicylaldehyde was synthesized as reported elsewhere.23![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CH2CH2–N or CN–CH2CH2–N), 3.83 (s, 6H, O–CH3), 4.07 (m, 2H, CN–CH2CH2–N or CN–CH2CH2–N), 4.26 (m, 1H, –NH), 4.50 (m, 4H, CN–CH2CH2–N or CN–CH2CH2–N), 6.86 (d, 2H, aryl, JH–H = 3.2 Hz), 7.32 (d, 2H, aryl, JH–H = 3.2 Hz), 9.38 (s, 2H, NCH–). 1H NMR (399.78 MHz, DMSO-d6, δ/ppm vs. TMS): 1.65 (s, 18H, –C(CH3)3), 3.40 (m, 2H, CN–CH2CH2–N or CN–CH2CH2–N), 3.75 (s, 6H, O–CH3), 4.02 (m, 2H, CN–CH2CH2–N or CN–CH2CH2–N), 4.49 (m, 4H, CN–CH2CH2–N or CN–CH2CH2–N), 6.51 (m, 1H, –NH), 7.04 (d, 2H, aryl, JH–H = 3.2 Hz), 7.14 (d, 2H, aryl, JH–H = 3.2 Hz), 9.57 (s, 2H, NCH–). IR (ATR, cm−1): 858 (OUO asymmetric stretching, ν3), 1636 (CN stretching, νCN). Elemental analysis (%) calcd for UO2(LNH) (C28H39N3O6U1): C, 44.74; H, 5.23; N, 5.59. Found: C 44.64; H, 5.29; N, 5.42. The obtained crystals were also suitable for the X-ray crystallography.N–CH2CH2–O or CN–CH2CH2–O), 4.65 (m, 4H, CN–CH2CH2–O or CN–CH2CH2–O), 6.91 (d, 2H, aryl, JH–H = 3.2 Hz), 7.33 (d, 2H, aryl, JH–H = 3.6 Hz), 9.50 (s, 2H, NCH–). 1H NMR (399.78 MHz, DMSO-d6, δ/ppm vs. TMS): 1.64 (s, 18H, –C(CH3)3), 3.77 (s, 6H, O–CH3), 4.53 (t, 4H, CN–CH2CH2–O or CN–CH2CH2–O, JH–H = 5.2 Hz), 4.66 (t, 4H, CN–CH2CH2–O or CN–CH2CH2–O, JH–H = 5.2 Hz), 7.09 (d, 2H, aryl, JH–H = 3.2 Hz), 7.17 (d, 2H, aryl, JH–H = 3.6 Hz), 9.75 (s, 2H, NCH-). IR (ATR, cm−1): 883 (OUO asymmetric stretching, ν3), 1637 (CN stretching, νCN). Elemental analysis (%) calcd for UO2(LO) (C28H38N2O7U1): C, 44.68; H, 5.09; N, 3.72. Found: C 44.79; H, 5.21; N, 3.65. The crystals suitable for X-ray diffraction were obtained by recrystallization from pyridine/hexane.N–CH2CH2–S or CN–CH2CH2–S), 3.83 (s, 6H, O–CH3), 3.74 (s, 4H, CN–CH2CH2–S or CN–CH2CH2–S), 5.33 (s, 1H, CH2Cl2), 6.89 (d, 2H, Aryl, JH–H = 3.2 Hz), 7.33 (d, 2H, aryl, JH–H = 3.6 Hz), 9.47 (s, 2H, NCH–). 1H NMR (399.78 MHz, DMSO-d6, δ/ppm vs. TMS): 1.64 (s, 18H, –C(CH3)3), 3.77 (s, 6H, O–CH3), 3.79 (br, 4H, CN–CH2CH2–S or CN–CH2CH2–S), 4.60 (br, 4H, CN–CH2CH2–S or CN–CH2CH2–S), 7.09 (d, 2H, aryl, JH–H = 3.2 Hz), 7.18 (d, 2H, aryl, JH–H = 2.8 Hz), 9.74 (s, 2H, NCH–). IR (ATR, cm−1): 880 (OUO asymmetric stretching, ν3), 1623 (CN stretching, νCN). Elemental analysis (%) calcd for UO2(LS)·0.5CH2Cl2 (C28H38N2O6S1U1·0.5CH2Cl2): C, 42.20; H, 4.85; N, 3.45. Found: C 42.24; H, 4.79; N, 3.40. The obtained crystals were also suitable for the X-ray crystallography.
N–CH2CH2–N or CN–CH2CH2–N), 3.83 (s, 6H, O–CH3), 4.07 (m, 2H, CN–CH2CH2–N or CN–CH2CH2–N), 4.26 (m, 1H, –NH), 4.50 (m, 4H, CN–CH2CH2–N or CN–CH2CH2–N), 6.86 (d, 2H, aryl, JH–H = 3.2 Hz), 7.32 (d, 2H, aryl, JH–H = 3.2 Hz), 9.38 (s, 2H, NCH–). 1H NMR (399.78 MHz, DMSO-d6, δ/ppm vs. TMS): 1.65 (s, 18H, –C(CH3)3), 3.40 (m, 2H, CN–CH2CH2–N or CN–CH2CH2–N), 3.75 (s, 6H, O–CH3), 4.02 (m, 2H, CN–CH2CH2–N or CN–CH2CH2–N), 4.49 (m, 4H, CN–CH2CH2–N or CN–CH2CH2–N), 6.51 (m, 1H, –NH), 7.04 (d, 2H, aryl, JH–H = 3.2 Hz), 7.14 (d, 2H, aryl, JH–H = 3.2 Hz), 9.57 (s, 2H, NCH–). IR (ATR, cm−1): 858 (OUO asymmetric stretching, ν3), 1636 (CN stretching, νCN). Elemental analysis (%) calcd for UO2(LNH) (C28H39N3O6U1): C, 44.74; H, 5.23; N, 5.59. Found: C 44.64; H, 5.29; N, 5.42. The obtained crystals were also suitable for the X-ray crystallography.N–CH2CH2–O or CN–CH2CH2–O), 4.65 (m, 4H, CN–CH2CH2–O or CN–CH2CH2–O), 6.91 (d, 2H, aryl, JH–H = 3.2 Hz), 7.33 (d, 2H, aryl, JH–H = 3.6 Hz), 9.50 (s, 2H, NCH–). 1H NMR (399.78 MHz, DMSO-d6, δ/ppm vs. TMS): 1.64 (s, 18H, –C(CH3)3), 3.77 (s, 6H, O–CH3), 4.53 (t, 4H, CN–CH2CH2–O or CN–CH2CH2–O, JH–H = 5.2 Hz), 4.66 (t, 4H, CN–CH2CH2–O or CN–CH2CH2–O, JH–H = 5.2 Hz), 7.09 (d, 2H, aryl, JH–H = 3.2 Hz), 7.17 (d, 2H, aryl, JH–H = 3.6 Hz), 9.75 (s, 2H, NCH-). IR (ATR, cm−1): 883 (OUO asymmetric stretching, ν3), 1637 (CN stretching, νCN). Elemental analysis (%) calcd for UO2(LO) (C28H38N2O7U1): C, 44.68; H, 5.09; N, 3.72. Found: C 44.79; H, 5.21; N, 3.65. The crystals suitable for X-ray diffraction were obtained by recrystallization from pyridine/hexane.N–CH2CH2–S or CN–CH2CH2–S), 3.83 (s, 6H, O–CH3), 3.74 (s, 4H, CN–CH2CH2–S or CN–CH2CH2–S), 5.33 (s, 1H, CH2Cl2), 6.89 (d, 2H, Aryl, JH–H = 3.2 Hz), 7.33 (d, 2H, aryl, JH–H = 3.6 Hz), 9.47 (s, 2H, NCH–). 1H NMR (399.78 MHz, DMSO-d6, δ/ppm vs. TMS): 1.64 (s, 18H, –C(CH3)3), 3.77 (s, 6H, O–CH3), 3.79 (br, 4H, CN–CH2CH2–S or CN–CH2CH2–S), 4.60 (br, 4H, CN–CH2CH2–S or CN–CH2CH2–S), 7.09 (d, 2H, aryl, JH–H = 3.2 Hz), 7.18 (d, 2H, aryl, JH–H = 2.8 Hz), 9.74 (s, 2H, NCH–). IR (ATR, cm−1): 880 (OUO asymmetric stretching, ν3), 1623 (CN stretching, νCN). Elemental analysis (%) calcd for UO2(LS)·0.5CH2Cl2 (C28H38N2O6S1U1·0.5CH2Cl2): C, 42.20; H, 4.85; N, 3.45. Found: C 42.24; H, 4.79; N, 3.40. The obtained crystals were also suitable for the X-ray crystallography.Methods
