
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc vacancy modulated quaternary metallic oxynitride GeZn1.7ON1.8: as a high-performance anode for lithium-ion storage
Jinli Yaoa,
Fukun Ma*b,
Yan-Jie Wang*b,
Yinzhe Zuo c and
Wei Yan*c
c and
Wei Yan*c
aDepartment of Research and Development, Meijin Energy Ltd, Beijing 100052, China. E-mail: mafukun666@126.com; wyj@dgut.edu.cn
bNew Energy and Advanced Functional Materials Group, School of Materials Science and Engineering, Dongguan University of Technology, Dongguan 523808, Guangdong, China
cCollege of Materials Science and Engineering, Fuzhou University, Fuzhou 350108, China. E-mail: weiyan@fzu.edu.cn
First published on 23rd September 2022
Abstract
The development of alternative anode materials to achieve high lithium-ion storage performance is crucial for the next-generation lithium-ion batteries (LIBs). In this study, a new anode material, Zn-defected GeZn1.7ON1.8 (GeZn1.7−xON1.8), was rationally designed and successfully synthesized by a simple ammoniation and acid etching method. The introduced zinc vacancy can increase the capacity by more than 100%, originating from the additional space for the lithium-ion insertion. This GeZn1.7−xON1.8 particle anode delivers a high capacity (868 mA h g−1 at 0.1 A g−1 after 200 cycles) and ultralong cyclic stability (2000 cycles at 1.0 A g−1 with a maintained capacity of 458.6 mA h g−1). Electrochemical kinetic analysis corroborates the enhanced pseudocapacitive contribution and lithium-ion reaction kinetics in the GeZn1.7−xON1.8 particle anode. Furthermore, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) analyses at different electrochemical reaction states confirm the reversible intercalation lithium-ion storage mechanism of this GeZn1.7−xON1.8 particle anode. This study offers a new vision toward designing high-performance quaternary metallic oxynitride-based materials for large-scale energy storage applications.
1. Introduction
Lithium-ion batteries (LIBs) have received particular attention as efficient energy storage and conversion devices, particularly for electric vehicles, portable powers, and stationary power plants.1–3 However, the energy densities of LIBs are still not satisfactory for the long recharge mileages.4,5 For further increasing the energy density of LIBs, both high capacity anode and cathode materials are required.6,7 Regarding LIB anode materials, the commercially available graphite acting as the active LIB anode material seems to have insufficient capacity and high rate capability.8,9 Besides, graphite anodes are susceptible to lithium metal dendrite formation, which can penetrate through the separator resulting in a short-circuit.10,11 Therefore, efforts are still ongoing to explore alternative anode materials with high capacities and cyclic stabilities to support the continuous requirement for next-generation LIBs.12 Among the different types of LIB anode materials, metal nitrides (e.g., Ti–O–N,13 Fe–O–N,14 W–O–N,15 and V–O–N16) have been recently studied as alternatives based on their advantages such as multiple electron transfers, negative redox voltages, and high theoretical capacities.17 They operate via a conversion mechanism at the particle surfaces, with the reduction of the metal nitride to the metal, presumably with the formation of amorphous lithium nitride (Li3N). Therefore, the irreversible crystalline-phase transformation will lead to low coulombic efficiencies, while pulverization will result in the loss of electrical contact for active materials and the repeated formation of the solid electrolyte interphase (SEI) layer. Nevertheless, the metal nitride-based anodes show sluggish charge transport and extreme volumetric variation during the electrochemical reaction.18 Furthermore, the undesired side reaction can give rise to inferior coulombic efficiency with the “dead lithium”.19,20 In order to realize fast charge transport efficiency, high capacity, and cyclic stability, new material structures with favorable electrical conductivity and structural stability need to be developed.21 As reported, metallic oxynitrides, a kind of hybrid anionic materials, have been lately explored as potential anode-active materials for higher energy/power densities of LIBs because of their excellent chemical/structural stability.22,23In metallic oxynitrides, as the electronegativity of N is less than that of O, the introduction of N 2p orbitals can decrease the position of the maximum valence bands (VBs) and enhance the electron conduction efficiency.24 For example, as a typical quaternary crystal with a hexagonal wurtzite structure, GeZn1.7ON1.8 possesses low conversion potential and high saturated electron mobility. The less electronegative N atom, when introduced, can result in the p-d repulsion between the Zn 3d and N 2p orbitals with enhanced electron transfer efficiency.24 Consequently, GeZn1.7ON1.8 exhibits high conductivity and fast ionic diffusion than those of traditional metal nitrides, and its excellent structural and chemical stability also favor its usage in violent electrochemical reaction environments.25 Therefore, GeZn1.7ON1.8 is considered to be the promising alternative anode-active material with excellent structural stability in electrochemical reactions for the lithium-ion storage, which is even superior to the conventional metal nitride-based anode materials. However, when it comes to practical applications, issues such as unsatisfied lithium-ion transport kinetics and cycling capacity have been identified, which resulted from their insufficient conductance and active sites. Furthermore, the SEI film grown during the electrochemical reaction could easily lead to an inferior coulombic efficiency and dead lithium near the anode surface. Based on previous reports, morphological and structural design strategies could be adopted to improve the charge transport efficiency, capacities and cyclic stability of the anode-active materials for higher electrochemical performance.26 Unfortunately, although some progress has been made, the ion diffusion efficiency and conductance of the GeZn1.7ON1.8-based anode could still not be intrinsically improved, and its rate capability and cycling capacity are still unsatisfactory and need to be further improved. To address these issues and improve lithium-ion storage performance, defect engineering has been proposed to be an effective method to regulate the electron density and ion diffusion efficiency of the anode-active materials for higher energy/power densities.27,28 With respect to this, zinc vacancies have been identified to be able to provide a larger interface for lithium-ion and electron insertion/extraction, activating more reactive sites and extending faster transport paths, thus greatly improving the electrochemical performance of the materials.29 However, the prepared Zn-defected GeZn1.7ON1.8 particle anodes for obtaining the best electrochemical performance have not been optimized thus far.
In this study, GeZn1.7−xON1.8 particles were successfully synthesized using ammoniation and the acid etching method. XRD, EDS, EPR and XPS results further confirmed the successful synthesis of GeZn1.7−xON1.8 particles. The improvement in lithium-ion storage performance using these materials as anodes was validated by electrochemical measurements. Compared with the pristine GeZn1.7ON1.8 particle anode, the GeZn1.7−xON1.8 particle anode exhibits a higher superior reversible capacity, rate capability, and ion diffusion coefficient as indicated by the systematic electrochemical measurements and kinetic analysis. The XRD and XPS analyses were used to reveal the reversible lithium-ion storage mechanism for fundamental understanding. As a result, a GeZn1.7−xON1.8 particle anode shows a reversible specific capacity of 878.6 mA h g−1 after 200 cycles at 0.1 A g−1. The defect engineering employed in this study is significant for the rational regulation of charge transfer and high-performance lithium-ion storage of anode materials.
2. Experimental section
2.1. Material synthesis
For the material synthesis as shown in Fig. 1, GeO2 and ZnO (Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ge = 1:1) powders were thoroughly ground and mixed in an agate mortar and then placed in a porcelain boat in the middle of a horizontal high-temperature tube furnace. The quartz reactor was pumped (≤1 mTorr) and then purged with nitrogen (N2). Nitrogen was used as a protective atmosphere to prevent the oxidation of the raw material at high temperatures. The nicely mixed GeO2 and ZnO were further treated at 850 °C in NH3 atmosphere for 2 h. After cooling down to room temperature, GeZn1.7ON1.8 particles were obtained on porcelain boats. GeZn1.7−xON1.8 particles were obtained via the corrosion of GeZn1+xON in 2.0 mol L−1 hydrochloric acid. After the corrosion process, the obtained GeZn1.7−xON1.8 particles were collected by washing several times with ethanol and ultrapure water.
