DOI:
10.1039/D2RA04624E
(Paper)
RSC Adv., 2022,
12, 24427-24438
Highly active and stable surface structure for oxygen evolution reaction originating from balanced dissolution and strong connectivity in BaIrO3 solid solutions†
Received
25th July 2022
, Accepted 22nd August 2022
First published on 26th August 2022
Abstract
Catalysts for the oxygen evolution reaction (OER) are receiving great interest since OER remains the bottleneck of water electrolyzers for hydrogen production. Especially, OER in acidic solutions is crucial since it produces high current densities and avoids precipitation of carbonates. However, even the acid stable iridates undergo severe dissolution during the OER. BaIrO3 has the strongest IrO6 connectivity and stable surface structure, yet it suffers from lattice collapse after OER cycling, making it difficult to improve the OER durability. In the present study, we have successfully developed an OER catalyst with both high intrinsic activity and stability under acidic conditions by preventing the lattice collapse after repeated OER cycling. Specifically, we find that the substitution of Ir-site with Mn for BaIrO3 in combination with OER cycling leads to a remarkable activity enhancement by a factor of 28 and an overall improvement in stability. This dual enhancement of OER performance was accomplished by the novel strategy of slightly increasing the Ir-dissolution and balancing the elemental dissolution in BaIr1−xMnxO3 to reconstruct a rigid surface with BaIrO3-type structure. More importantly, the mass activity for BaIr0.8Mn0.2O3 reached ∼73 times of that for IrO2, making it a sustainable and promising OER catalyst for energy conversion technologies.
1. Introduction
The oxygen evolution reaction (OER) is a crucial reaction for emerging energy conversion technologies such as electrochemical water splitting1,2 and rechargeable metal–air batteries.3,4 Since OER remains to be the bottleneck of water electrolyzers for hydrogen production that can be operated by renewable energy sources producing high current densities and avoiding precipitation of carbonates (CO2 poisoning), OER catalysts in acidic solutions are receiving great interest.5,6 Recent discoveries of transition metal oxide-based catalysts7–16 in both acidic and alkaline solutions have greatly advanced our understanding about the OER, revealing new OER mechanisms17–19 and numerous OER descriptors.20–22 However, particularly for catalysts in acidic solutions, it remains difficult to find the descriptors for OER and the underlying mechanism due to the dynamic nature of the oxide surface, which undergoes severe dissolution and amorphization during the OER. Such surface dissolution and amorphization of OER catalysts usually leads to the decrease of intrinsic OER activity or low stability, even for acid stable iridium and ruthenium metal oxide-based catalysts.23–27 In addition, the kinetics of the OER becomes even more sluggish because of the multistep electron transfer process,4 and the possible OER mechanisms are still under discussion. Thus, finding OER catalysts with both high activity and stability in acidic solutions is a crucial and challenging task. Accordingly, catalyst discovery in acidic solutions must begin with the most stable iridium-based oxide (iridate) catalysts before moving on to the generally less stable 3d transition metal oxide catalysts.
Recently, Song et al.27 systematically studied the OER performance of three groups of iridates in 0.5 M H2SO4 aqueous solution, revealing the relationship between IrO6 octahedral connectivity and OER stability. Among the three groups of iridates, BaIrO3 with strong IrO6 connectivity (dominated by the face-sharing octahedra) exhibited the highest OER stability, while SrIrO3 with intermediate IrO6 connectivity (a mixture of face-sharing and corner-sharing octahedra) exhibited moderate OER stability and Ba4Ir3O10 with weak IrO6 connectivity (dominated by the corner-sharing clusters) exhibited the lowest OER stability. Furthermore, the recent OER study about BaIrO3 synthesized by solution calcination has demonstrated its excellent OER performance due to the high valence of Ir (Ir5+) in the IrOx surface layer.28 However, despite being one of the most stable OER catalysts, BaIrO3 (with strong IrO6 connectivity) experiences a decrease in intrinsic OER activity (OER current density normalized to the electrochemically active surface area) with cycling,27,28 and the OER activity per oxide begins to decrease after repeated cycling due to the substantial dissolution of IrO6 units. This dissolution of IrO6 units originates from the lattice collapse (specifically at the surface amorphous layer) of BaIrO3 particles and leads to the loss of intrinsic OER activity (such lattice collapse takes place at a much earlier stage of OER for SrIrO3 due to its weaker IrO6 connectivity). In other words, the lattice collapse of the surface amorphous layer occurs sooner or later because its chemical bonding (of the IrO6 units) is weaker than that of the crystalline layer. Therefore, the lattice collapse of iridate particles27 after repeated OER cycling is a challenging issue to overcome, and further reduction of Ir-dissolution may not be a viable option since BaIrO3 has the strongest IrO6 octahedral connectivity (dominated by the face-sharing octahedra) among iridates.
In the present study, we have prevented the lattice collapse of BaIrO3 catalyst by substituting the Ir-site with Mn and succeeded in developing an OER catalyst with both high intrinsic activity and stability. This was achieved by the novel strategy of slightly increasing the Ir-dissolution and balancing the elemental dissolution in BaIr1−xMnxO3 to reconstruct a rigid BaIrO3-type structure at the surface amorphous layer (Mn is at the same crystallographic site as Ir, and it will induce the Ir-dissolution since Mn is easier to dissolve during the OER under acidic conditions). Specifically, the OER activities of the isostructural BaIr1−xMnxO3 (0 ≤ x ≤ 0.3) solid solutions were systematically studied, and a remarkable activity enhancement by a factor of 28 and an overall improvement in stability was achieved by the balanced elemental dissolution and strong IrO6 octahedral connectivity in BaIr1−xMnxO3. In concrete, both high intrinsic OER activity and high OER stability in acidic solutions was enabled by the robust and thin surface layer of BaIr1−xMnxO3, which was formed by the good balance between Ba- and Ir-dissolution, and the strong IrO6 connectivity of BaIrO3-type structure. Thus, our study suggests that BaIr1−xMxO3 solid solutions (M: 3d transition metal) provide a promising venue for the development of highly active and durable OER catalysts in acidic environment.