The 1H NMR spectra were recorded by using JEOL ECX-400 (1H: 399.78 MHz) NMR spectrometer. The chemical shifts of 1H NMR were referenced to TMS (δ = 0 ppm). The IR measurements were performed by JASCO FT/IR4700 equipped with a diamond ATR attachment. Elemental analyses were carried out by Yanaco MT-6 CHN elemental analyzer. Cyclic voltammetry (CV) measurements of UO2(LX) (1 mM) dissolved in DMSO containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) were performed at 295 K under a dry Ar atmosphere by using BAS ALS660B electrochemical analyzer. A three-electrode system consisted of a Pt disk working electrode (diameter: 1.6 mm, surface area: 0.020 cm2), a Pt wire counter electrode, and an Ag0/+ reference electrode (0.1 M TBAP + 1 mM AgNO3/CH3CN). A ferrocene/ferrocenium ion redox couple (Fc0/+) was taken as an external standard redox system. All samples were prepared under an inert Ar atmosphere. Dissolved oxygen gas in each sample solution was expelled by purging Ar gas for at least 10 min prior to starting the CV experiments. UV-vis-NIR spectroelectrochemical measurements in DMSO were performed with a JASCO V-770 spectrophotometer equipped with an optically transparent thin layer electrode (OTTLE) cell at 295 K.24–27 Its optical path length was 1.0 × 10−2 cm, which was calibrated spectrophotometrically.22 The three-electrode system was the same as that in the above electrochemical experiments with a replacement of the working electrode by a Pt gauze (80 mesh). The potential applied on OTTLE was controlled by BAS ALS660B. The absorption spectrum at each potential step was recorded after equilibration of the electrochemical reaction at the applied potential on the working electrode, which completed within 3 min. The sample solution in the OTTLE cell was prepared in a similar manner to that for the CV measurements.Crystallographic analysis
The X-ray diffraction data of the well-shaped single crystals of UO2(LNH)·(CH2Cl2), UO2(LO)·(C5H5N) and UO2(LS)·(CH2Cl2) were collected by a Rigaku XtaLAB mini II equipped with hybrid pixel array detector and graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Each sample was mounted on a MiTeGen Dual Thickness MicroMounts, and located in the temperature-controlled N2 gas flow. Intensity data were collected by taking oscillation photographs. Reflection data were corrected for both Lorentz and polarization effects. The structures were solved by the direct method and refined anisotropically by the SHELX program suite28 for non-hydrogen atoms by full-matrix least-squares calculations. Each refinement was continued until all shifts were smaller than one-third of the standard deviations of the parameters involved. Hydrogen atoms were located at the calculated positions. All hydrogen atoms were constrained to ideal geometry with C–H = 0.95 Å. The thermal parameters of all hydrogen atoms were related to those of their parent atoms by U(H) = 1.2Ueq(C). All calculations were performed by using the Olex2 crystallographic software program package.29 Crystallographic data of all complexes were summarized in Table S1,† and deposited with Cambridge Crystallographic Data Centre as supplementary publication no: CCDC 2177295 (UO2(LO)·(C5H5N)), 2177296 (UO2(LNH)·(CH2Cl2)), and 2177297 (UO2(LS)·(CH2Cl2)).UV-vis titration
Sample solutions of H2LX (X = NH, O, S, 0.1 mM) were prepared by mixing 3-tert-butyl-5-methoxysalicylaldehyde and the corresponding diamines in ethanol. The formation of H2LX (X = NH, O, S) was checked by 1H NMR spectroscopy (see Fig. S1†). Triethylamine (NEt3, 0.4 mM) was added as a H+ scavenger after the formation of UO2(LX). The total concentration of UO22+ was stepwise increased up to 0.12 mM (0 ≤ [UO22+]/[LX2−] ≤ 1.2) by adding a feed solution of UO2(NO3)3·6H2O (10 mM) in ethanol. The UV-vis absorption spectrum at each increment step was recorded by JASCO V-770 spectrophotometer. During the whole titration experiment, temperature of the sample solution was kept at 293 K in a thermostat cell holder equipped with the spectrophotometer. The obtained titration series of the UV-vis absorption spectra was analyzed by HypSpec (version 1.1.33)30 to determine conditional stability constants of UO2(LX) (X = NH, O, S) under the presence of 0.4 mM NEt3 in ethanol.Theoretical calculations
Density functional theory (DFT) calculations were performed using Gaussian 16 program (Revision B.01)31 for characterization of UO2(LX) and the one-electron reduced complexes, [UO2(LX)]− (X = NH, O, S). The atomic coordinates of UO2(LX) were taken from those experimentally-determined and were used for structure optimization. Hybrid DFT functional B3LYP32 was employed and solvent was modelled through a conductor-like polarized continuum model (CPCM) for DMSO (dielectric ε = 46.7).33 For uranium, Stuttgart-type small-core effective core potential (ECP) and corresponding basis set has been used.34 The most diffuse basis functions on uranium with the exponent 0.005 (all s, p, d, and f type functions) were omitted as in previous studies.35–39 The 6-311G(d) basis sets were used for other elements (C, H, N, O, S). Vibrational frequency calculations at the same level of theory confirmed that no imaginary frequency was found to be present. Single-point calculations for energetic analysis were performed using the same condition. NBO analysis were carried out by using the NBO 5.0 program.40 The molecular structures of [UO2(LX)]− were taken from those of UO2(LX) determined experimentally and were optimized after addition of a single negative charge and doublet spin degeneracy to assume the one-electron reduction using the same condition. The Mulliken spin-density plots were illustrated by GaussView 6.1.41Results and discussion
Synthesis and structure determination of UO2(LX) (X = NH, O, S)
Each ligand was synthesized through a condensation reaction between a 3-tert-butyl-5-methoxysalicylaldehyde and the corresponding diamine in ethanol, and further reacted with one equivalent of UO2(NO3)2·6H2O to afford UO2(LX). These complexes were yielded as red microcrystalline solids, which were recrystallized from appropriate solvent mixtures to obtain single crystals suitable for X-ray structure determination. The IR peaks of [OUO]2+ asymmetric stretching (ν3) and CN stretching (νCN) of UO2(LX) were observed at around 860–880 and 1630 cm−1, respectively. The elemental analysis for UO2(LX) well-agreed with the expected chemical formulae of them.