:Ge = 1:1) powders were thoroughly ground and mixed in an agate mortar and then placed in a porcelain boat in the middle of a horizontal high-temperature tube furnace. The quartz reactor was pumped (≤1 mTorr) and then purged with nitrogen (N2). Nitrogen was used as a protective atmosphere to prevent the oxidation of the raw material at high temperatures. The nicely mixed GeO2 and ZnO were further treated at 850 °C in NH3 atmosphere for 2 h. After cooling down to room temperature, GeZn1.7ON1.8 particles were obtained on porcelain boats. GeZn1.7−xON1.8 particles were obtained via the corrosion of GeZn1+xON in 2.0 mol L−1 hydrochloric acid. After the corrosion process, the obtained GeZn1.7−xON1.8 particles were collected by washing several times with ethanol and ultrapure water.
 | ||
| Fig. 1 Schematic of the preparation process of GeZn1.7−xON1.8 particles. | ||
2.2. Material characterizations
The morphology of the samples was examined using scanning electron microscopy (SEM, Hitachi S-4800) and a transmission electron microscopy (TEM, Tecnai 20U dual microscope). The scanning of energy dispersive spectroscopy (EDS) elements was performed using the same instrument equipped with a detector (7593-H, Horiba). X-ray diffraction (XRD) patterns were obtained using a diffractometer with Cu-Kα radiation (λ = 0.15443 nm, X'pert Pro MPD). X-ray photoelectron spectroscopy (XPS) was inspected using a Thermo ESCALAB 250.2.3. Battery preparation and electrochemical measurements
The electrochemical behavior of pristine GeZn1.7ON1.8 particles and GeZn1.7−xON1.8 particles was studied by assembling CR2016 type coin-shaped batteries. In the anode preparation process, 80 wt% of active material, 10 wt% of conductive carbon (Ketjen black) and 10 wt% of polyvinylidene fluoride (PVDF) were mixed in N-methyl-2-pyrrolidone as a binder, and then the fully mixed active material was adhered to the copper foil and then dried at 80 °C under vacuum overnight prior to use. The active material had an average loading density of about 1.5 mg and a diameter of 16 mm. Lithium metal was used as the counter and reference electrodes. The electrolyte solution was LiPF6 (1 M) in vinyl carbonate/dimethyl carbonate/diethyl carbonate (1:1:1 vol%). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were conducted using a CHI660D electrochemical workstation. The galvanostatic charge/discharge curves were recorded using a Neware CT-3008W battery system.
3. Results and discussion
As shown in Fig. 2a–c, the GeZn1.7−xON1.8 particles agglomerated after calcination in an ammonia atmosphere at high temperatures. The low-magnification TEM image (Fig. 2d) displays homogeneous GeZn1.7−xON1.8 particles prepared by the ammoniation and acid etching method, and the average size of GeZn1.7−xON1.8 particles is ∼200 nm. The regular atomic arrangement can be observed in the HRTEM image (Fig. 2e), which shows the well-crystallized structure of GeZn1.7−xON1.8 particles. The high-resolution lattice stripes with an interplanar distance of 0.27 nm can be indexed to the (100) crystal plane of GeZn1.7ON1.8 (Fig. 2f),30 and it is expected that the high crystallinity of the GeZn1.7−xON1.8 particles is important for the high-rate performance energy storage device. The SEM image and the corresponding EDS elemental mapping results (Fig. 2g–k) demonstrate the uniform distribution of Ge, Zn, O, and N elements in GeZn1.7−xON1.8 particles, indicating that GeZn1.7−xON1.8 particles were successfully synthesized. | ||
| Fig. 2 (a) Low-magnification SEM image, (b) and (c) high-magnification SEM images, (d) TEM image, and (e) and (f) HRTEM images of Zn-defected GeZn1.7ON1.8 particles. (g)–(k) SEM image and the corresponding EDS elemental mapping of Zn-defected GeZn1.7ON1.8 particles. | ||
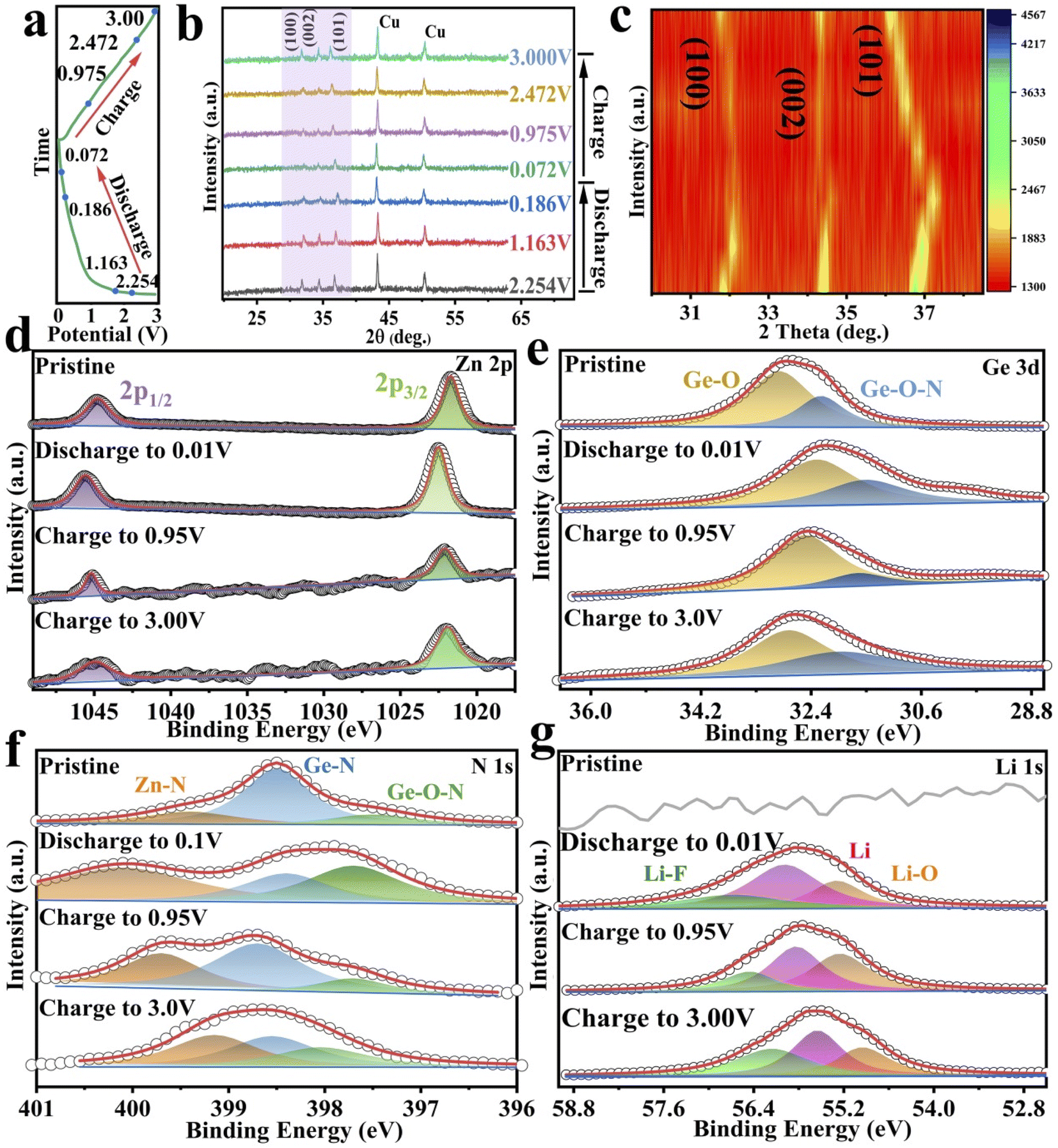