2. Experimental
2.1 Sample synthesis and characterization
Polycrystalline samples of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) were synthesized by solid state reaction from BaCO3 (Wako Pure Chemical Industries Ltd.), IrO2 (Wako Pure Chemical Industries Ltd.) and Mn3O4. The purpose of this study is to compare the OER activity of BaIr1−xMnxO3 with that of BaIrO3 within the robust structural regime of BaIrO3. Therefore, we focused on the composition (x = 0, 0.1, 0.2, and 0.3) that BaIr1−xMnxO3 adopts the crystal structure isostructural with BaIrO3 (in the space group of C12/m1). Mn3O4 powder was prepared from MnCO3 (Kojundo Chemical Laboratory) by heating in air at 1000 °C for 18 h. Well-ground mixtures of the starting materials in the stoichiometric ratio were heated in air at 900 °C for 12 h, followed by regrinding and heating at 1000 °C for a total of 73 h as in Zhao et al.29
The phase purity and lattice constants of the BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) powders were examined by powder X-ray diffraction (XRD) (Rigaku; RINT2500Ultra18) using Cu Kα radiation (λ = 1.540598 Å) at RT, and the structures were refined via the Rietveld method starting from the crystal structure of BaIrO3 in JCPDS #01-080-1880 using the GSAS Rietveld refinement software.30 Also, the synchrotron X-ray powder diffraction profile of BaIrO3 was obtained at the TPS19A beamline, National Synchrotron Radiation Research Center (NSRRC) with 15 keV beam and the MYTHEN detector in order to check the validity of the zero position and the lattice constants obtained by powder X-ray diffraction. Scanning electron microscopy with energy-dispersive X-ray analysis (SEM/EDX) was conducted using a JEOL JSM-6701F at an acceleration voltage of 20 kV. High-resolution transmission electron microscopy (HRTEM) measurements were conducted using a JEOL JEM-2100. The valence state and the chemical bonding of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) samples were examined by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra collected in transmission mode at Aichi synchrotron BL5S1. After the data collection, XANES and EXAFS spectra were analyzed with the software of Ifeffit Athena and Artemis.31
2.2 Electrochemical measurements
The catalyst inks for the electrochemical measurements were prepared referring to methods reported by Suntivich et al.12 and Grimaud et al.13 K+ ion-exchanged Nafion® was used as an immobilizing binder, enabling smooth transport of dissolved O2 to the surface of the catalysts. A 3.33 wt% K+ ion-exchanged Nafion® suspension was first obtained from a mixture of 5 wt% proton-type Nafion® suspension (Sigma-Aldrich) and a 0.1 M KOH aqueous solution in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio. This process increased the pH of the initial 5 wt% proton-type Nafion® suspension from 1–2 to 11. The catalyst inks were prepared from a mixture of the as-prepared samples (25 mg), acetylene black (AB, Strem Chemicals Inc., 5 mg) and 3.33 wt% K+ ion-exchanged Nafion® suspension (1.5 mL). The total ink volumes were adjusted to 5 mL by the addition of tetrahydrofuran (Sigma-Aldrich), to give final concentrations of 5 mg sample per mL, 1 mg AB per mL, and 1 mg Nafion per mL in the ink. A sample of the ink (6.4 μL) was then drop-casted on a rotating-ring disk electrode composed of a glassy carbon (GC) disk (0.2 × 0.2 × π cm2) and a Pt ring (BAS Inc., Japan), which was used as the working electrode after mirror polishing with 0.05 μM alumina slurry (BAS Inc., Japan). The catalyst layer on the GC disk was dried overnight in vacuum at room temperature, and was composed of 0.25 mg sample per cmdisk2, 0.05 mg AB per cmdisk2 and ∼0.05 mg Nafion per cmdisk2.
:1 (v/v) ratio. This process increased the pH of the initial 5 wt% proton-type Nafion® suspension from 1–2 to 11. The catalyst inks were prepared from a mixture of the as-prepared samples (25 mg), acetylene black (AB, Strem Chemicals Inc., 5 mg) and 3.33 wt% K+ ion-exchanged Nafion® suspension (1.5 mL). The total ink volumes were adjusted to 5 mL by the addition of tetrahydrofuran (Sigma-Aldrich), to give final concentrations of 5 mg sample per mL, 1 mg AB per mL, and 1 mg Nafion per mL in the ink. A sample of the ink (6.4 μL) was then drop-casted on a rotating-ring disk electrode composed of a glassy carbon (GC) disk (0.2 × 0.2 × π cm2) and a Pt ring (BAS Inc., Japan), which was used as the working electrode after mirror polishing with 0.05 μM alumina slurry (BAS Inc., Japan). The catalyst layer on the GC disk was dried overnight in vacuum at room temperature, and was composed of 0.25 mg sample per cmdisk2, 0.05 mg AB per cmdisk2 and ∼0.05 mg Nafion per cmdisk2.
Electrochemical measurements were conducted using a rotating ring disk electrode rotator (RRDE-3A, BAS Inc., Japan) at 1600 rpm, in combination with a bipotentiostat (BAS Inc., Japan). In addition, a Pt wire counter electrode, and an Hg/Hg2SO4 reference electrode (BAS Inc., Japan) filled with 0.5 M H2SO4 aqueous solution (Wako, Ltd., Japan) were used. Electrochemical measurements were conducted with O2 saturation (30 min bubbling O2 gas through the solution). During OER current measurements for each sample, the potential of the sample-modified GC was controlled from 0.53 to 0.95 V vs. Hg/Hg2SO4 with an internal solution of 0.5 M H2SO4 (1.228–1.648 V vs. RHE) at 10 mV s−1. For all measurements, the OER current density was iR-corrected (R = ∼8 Ω) using the measured solution resistance, and capacitance-corrected by averaging the anodic and cathodic scans12 to remove the non-faradaic current contribution. The current densities (mA cmoxide−2 and mA cmdisk−2) were obtained by dividing the OER current with the oxide BET surface area and the glassy carbon disk electrode area. The electrochemical impedance spectra (EIS) were measured in the frequency range between 200 kHz and 100 mHz at the amplitude of 10 mV.
3. Results and discussion
3.1 OER activity of BaIr1−xMnxO3
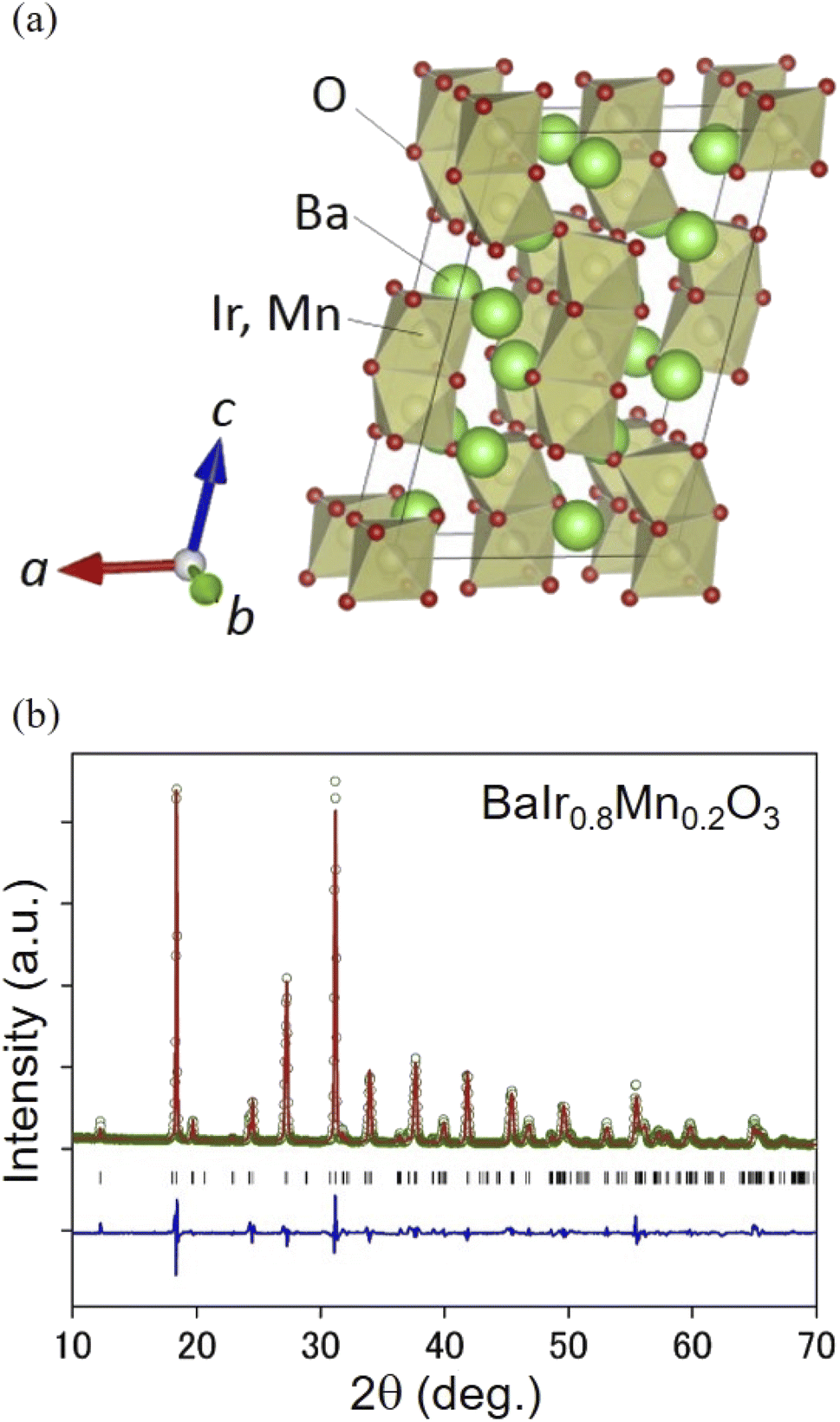
The OER activity of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) was studied in 0.5 M H2SO4 aqueous solutions. We selected this range of Mn content to exclude any structural effect on OER performance and to preserve the strong IrO6 octahedral connectivity of the OER stable BaIrO3 (ref. 27 and 32) denoted as ‘9M’29 (that is a monoclinically distorted version of BaRuO3 (ref. 33)). We confirmed by X-ray diffraction that BaIr1−xMnxO3 adopts a monoclinic structure with the space group of C12/m1 (Fig. 1a and b), which is isostructural with BaIrO3 (also see the XRD profiles in Fig. S1† and crystallographic information in Table S1 and Fig. S2†). BaIr1−xMnxO3 particles are in the size of 1–3 μm (see the SEM images in Fig. S3 and S4†). Mn content x was determined by EDX (Fig. S3†) and the relationship between Mn content and lattice volume (Fig. S2b†), and its validity was tested with the Ir and Mn valence confirmed by the X-ray absorption near-edge structure (XANES) spectra (Fig. S5a and S7a†).