The molecular structures of UO2(LX) were determined by single crystal X-ray diffraction (SCXRD). The resulting molecular structures of UO2(LX) are shown in Fig. 2 and S2.† The selected bond lengths of them are summarized in Table 1. As a general trend, UO22+ in UO2(LX) is five-coordinated in its equatorial plane to give a pentagonal bipyramidal coordination geometry as expected in Fig. 1(b), which is typically found in UO22+ complexes.21,22
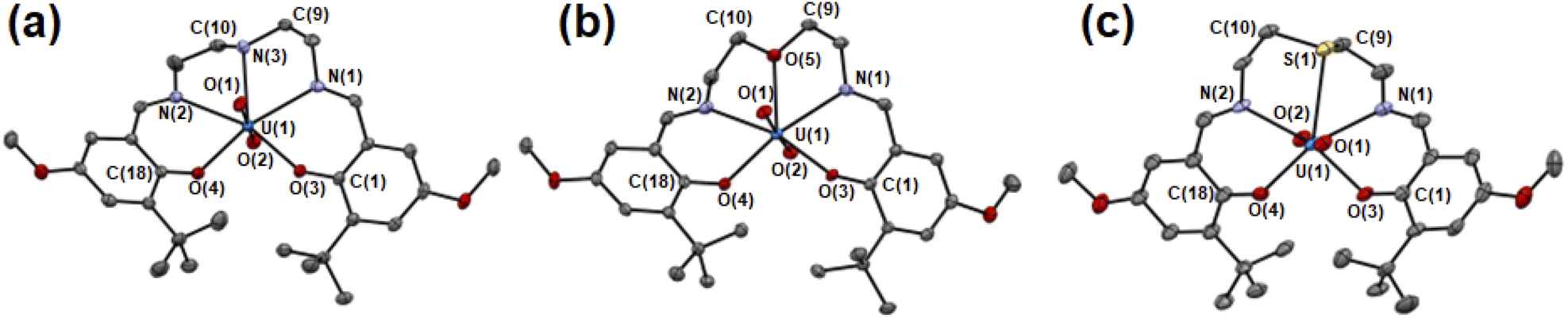 | ||
| Fig. 2 ORTEP views of UO2(LNH) (a), UO2(LO) (b), UO2(LS) (c). Ellipsoids are at 50% probability level. Hydrogen atoms and solvent molecules were omitted for clarify. | ||
| UO2(LNH) | UO2(LO) | UO2(LS) | |
|---|---|---|---|
| U–O(1) | 1.793(2) | 1.792(2) | 1.776(5) |
| U–O(2) | 1.787(2) | 1.790(2) | 1.778(5) |
| U–O(3) | 2.238(2) | 2.239(2) | 2.241(6) |
| U–O(4) | 2.239(2) | 2.239(2) | 2.231(5) |
| U–N(1) | 2.535(3) | 2.523(3) | 2.543(7) |
| U–N(2) | 2.554(2) | 2.531(3) | 2.542(6) |
| U–X | 2.594(5) | 2.581(3) | 2.981(2) |
| C(1)–O(3) | 1.342(3) | 1.332(3) | 1.34(1) |
| C(18)–O(4) | 1.333(3) | 1.334(3) | 1.34(1) |
The UOax bond lengths of UO22+ in UO2(LX) (U(1)–O(1), U(1)–O(2)) are 1.78–1.79 Å, which is similar to those in UO22+ complexes reported previously.21,22 Herein, we introduced tert-butyl groups at the ortho-positions of the phenolate moieties in each system to control the structure of the ligand after coordination to UO22+. To avoid steric collision between these bulky groups in UO2(LX), two phenolate moieties are forced to be present in the opposite sides of the equatorial plane of UO22+ (Fig. 2 and S2†). Such a twisted structure of a planar pentadentate ligand was also observed in the saldien-type ligands (e.g., Fig. 1(a)) we reported previously.21,22
The bond angles around X are strongly affected by the difference in X. The mean bond angle of C(9)–N(3)–C(10), C(9)–N(3)–U(1) and C(10)–N(3)–U(1) in UO2(LNH) is 111°, which is close to the ideal value (109.5°) of the tetrahedral coordination around N, showing its sp3 character. Note that the N–H moiety in UO2(LNH) forms a hydrogen bond with the axial O of UO22+ of the neighboring complexes (D⋯A: 3.15 Å, D–H: 1.00 Å, H⋯A: 2.210 Å, D–H⋯A: 154.9°). Nevertheless, there seems to be little effect on the structure of this UO22+ complex, because the similar bond angles around N was also observed in UO2(tBu,MeO–Mesaldien) (109°, see Fig. 1). The mean bond angles around X in UO2(LO) and UO2(LS) are 120° and 98°. Moreover, the deviation of X from the mean planes defined by U1, N1, N2, O3, and O4 in UO2(LS) (0.489 Å) is larger than those of UO2(LNH) (0.261 Å) and UO2(LO) (0.116 Å). Such a difference would be related to the bonding nature and steric factor of these X atoms. However, it is difficult at this moment to clearly describe in detail how the hardness/softness of X affects such a structural trend. Hence, we decided to focus on the bond lengths around X as another structural parameter directly affected by the coordination strength.
In the UO2(LX) complexes studied here, the bond lengths between U and the phenolic O (U(1)–O(3), U(1)–O(4)) are 2.23–2.24 Å regardless of difference in X. This is also the case for those between U and the imino N (U(1)–N(1), U(1)–N(2), 2.52–2.55 Å). In contrast, the U(1)–X bond lengths depend on X. The U(1)–N(3) distance of UO2(LNH) is 2.594(5) Å, which is slightly longer than the corresponding interaction in UO2(LO) (U(1)–O(5) = 2.581(3) Å). These bond lengths are commonly found in the previous reports.21,22 The U(1)–S(1) distance in UO2(LS) is significantly longer than the others. However, the U(1)–S(1) distance of UO2(LS) is still shorter than the sum of van der Waals radii of U and S (2.3 Å + 1.8 Å = 4.1 Å),42 suggesting that chemical bonding interaction is certainly present between U(1) and S(1) in this complex. Indeed, the U(1)–S(1) distance of UO2(LS) (2.981(2) Å) is close to the U–S bond lengths in UO22+–thioether complexes reported previously (2.96–3.02 Å).2,43,44 The observed structural parameters of the U–X interactions in UO2(Lx) are quite common in uranyl complexes having X atom coordination reported so far.2,21,22,43,44 Therefore, UO2(LX) studied here are suitable for exploring impacts of X in the coordination chemistry of UO22+.