The crystallographic structure of GeZn1.7−xON1.8 particles was further analyzed by the X-ray diffraction (XRD) technique. As shown in Fig. 3a, the GeZn1.7−xON1.8 particles maintain the hexagonal wurtzite structure, which can be easily indexed with GeZn1.7ON1.8 (PDF#24-1443). After acid etching, the GeZn1.7−xON1.8 particles maintain the same lattice structure as the pristine GeZn1.7ON1.8 particles with a slight low-angle shift of the (101) peak, indicating the expanded crystal plane spacing after acid etching.31 The high-resolution XPS spectra of Zn 2p (Fig. 3b) could be deconvoluted into two distinct peaks at 1022.5 eV and 1045.7 eV, depending on the location of Zn 2p1/2 and Zn 2p2/3, respectively.32 The binding energy difference (23.1 eV) between Zn 2p1/2 and Zn 2p2/3 shows the Zn2+ oxidation state in the GeZn1.7−xON1.8 particles.33 It can be clearly observed that the Zn 2p binding energy of GeZn1.7−xON1.8 particles shifts to a lower binding energy compared to that of pristine GeZn1.7ON1.8 particles, which is attributed to the change in the surface electron density in the presence of defects states.34 It has been reported that the change in the binding energy could be related to the change in the surface electron density, while zinc vacancies increase the surface electron density.35 In Fig. 3c, the high-resolution XPS spectra of N 1s show the Ge–O–N, Ge–N, and Zn–N bonds at 397.6, 398.3, and 399.6 eV, respectively.36 As reported, the Ge–O–N bond can improve the reaction kinetics and lithium-ion storage performance of the GeZn1.7−xON1.8 particle anode.37 As shown in Fig. 3d, the high-resolution XPS spectra of Ge 3d show the Ge–O and Ge–O–N bonds at 32.8 and 32.2 eV, respectively.38,39 The above-mentioned characterizations demonstrate that the GeZn1.7−xON1.8 particles were successfully synthesized.
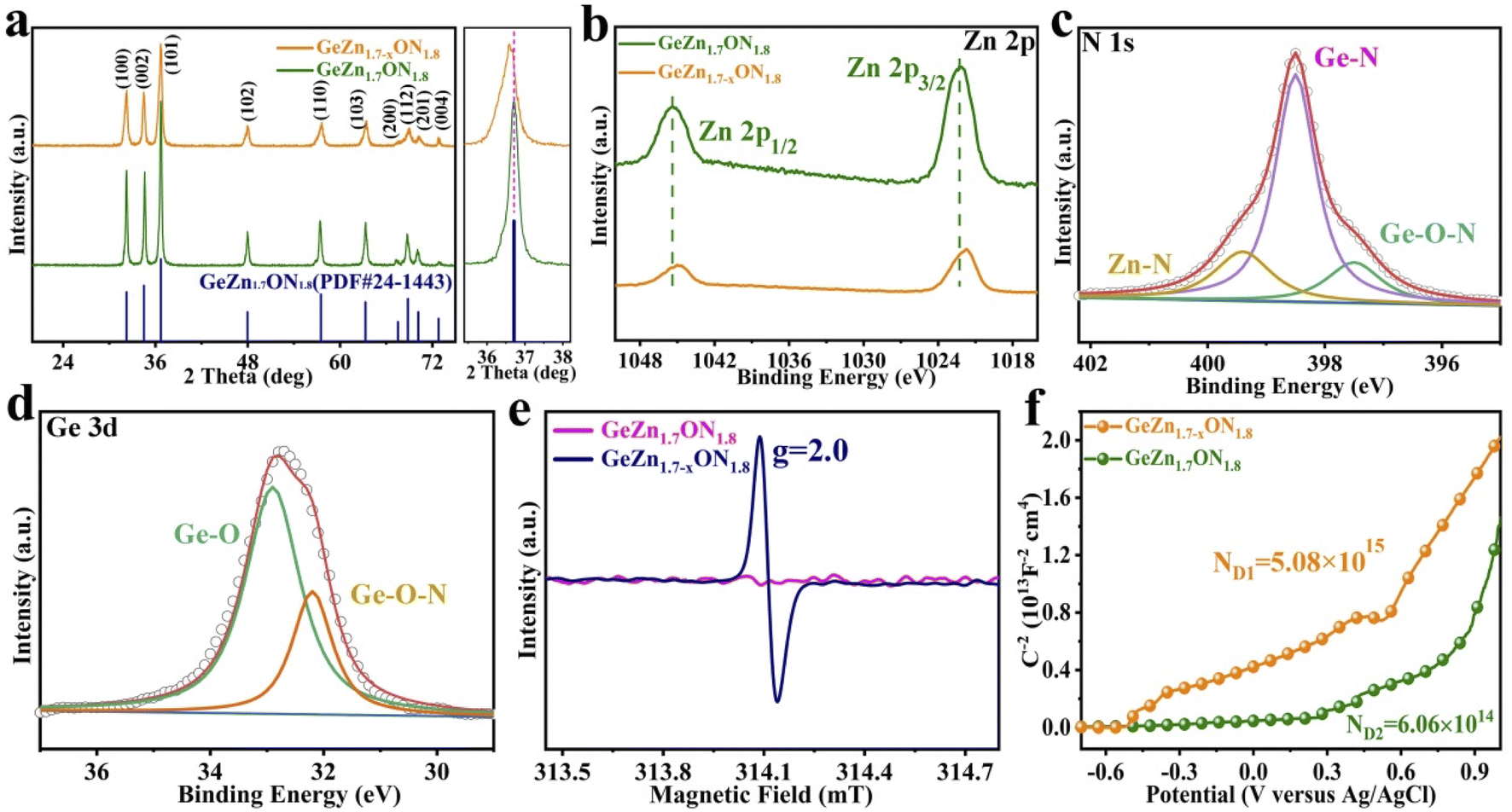 | ||
| Fig. 3 (a) XRD patterns of GeZn1.7−xON1.8 and GeZn1.7ON1.8 particles, the main diffraction peak is enlarged between 35.5° and 38.2°. The corresponding high-resolution XPS spectra for (b) Zn 2p, (c) N 1s, and (d) Ge 3d of GeZn1.7−xON1.8 particles. (e) EPR spectra and (f) Mott–Schottky plots of GeZn1.7−xON1.8 and GeZn1.7ON1.8 particles. | ||
To obtain an in-depth understanding of the chemical environment and unravel Zn vacancies, the electron paramagnetic resonance (EPR) spectroscopy analysis was performed. As shown in Fig. 3e, no signal was observed in GeZn1.7ON1.8 particles, while a prominent signal (g = 2.0) appears at 314.1 mT in GeZn1.7−xON1.8 particles, which clearly indicate that this material possesses numerous Zn vacancies.40 Zinc vacancies can be considered to be the “electron acceptors”, and the significant signal in GeZn1.7−xON1.8 particles can be attributed to the spin resonance of the electrons captured by zinc vacancies.35 Mott–Schottky plots were measured in 0.1 M Na2SO4 buffer at a frequency of 10 kHz in order to investigate the charge-carrier density changes of GeZn1.7ON1.8 and GeZn1.7−xON1.8 particles. As shown in Fig. 3f, the positive slope in the linear region reveals the n-type semiconductor properties of GeZn1.7ON1.8 and GeZn1.7−xON1.8 particles. The charge-carrier density (Nd) was calculated according to ND = (2/e0εε0)/[d(1/C2)/dV]−1, where e0 is the electron charge (1.6 × 10−19 C), ε is the dielectric constant, ε0 is the permittivity of vacuum (8.85 × 10−12 F m−1), and V is the applied potential.41 The calculated carrier concentrations are 50.86 × 1014 and 6.06 × 1014 cm−3 for GeZn1.7−xON1.8 and GeZn1.7ON1.8, respectively, indicating the improved electronic conductivity after the introduction of Zn vacancies. These results confirm the successful construction of GeZn1.7−xON1.8 particles with the efficient electron transport, which is indispensable to activate the electrochemical response with a high-rate performance energy storage device.