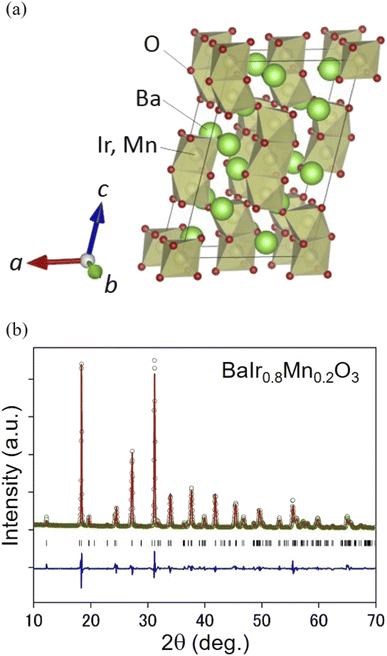 |
| | Fig. 1 (a) Schematic representation of the crystal structure of BaIr1−xMnxO3. This image was drawn using the VESTA software.38 BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) adopts a monoclinic structure with the space group of C12/m1, which is characterized by the ionic coordination denoted as ‘9M’29 (monoclinically distorted version of BaRuO3 with the ionic coordination of 9R33) with a zigzag network of three face-sharing IrO6 (MnO6) octahedra forming columns. (b) The representative XRD profile and Rietveld refinement result of BaIr1−xMnxO3 (x = 0.2). The green circles and the red line indicate the observed and calculated profile, respectively. The blue line displays the difference between the observed and calculated profile. The black tick marks indicate the Bragg reflection positions of BaIr0.8Mn0.2O3. Wavelength λ = 1.540598 Å. | |
Capacitance-corrected voltammograms (0.53 to 0.95 V vs. Hg/Hg2SO4 filled with 0.5 M H2SO4) for BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) are shown in Fig. 2a, while the corresponding cyclic voltammograms are depicted in Fig. S9a.† For the majority of catalysts, including SrIrO3,27 the lattice collapse and the elemental dissolution is severe during the OER, and the OER current density begins to decrease after repeated cycling due to the loss of intrinsic OER activity (current density normalized to the electrochemically active surface area) even when the electrochemically active surface area (ECSA) increases due to surface amorphization. In contrast, the OER current density of BaIrO3 continues to increase over a long cycle (200 to 1000 OER cycles depending on the potential range) as a result of the intense chemical bonding (enabled by the strong IrO6 connectivity) that minimizes the loss of intrinsic OER activity and the sharp increase of ECSA that originates from the thick IrOx amorphous layer.27 Thus, in this paper, we will evaluate the BaIr1−xMnxO3 catalyst that has experienced 1000 OER cycles as a separate OER catalyst (from pristine catalyst) because the final surface structure after OER is more crucial than the initial surface structure for BaIr1−xMnxO3. For simplicity, each BaIr1−xMnxO3 catalyst after 1000 OER cycles will be referred to as ect-BaIr1−xMnxO3 (abbreviation for electrochemically treated BaIr1−xMnxO3). It is clear from Fig. 2a and S11† that neither the OER activity per oxide surface area (see the details of BET surface area in Table S2†) nor the OER activity per geometrical electrode area (see also Fig. S9b†) for BaIr1−xMnxO3 varies monotonically with the Mn content. Instead, the OER activities of the BaIr1−xMnxO3 solid solution (0.1 ≤ x ≤ 0.2) are significantly higher than those of BaIrO3 for both pristine and electrochemically treated samples. This is also the case for other parameters describing the OER performance of BaIr1−xMnxO3 (see the relationship between Mn content and OER descriptors in Fig. S16†). As evident from the cyclic voltammograms in Fig. S9a,† the double-layer capacitance of BaIrO3 shows a sharp increase after 1000 OER cycles, while that of BaIr1−xMnxO3 does not show a rapid increase after 1000 OER cycles. Therefore, it is important to calculate the OER activity normalized to the ECSA for the comparison of intrinsic OER activity and the evaluation of OER cyclic stability over the long term. Since the chemical composition is similar in pristine and electrochemically treated samples, ECSA is proportional to the double-layer capacitance within each chemical composition of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3). For this reason, we evaluate the intrinsic OER activity of BaIr1−xMnxO3 in Fig. 2b by normalizing to the relative ECSA (that is proportional to the double-layer capacitance in Fig. S10c†), which is defined by the following ratio: ECSA of electrochemically treated BaIr1−xMnxO3/ECSA of pristine BaIr1−xMnxO3. Furthermore, we should limit the comparison of intrinsic OER activity in Fig. 2b to potentials below 1.57 V vs. RHE since the coverage of the catalytic surface by O2 bubbles cannot be ignored above 1.57 V vs. RHE (the electrode potential with 20 mA cmgeo−2 for electrochemically treated BaIrO3) for the electrochemically treated samples, and dividing the current density above 1.57 V vs. RHE with the ECSA calculated from the potential range below 1.57 V vs. RHE will underestimate the intrinsic OER activity. In particular, when the current density is normalized to the relative ECSA, the intrinsic OER activity of electrochemically treated samples for BaIr0.8Mn0.2O3 and BaIr0.7Mn0.3O3 below 1.57 V vs. RHE is comparable to that of pristine samples (Fig. 2b). On the other hand, electrochemically treated BaIrO3 shows a 50% decrease in intrinsic OER activity relative to pristine sample at 1.57 V vs. RHE due to the lattice collapse of the surface structure.27 Similarly, electrochemically treated BaIr0.9Mn0.1O3 shows a 30% decrease in intrinsic OER activity relative to pristine sample at the same potential (Fig. 2b). Therefore, the remarkably high OER activity of electrochemically treated BaIr0.8Mn0.2O3 originates from the well maintained intrinsic OER activity during 1000 OER cycles. At an applied potential of 1.58 V vs. RHE, a current density of 85 mA cm−2 (Fig. 3a) was recorded for electrochemically treated BaIr0.8Mn0.2O3 with a small charge transfer resistance of ∼16 Ω in Table S3d† (this value is obtained by fitting the electrochemical impedance spectra of Fig. S13a,† and it is smaller than the value of BaIrO3 in Tables S3a and S3c†). This indicates that the observed high current density comes from the OER. The observed current