It could be misleading to discuss the strengths of the U–X bonding interactions solely on the basis of the observed bond lengths, because the sizes of N, O and S are different from each other. The bond strengths between two atoms can be normalized by reduction in an interaction distance (RUX) derived from the sum of van der Waals radii and an actual bond length between U and X as shown in eqn (1).45–47
| RUX = (dUX)/(rU + rX) | (1) |
Thermodynamic stability of UO2(LX) (X = NH, O, S)
In the crystal structures of UO2(LX) (X = NH, O, S), the U–X bond strength depends on the difference in X. Therefore, there would also be some impact on the thermodynamic stability of UO2(LX). To confirm this issue, we investigated the complexation of UO22+ and LX2− in ethanol by spectrophotometric titration. Fig. 3 shows the UV-vis absorption spectra recorded at different total concentration ratios between UO22+ and LX2− represented by CU/CL. Note that these titration experiments were conducted under the presence of 0.4 mM NEt3 employed as a H+ scavenger after the formation of UO2(LX).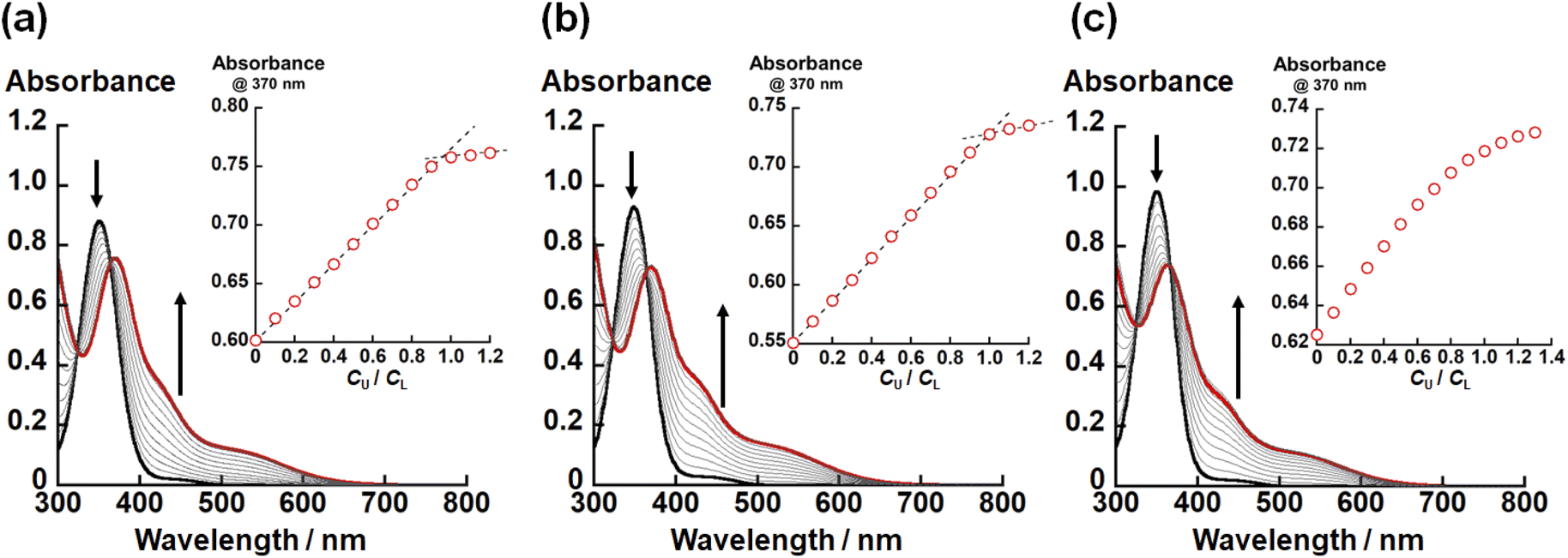 | ||
| Fig. 3 UV-vis absorption spectra of the ethanol solutions of (a) LNH2−, (b) LO2− and (c) LS2− at different total UO22+ concentrations under the presence of 0.4 mM NEt3. Total concentration of LX2−: 0.1 mM. Black and red lines are spectra of CU/CL = 0 and 1.0, respectively. Insets: plots of absorbance at 370 nm with an increase in CU/CL. | ||
In all titration series shown in Fig. 3, the absorbance at 370 nm and 425 nm increased with an increase in CU/CL. Simultaneously, the absorption intensity at 350 nm decreased. The isosbestic points were clearly observed, indicating that the complexation equilibrium between UO22+ and LX2− only takes place in each system. As shown in the insets of Fig. 3(a) and (b), the absorbance at 370 nm tends to be saturated at CU/CL = 1.0, indicating that UO2(LNH) and UO2(LO) are almost quantitatively formed. On the other hand, such a trend is equivocal for X = S (Fig. 3(c)), implying that the weaker coordination of LS2−.
To estimate the conditional stability constants (βX = [UO2(LX)]/([UO22+][LX2−])) of UO2(LX) (X = NH, O, S) in ethanol containing NEt3 (0.4 mM), the spectral series of Fig. 3 were analyzed by HypSpec program.30 As a result, logβX of UO2(LX) for X = NH, O, and S are estimated to be 10 ± 1, 7.24 ± 0.02, and 5.2 ± 0.1, respectively. Since all the coordinating atoms except for X are common in the studied systems, the difference in logβX observed here can be ascribed to the difference in affinity of X with UO22+. As widely-accepted in the HSAB principle, the hardness of X atoms follows O > NH > S.14,15 However, logβX of UO2(LX) decrease in the order of UO2(LNH) > UO2(LO) > UO2(LS), which is difficult to be rationalized only by the HSAB principle. To understand this trend, we focus on difference in basicity of X in LX2−. The pKa values of protonated diethylamine ((CH3CH2)2NH2+), dimethyl ether ((CH3)2OH+), and dimethyl thioether ((CH3)2SH+) are 11.0 (ref. 48), −3.8 (ref. 48), and −5.4 (ref. 48), respectively, which is exactly in line with the order of logβX of UO2(LX) described above. Therefore, the basicity of X atom would also provide some contribution to the thermodynamic stability of UO2(LX). At this moment, it is still too early to verify linear free energy relationship between logβX and pKa.