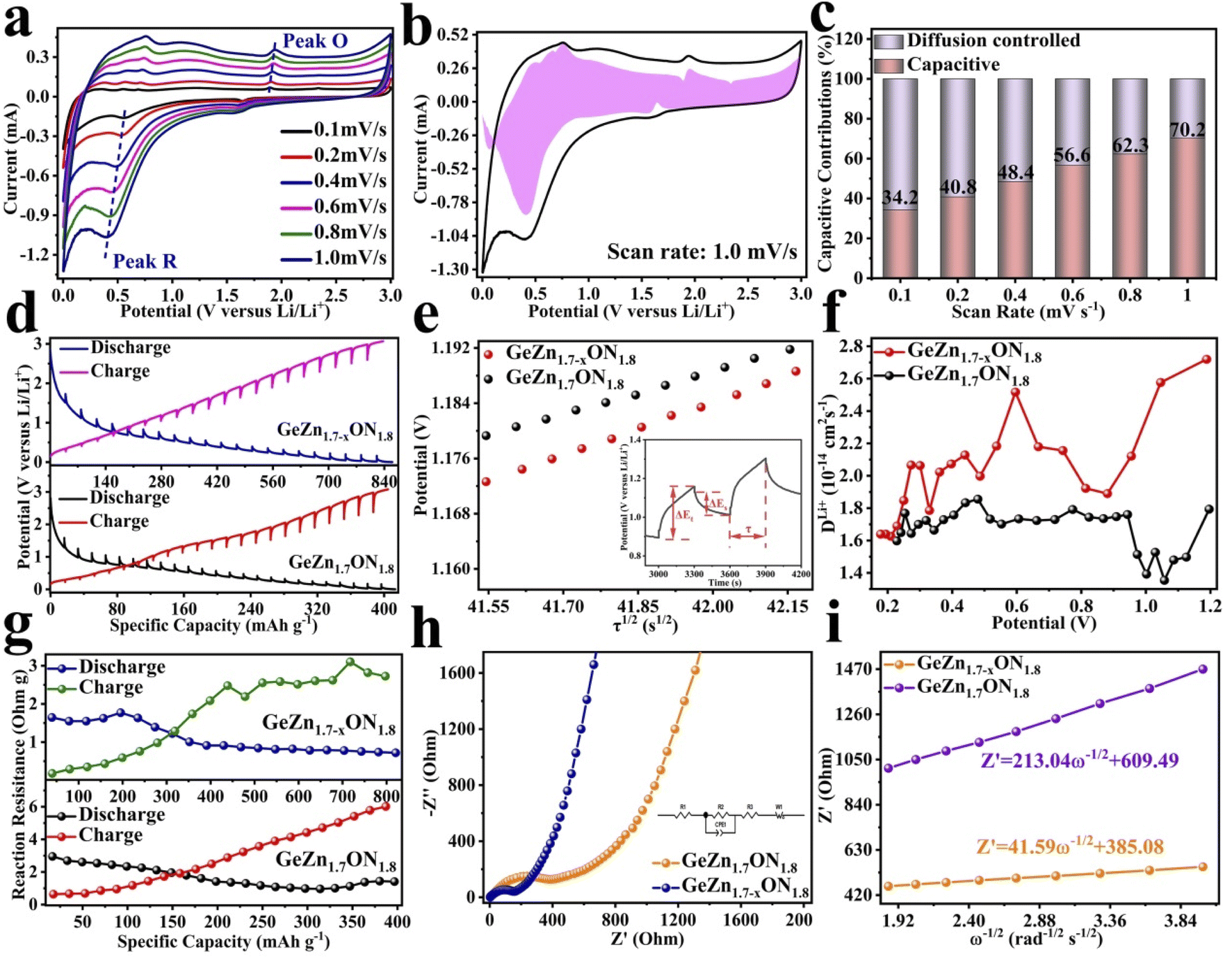
The CV curves were first performed to gain a better understanding of the electrochemical reaction mechanism of the GeZn1.7−xON1.8 particles at 0.1 mV s−1 from 0.01 to 3.0 V (vs. Li/Li+). As shown in Fig. 4a, during the first discharge process, the irreversible broad peak at ∼1.6 V could be attributed to the formation of the solid electrolyte interface (SEI) film.42 The irreversible SEI peak disappears in the subsequent discharge process. The broaden peak centered at 0.5–0.8 V can be attributed to the reversible lithium-ion insertion with the formation of LixGeZn1.7−xON1.8.43 In the following charge process, the peaks at 1.1 and 2.0 V are ascribed to the lithium-ion desertion from the GeZn1.7−xON1.8 anode.44 The insertion/desertion peaks are stable in the second to fifth CV cycles, reflecting the reversible lithiation/delithiation mechanism of the GeZn1.7−xON1.8 anode. In the subsequent cycles, the overlapped CV curves and the stable insertion/extraction peaks indicate the excellent stability of the discharge and charge processes. More importantly, the lithiation and delithiation peaks are entirely different from the reported metal-based anode.45 Therefore, the lithiation mechanism of the GeZn1.7−xON1.8 anode does not involve the alloying and conversion reaction. The similar CV curves of the GeZn1.7−xON1.8 and pristine GeZn1.7ON1.8 anodes reveal that the Zn vacancy shows limited influence on the lithiation mechanism. As shown in Fig. 4b, the GeZn1.7−xON1.8 particle anode shows typical charge/discharge curves with high reversibility. The galvanostatic charge/discharge profiles demonstrate that the initial discharge and charge capacities are 988.4 and 889.2 mA h g−1, respectively, with the initial coulombic efficiency of 89.9%. The initial irreversible capacity loss might result from the SEI layer and electrolyte decomposition, which leads to irreversible capacity loss.46 With the increase in the cycle number, the coulombic efficiency increases gradually and maintains 100% after 12 cycles. The overlapped cycle curves imply the excellent stability of the electrochemical reaction. In the following cycles, the overlapping discharge/charge profiles imply the good structural stability and reversible electrochemical reaction, matching well with the CV results. As shown in Fig. 4c, the reversible capacity gradually decreases in the first 30 cycles and maintains a highly stable reversible capacity in the subsequent cycles. This phenomenon can be associated with the lithiation-induced reactivation process, which is commonly found in the nanostructure-based electrodes.47 After experiencing a quick activation process in the initial 30 cycles, the discharge capacity increases gradually. After 200 cycles, the GeZn1.7−xON1.8 particle anode still maintains charge/discharge capacities of 868.1 mA h g−1 and 858.5 mA h g−1, with almost 100% coulombic efficiency. For the GeZn1.7ON1.8 particle anode, after 200 cycles, the discharge capacity is 412 mA h g−1. Those results indicate the good reversibility of the GeZn1.7−xON1.8 particle anode. Therefore, the improved capacity can be ascribed to the enhanced ion/electron diffusion efficiencies and reduced lithium-ion diffusion barrier after acid etching. Fig. 4d shows the rate performances of both the GeZn1.7−xON1.8 and GeZn1.7ON1.8 particle anodes at different current densities. The Co–GeZn1.7ON1.8 particle anode delivers reversible capacities of 844.2, 727.7, 561.4, 454.8, 357.5, and 265.7 mA h g−1 at 0.1, 0.2, 0.5, 1.0, 2.0, and 4.0 A g−1, respectively. It is clear that the GeZn1.7−xON1.8 particle anode still maintains a good reversible capacity of 189.4 mA h g−1 at 5.0 A g−1, indicating the excellent rate capacity. When the current density reverts back to 0.1 A g−1, the capacity can be recovered, showing the good reversibility and stability of the GeZn1.7−xON1.8 particle anode. The charge/discharge curves (Fig. 4e) also reveal the reversibility of the electrochemical reaction at different current densities. The enhanced rate performance can be attributed to the reduced lithium-ion diffusion barrier and excellent ion diffusion efficiency after acid etching. After 2000 cycles (Fig. 4g), the GeZn1.7−xON1.8 particle anode still shows a stable specific capacity of 458.6 mA h g−1 at 1.0 A g−1, and its capacity retention ratio is about 76.8% (Fig. 4f). The specific capacity of GeZn1.7ON1.8 particle anode is only about 156 mA h g−1, and its capacity retention ratio is approximately 42.5%. This result indicates that the GeZn1.7−xON1.8 particle anode can maintain high cyclic stability under high current density conditions.