density for electrochemically treated BaIr0.8Mn0.2O3 is ∼28 times higher than that for pristine BaIrO3, ∼3.5 times higher than that for electrochemically treated BaIrO3, ∼46 times higher than that for SrIrO3, and ∼45 times higher than that for IrO2 (we note that BaIrO3 is one of the most stable OER catalysts with high current density, and the initial OER activity of SrIrO3 is comparable with that of pristine BaIrO3 (ref. 27)). As shown in Fig. 2b, this trend is maintained even when the current density is normalized to the relative ECSA. In other words, the comparison of OER activity for BaIr1−xMnxO3 (Fig. 2a and 3a) is not biased by the surface area or by the number of accessible active sites. Furthermore, Table S4† demonstrates that the electrochemically treated BaIr0.8Mn0.2O3 and BaIr0.9Mn0.1O3 show 60–100 times higher turnover frequency (TOF) than that of IrO2, and 10–20 times higher TOF than that of SrIrO3 (here, TOF was calculated by TOF = J/4Fn, where J is the OER current at 1.58 V vs. RHE, F is the Faraday constant, and n is the number of moles of the Ir atom on the working electrode). Fig. 3b shows the iR-corrected Tafel plots obtained under steady-state conditions for pristine and electrochemically treated BaIr1−xMnxO3. BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) exhibited Tafel slopes of 36–44 mV dec−1 for electrochemically treated samples, and 48–57 mV dec−1 for pristine samples. The smaller Tafel slope in Fig. 3b and the lower onset potential in Fig. 2a and S11† demonstrate that both pristine and electrochemically treated BaIr0.8Mn0.2O3 and BaIr0.9Mn0.1O3 exhibit higher OER activity than BaIrO3 and other iridates, such as IrO2 and SrIrO3. Consequently, we have demonstrated that electrochemically treated BaIr0.8Mn0.2O3 and BaIr0.9Mn0.1O3 show excellent overall OER performance. Next, we will investigate the reason for the extremely high OER activity of BaIr0.8Mn0.2O3 and the high cyclic stability of BaIr1−xMnxO3 (0.1 ≤ x ≤ 0.3). We note that the trend of OER activity in BaIr1−xMnxO3 (depicted in Fig. 2a, b, and S16†) cannot be explained by the difference of Ir–O bond distance (see the Ir–O bond distance of Table S1† obtained by the EXAFS fitting in Fig. S6,† and the successful Mn K-edge EXAFS fitting). Instead, the relatively longer Ir–O bond distance of electrochemically treated BaIrO3 suggests the existence of a weaker Ir–O bonding at the BaIrO3 surface after repeated OER cycling, which is consistent with the thick amorphous layer observed in the transmission electron microscopy image of Fig. 4b (that will be explained in detail in the next paragraph). Therefore, the highest OER activity at x ∼ 0.2 and the high overall OER activity of BaIr1−xMnxO3 (0.1 ≤ x ≤ 0.3) must be explained by a descriptor other than Ir–O covalency or orbital hybridization (Table S1†).
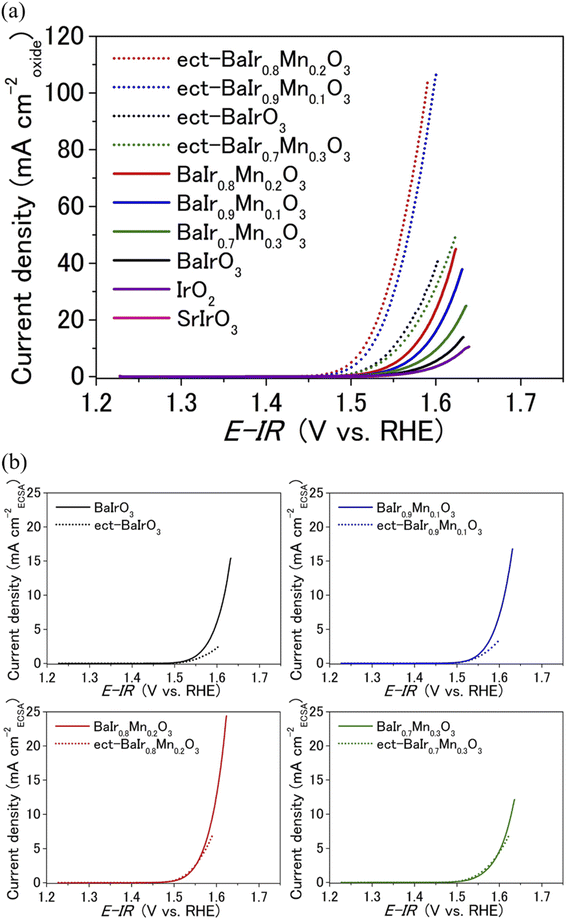 |
| | Fig. 2 (a) Linear sweep voltammograms (LSV) for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) in 0.5 M H2SO4 aqueous solution normalized to the oxide surface area in Table S2.† LSV of SrIrO3 is overlapping with that of IrO2. (b) LSV for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) normalized to the relative ECSA determined from the slope of Fig. S10c.† | |
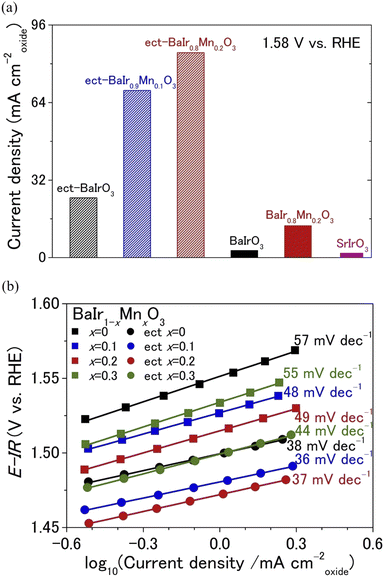 |
| | Fig. 3 (a) The representative OER current density at 1.58 V vs. RHE for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 in 0.5 M H2SO4 aqueous solution. The current density of SrIrO3 is shown for comparison. By the way, the current density of IrO2 is ∼1.9 mA cmoxide−2 at 1.58 V vs. RHE, which is obtained from Fig. 2a. (b) Tafel plots for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) collected under steady-state conditions. | |
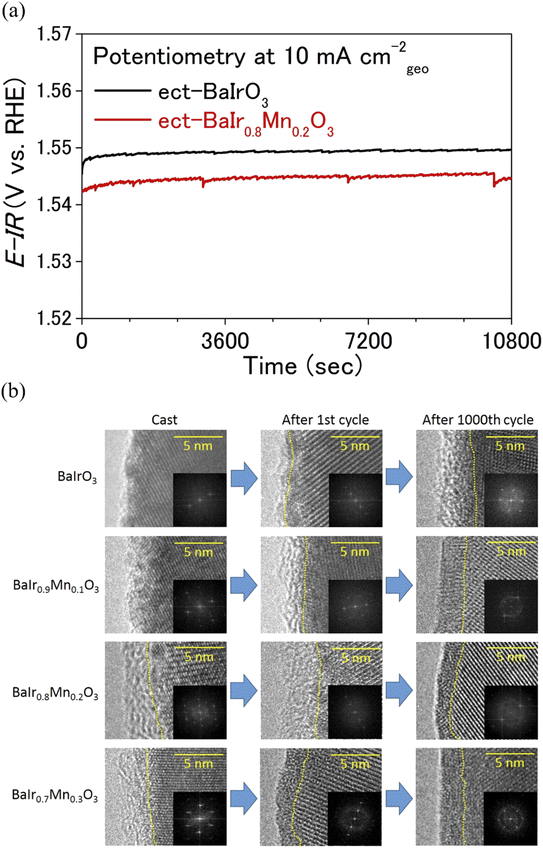 |
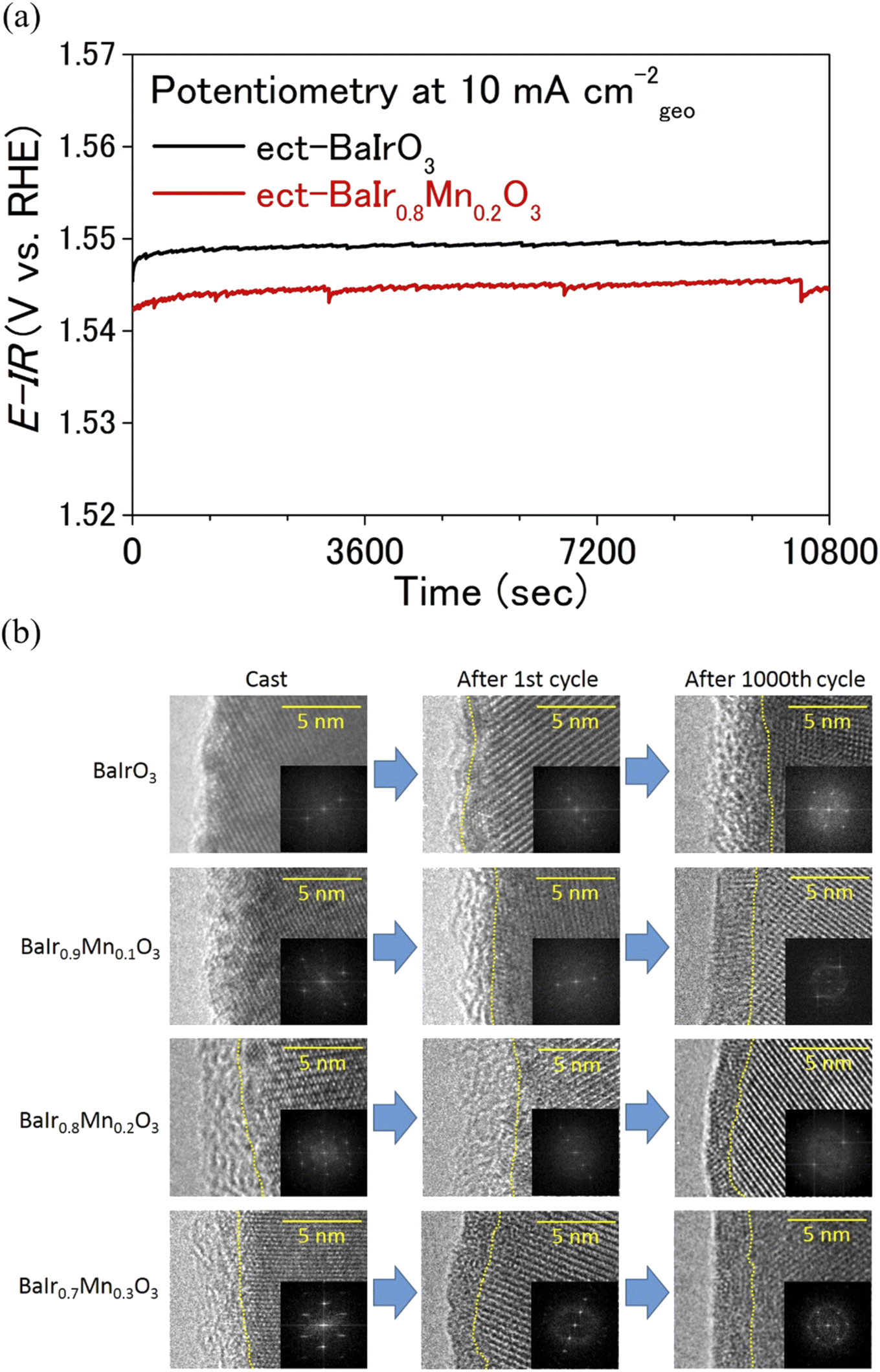
| | Fig. 4 (a) Chronopotentiometric curves at 10 mA cmgeo−2 (current density normalized to the geometrical electrode area) for electrochemically treated (ect-) BaIr0.8Mn0.2O3 and BaIrO3. (b) The OER catalytic surface of BaIr1−xMnxO3. HRTEM and fast Fourier transform (FFT) images of as-cast, after 1st and 1000 OER cycles for BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) are shown. The boundaries between the crystalline layer and the amorphous layer are divided by yellow dotted lines. | |
3.2 OER stability of BaIr1−xMnxO3
Extended electrochemical studies were conducted to investigate the catalytic stability of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3). The chronopotentiometric curve depicted in Fig. 4a demonstrates that the OER activity of BaIr0.8Mn0.2O3 is stable in 0.5 M H2SO4 with the polarization potential being nearly constant (at a geometrical current density of 10 mA cm−2) for 3 h, while remaining lower than that of BaIrO3. Moreover, it is evident from the inductively coupled plasma optical emission spectroscopy (ICP-OES) result (see the elemental concentration in Table S5†) after 1000 OER cycles that the bulk dissolution during the OER is negligible and BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3) is an exceptionally stable OER catalyst. This high OER stability of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) was further probed using high-resolution transmission electron microscopy (HRTEM) for both pristine and electrochemically treated samples (including the samples after the first OER cycle). Fig. 4b shows the HRTEM and fast Fourier transform (FFT) images of BaIr1−xMnxO3 before and after OER measurements (also see the expansion of FFT images in Fig. S14†). Before discussing the catalytic stability in detail, we note that, as reported by Yagi et al.14 and May et al.,34 