To further elucidate the nature of U–X bonds in UO2(LX), we carried out DFT calculations of UO2(LX), followed by the natural bond orbital (NBO) analysis. The molecular structures of UO2(LX) were taken from those of UO2(LX) determined by X-ray crystallography, and were optimized with B3LYP method.32 The optimized structures of UO2(LX) are shown in Fig. S4,† and selected bond lengths are summarized in Table S2.† All the bond distances well agree with those determined crystallographically (Table 1). Table S3† summarizes natural charges and Wiberg bond indices (WBI)17,18,40 of center U and coordinating atoms in the optimized structures.
No significant differences were found in the natural charge on the axial and equatorial coordinating atoms except for X. Both N(3) in UO2(LNH) (−0.646) and O(5) in UO2(LO) (−0.565) have negative natural charges, indicating that the center U and X atoms interact electrostatically. In contrast, the natural charge of S(1) in UO2(LS) is positive (+0.326), implying that the electrostatic attraction between U and S is little expectable despite significant penetration between these atoms in UO2(LS) within the sum of van der Waals radii as described above. To provide a rationale for the U–S bonding interaction experimentally observed, bond orders of U–X interactions were estimated in terms of WBI. As a result, some covalency was detected in the U–S bond of UO2(LS) as pronounced by WBI = 0.471, which is significantly larger than those of the other U–X bonds (WBI = 0.277–0.345). Therefore, the bonding interaction between U and S of UO2(LS) is rather covalent, while it is somewhat weakened by the electrostatic repulsion between these positively charged atoms. The significant covalency of the U–S interaction compared with the electrostatic characters in U–NH and U–O would be a typical manifestation of the HSAB principle. In connection with this, N is usually considered to be softer than O, while the stability of UO2(LNH) is greater than UO2(LO) despite the hardness of UO22+. The stronger basicity of NH provides an additional effect to strengthen the U–NH bond compared with that of U–O.
Electrochemistry and spectroelectrochemistry of UO2(LX) (X = NH, O, S)
As mentioned above, X strongly affects the thermodynamic stability of UO2(LX). Recently, we have reported that the redox potential of UO2(R1,R2–Mesaldien) (Fig. 1(a)) is significantly governed by substitution at R1 and R2 positions. Therefore, we expect that the difference in X may also vary the redox potentials of UO2(LX). To clarify this point, the electrochemical measurements of UO2(LX) in DMSO were carried out. Fig. 4 shows the obtained cyclic voltammograms of UO2(LX), where a couple of cathodic (Epc) and anodic peaks (Epc) has been observed. These redox waves are reproducible even in multiple scanned cyclic voltammograms recorded at the potential sweep rate (ν) of 100 mV s−1, indicating that the reduction product at Epc undergoes no successive reactions, and is fully reoxidized to UO2(LX) at Epa (Fig. S6†). The peak potential separation (Epc − Epa) tends to increase (111–490 mV) with increasing v from 50 mV s−1 to 500 mV s−1 (Fig. S7 and Tables S4–S6†), implying that these redox systems of UO2(LX) are quasireversible. Even after careful survey of the DFT calculations described later, we, however, could not find any critical rationales for the differences in the electrochemical reversibility of these systems. Anyway, the peak potential separations (Epa − Epc, see Table S6 in ESI†) of UO2(LS) were also much greater than the theoretical value of a reversible system (59 mV). Therefore, all the systems studied here are regarded to be electrochemically irreversible. Although we do not have unequivocal explanation for the above points at this moment, solvation structures around these uranyl complexes could be largely modified through the electron transfer. Note that all the redox reactions are chemically reversible as demonstrated by occurrence of the isosbestic points in the spectroelectrochemical experiments shown in Fig. 5, S8 and S9.† The diffusion coefficients (Do) of UO2(LNH), UO2(LO), and UO2(LS) in these systems at 295 K were estimated as 1.6 × 10−6, 1.8 × 10−6 and 8.1 × 10−7 cm2 s−1, respectively, where the redox reactions observed in Fig. 4 were assumed to be electrochemically irreversible.49 As summarized in Tables S4–S6,† the formal potential E°′ (=(Epc + Epa)/2) of each UO2(LX) is around −1.60 V vs. Fc0/+ with regardless of v, and also seems not to be largely affected by X. The E°′ value of UO2(LNH) well agrees with that of its analogue, UO2(tBu,MeO–Mesaldien), we reported previously (−1.60 V vs. Fc0/+ in DMSO).21,22 Therefore, the coordinating LX2− would not have strong contribution to the redox events of UO2(LX). From these results, we assume that the redox centers of all UO2(LX) are the UO22+ moiety. However, cyclic voltammograms does not provide any detailed information about the reductant of UO2(LX). Hence, we carried out the spectroelectrochemical measurements and theoretical calculations to further understand the redox chemistry of UO2(LX).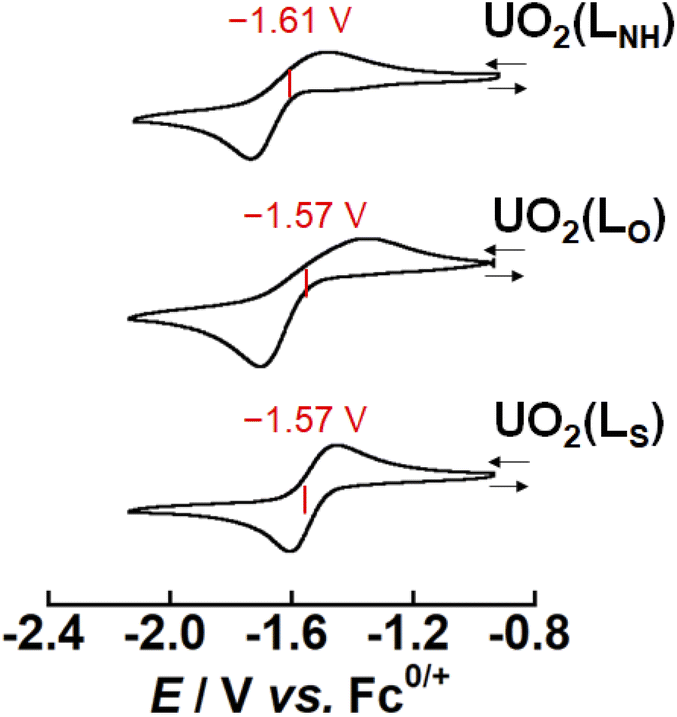 | ||
| Fig. 4 Cyclic voltammograms for the redox couples of UO2(LX) (X = NH, O, S) in DMSO at 295 K. Concentration of the complex was adjusted to 1 mM and tetra-n-butylammonium perchlorate (0.1 M) was used as a supporting electrolyte. Scan rates are 100 mV s−1. | ||
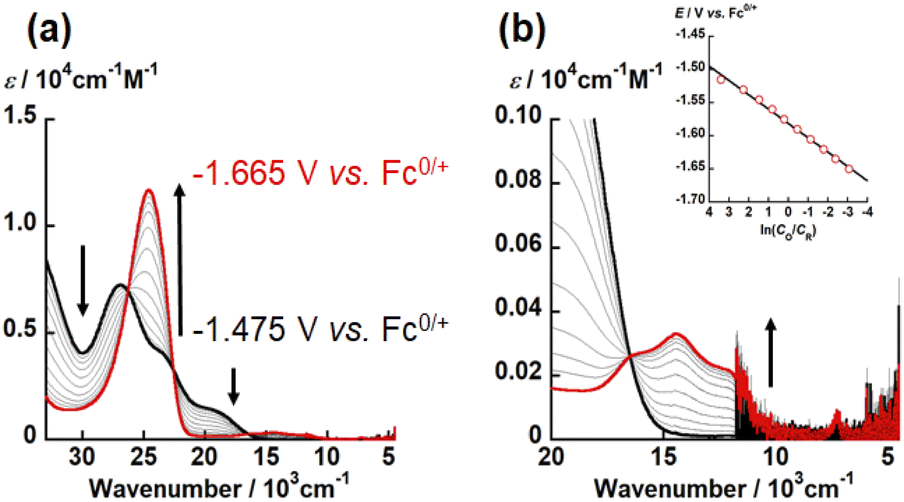 | ||
| Fig. 5 UV-vis-NIR spectral change of electrochemical reduction of UO2(LO) recorded at different applied potentials from −1.475 V to −1.665 V vs. Fc0/+ (potential step: 0.015 V) in DMSO with 0.1 M TBAP at 295 K. Black and red bold curves represent absorption spectra of UO2(LO) and [UO2(LO)]−, respectively. Wavenumber regions: (a) 33333–4500 cm−1, (b) 20 000–4500 cm−1. Inset: Nernstian plot calculated from absorbance at 24630 cm−1. | ||
To determine the electron stoichiometry (n) in the reduction of UO2(LX), the spectroelectrochemical measurements were performed. The UV-vis-NIR spectra of each system were recorded at different potentials (E). Fig. 5, S8 and S9† show the obtained spectral variations at X = O, NH, and S, respectively. As a general trend, the absorption bands of UO2(LX) around 30000 and 20000 cm−1 gradually decreased with decreasing E, while new absorption bands appeared around 25000 and 15000 cm−1. Moreover, isosbestic points were clearly observed, indicating that the redox equilibria of UO2(LX) only take place in the current potential ranges. Using the absorbance at 24630 or 24876 cm−1, the concentration ratio (CO/CR) between the oxidant (UO2(LX)) and its reductant at each E was calculated. The relationship between CO/CR and E should follow the Nernstian equation, eqn (2).
| E = E°′ + (RT/nF)ln(CO/CR) | (2) |
485 C mol−1), respectively. The slope and intercept of the linear relationship between E and ln(CO/CR) (insets of Fig. 5(b), S8(b) and S9(b)†) allow to determine n and E°′ of the redox reactions of UO2(LX). The estimated n values of UO2(LX) are close to unity (Table S7†), indicating that the reduction of UO2(LX) affords [UO2(LX)]−. The E°′ values estimated from the spectroelectrochemical measurements (Table S7†) agree with those observed in the CV measurements (Tables S4–S6†).
UV-vis-NIR spectra of UO2(LX) and [UO2(LX)]− in DMSO were summarized in Fig. 6. The spectral features of all UO2(LX) are quite similar to each other. All UO2(LX) showed characteristic bands around 28000 and 24000 cm−1. These absorption bands were also observed in UO22+ complexes with Schiff base ligands, and can be assigned to the π–π* transition bands of Schiff base ligands.21,22 Therefore, the difference in X leads to no significant differences in the electronic structures of UO2(LX).
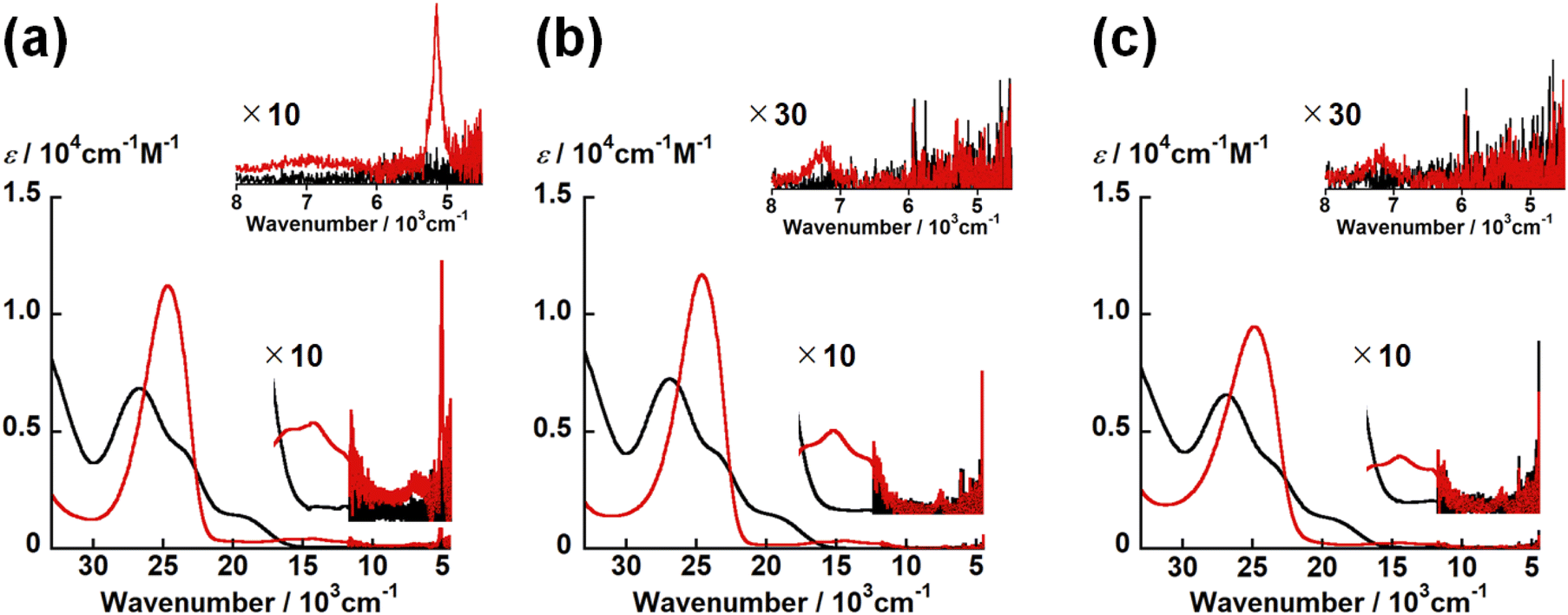 | ||
| Fig. 6 UV-vis-NIR spectrum of UO2(LX) (black) and one-electron reduced complexes [UO2(LX)]− (red) in DMSO containing 0.1 M tetra-n-butylammonium perchlorate at 295 K. [UO2(LNH)]−/0 (a), [UO2(LO)]−/0 (b) and [UO2(LS)]−/0 (c). Inset: expended views of NIR region. | ||
Even after the reduction, [UO2(LX)]− with different X commonly have the intense bands at around 25000 cm−1 with ε ∼ 104 M−1 cm−1 and weak bands at 16400, 14500, 12200 and 7200 cm−1 with ε ∼ 102 M−1 cm−1 (Fig. 6). Note that [UO2(LNH)]− has a characteristic band at 5200 cm−1, although this absorption is not clearly observed in [UO2(LO)]− and [UO2(LS)]− (Fig. 6). The absorption bands at around 25000, 16400, 14500, 12200 and 7200 cm−1 are generally observed in UVO2+ complexes with Schiff base ligands as reported previously.21,22 The intense absorption at 25000 cm−1 is assigned to a π–π* transition in the Schiff base ligands and/or a ligand-to-metal charge transfer (LMCT).21,22 In accordance with TD-DFT calculation,21,22 the absorption band at 16400 cm−1 is attributable to a metal-to-ligand charge transfer (MLCT) from a 5fδu orbital of the U5+ center to the π* orbital of the coordinating ligand. Finally, those at 14500, 12200 and 7200 cm−1 are ascribed to the f–f transitions arising from the 5f1 electron configuration of U5+.21,22,35 [UO2(LNH)]− only exhibited the absorption band at 5200 cm−1 attributable to another f–f transition,21,22,35 while this is not the case for the others studied here. As a matter of fact, this transition is not always clearly observable as we reported previously.22 To theoretically support occurrence of UVO2+ in each [UO2(LX)]−, we further performed DFT calculations of [UO2(LX)]−.
Initially, the molecular structures of [UO2(LX)]− in DMSO were taken from those of UO2(LX) determined by the X-ray crystal structure and were optimized after addition of a single negative charge and doublet spin degeneracy to assume the one-electron reduction. The optimized structures of [UO2(LX)]− were shown in Fig. S10† and the selected structural parameters are summarized in Table S2.†
The UOax bond lengths of [UO2(LX)]− are 1.86 Å, which are ca. 0.08 Å longer than those of the corresponding UO2(LX) determined by SCXRD (Table 1). These UOax bond lengths are very similar to those of the UVO2+ complexes with bis(phenolate) ligands (1.851(7)–1.868(8) Å).50 The U–O bond lengths between U atom and phenolic O of [UO2(LX)]− are ca. 0.15 Å longer than those of the corresponding UO2(LX) (Table 1). These U–O bond elongations indicate that these bond strengths are weakened by a decrease in the positive charge of U through the reduction from UVIO22+ to UVO2+. Actually, an unpaired electron of [UO2(LX)]− is exclusively localized in the center U as shown in the Mulliken spin density surfaces (Fig. 7), clearly indicating that these reduced complexes are of UVO2+ regardless of difference in X. Consequently, the X moiety does not largely affect the redox chemistry of [UO2(LX)]−/0.
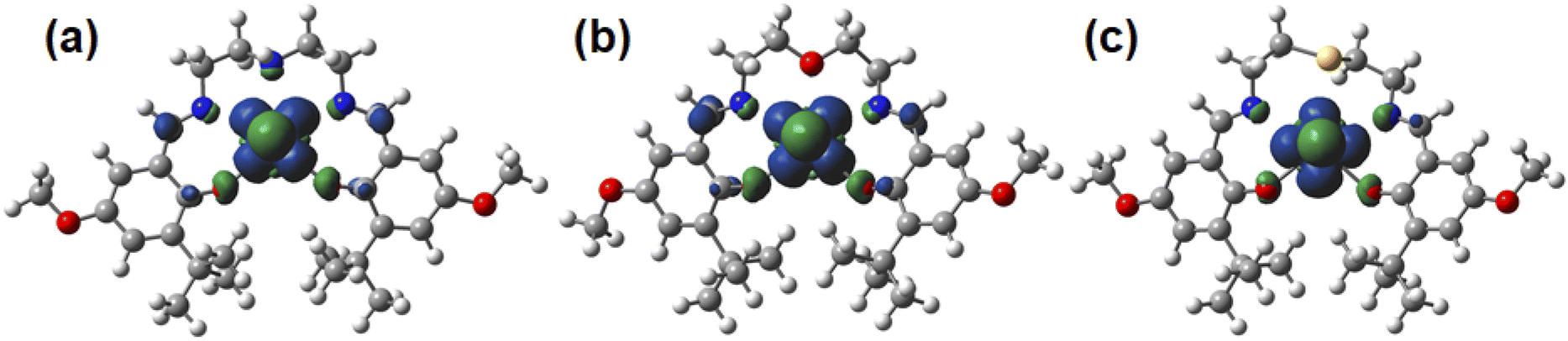 | ||
| Fig. 7 Spin-density plots of [UO2(LNH)]− (a), [UO2(LO)]− (b), [UO2(LS)]− (c). Spin density values of U atom are 1.09 for [UO2(LNH)]−, 1.09 for [UO2(LO)]−, 1.12 for [UO2(LS)]−. | ||
Conclusions
In this study, UO2(tBu,MeO–saldien–X) (UO2(LX); X = NH, O, S) were synthesized and structurally characterized to discuss impacts of the heteroatoms (X) to the coordination chemistry of UO22+. The crystal structures of UO2(LX) showed the five-coordinated UO22+ with LX2− in the equatorial plane. The UOax bond length of UO22+ and the bond length between U and phenolic O are not affected by the difference in X. On the other hand, the U–X bond length increases in the order of UO2(LO) < UO2(LNH) < UO2(LS). After taking into account the differences in the atomic size of X, the normalized U–X bond strength in UO2(LX) was found to follow U–O ≈ U–NH > U–S. While the U–O and U–NH bond strengths are similar to each other, the weaker U–S interaction can be explained by the HSAB principle. The logarithmic conditional stability constant (logβX) of UO2(LX) in ethanol containing 0.4 mM NEt3 decreases in the order of UO2(LNH) (logβNH = 10) > UO2(LO) (logβO = 7.24) > UO2(LS) (logβS = 5.2). This trend cannot be explained only by the HSAB principle, but rather follows the order of basicity of X. The theoretical calculations of UO2(LX) suggested that the ionic character of U–X bonds decreases in the order of U–NH > U–O > U–S. In contrast, the covalency increases as U–O < U–NH < U–S. No significant differences were found in the electrochemistry of UO2(LX) with different X in terms of E°′ and U-centered redox reaction. As demonstrated in this work, a UO22+-ligand bond strength does not always follow the HSAB principle, but is also affected by other factors such as Lewis basicity and balance between ionic and covalent interactions of donating atoms to the center metal. These points should be more carefully considered to design molecular structures of ligands suitable for hydrometallurgical separations of metal ions of interest.
Author contributions
T. T. devised the main conceptual ideas, carried out all experiments, and wrote the manuscript in consultation with K. T. K. T. supervised this project, discussed all experimental results with T. T., and edited the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Dr Satoru Tsushima of Institute of Resource Ecology, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), for supporting the DFT calculation. This work was supported in part by Grants-in-Aid for Scientific Research (No. 20H02663 to K. T., and 21J11942 to T. T.), JAEA Nuclear Energy S&T and Human Resource Development Project through concentrating wisdom Grant Number JPJA19P19209861.Notes and references
- U. Casellato, M. Vidali and P. A. Vigato, Coord. Chem. Rev., 1979, 28, 231–277 CrossRef CAS.
- M. Ephritikhine, Coord. Chem. Rev., 2016, 319, 35–62 CrossRef CAS.
- B. J. Thamer, J. Am. Chem. Soc., 1957, 79, 4298–4305 CrossRef CAS.
- E. Bullita, P. Guerriero, S. Tamburini, P. A. Vigato, J. C. Dupuy, M. Prevost, R. Bonora and L. Marchesini, Mater. Chem. Phys., 1992, 31, 181–198 CrossRef CAS.
- S. Chattaraj, A. Bhattacharyya and B. Sadhu, Inorg. Chem., 2021, 60, 15351–15363 CrossRef CAS PubMed.
- K. E. Gutowski, V. A. Cocalia, S. T. Griffin, N. J. Bridges, D. A. Dixon and R. D. Rogers, J. Am. Chem. Soc., 2007, 129, 526–536 CrossRef CAS PubMed.
- X. Li, Z. Zhang, L. R. Martin, S. Luo and L. Rao, Inorg. Chem., 2018, 57, 7684–7693 CrossRef CAS PubMed.
- B. G. Vats, A. Bhattacharyya, K. Sanyal, M. Kumar, J. S. Gamare and S. Kannan, Inorg. Chem., 2021, 60, 17529–17536 CrossRef CAS PubMed.
- C. Berger, C. Marie, D. Guillaumont, C. Tamain, T. Dumas, T. Dirks, N. Boubals, E. Acher, M. Laszczyk and L. Berthon, Inorg. Chem., 2020, 59, 1823–1834 CrossRef CAS PubMed.
- Y. Zhang, W. Duan, Y. Yang, T. Jian, Y. Qiao, G. Ren, N. Zhang, L. Zheng, W. Yan, J. Wang, J. Chen, S. G. Minasian and T. Sun, Inorg. Chem., 2022, 61, 92–104 CrossRef CAS PubMed.
- Y. Sasaki and S. Tachimori, Solvent Extr. Ion Exch., 2002, 20, 21–34 CrossRef CAS.
- K. Takao and Y. Ikeda, Eur. J. Inorg. Chem., 2020, 36, 3443–3459 CrossRef.
- T. Mizumachi, M. Sato, M. Kaneko, T. Takeyama, S. Tsushima and K. Takao, Inorg. Chem., 2022, 61, 6175–6181 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- K. Takao, S. Takao, Y. Ikeda, G. Bernhard and C. Hennig, Dalton Trans., 2013, 42, 13101–13111 RSC.
- J. Narbutt, A. Wodyńskia and M. Pecul, Dalton Trans., 2015, 44, 2657–2666 RSC.
- L. Xu, Y. Hao, X. Yang, Z. Wang, C. Xu, N. E. Borisova, M. Sun, X. Zhang, L. Lei and C. Xiao, Chem.–Eur. J., 2021, 27, 10717–10730 CrossRef CAS PubMed.
- T. Kimura, M. Kaneko, M. Watanabe, S. Miyashita and S. Nakashima, Dalton Trans., 2018, 47, 14924–14931 RSC.
- M. Kaneko and M. Watanabe, J. Radioanal. Nucl. Chem., 2018, 316, 1129–1137 CrossRef CAS.
- K. Takao, M. Kato, S. Takao, A. Nagasawa, G. Bernhard, C. Hennig and Y. Ikeda, Inorg. Chem., 2010, 49, 2349–2359 CrossRef CAS PubMed.
- T. Takeyama, S. Tsushima and K. Takao, Inorg. Chem., 2021, 60, 11435–11449 CrossRef CAS PubMed.
- H. Shi and Y. Yin, Inorg. Chim. Acta, 2014, 421, 446–450 CrossRef CAS.
- T. P. DeAngelis and W. R. Heineman, J. Chem. Educ., 1976, 53, 594–597 CrossRef CAS.
- W. R. Heineman, J. Chem. Educ., 1983, 60, 305–308 CrossRef CAS.
- X. Q. Lin and K. M. Kadish, Anal. Chem., 1985, 57, 1498–1501 CrossRef CAS PubMed.
- A. Endo, I. Mochida, K. Shimizu and G. P. Sato, Anal. Sci., 1995, 11, 457–459 CrossRef CAS.
- G. M. Sheldrick, SHELXL Version 2018/3, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Sabatini, A. Vacca and P. Gans, Coord. Chem. Rev., 1992, 120, 389–405 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Ikeda, C. Hennig, S. Tsushima, K. Takao and Y. Ikeda, Inorg. Chem., 2007, 46, 4212–4219 CrossRef CAS PubMed.
- K. Takao, S. Tsushima, T. Ogura, T. Tsubomura and Y. Ikeda, Inorg. Chem., 2014, 53, 5772–5780 CrossRef CAS PubMed.
- P. Macak, S. Tsushima, U. Wahlgren and I. Grenthe, Dalton Trans., 2006, 3638–3646 RSC.
- S. Tsushima, A. Rossberg, A. Ikeda, K. Muller and A. C. Scheinost, Inorg. Chem., 2007, 46, 10819–10826 CrossRef CAS PubMed.
- T. Takeyama, S. Iwatsuki, S. Tsushima and K. Takao, Dalton Trans., 2022, 51, 6576–6586 RSC.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, Gauss View, ver. 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 1031–1046 Search PubMed.
- U. Casellato, S. Sitran, S. Tamburini, P. A. Vigato and R. Graziani, Inorg. Chim. Acta, 1986, 114, 111–117 CrossRef CAS.
- S. Sitran, D. Fregona, U. Casellato, P. A. Vigato, R. Graziani and G. Faraglia, Inorg. Chim. Acta, 1987, 132, 279–288 CrossRef CAS.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- A. M. S. Riel, M. J. Jessop, D. A. Decato, C. J. Massena, V. R. Nascimento and O. B. Berryman, Acta Crystallogr., 2017, B73, 203–209 Search PubMed.
- Organic chemistry data, ACS Division of Organic Chemistry, https://organicchemistrydata.org/hansreich/resources/pka/, (accessed August 2022) Search PubMed.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- G. Nocton, P. Horeglad, V. Vetere, J. Pecaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of UO2(LX), crystal structure of H2LS, electrochemical data, spectroelectrochemical data, and optimized structures and structural parameters of [UO2(LX)]−/0. CCDC 2177295 for UO2(LO)·(C5H5N), 2177296 for UO2(LS)·(CH2Cl2) and 2177297 for UO2(LNH)·(CH2Cl2). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04639c |
| This journal is © The Royal Society of Chemistry 2022 |