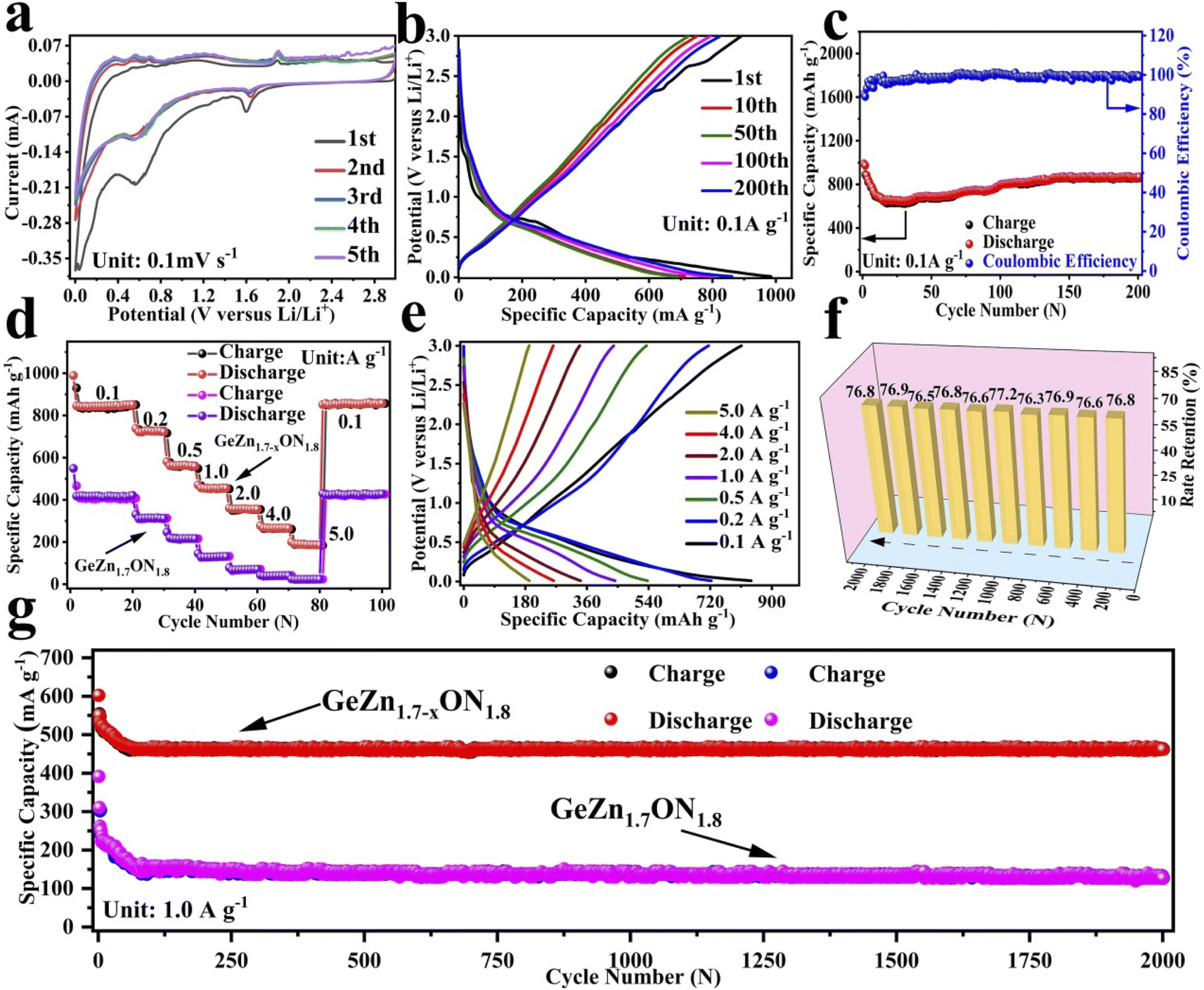 | ||
| Fig. 4 Electrochemical properties of the GeZn1.7−xON1.8 particle anode: (a) CVs at 0.1 mV s−1. (b) Charge/discharge curves and (c) cycle property at 0.1 A g−1. (d) Rate capabilities from 0.1 to 5.0 A g−1 and (e) the corresponding galvanostatic charge/discharge profiles from 0.1 to 5.0 A g−1. (f) Long cycle retention histogram at 1.0 A g−1. (g) Cycling property comparison at 1.0 A g−1. | ||
The CV curves in Fig. 5a show a similar shape at different scan rates, indicating the negligible polarization of the electrochemical reaction.48 Based on i = avb (where i and v are the current density and scan rate, respectively), the calculated b values (0.5 ≤ b ≤ 1) reveal the coexistence of the capacitive and diffusive contributions during the charge/discharge processes.49 The pseudo-capacitance contribution can be determined precisely by capacitive effects (k1v) and diffusion-controlled insertion (k2v1/2). As shown in Fig. 5b, more than 70% of the total lithium-ion storage comes from the capacitive process at 1.0 mV s−1, and the charge diffusion process is mainly concentrated at low-voltage regions (≤0.5 V).50 As shown in Fig. 5c, the contribution of the pseudo-capacitance progressively increases and diffusion contribution decreases with the increase in the scan rate, and this result is in good agreement with the previous report.51 Galvanostatic intermittent titration technique (GITT) analysis was further performed to analyze the charge transfer behavior of the GeZn1.7−xON1.8 particle anode. As shown in Fig. 5d, the negligible overpotential indicates the fast charge transfer efficiency and stable reaction resistance during the electrochemical reaction.52 Compared with the GeZn1.7ON1.8 particle anode, the GeZn1.7−xON1.8 particle anode exhibits a higher capacity and lower reaction resistance, indicating its improved lithium-ion diffusion kinetics. Based on Fick's second law (Fig. 5e), the lithium-ion diffusion coefficient 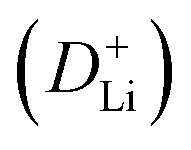 can be determined from the linear relationship between the voltage and square root of the pulse duration (τ1/2). As shown in Fig. 5f, a similar overall trend of
can be determined from the linear relationship between the voltage and square root of the pulse duration (τ1/2). As shown in Fig. 5f, a similar overall trend of 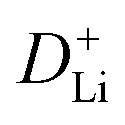 manifests the same lithium-ion diffusion behavior and electrochemical reaction mechanism, and the higher lithium-ion diffusion coefficient means the higher charge transfer kinetics. The stable ion diffusion process reveals that no conversion and alloying reactions occur in the charge/discharge processes.53 The in situ response resistance was further calculated based on the closed-circuit voltage and the quasi-open circuit voltage. As shown in Fig. 5g, the reaction resistance of the GeZn1.7−xON1.8 particle anode is lower, indicating the improved conductivity and lithium-ion diffusion kinetics after acid etching. Electrochemical impedance spectroscopy (EIS) analysis was performed to further investigate the lithium-ion diffusion behavior. As shown in Fig. 5h, the semicircles in the high-to-medium frequency region represent the charge-transfer resistance (Rct) and impedance of the SEI layer (RSEI), and the inclined line in the low-frequency region represents the mass-transfer resistances (Warburg impedance, Rw).54 After the equivalent circuit model fitting (inset in Fig. 5h), the Rct value of the GeZn1.7−xON1.8 particle anode (162.4 Ω) is smaller than that of the GeZn1.7ON1.8 (345.3 Ω) particle anode, indicating the high electrical conductivity of acid etching. In the inclined region, the higher slope manifests better lithium-ion mobility. According to Zreal = Re + Rct + σω−1/2, in Fig. 5i, the calculated Warburg factor (σ) of the GeZn1.7−xON1.8 particle (41.59) is much lower than that of the GeZn1.7ON1.8 particle (213.04), and the calculated