the catalytic activity of an OER catalyst is determined by the ∼10 nm layer of the catalytic surface. For the as-cast sample, BaIrO3 possesses a highly-crystalline surface structure. However, after the first OER cycle, a 1–2 nm amorphous layer is observed at the catalytic surface, and after 1000 OER cycles, the amorphous layer finally develops to ∼5 nm thickness (Fig. 4b), which eventually leads to the lattice collapse of the surface structure.27 In contrast, BaIr1−xMnxO3 solid solutions (x = 0.1, 0.2, and 0.3) show 2–3 nm amorphous layer already after the first OER cycle, but the thickness of the amorphous layer decreases (in the case of BaIr0.8Mn0.2O3) or remains nearly constant even after 1000 OER cycles. Furthermore, the amorphous layer of the BaIr1−xMnxO3 solid solution (x = 0.1, 0.2, and 0.3) in Fig. 4b shows somewhat crystalline stripes after 1000 OER cycles (which is not observed after the first OER cycle for BaIr0.8Mn0.2O3 and BaIr0.9Mn0.1O3), indicating the existence of a short-range atomic order at the outermost surface and the reconstruction of the catalytic surface (that is likely to be Ba1−zIr1−yMnyOx units originating from the highly crystalline BaIr1−xMnxO3 layer beneath the amorphous layer). This observation of short-range atomic order (or the existence of long-range atomic order to some extent) suggests that the lattice collapse and subsequent loss of OER activity are prevented in the BaIr1−xMnxO3 solid solution (x = 0.1, 0.2, and 0.3) unlike the case for BaIrO3 (BaIrO3 suffers from lattice collapse after repeated OER cycling as reported by Song et al.27). We noted that these observations are in good agreement with the change of ECSA, which originates primarily from the ECSA of the amorphous layer at the catalytic surface. Based on Fig. S10c,† the relative ECSA after 1000 OER cycles is ∼19 for BaIrO3, while it is ∼14 for BaIr0.9Mn0.1O3, ∼8.1 for BaIr0.8Mn0.2O3, and ∼3.5 for BaIr0.7Mn0.3O3. With increasing Mn content, the relative ECSA continues to decrease, indicating that not only does the thickness of the amorphous layer decrease, but also the atomic order within the amorphous layer improves (since the more crystalline surface has less ECSA). This change of ECSA is well reflected in the somewhat crystalline stripes of the thinner (relative to BaIrO3) amorphous layers in BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3) (the feature of crystalline stripes is particularly evident for x = 0.2 and 0.3). We note that the amorphization mechanism of the SryIrOx surface layer demonstrated for the SrIrO3 thin film grown on a DyScO3 (110) orthorhombic substrate along the (001) pseudo-cubic orientation is another example of a non IrOx unit at the catalytic surface,35 while all of the BaIr1−xMnxO3 samples in this study were obtained by solid state synthesis. We also note that Mn and Ir occupy the same crystallographic site, and Mn-dissolution will induce Ir-dissolution because Mn is easier to dissolve under acidic conditions during the OER (yet Mn is among the OER stable elements under acidic conditions when it forms an oxide, and manganese oxides show higher stability than any other 3d transition metal oxides).36 For this reason, the crystalline stripes after 1000 OER cycles (Fig. 4b) and the high OER stability for the BaIr1−xMnxO3 solid solution (x = 0.1, 0.2, and 0.3) in Fig. 5b and 4a may well be the result of a balanced surface dissolution of Ba- and Ir-site (Mn-site) that enables the formation of Ba1−zIr1−yMnyOx units (instead of the Ir-rich IrOx units) in combination with the strong IrO6 connectivity of BaIr1−xMnxO3 (that protects the catalytic surface from suffering excessive Ir-site dissolution and therefore minimizes the particle size reduction during the OER). The well sustained intrinsic OER activity observed for electrochemically treated BaIr0.8Mn0.2O3 and BaIr0.7Mn0.3O3 in Fig. 2b below 1.57 V vs. RHE is in good agreement with this surface reconstruction (observed as crystalline stripes), while the 30% decrease of intrinsic OER activity observed for electrochemically treated BaIr0.9Mn0.1O3 may suggest that the surface reconstruction only proceeds partially when the surface dissolution of Ir is insufficient (due to the lack of Mn) and does not balance with the surface dissolution of Ba. Here, we demonstrate the scenario of the surface structure of BaIr1−xMnxO3 after repeated OER cycling in Fig. 5a, which is consistent with the high intrinsic OER activity and high stability of the BaIr1−xMnxO3 solid solution (x = 0.1, 0.2, and 0.3) after 1000 OER cycles shown in Fig. 2b and 5b. We stress that the fresh (meaning that the chemical composition is similar to the initial BaIr1−xMnxO3) and robust amorphous layer that is formed at the catalytic surface is the key for the high intrinsic OER activity and high stability of BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3). This is in contrast to SrIrO3, in which the peeling off of the surface structure (just like the skin of an onion) leads to the loss of intrinsic OER activity due to the particle size reduction caused by the weaker IrO6 connectivity (that is a mixture of face-sharing and corner-sharing octahedra) at amorphous layer. In concrete, BaIrO3 and BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3) possesses a zigzag network of three face-sharing IrO6 octahedra forming columns that is characteristic for the 9M ionic coordination, while SrIrO3 possesses two face-sharing IrO6 octahedra separated by single IrO6 octahedra characteristic for the 6M ionic coordination (Fig. 5a), and thereby the IrO6 connectivity is weaker for SrIrO3. After repeated OER cycling, the surface structure of IrO2 resembles that of SrIrO3, and it is reported that the surface structure of IrO2 belongs to the same group as that of SrIrO3.27 Moreover, the increase of Ir-dissolution in BaIr1−xMnxO3 compared to BaIrO3 may appear to have a negative effect at first glance, but the peeling off
of the surface structure by the balanced dissolution of Ba and Ir is easily relaxed after repeated OER cycling due to the strong IrO6 connectivity, and the surface structure will be reconstructed in contrast to the case of BaIrO3 (in which an Ir-rich amorphous layer keeps on growing at the catalytic surface due to the unbalanced dissolution of Ba and Ir) and SrIrO3 (in which the peeling off of the surface structure simply leads to the particle size reduction due to the weaker IrO6 connectivity). Therefore, our new strategy of slightly increasing the Ir-dissolution and balancing the elemental dissolution in BaIr1−xMnxO3 for reconstructing a rigid BaIrO3-type structure at the amorphous layer seems to be successful. The importance of the robust BaIrO3-type structure for the reconstruction of the surface layer is strongly supported by the low cyclic stability of x = 0.4 (see the severe decrease of current density after 1000 OER cycles in Fig. S12b†). As shown in the crystal structure of Fig. S12a,† as soon as the monoclinic distortion in BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3) is lost and the structure transforms into the BaRuO3-type structure for BaIr0.6Mn0.4O3 (i.e., the chemical pressure decreases with the decrease of average ionic radius at the octahedral site and the crystal structure becomes less rigid), the intrinsic OER activity of BaIr0.6Mn0.4O3 in Fig. S12c† shows a sharp decrease (at a much lower potential than x = 0.1, 0.2, and 0.3) after 1000 OER cycles. In other words, not only the balanced elemental dissolution but also the rigid BaIrO3-type structure of BaIr1−xMnxO3 is essential for the reconstruction of the robust amorphous layer at the catalytic surface.
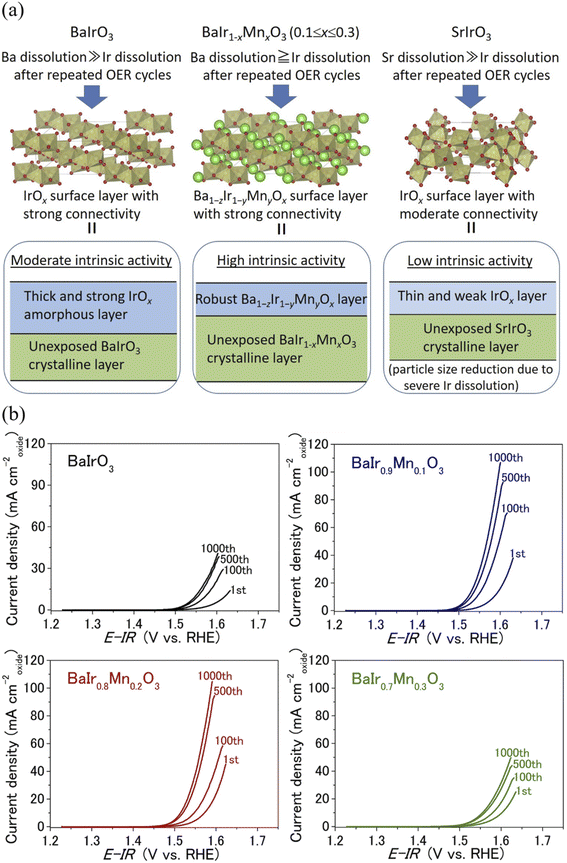 |
| | Fig. 5 (a) Schematic representation of the surface structure in BaIr1−xMnxO3 after repeated OER cycling. The surface structure image was drawn using the VESTA software.38 The surface structure of SrIrO3 is shown for comparison. (b) The cycle dependence of the linear sweep voltammograms for BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3). | |
3.3 Chemical state and further OER cycling of BaIr1−xMnxO3
Next, we will examine the surface structure scenario depicted in Fig. 5a from the standpoint of the chemical state of BaIr1−xMnxO3. The chemical state of BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3) was probed using X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra for pristine and electrochemically treated samples (by 1000 OER cycles). As shown in the XANES spectra of Fig. 6a, for pristine samples, the valence of Ir remains 4+ throughout the system, while the valence of Mn is 4+ for x = 0.1 and a mixture of 3+ and 4+ for x = 0.2 and 0.3 (that is supported by the visible energy shift of the Mn absorption edge with respect to BaMnO3 in Fig. S7a†). The presence of Mn3+ for x = 0.2 and 0.3 implies that Mn3+-induced Ir dissolution (Mn–O bonding becomes weaker with the presence of Mn3+) enables a good balance with the Ba-site dissolution of BaIr1−xMnxO3 (0.1 ≤ x ≤ 0.3) and leads to the emergence of Ba1−zIr1−yMnyOx units at the surface layer, which possesses higher OER activity and stability than the Ir-rich IrOx units formed at the BaIrO3 surface (that experiences lattice collapse during the OER). This is consistent with the scenario depicted in Fig. 5a and suggests that Mn3+ drives the balanced dissolution of Ba and Ir in BaIr1−xMnxO3 (0.1 ≤ x ≤ 0.3). Simultaneously, too high concentrations of oxygen vacancies for BaIr1−xMnxO3 at high Mn content (caused by the high concentration of Mn3+) may slightly increase the number of active sites for OER, but it will also induce increased Ir-dissolution and result in the loss of initial OER activity. This is likely to be the reason for the lower initial activity of x = 0.3 compared with x = 0.1 and x = 0.2 in Fig. 2a. In contrast, x = 0.2 demonstrates the highest OER activity and stability owing to the combination of a good balance of elemental dissolution of Ba and Ir from the surface Ba1−zIr1−yMnyOx units and the strong IrO6 connectivity originating from the BaIrO3-type structure. In other words, BaIr0.8Mn0.2O3 realizes the ideal case for the surface structure of BaIr1−xMnxO3 (Fig. 5a). Next, we will examine the chemical state of Ir. The Ir LIII-edge white line of electrochemically treated BaIr1−xMnxO3 (Fig. 6b) shows either a stronger peak intensity or a slight energy shift with respect to pristine BaIr1−xMnxO3 (Fig. S5a†). Therefore, Ir4+ is partially oxidized to Ir5+ during the 1000 OER cycles. This is also consistent with the previously reported enhancement of OER activity by the high-valence Ir5+ of IrOx formed as a BaIrO3 surface layer.28 As shown in the EXAFS spectra of Fig. 6c (also see the detailed version in Fig. S5b†), the pronounced peak between 2–3 Å ascribed to the Ir–Ir metal–metal bonding for Ir metal is present for BaIrO3, while it is not present for BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3). This suggests that the Ir-rich IrOx units (in good consistency with the previous EXAFS study about iridium oxide OER catalysts37) are formed at the surface amorphous layer of electrochemically treated BaIrO3, while the robust Ba1−zIr1−yMnyOx framework is formed at the surface amorphous layer of electrochemically treated BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3). This is also in good agreement with the scenario presented in Fig. 5a and suggests that the Ir-rich IrOx unit is not formed at the surface layer of BaIr1−xMnxO3 (x = 0.1, 0.2, and 0.3). We have also looked into the redox behavior of the BaIr1−xMnxO3 solid solution (x = 0.1, 0.2, and 0.3). Fig. 6d and (the more detailed) Fig. S15† show cyclic voltammograms for detecting the redox behavior of the Ir-site in both pristine and electrochemically treated BaIr1−xMnxO3. The Ir oxidation peak of BaIr1−xMnxO3 appears at a lower or nearly identical potential to that of BaIrO3 (the first oxidation peak corresponds to the oxidation from Ir3+ to Ir4+, and the second oxidation peak corresponds to the oxidation from Ir4+ to Ir5+), indicating that Mn is a redox-inactive element during the OER for the BaIr1−xMnxO3 solid solution as in the case of Zr for SrIr2/3Zn1/3O3.10 In other words, Ir4+ is partially oxidized to Ir5+ during the OER mainly due to the Ir-site dissolution in BaIr1−xMnxO3 (and not because of the valence change of Mn). The excellent OER performance of electrochemically treated (ect-) BaIr0.8Mn0.2O3 was further examined by the extended OER stability test over 1000 cycles. As shown in Fig. 7a, the current density of electrochemically treated BaIr0.8Mn0.2O3 does not decrease even after additional 1000 OER cycles, instead it shows slight increase due to an increase in electrochemical surface area (Fig. S17a and b†) caused by the slight progression of amorphization. Furthermore, the intrinsic OER activity of electrochemically treated BaIr0.8Mn0.2O3 in Fig. S17c† (the current density normalized to the relative ECSA with respect to pristine BaIr0.8Mn0.2O3, which is determined from Fig. S17b†) remains high after additional 1000 OER cycles and it is comparable with the initial activity below 1.57 V vs. RHE. Therefore, electrochemically treated BaIr0.8Mn0.2O3 demonstrates not only high OER stability during the potentiometric measurement, but also high cyclic stability owing to the high intrinsic OER activity even after 1000 OER cycles. More importantly, Fig. 7b shows that the OER activity per Ir mass at 1.58 V vs. RHE for electrochemically treated BaIr0.8Mn0.2O3 eventually becomes ∼25 times higher than that for pristine BaIrO3, and ∼73 times higher than that for IrO2. This excellent mass activity of electrochemically treated BaIr0.8Mn0.2O3 has important implications, as the Ir content of commercial IrO2 can be drastically reduced without compromising OER performance. In conclusion, electrochemically treated BaIr0.8Mn0.2O3 is not only a sustainable but also a highly active and an extremely stable OER catalyst benefitting from the reconstruction of the robust amorphous layer at the catalytic surface, which originates from the good balance between Ba- and Ir-dissolution, and the strong IrO6 connectivity of the BaIrO3-type structure.
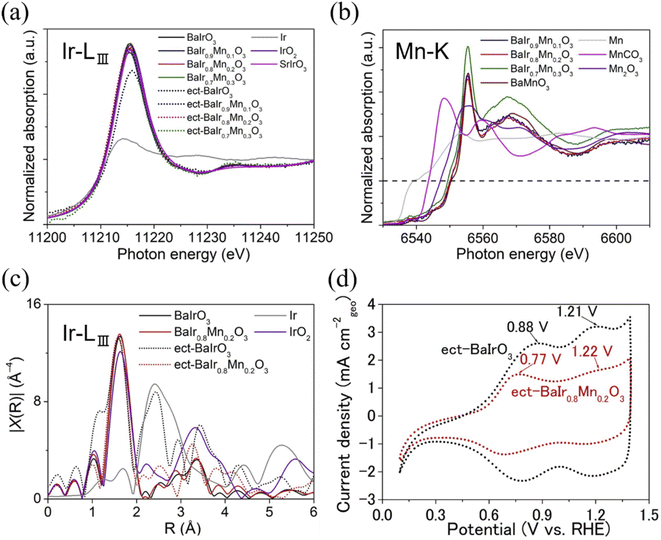 |
| | Fig. 6 (a) Normalized Ir LIII-edge X-ray absorption near-edge structure (XANES) spectra for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3). Ir LIII-edge XANES spectra of Ir, IrO2 and SrIrO3 were collected for comparison. (b) Normalized Mn K-edge XANES spectra for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 (x = 0, 0.1, 0.2, and 0.3). Mn K-edge XANES spectra of Mn, MnCO3, Mn2O3, and BaMnO3 were collected as standard materials. For Mn K-edge XANES spectra, the dashed line is a guide for the energy position with the half height of the normalized edge height. (c) The representative Fourier transformed k3-weighted Ir LIII-edge EXAFS for pristine and electrochemically treated (ect-) BaIr1−xMnxO3 in the representation form of |X(R)|. (d) The representative cyclic voltammograms for electrochemically treated (ect-) BaIr1−xMnxO3 measured from −0.6–0.7 V vs. Hg/Hg2SO4 at the scan rate of 100 mV s−1. | |
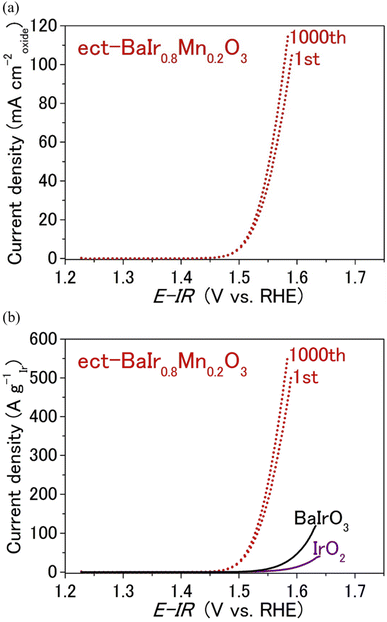 |
| | Fig. 7 (a) The cycle dependence of the linear sweep voltammograms for electrochemically treated (ect-) BaIr0.8Mn0.2O3 in 0.5 M H2SO4 aqueous solution normalized to the oxide surface area. (b) The cycle dependence of the linear sweep voltammograms for electrochemically treated (ect-) BaIr0.8Mn0.2O3 normalized to the mass of Ir. The mass activity of BaIrO3 and IrO2 are shown for comparison. | |
4. Conclusion
In conclusion, we have prevented the lattice collapse after repeated OER cycling, which is an issue even for the acid stable iridates, by substituting the Ir-site of BaIrO3 with Mn and successfully enhanced both the intrinsic OER activity and catalytic stability. Specifically, we find that BaIr0.8Mn0.2O3 in combination with OER cycling shows remarkable activity enhancement by a factor of 28 and an overall improvement in stability. The trade-off relationship of activity and stability in OER catalysts is solved by the novel strategy of slightly increasing the Ir-dissolution and balancing the elemental dissolution in BaIr1−xMnxO3 for reconstructing a rigid BaIrO3-type structure at the surface amorphous layer. Our finding suggests that both high intrinsic OER activity and high OER stability originate from the robust and thin surface layer of BaIr1−xMnxO3 that is constructed by the good balance between Ba- and Ir-dissolution, and the strong IrO6 connectivity of the BaIrO3-type structure. Moreover, the mass activity of BaIr0.8Mn0.2O3 reached ∼73 times of that for IrO2, making it a sustainable and promising OER catalyst for energy conversion technologies. The electrochemically treated BaIr0.8Mn0.2O3 has not only broken the trade-off relationship of activity and stability in OER catalysts but also drastically reduced the Ir content of commercial IrO2 without compromising the OER performance.
Author contributions
S. H. mainly conceived, designed and wrote this paper. S. Y. partially designed, co-wrote and contributed to this paper. O. H. and Y. S. synthesized the catalysts. S. H., O. H. and Y. S. conducted the electrochemical measurements. S. Y. and W. L. conducted the BET surface area measurements. E.-P. L. and W.-T. C. collected the synchrotron XRD profiles. A. M. conducted the ICP-OES measurements. M. N. and A. M. collected the XAS measurements. All authors discussed the results and commented on the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was mainly supported by a Grant-in-Aid for Scientific Research (20H02831 and 22H04497) from the Japan Society for the Promotion of Science, the Ministry of Education, Culture, Sports, Science and Technology of Japan. W.-T. C. thanks the supports by the Ministry of Science and Technology of Taiwan (MOST 108-2112-M-002-025-MY3), the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education of Taiwan (108L900803), and the awarded Synchrotron XRD experiment by NSRRC (2020-1-174).
References
- T. E. Mallouk, Nat. Chem., 2013, 5, 362–363 CrossRef CAS PubMed.
- F. M. Sapountzi, J. M. Gracia, C. J. Weststrate, H. O. A. Fredriksson and J. W. Niemantsverdriet, Prog. Energy Combust. Sci., 2017, 58, 1–35 CrossRef.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. C. Lu, Z. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. S. Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- O. Diaz-Morales, S. Raaijman, R. Kortlever, P. J. Kooyman, T. Wezendonk, J. Gascon, W. T. Fu and M. T. M. Koper, Nat. Commun., 2016, 7, 12363 CrossRef CAS PubMed.
- L. Yang, G. Yu, X. Ai, W. Yan, H. Duan, W. Chen, X. Li, T. Wang, C. Zhang, X. Huang, J.-S. Chen and X. Zou, Nat. Commun., 2018, 9, 5236 CrossRef CAS PubMed.
- J. Chen, P. Cui, G. Zhao, K. Rui, M. Lao, Y. Chen, X. Zheng, Y. Jiang, H. Pan, S. X. Dou and W. Sun, Angew. Chem., Int. Ed., 2019, 58, 12540–12544 CrossRef CAS PubMed.
- X. Liang, L. Shi, R. Cao, G. Wan, W. Yan, H. Chen, Y. Liu and X. Zou, Adv. Mater., 2020, 32, 2001430 CrossRef CAS PubMed.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- S. Hirai, S. Yagi, W.-T. Chen, F.-C. Chou, N. Okazaki, T. Ohno, H. Suzuki and T. Matsuda, Adv. Sci., 2017, 4, 1700176 CrossRef PubMed.
- J. G. Lee, J. Hwang, H. J. Hwang, O. S. Jeon, J. Jang, O. Kwon, Y. Lee, B. Han and Y. G. Shul, J. Am. Chem. Soc., 2016, 138, 3541–3547 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- Y. Y. Tian, S. Wang, E. Velasco, Y. P. Yang, L. J. Cao, L. J. Zhang, X. Li, Y. C. Lin, Q. J. Zhang and L. Chen, iScience, 2020, 23, 100756 CrossRef CAS PubMed.
- W. T. Hong, R. E. Welsch and Y. Shao-Horn, J. Phys. Chem. C, 2016, 120, 78–86 CrossRef CAS.
- S. Hirai, S. Yagi, A. Seno, M. Fujioka, T. Ohno and T. Matsuda, RSC Adv., 2016, 6, 2019–2023 RSC.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- M. Retuerto, L. Pascual, O. Piqué, P. Kayser, M. A. Salam, M. Mokhtar, J. A. Alonso, M. Peña, F. Calle-Vallejo and S. Rojas, J. Mater. Chem. A, 2021, 9, 2980–2990 RSC.
- J. Edgington, N. Schweitzer, S. Alayoglu and L. C. Seitz, J. Am. Chem. Soc., 2021, 143, 9961–9971 CrossRef CAS PubMed.
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager III, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 CrossRef CAS PubMed.
- C. W. Song, J. Lim, H. B. Bae and S.-Y. Chung, Energy Environ. Sci., 2020, 13, 4178–4188 RSC.
- N. Li, L. Cai, C. Wang, Y. Lin, J. Huang, H. Sheng, H. Pan, W. Zhang, Q. Ji, H. Duan, W. Hu, W. Zhang, F. Hu, H. Tan, Z. Sun, B. Song, S. Jin and W. Yan, J. Am. Chem. Soc., 2021, 143, 18001–18009 CrossRef CAS PubMed.
- J. G. Zhao, L. X. Yang, Y. Yua, F. Y. Li, R. C. Yu and C. Q. Jin, J. Solid State Chem., 2010, 183, 720–726 CrossRef CAS.
- A. C. Larson and R. B. von Dreele, Los Alamos National Laboratory Report, LAUR, 2000, pp. 86–748 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- T. Siegrist and B. L. Chamberland, J. Less-Common Met., 1991, 170, 93–99 CrossRef CAS.
- P. C. Donohue, L. Katz and R. Ward, Inorg. Chem., 1965, 4, 306–310 CrossRef CAS.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y. L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS.
- G. Wan, J. W. Freeland, J. Kloppenburg, G. Petretto, J. N. Nelson, D.-Y. Kuo, C.-J. Sun, J. Wen, J. T. Diulus, G. S. Herman, Y. Dong, R. Kou, J. Sun, S. Chen, K. M. Shen, D. G. Schlom, G.-M. Rignanese, G. Hautier, D. D. Fong, Z. Feng, H. Zhou and J. Suntivich, Sci. Adv., 2021, 7, eabc7323 CrossRef CAS PubMed.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS PubMed.
- G. Kwon, S. H. Chang, J. E. Heo, K. J. Lee, J.-K. Kim, B.-G. Cho, T. Y. Koo, B. J. Kim, C. Kim, J. H. Lee, S.-M. Bak, K. A. Beyer, H. Zhong, R. J. Koch, S. Hwang, L. M. Utschig, X. Huang, G. Hu, G. W. Brudvig, D. M. Tiede and J. Kim, ACS Catal., 2021, 11, 10084–10094 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shunsuke Yagi
*a,
Shunsuke Yagi