manifests the same lithium-ion diffusion behavior and electrochemical reaction mechanism, and the higher lithium-ion diffusion coefficient means the higher charge transfer kinetics. The stable ion diffusion process reveals that no conversion and alloying reactions occur in the charge/discharge processes.53 The in situ response resistance was further calculated based on the closed-circuit voltage and the quasi-open circuit voltage. As shown in Fig. 5g, the reaction resistance of the GeZn1.7−xON1.8 particle anode is lower, indicating the improved conductivity and lithium-ion diffusion kinetics after acid etching. Electrochemical impedance spectroscopy (EIS) analysis was performed to further investigate the lithium-ion diffusion behavior. As shown in Fig. 5h, the semicircles in the high-to-medium frequency region represent the charge-transfer resistance (Rct) and impedance of the SEI layer (RSEI), and the inclined line in the low-frequency region represents the mass-transfer resistances (Warburg impedance, Rw).54 After the equivalent circuit model fitting (inset in Fig. 5h), the Rct value of the GeZn1.7−xON1.8 particle anode (162.4 Ω) is smaller than that of the GeZn1.7ON1.8 (345.3 Ω) particle anode, indicating the high electrical conductivity of acid etching. In the inclined region, the higher slope manifests better lithium-ion mobility. According to Zreal = Re + Rct + σω−1/2, in Fig. 5i, the calculated Warburg factor (σ) of the GeZn1.7−xON1.8 particle (41.59) is much lower than that of the GeZn1.7ON1.8 particle (213.04), and the calculated 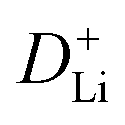 values of the GeZn1.7ON1.8 and GeZn1.7−xON1.8 particle anodes are 1.78 × 10−14 and 2.36 × 10−14 cm2 s−1, respectively. These results indicate the enhanced lithium-ions diffusion efficiency after acid etching.
values of the GeZn1.7ON1.8 and GeZn1.7−xON1.8 particle anodes are 1.78 × 10−14 and 2.36 × 10−14 cm2 s−1, respectively. These results indicate the enhanced lithium-ions diffusion efficiency after acid etching.
 | ||
| Fig. 5 Kinetic analysis of GeZn1.7−xON1.8: (a) CVs curves from 0.1 to 1.0 mV s−1. (b) Pseudocapacitive contribution shown by pink region at 1.0 mV s−1. (c) Pseudocapacitive contribution ratio from 0.1 to 1.0 mV s−1. (d) Potential response in the GITT profiles. (e) Relationship between potential and τ1/2 (inset: titration profile). (f) Lithium-ion diffusion coefficients and (g) reaction resistances calculated by GITT profiles. (h) Nyquist plots and (i) relationship between Z′ and ω−1/2. | ||
To better understand the lithium-ion storage mechanism, XRD and XPS analyses were performed in different electrochemical states (Fig. 6a). As shown in Fig. 6b and c, the diffraction peaks at 43.3° and 50.4° (marked with asterisk) come from the copper foil. The diffraction peaks of GeZn1.7−xON1.8 are observed in the whole electrochemical reaction, and no additional diffraction peaks about Ge or Zn appear, showing the reversible lithium-ion storage mechanism and good structural stability. To further analyze the structural changes, the XRD profiles are expanded in the form of intensity isopleth maps in the characteristic peak region (from 30° to 40°), as shown in Fig. 6c. In the discharged states, the characteristic diffraction peaks (at 32.3°, 34.5°, and 36.7°) shift toward a higher angle direction as the diffraction intensity is decreased, and the characteristic diffraction peaks are gradually broadened with the lattice expansion, revealing the reversible lithium-ion storage mechanism. The decreased diffraction intensity of the characteristic peaks may be due to the reduced crystallinity after lithium-ion insertion.55 In the charge states, the characteristic peaks shift toward the opposite direction with the lithium-ion desertion reaction. The XRD analysis clearly shows reversible lithium-ion storage mechanism of the GeZn1.7−xON1.8 particles.
 | ||
| Fig. 6 Lithium-ion storage mechanism study of the GeZn1.7−xON1.8 particle anode. (a) Charge/discharge profile and the corresponding (b) XRD patterns. The shallow region represents (c) the intensity contour map of characteristic diffraction peaks from 30° to 40°. (d)–(g) XPS analysis of the GeZn1.7−xON1.8 particle anode at different charge/discharge states. | ||
The electrochemical charge storage mechanism was further confirmed by XPS analysis in different electrochemical states. For the Zn 2p spectrum before discharge (Fig. 6d), the main peaks at 1022.5 and 1045.7 eV correspond to Zn 2p3/2 and Zn 2p1/2.56 At the complete discharge state (0.01 V), the Zn 2p3/2 and Zn 2p1/2 peaks move to 1023.4 and 1046.5 eV, respectively, indicating similar bonding conditions during the discharge.36 In addition, in the fully discharged state, the electronic states of Zn 2P3/2 and Zn 2P1/2 are significantly different from metal Zn0, which reveals the reversible lithium-ion storage mechanism of GeZn1.7−xON1.8 particles.57 The Ge 3d spectrum (Fig. 6e) shows that the Ge–O and Ge–O–N core energy levels of pristine GeZn1.7−xON1.8 are located at 32.8 and 32.2 eV, respectively.38,39 During the discharge process, the Ge 3d spectrum shifts to a lower energy, while the Ge 3d spectrum shifts to a higher energy during the charging process. Compared with the pristine state, the lower binding energy shift of the fully charged Ge 3d spectrum is attributable to the existence of lithium-ions in the interlayers and on the surface of the anode and also due to the SEI film formation.58 This periodical change in the Ge 3d spectrum also reveals the reversible lithium-ion storage mechanism of the GeZn1.7−xON1.8 particle. Moreover, the high-resolution N 1s spectra at different charging/discharging states were further analyzed (Fig. 6f). Before discharge, the major peaks at 399.6, 398.3, and 397.6 eV could be assigned to Zn–N, Ge–N, and Ge–O–N bonds, respectively.36 During discharge, the N 1s spectrum shifts toward higher energies, while during charging, the N 1s spectrum shifts toward lower energies. The binding energy shift of the fully charged N 1s spectrum is higher than that of the pristine state. The N 1s spectrum with minor intensity changes in the coordination further confirms that the lithium-ion storage mechanism involves lithiation/delithiation reactions. The peak at 55.5 eV could be attributed to lithium alkyl carbonate in the discharge product as shown in the Li 1s spectrum (Fig. 6g).59 The Li–O bond at 54.8 eV could be attributed mainly to the oxidizing electrolyte in air. The Li–F bond (56.6 eV) exists in the overall lithiation/delithiation processes.60,61 Based on all the analyses, the overall reaction mechanism of the GeZn1.7−xON1.8 particle anode during the lithiation/delithiation processes can be expressed as eqn (1):
| GeZn1.7−xON1.8 + xLi+ + xe− ↔ LixGeZn1.7−xON1.8 | (1) |
4. Conclusion
In summary, GeZn1.7−xON1.8 particles have been successfully prepared by an ammoniation and acid etching method as confirmed by XRD, EDS, EPR, and XPS characterization techniques. The Mott–Schottky testing demonstrated an efficient electron transport in GeZn1.7−xON1.8 particle anodes. Systematic electrochemical analyses revealed that the GeZn1.7−xON1.8 particle anode possessed a higher rate capacity and excellent cyclic stability compared with the pristine GeZn1.7ON1.8 particle anode. Electrochemical kinetics analysis further proved the excellent electrochemical behavior of the GeZn1.7−xON1.8 particle anodes. Furthermore, the XRD and XPS results obtained at different electrochemical reaction states show that the good cyclic stability could be attributed to favorable structural tolerance and reversible lithium-ion storage mechanism. This study provides a feasible way to understand high-performance quaternary metal oxynitride anodes prepared via rational design defect engineering.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Foundation of Chinese Academy of Sciences Science and Technology Service Network Program-Dongguan Special Project (No. 2137003); the High-level Talents (GB200902-30, No. 196100041018 and GB 200902-43, No. 196100041009), the Foundation of Regular Research Team (TDYB2019007, No. 196100043028 and No. 2062011047), the Foundation of Young Research Team (TDQN2019004, No. 193500008008) at Dongguan University of Technology, the Guangdong Basic and Applied Basic Research Foundation (2019A1515110933, 2019A1515110554, 2020A1515111086, 2020A1515110219, and 2020A1515110770), China Postdoctoral Science Foundation (Grant No. 2021M700915), and the Foundation of Doctor’s Workstation of Mcnair New Power Co., Ltd (GC200104-40, No. 186100030017).References
- J. Ni, Y. Zhao, L. Li and L. Mai, Nano Energy, 2015, 11, 129–135 CrossRef CAS.
- J. R. Rodriguez, Z. Qi, H. Wang, M. Y. Shalaginov, C. Goncalves, M. Kang, K. A. Richardson, J. Guerrero-Sanchez, M. G. Moreno-Armenta and V. G. Pol, Nano Energy, 2020, 68, 104326 CrossRef CAS.
- W. Li, X. Li, J. Yu, J. Liao, B. Zhao, L. Huang, A. Ali, H. Zhang, J. H. Wang, Z. Guo and M. Liu, Nano Energy, 2019, 61, 594–603 CrossRef CAS.
- C. Sun, Y.-J. Wang, H. Gu, H. Fan, G. Yang, A. Ignaszak, X. Tang, D. Liu and J. Zhang, Nano Energy, 2020, 77, 105092 CrossRef CAS.
- T. Wang, C. Sun, M. Yang, L. Zhang, Y. Shao, Y. Wu and X. Hao, Electrochim. Acta, 2018, 259, 1–8 CrossRef CAS.
- T. Wang, J. Shen, M. Yang, C. Cheng, Y. Zhang, C. Sun and L. Zhang, Ceram. Int., 2021, 47, 21610 CrossRef CAS.
- T. Wang, C. Sun, M. Yang, G. Zhao, S. Wang, F. Ma, L. Zhang, Y. Shao, Y. Wu, B. Huang and X. Hao, J. Alloys Compd., 2017, 716, 112–118 CrossRef CAS.
- C. Sun, X. Tang, Z. Yin, D. Liu, Y.-J. Wang, G. Yang, A. Ignaszak and J. Zhang, Nano Energy, 2020, 68, 104376 CrossRef CAS.
- C. Sun, M. Yang, T. Wang, Y. Shao, Y. Wu and X. Hao, ACS Appl. Mater. Interfaces, 2017, 9, 26631–26636 CrossRef CAS.
- C. Sun, M. Yang, T. Wang, Y. Shao, Y. Wu and X. Hao, ACS Appl. Mater. Interfaces, 2018, 10, 2574–2580 CrossRef CAS PubMed.
- T. Wang, G. Zhao, C. Sun, L. Zhang, Y. Wu, X. Hao and Y. Shao, Adv. Mater. Interfaces, 2017, 4, 1601187 CrossRef.
- M. Yang, C. Sun, T. Wang, F. Chen, M. Sun, L. Zhang, Y. Shao, Y. Wu and X. Hao, ACS Appl. Energy Mater., 2018, 1, 4708–4715 CrossRef CAS.
- W. H. Choi, C. H. Lee, H.-e. Kim, S. U. Lee and J. H. Bang, Nano Energy, 2020, 74, 104829 CrossRef CAS.
- B. Long, H. Yang, F. Wang, Y. Mao, M.-S. Balogun, S. Song and Y. Tong, Electrochim. Acta, 2018, 284, 271–278 CrossRef CAS.
- B. Zou, W. Zhang, Y. Cui, S. Li, G. Li, X. Liu, D. H. L. Ng, J. Qiu and J. Lian, J. Mater. Chem. A, 2022, 10, 7391–7398 RSC.
- B. Long, M.-S. Balogun, L. Luo, Y. Luo, W. Qiu, S. Song, L. Zhang and Y. Tong, Small, 2017, 13, 1702081 CrossRef.
- K. Xie, J. Wang, S. Yu, P. Wang and C. Sun, Arabian J. Chem., 2021, 14, 103161 CrossRef CAS.
- A. Achour, J. B. Ducros, R. L. Porto, M. Boujtita, E. Gautron, L. Le Brizoual, M. A. Djouadi and T. Brousse, Nano Energy, 2014, 7, 104–113 CrossRef CAS.
- X. Yang, X. Li, K. Adair, H. Zhang and X. Sun, Electrochem. Energy Rev., 2018, 1, 239–293 CrossRef CAS.
- L. Bai, Y. Zhang, W. Tong, L. Sun, H. Huang, Q. An, N. Tian and P. K. Chu, Electrochem. Energy Rev., 2020, 3, 395–430 CrossRef CAS.
- C. Sun, F. Chen, X. Tang, D. Zhang, K. Zheng, G. Zhu, U. Bin Shahid, Z. Liu, M. Shao and J. Wang, Nano Energy, 2022, 99, 107369 CrossRef CAS.
- W. Cha, S. Kim, P. Selvarajan, J. M. Lee, J. M. Davidraj, S. Joseph, K. Ramadass, I. Y. Kim and A. Vinu, Nano Energy, 2021, 82, 105733 CrossRef CAS.
- S. Wang, C. Sun, Y. Shao, Y. Wu, L. Zhang and X. Hao, Small, 2017, 13, 1603330 CrossRef PubMed.
- Y. Wu, P. Lazic, G. Hautier, K. Persson and G. Ceder, Energy Environ. Sci., 2013, 6, 157–168 RSC.
- C. Sun, F. Ma, L. Cai, A. Wang, Y. Wu, M. Zhao, W. Yan and X. Hao, Sci. Rep., 2017, 7, 6617 CrossRef PubMed.
- Z. Li, K. Gao, Y. Han, S. Ding, Y. Cui, M. Hu, J. Zhao, M. Zhang, A. Meng, J. Yun, Z. Liu, D.-W. Wang and C. Sun, J. Energy Chem., 2022, 67, 46–54 CrossRef.
- G. Ali, J.-H. Lee, S. H. Oh, H.-G. Jung and K. Y. Chung, Nano Energy, 2017, 42, 106–114 CrossRef CAS.
- H. He, D. Huang, Y. Tang, Q. Wang, X. Ji, H. Wang and Z. Guo, Nano Energy, 2019, 57, 728–736 CrossRef CAS.
- G. Huang, Y. Yang, H. Sun, S. Xu, J. Wang, M. Ahmad and Z. Xu, J. Alloys Compd., 2017, 724, 1149–1156 CrossRef CAS.
- C. Schliehe and C. Giordano, Nanoscale, 2013, 5, 3235–3239 RSC.
- L. Pan, S. Wang, W. Mi, J. Song, J.-J. Zou, L. Wang and X. Zhang, Nano Energy, 2014, 9, 71–79 CrossRef CAS.
- S. Li, Y. Xiao, X. Wang and M. Cao, Phys. Chem. Chem. Phys., 2014, 16, 25846–25853 RSC.
- L. Huang, G. H. Waller, Y. Ding, D. Chen, D. Ding, P. Xi, Z. L. Wang and M. Liu, Nano Energy, 2015, 11, 64–70 CrossRef CAS.
- Y. Xu, M. Zhou, C. Zhang, C. Wang, L. Liang, Y. Fang, M. Wu, L. Cheng and Y. Lei, Nano Energy, 2017, 38, 304–312 CrossRef CAS.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed.
- Y. Han, C. Sun, K. Gao, S. Ding, Z. Miao, J. Zhao, Z. Yang, P. Wu, J. Huang, Z. Li, A. Meng, L. Zhang and P. Chen, Electrochim. Acta, 2022, 408, 139931 CrossRef CAS.
- D. Choi, G. E. Blomgren and P. N. Kumta, Adv. Mater., 2006, 18, 1178–1182 CrossRef CAS.
- Y. Zhu, N. Jain, M. K. Hudait, D. Maurya, R. Varghese and S. Priya, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2014, 32, 011217 Search PubMed.
- K. S. Agrawal, V. S. Patil, A. G. Khairnar and A. M. Mahajan, Appl. Surf. Sci., 2016, 364, 747–751 CrossRef CAS.
- A. B. Djurišić, W. C. H. Choy, V. A. L. Roy, Y. H. Leung, C. Y. Kwong, K. W. Cheah, T. K. Gundu Rao, W. K. Chan, H. Fei Lui and C. Surya, Adv. Funct. Mater., 2004, 14, 856–864 CrossRef.
- Z. Ma, O. Linnenberg, A. Rokicinska, P. Kustrowski and A. Slabon, J. Phys. Chem. C, 2018, 122, 19281–19288 CrossRef CAS.
- M. M. Rahman, I. Sultana, T. Yang, Z. Chen, N. Sharma, A. M. Glushenkov and Y. Chen, Angew. Chem., Int. Ed., 2016, 128, 16293–16297 CrossRef.
- Q. Peng, Y. Lei, Z. Tang, C. Sun, J. Li, G. Wu, T. Wang, Z. Yin and H. Liu, Electrochim. Acta, 2020, 350, 136380 CrossRef CAS.
- K. Zhang, H. Wang, X. He, Z. Liu, L. Wang, L. Gu, H. Xu, P. Han, S. Dong and C. Zhang, J. Mater. Chem., 2011, 21, 11916–11922 RSC.
- Y. Wu, L. Huang, X. Huang, X. Guo, D. Liu, D. Zheng, X. Zhang, R. Ren, D. Qu and J. Chen, Energy Environ. Sci., 2017, 10, 1854–1861 RSC.
- D. Kundu, F. Krumeich, R. Fotedar and R. Nesper, J. Power Sources, 2015, 278, 608–613 CrossRef CAS.
- C. Yan, C. Lv, Y. Zhu, G. Chen, J. Sun and G. Yu, Adv. Mater., 2017, 29, 1703909 CrossRef PubMed.
- X. Xia, D. Chao, Y. Zhang, J. Zhan, Y. Zhong, X. Wang, Y. Wang, Z. X. Shen, J. Tu and H. J. Fan, Small, 2016, 12, 3048–3058 CrossRef CAS PubMed.
- P. He, Y. Quan, X. Xu, M. Yan, W. Yang, Q. An, L. He and L. Mai, Small, 2017, 13, 1702551 CrossRef PubMed.
- C. Chen, Y. Wen, X. Hu, X. Ji, M. Yan, L. Mai, P. Hu, B. Shan and Y. Huang, Nat. Commun., 2015, 6, 6929 CrossRef CAS PubMed.
- G. Fu, Z. Cui, Y. Chen, L. Xu, Y. Tang and J. B. Goodenough, Nano Energy, 2017, 39, 77–85 CrossRef CAS.
- J. Zhou, Z. Jiang, S. Niu, S. Zhu, J. Zhou, Y. Zhu, J. Liang, D. Han, K. Xu, L. Zhu, X. Liu, G. Wang and Y. Qian, Chem, 2018, 4, 372–385 CAS.
- S. Wang, S. Liu, X. Li, C. Li, R. Zang, Z. Man, Y. Wu, P. Li and G. Wang, Chem.–Eur. J., 2018, 24, 3873–3881 CrossRef CAS PubMed.
- S. S. Zhang, J. Electrochem. Soc., 2012, 159, A920–A923 CrossRef CAS.
- X. Xu, C. Niu, M. Duan, X. Wang, L. Huang, J. Wang, L. Pu, W. Ren, C. Shi, J. Meng, B. Song and L. Mai, Nat. Commun., 2017, 8, 460 CrossRef PubMed.
- J. Chu, W. A. Wang, J. Feng, C.-Y. Lao, K. Xi, L. Xing, K. Han, Q. Li, L. Song, P. Li, X. Li and Y. Bao, ACS Nano, 2019, 13, 6906–6916 CrossRef CAS PubMed.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS.
- F. W. Fenta, B. W. Olbasa, M.-C. Tsai, M. A. Weret, T. A. Zegeye, C.-J. Huang, W.-H. Huang, T. S. Zeleke, N. A. Sahalie, C.-W. Pao, S.-h. Wu, W.-N. Su, H. Dai and B. J. Hwang, J. Mater. Chem. A, 2020, 8, 17595–17607 RSC.
- E. Radvanyi, W. Porcher, E. De Vito, A. Montani, S. Franger and S. J. S. Larbi, Phys. Chem. Chem. Phys., 2014, 16, 17142–17153 RSC.
- P. Guan, L. Liu and X. Lin, J. Electrochem. Soc., 2015, 162, A1798–A1808 CrossRef CAS.
- R. Younesi, P. Norby and T. Vegge, ECS Electrochem. Lett., 2014, 3, A15–A18 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |