
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the reactivity of aminomethylene derivatives of resorc[4]arene: amine group transfer, conformational analysis, reaction mechanism†
Waldemar Iwanek
Bydgoszcz University of Science and Technology, Faculty of Chemical Technology and Engineering, Seminaryjna 3, 85-326 Bydgoszcz, Poland. E-mail: Waldemar.Iwanek@pbs.edu.pl
First published on 27th September 2022
Abstract
Using the example of aminomethylene derivatives of resorc[4]arene and their Michael reaction with 4-hydroxycoumarin, the possibility of transferring an amine molecule from substrate to product is demonstrated. The conformation of the aminocoumarin derivatives of resorc[4]arene formed is controlled by the polarity of the solvent. For one of the products, conformational analysis was performed by kinetic sampling using metadynamics (MTD). The energies of the final set of conformers were calculated by DFT (r2scan-3c). A reaction mechanism based on multiscale (ONIOM) Nudged Elastic Band (NEB-TS) reaction profile calculations is discussed.
Introduction
Currently, there has been an increase in the number of studies in the literature demonstrating the use of resorc[4]arenes, particularly in the organocatalytic1 and biological2 fields as well as in the creation of efficient molecular switches.3 However, the possibilities of synthesising structurally ‘interesting’ resorc[4]arenes are still open. In this context, a worthwhile task seemed to be the synthesis of new resorc[4]arenes with an interesting molecular architecture, containing in their structure various heterocyclic units linked to each other by a system of directional intramolecular hydrogen bonds. By selecting appropriate proton donor/acceptor substituents in the attached heterocyclic units, the type of solvent and the temperature, we can influence the selectivity and rigidity of formation of specific resorc[4]arene conformers.4 Conformational dynamics controlled in this way are of interest from the point of view of, among other things, modelling and mimicking enzymes, controlling substrate complexation processes and creating conformationally and spectroscopically variable molecular switches.The Mannich reaction of resorc[4]arene with formaldehyde and secondary amines is one of the basic reactions for the formation of resorc[4]arene derivatives with enlarged cavity.5 The products of this reaction are often formed at room temperature with high yields. They have been the subject of papers including studies of their receptor properties towards anions.6 The presence of a methylene amine group and a hydroxyl group in the ortho position in the resorc[4]arene molecule opens up the possibility of their reaction via two pathways: (1) through the departure of the amino group with the formation of a reactive transition product, which is the o-quinomethine derivative of resorc[4]arene;7 (2) the formation of a coordination bond with the boron atom, with the formation of new boron-cyclic rings in the resorc[4]arene molecule.8
Enzymatically catalysed functional group transfer reactions are fundamental to the functioning of living organisms. One such process is the transamination reaction of amino acids, catalysed by enzymes called transaminases.9 In the work presented here, I present the possibility of amine group transfer in the thermal reaction of aminomethylene derivatives of resorc[4]arene with 4-hydroxycoumarin. The possibility of a diverse arrangement of substituents in the resorc[4]arene molecule generates a potentially large number of conformers for this molecule. The NMR spectra observed are of greater or lesser complexity depending on the solvent. This prompted the author to carry out a conformational analysis of the morpholine–coumarin derivative of resorc[4]arene by metadynamics using the semi-empirical DFT methods of tight binding GFNn−xTB developed by S. Grimme's team.10 In order to better understand the mechanism of the ongoing reaction, multiscale calculations of the energy profile of the reaction by NEB-TS11 between the morpholine derivative of resorc[4]arene and 4-hydroxycoumarin were performed.
The results presented in this work open up and demonstrate the possibilities for the synthesis of new resorc[4]arene derivatives via an amine group transfer reaction and describe their solvent-controlled conformational changes. In the author's opinion, this is an interesting example of a reaction that has so far (to the author's knowledge) not been reported in the literature. It can greatly simplify the procedure for the synthesis of such compounds. Furthermore, no literature example was found showing this type of reaction using the known and structurally relatively simple Betti bases.12
Results and discussion
Synthesis of 4-aminocoumarin derivatives of resorc[4]arene
Aminomethylene derivatives of resorc[4]arene (1) were synthesised with formaldehyde and selected secondary amines in ethanol at room temperature.5 They were then used to thermally react with 4-hydroxycoumarin by performing the reaction in chloroform for 30 minutes at 160 °C using a Monowave 50 reactor. After evaporation of the chloroform, the product was isolated by adding ethyl acetate or methanol. The precipitate was washed with a suitable solvent under reduced pressure. After crystallisation, the resulting precipitate was subjected to spectroscopic analysis. The yields of the reaction in question, shown in Table 1, range from 58–78%. A collective diagram for the synthesis of 4-aminocoumarin derivatives of resorc[4]arene (2) is shown in Scheme 1.| Aminocoumarin derivatives of resorc[4]arene | Reaction yield (%) |
|---|---|
| 2a | 58 |
| 2b | 67 |
| 2c | 78 |
| 2d | 68 |
| 2e | 64 |
| 2f | 69 |
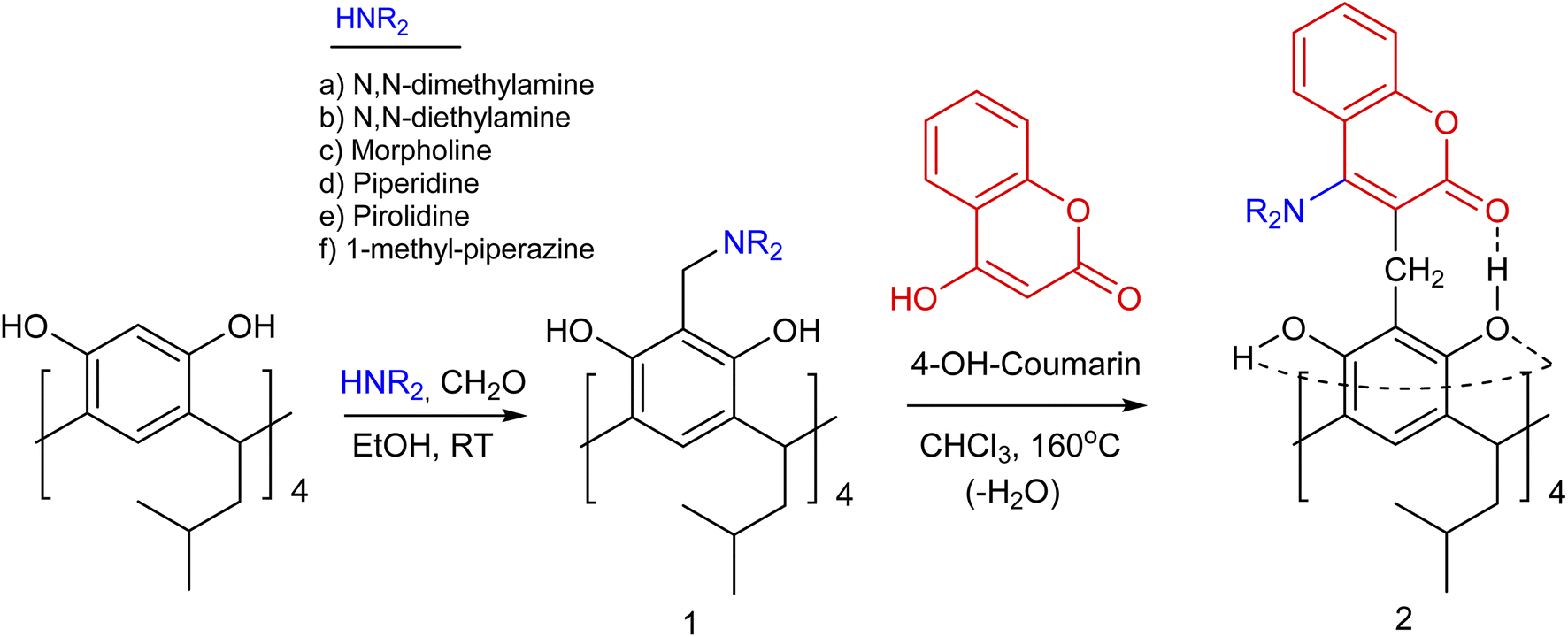 | ||
| Scheme 1 The synthesis of 4-amino coumarin derivatives of resorc[4]arene (2). | ||
In an earlier paper,13 the possibility of synthesising 4-hydroxycoumarin derivatives of resorc[4]arene was shown using the Michael reaction of the o-quinomethine derivative of resorc[4]arene, which was generated thermally from the methoxy derivative of resorc[4]arene. The resulting 4-hydroxycoumarin derivative of resorc[4]arene exhibited a variable ‘in-out’ conformation depending on the reaction environment.
In the example described in this work, a series of aminomethylene derivatives of resorc[4]arene were used for the reaction, which did not yield 4-hydroxycoumarin derivatives of resorc[4]arene, but 4-aminocoumarin derivatives of resorc[4]arene, when reacted with 4-hydroxycoumarin. During the reaction, there was a transfer of the amino group from the aminomethylene derivative of resorc[4]arene to the reaction product. The mechanism of this transfer is discussed in more detail in the section ‘Reaction Mechanism’.
The resulting products, depending on the solvent, are characterised by NMR spectra of greater or lesser complexity. Structural and conformational analysis will be discussed below using the structure of the morpholine–coumarin derivative of resorc[4]arene as an example (2c). The 1H-NMR spectrum of compound 2c in DMSO is shown in Fig. 1, together with the assigned chemical shifts of the proton signals. It indicates that the molecule exhibits C4 symmetry, which greatly simplifies its analysis. The morpholine–coumarin substituents locate outside the cavity of the resorc[4]arene skeleton to form a conformer of the type “all-out”. Slightly broadened signals on the spectrum indicate the rather large dynamics of this molecule.
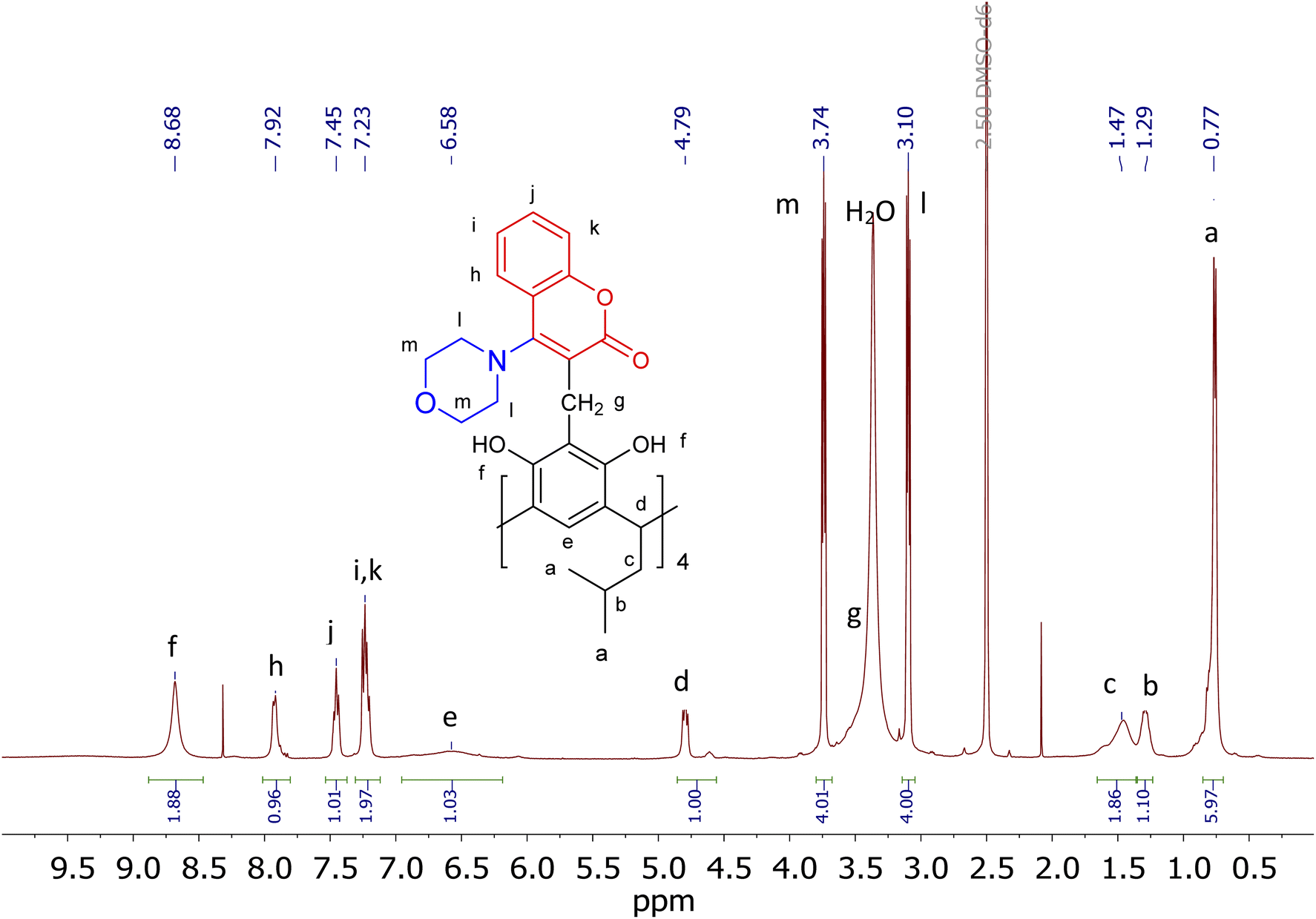 | ||
| Fig. 1 1H-NMR spectrum (400 MHz) of derivative 2c in DMSO. | ||
The ability of the morpholine–coumarin substituent to rotate along the CH2–C-fragment coumarin bond in derivative 2c means that they can locate above the rim of the resorc[4]arene or below it, giving rise to 2 theoretically possible ‘all-out’ conformers. The first of these, for which the morpholine moiety is located above the upper rim (up) of the resorc[4]arene and the coumarin moiety below (down) was designated 2c-all-out-Mup-Cdown (M = morpholine, C = coumarin). While the second conformer, in which the morpholine fragment is located below the lower rim and the coumarin part above respectively 2c-all-out-Mdown-Cup.
Calculated with r2scan-3c,14 the energies of the 2c-all-out-Mup-Cdown and 2c-all-out-Mdown-Cup conformers in DMSO using the CPCM model of the solvent15 indicate that the more thermodynamically stable conformer in DMSO and in CHCl3 is the 2c-all-out-Mup-Cdown conformer. The calculated energy differences are summarised in Table 2, while the optimised structures of the corresponding conformers are shown in Fig. 2.
| r2scan-3c/CPCM | ||
|---|---|---|
| Conformer | DMSO kcal mol−1 | CHCl3 kcal mol−1 |
| 2c-all-out-Mup-Cdown | 0 | 0 |
| 2c-all-out-Mdown-Cup | 17.22 | 17.05 |
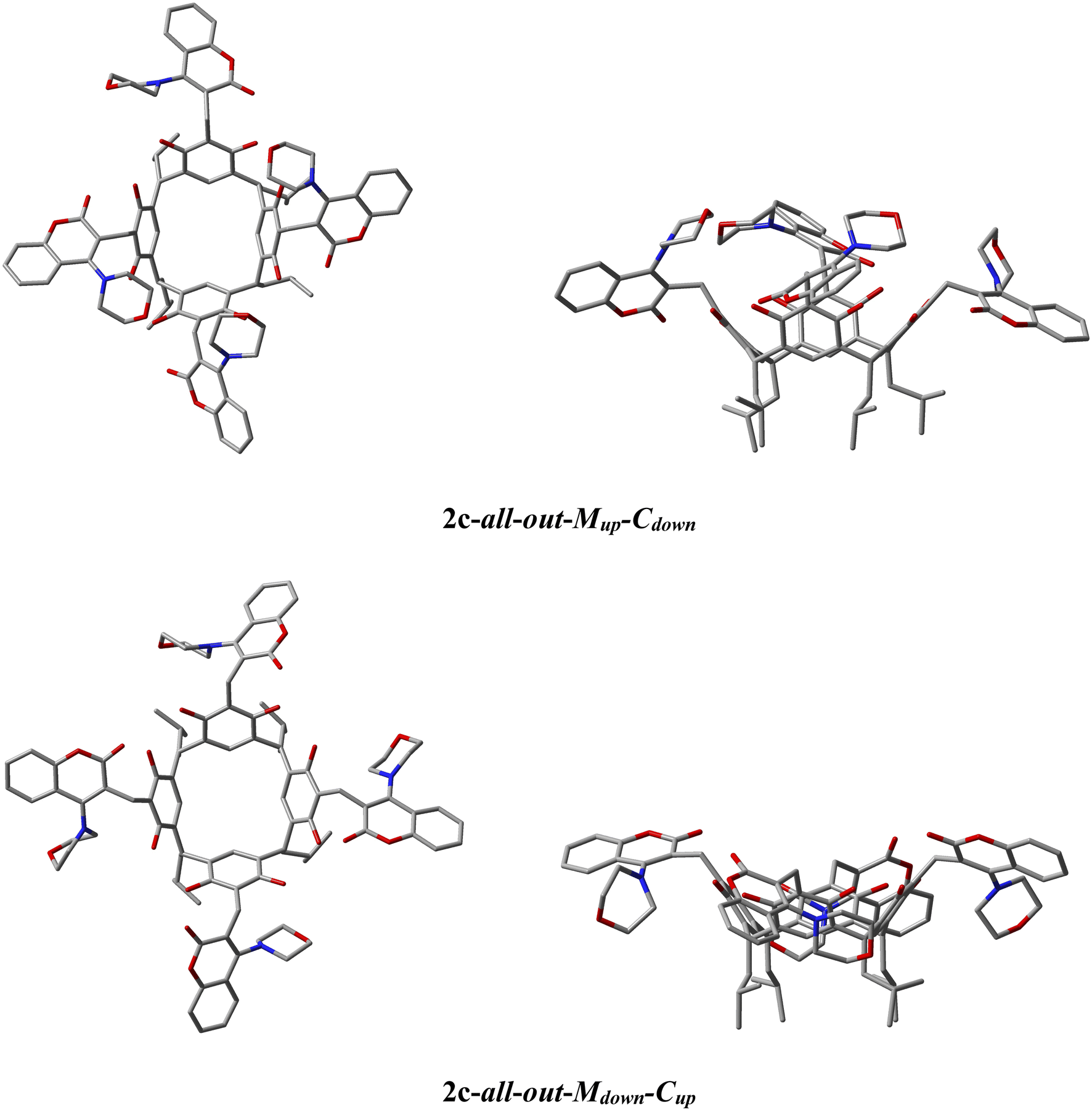 | ||
| Fig. 2 The r2scan-3c/CPCM(DMSO)-optimised conformer geometries, respectively: 2c-all-out-Mup-Cdown and 2c-all-out-Mdown-Cup (M = morpholine, C = coumarine) left side of structure in top view, right side in side view. Hydrogen atoms have been omitted for better visualisation. | ||
2D spectra do not show strong evidence of a correlation between coumarin's and resorc[4]arene's moieties. However, an interaction of the protons of the hydroxyl groups OH(f) with the protons of the morpholine groups CH2(m,l) can be observed (ESI†).
The proton signals of derivative 2c in chloroform, shown in Fig. 3, are strongly broadened. This demonstrates the high conformational dynamics of this derivative in chloroform. A different image of the 1H NMR spectrum can be observed in acetone (Fig. 3, brown colour). In this solvent, the proton signals are most clearly separated, but signals indicating the presence of a mixture of conformers can be observed on the spectrum.
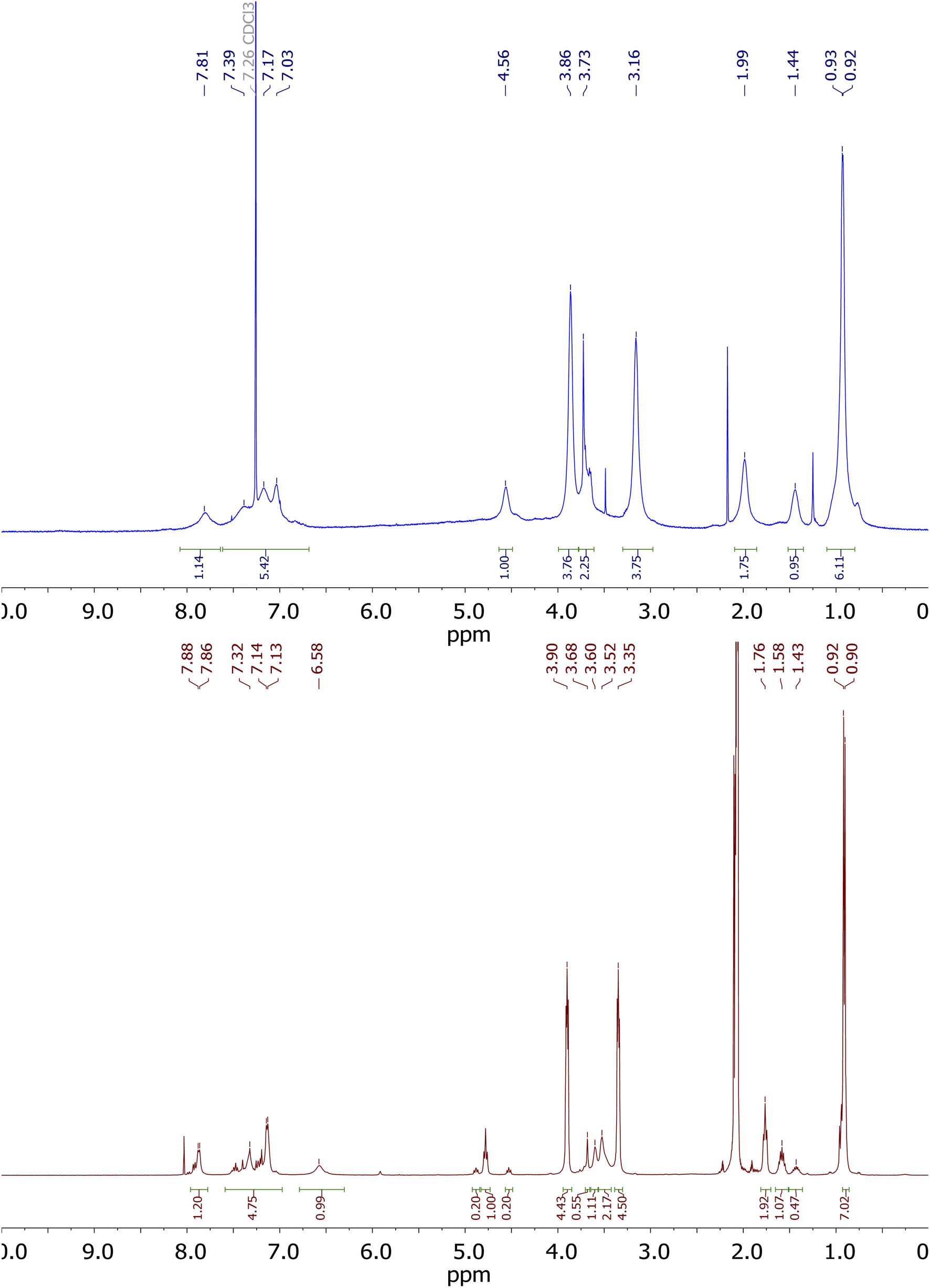 | ||
| Fig. 3 1H-NMR spectrum (400 MHz) of derivative 2c in CDCl3 (blue) and acetone-d6 (brown). | ||
Conformational analysis of derivative 2c in acetone
The search for the molecular conformations of derivative 2c present in acetone was performed by GFNn−xTB (semi-empirical DFT method) based on the xTB programme16 using molecular metadynamics (MTD).17 Kinetic sampling was performed at 433 K in CHCl3 (simulating the experimental conditions of the reaction) using GFN0−xTB18 and the implicit solvation model ALPB18 to calculate the ensemble energy of the generated structures. The total simulation time was 100 ps, writing to the trajectory file was set to 50 fs with a step size of 1 fs. The number of frames generated in the total trajectory was 2000. CREST19 was then used to optimise the dynamics trajectory in the gaseous state using GFN2−xTB with an accuracy level of 5 × 10−6 Econv/Eh. The optimised structures were sorted (default values) into an energy window of 4 kcal mol−1 yielding the 99 structures with the lowest energy. These structures were again optimised by GFN2−xTB in acetone (tier 1 × 10−7 Econv/Eh) using the ALPB solvation model. The optimised conformer structures were segregated within an energy window of 1 kcal mol−1 with an energy threshold of 0.25 kcal mol−1 and a geometry threshold of 0.5 A to distinguish them, yielding a final 5 conformers. These conformers were subjected to higher-level DFT calculations with the r2scan-3c function in acetone using the CPCM solvation model. The r2scan-3c functional is specifically recommended for calculating the energy of conformational changes of organic compounds.20 The ORCA 5.0.2 package21 was used for the calculations. The energies of the calculated conformers and their populations at 298 K are summarised in Table 3. Below, Fig. 4 shows the geometrically optimised structures of derivative 2c conformers (1–3).| Conformers of 2c | r2scan-3c/CPCM (acetone) kcal mol−1 | Conformer population 298 K/% |
|---|---|---|
| 1-all-out-Mup-Cdown | 0 | 62.22 |
| 2-out-3-Mup-Cdown-1-Mdown-Cup | 0.63 | 21.30 |
| 3-all-out-Mup-Cdown | 0.79 | 16.46 |
| 4-all-out-Mup-Cdown | 5.13 | 0.01 |
| 5-all-out-Mup-Cdown | 5.19 | 0.01 |
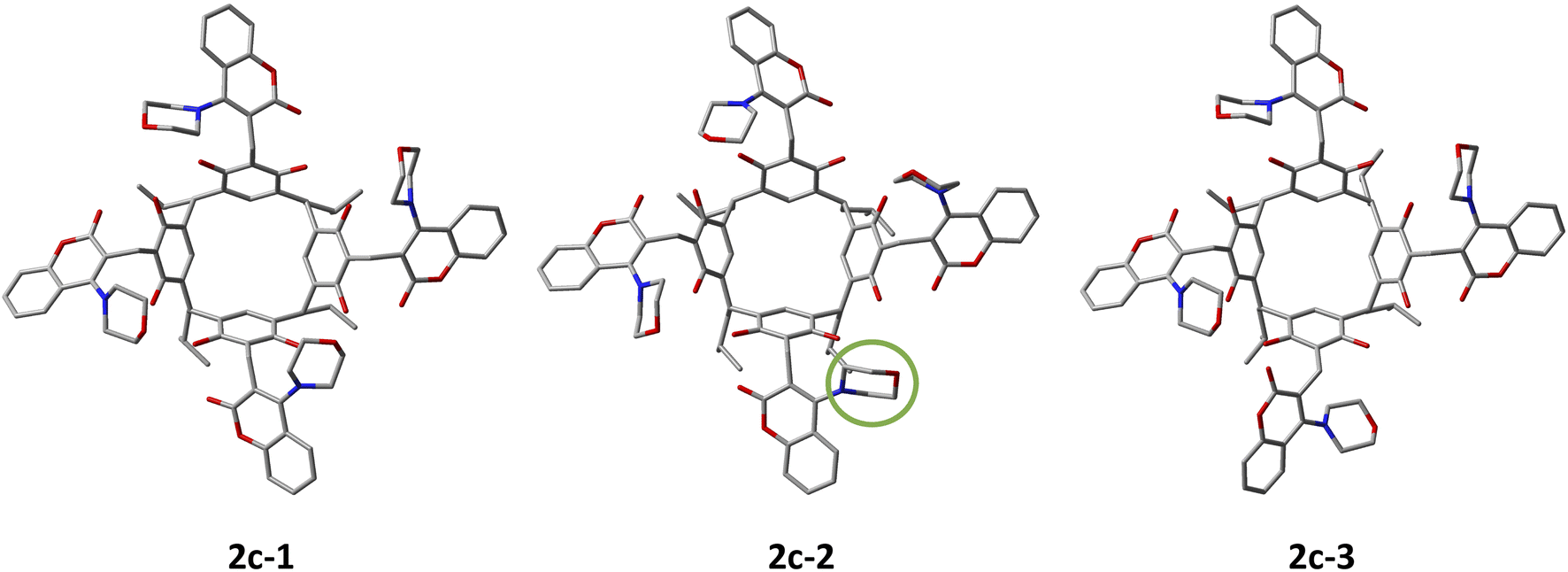 | ||
| Fig. 4 The geometrical structures of conformers 2c-1–3 optimised by the r2scan-3c functional in acetone are shown in Table 2. In the case of conformer 2c-2, the morpholine fragment that proceeds below the rim of the resorc[4]arene (Mdown) is shown in green. Hydrogen atoms have been omitted for better visualisation of the conformers. | ||
The population of conformers calculated theoretically at 298 K corresponds well with the results of the integration of the methine proton signal CH(d) in acetone, which are respectively: 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14:14. It is worth noting that the 2 conformers with the lowest energy (1–3) of the 5 conformers computed are all-out-Mup-Cdown-conformers with a slight rearrangement of the morpholine–coumarin fragments. In the case of conformer 2c-2 (out-3-Mup-Cdown-1-MdownCup), we have a case where one of the morpholine fragments is located below the rim of the resorc[4]arene, while the coumarin fragment is above. The low energy barrier of the conformational changes of conformers 1–3 at 298 K explains their high mobility, as manifested by broadened signals on 1H-NMR spectra in DMSO, particularly in CHCl3.
:14:14. It is worth noting that the 2 conformers with the lowest energy (1–3) of the 5 conformers computed are all-out-Mup-Cdown-conformers with a slight rearrangement of the morpholine–coumarin fragments. In the case of conformer 2c-2 (out-3-Mup-Cdown-1-MdownCup), we have a case where one of the morpholine fragments is located below the rim of the resorc[4]arene, while the coumarin fragment is above. The low energy barrier of the conformational changes of conformers 1–3 at 298 K explains their high mobility, as manifested by broadened signals on 1H-NMR spectra in DMSO, particularly in CHCl3.
Reaction mechanism
The aminocoumarin derivatives of resorc[4]arene in question are molecules containing approximately 160 atoms in their structure. Moreover, studies of reaction mechanisms using the NEB-TS method implemented in the ORCA package require the calculation of a large number of cycles (usually around several hundred) and are computationally expensive. This led the author to use the ONIOM multiscale method,22 in which two fragments of a molecule are calculated at different levels of DFT theory/methods (QM/XTB2). In the present case, the reaction of one morpholine–resorcinol unit (yellow fragment in Fig. 5) in the morpholine derivative of resorc[4]arene with 4-hydroxycoumarin was calculated at a higher level of DFT theory (QM = B97-3c), while the rest of the molecule was calculated at a lower level of DFT theory (XTB2 = GFN2). This simplified structure was subjected to NEB-TS calculations (25 images) in the gas phase using the LOOSE-NEB-TS command. Fig. 5 shows the energy profile of the reaction along with highlighted images of the reactants (1) and transition state (TS) structures respectively. In the case of the reactant, the yellow colour indicates the section optimised at a higher level of theory (B97-3c). The numbers (1–6), on the other hand, represent the structures shown in Scheme 2, describing the reaction mechanism.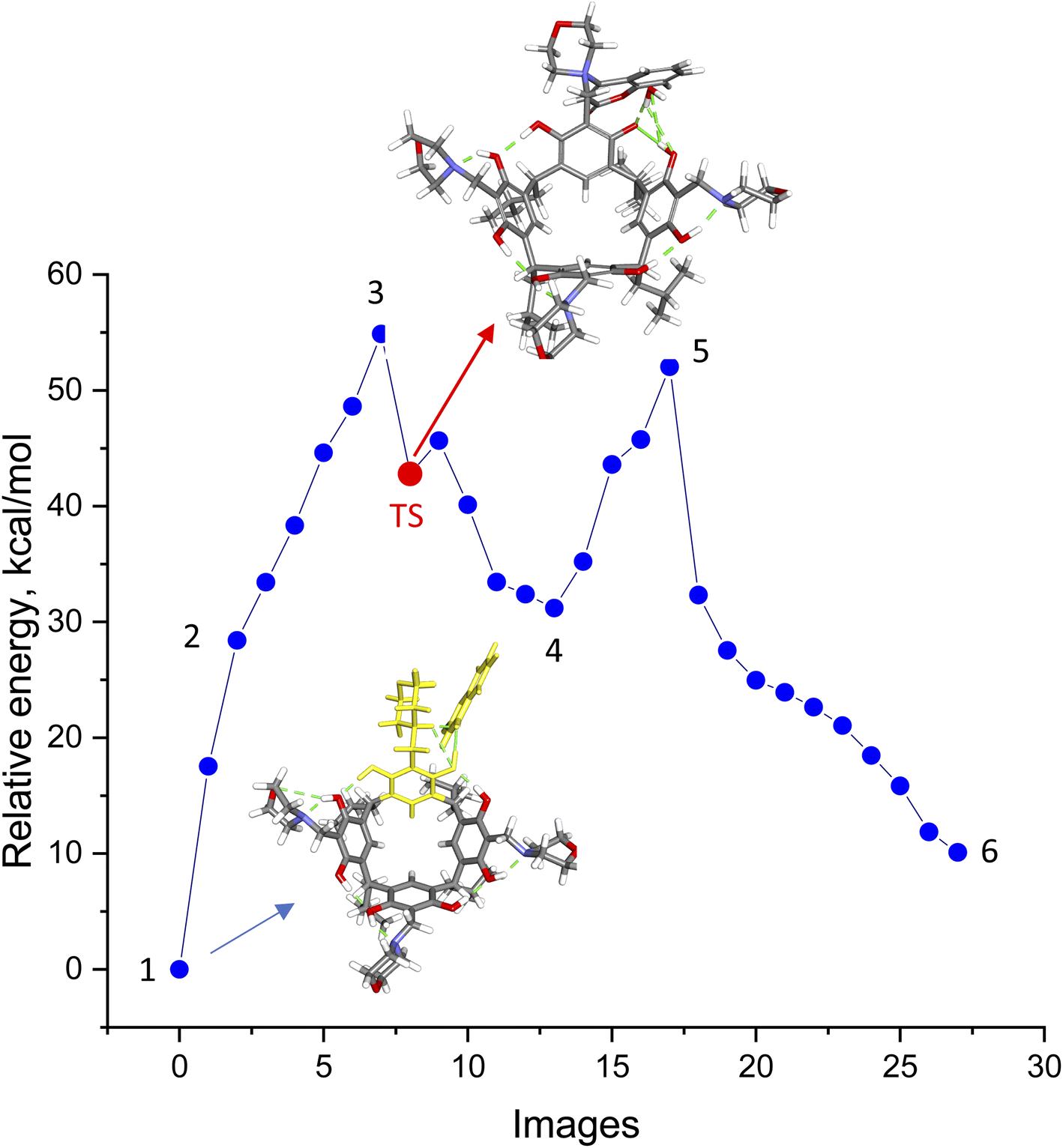 | ||
| Fig. 5 Energy plot of individual images calculated using the LOOSE-NEB-TS method with ONIOM QM/XTB2 (B97-3c/xTB-GFN2) in the gas phase. The figure highlights images for the reactants (1) and transition state (TS) along with their respective molecular structures. The dashed green line illustrates hydrogen bonds. In the case of regents (1), yellow is used to indicate the fragments of the molecules optimised at a higher level of theory. | ||
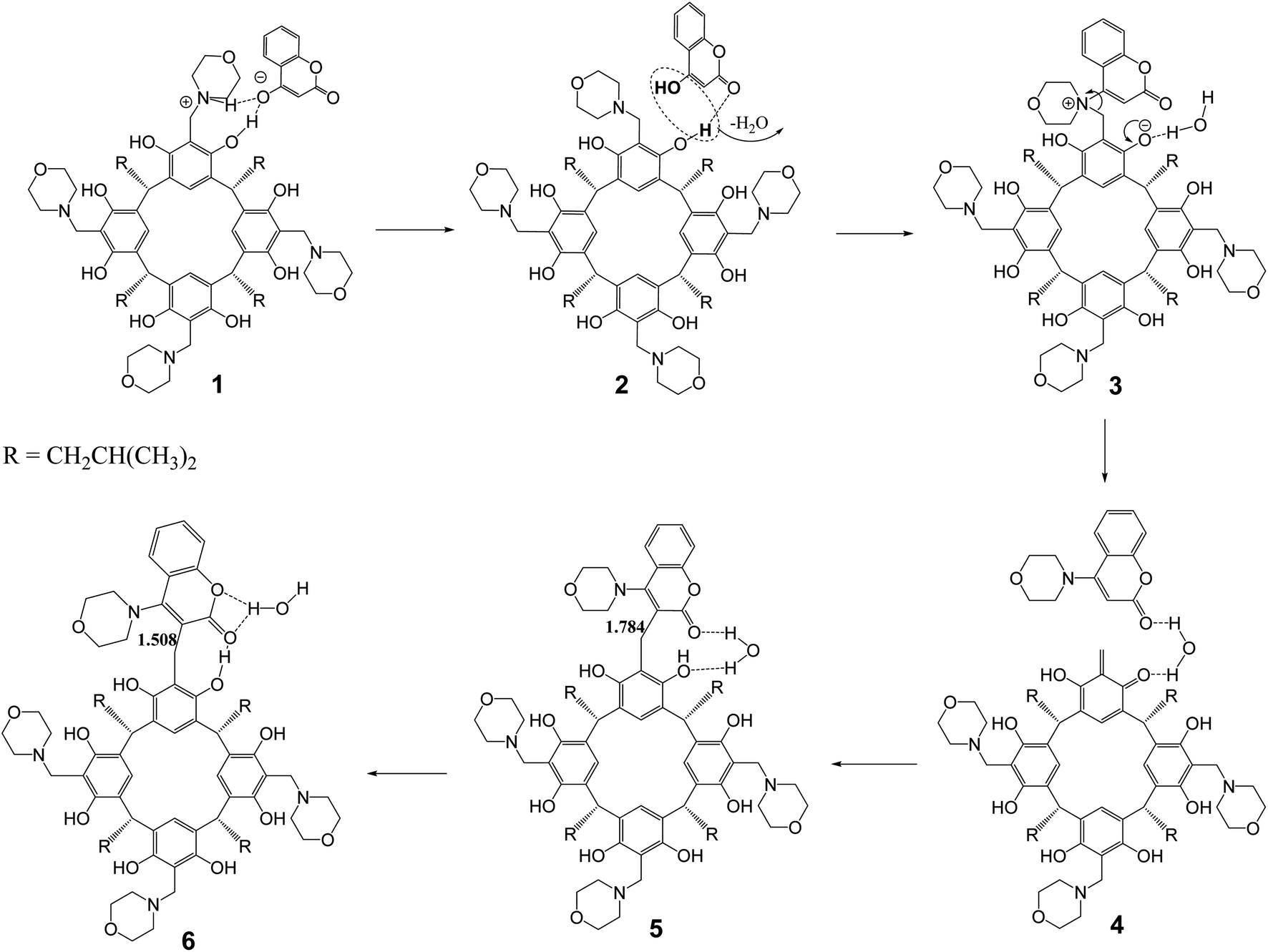 | ||
| Scheme 2 Proposed reaction mechanism for the formation of the 2c product based on NEB-TS calculations. | ||
The energy profile of the reaction shows that the reaction in question is a two-step reaction, where the rate-determining step is the formation of the transition state (TS) in the first reaction step. Its optimised structure indicates that it is a zwitterion with a positive charge on the nitrogen atom of the morpholine ring and an attached coumarin fragment and a negative charge located on the oxygen atom of the resorcinol ring. Data on reaction energy profile and transition state calculations, together with IRC calculations, are included in ESI.†
The proposed reaction mechanism (Scheme 2) based on the NEB-TS calculations is presented below. The optimized structure of the substrates (1) indicates that the energy minimum is achieved after proton transfer between the 4-hydroxycoumarin molecule and the morpholine unit in the resorc[4]arene. In the next stage, the proton is transferred back from the protonated morpholine unit in the resorc[4]arene to the 4-hydroxycoumarin with its simultaneous shift towards the resorc[4]arene hydroxyl group with the formation of the structure (2). In the next step, the water molecule is disconnected with the simultaneous formation of a dipolar morpholinocoumarin derivative of resorc[4]arene (3), in which the coumarin unit is attached to the morpholine unit in the 4 position. The dipolar nature of the structure (3) favors the formation of an o-quinomethine derivative of resorc[4]arene (4) with the simultaneous formation of 4-morpholinocoumarin. The interaction of the electrophilic carbon of the methine group of the o-chinomethine derivative of resorc[4]arene with electronically enriched C3 carbon of the 4-morpholinocoumarin leads to the formation of the reaction product (5). The length of the bond formed is 1.784 Å. The next steps of the reaction are the spatial reorganization of the created product to the most energy-stable product (6), in which the length of the formed bond is 1.508 Å.
The water molecule formed during the reaction plays an important role in the course of the reaction. It interacts through hydrogen bonds with the coumarin unit and the resorcinol unit in resorc[4]arene, as shown in the example of structures (3–6).
To calculate the height of the energy barrier, the M06-2X hybrid functional23 recommended for this purpose19 was used using the 6-311G** basis set. Single-point energy calculations for the structures shown in Fig. 5 in chloroform using the CPCM solvent model are shown in Table 4.
| B97-3c/CPCM(CHCl3) kcal mol−1 | M06-2X/6-311G**/CPCM(CHCl3) kcal mol−1 | |
|---|---|---|
| Reactant (1) | 0 | 0 |
| Transition state (TS) | 38.28 | 39.48 |
| Product (6) | 11.02 | 10.77 |
The energy barrier of the reaction in chloroform, calculated with the M06-2X/6-311G**/CPCM(CHCl3) hybrid functionals, is ΔE≠TS = 39.48 kcal mol−1, while the reaction energy is ΔEP–R = 10.77 kcal mol−1, respectively. Quite unexpectedly, the calculation results obtained using the B97-3c functional are very similar.
Experimantal teil
The NMR spectra were achieved using a Avance 400 ultra-shield spectrometer (Bruker, Karlsruhe, Germany). The mass spectra were recorded by electrospray ionisation (ESI) coupled with a TOF analyser (Bruker, Karlsruhe, Germany). The reaction was completed using a Monowave 50 reactor (Anton Paar, Graz, Austria). Reagents and solvents were obtained from Sigma-Aldrich, Fluka, and Merck and were used without purification.The semi-empirical GFN2−xTB method with the use of the S. Grimme program16 was also used for the calculations. The solvent effects in this program were accounted for by applying the implicit ALPB solvent model. MTD simulations were made using the CREST 2.12 program.
The geometry of compound 2c and its conformers was optimized with the r2scan-3c functional, using the ORCA 5.0.2 program suite. Solvent effects were considered within the SCRF theory using the polarized continuum model (CPCM) approach to model the interaction with the solvent.
Example procedure for the synthesis of amino-coumarin derivatives of resorcin[4]arene (2)
100 mg of the aminomethylene derivative of resorcin[4]arene (1) and 4 equivalents of 4-hydroxycoumarin were weighed into the reaction vessel, and then dissolved in 6 ml of chloroform. The reaction was carried out in closed reaction vessels, in which the pressure of chloroform at a temperature of 160 °C was 15 bar. After heating was stopped, the chloroform was evaporated on the rotary evaporator. Ethyl acetate was added to the residue to precipitate. Depending on the obtained derivative, the products were crystallized from ethyl acetate or from chloroform/methanol. The yields of the reactions were in the range of 58–78%.Conclusions
A novel, single-step, rapid and efficient synthesis of aminocoumarin derivatives of resorc[4]arene (2) is presented. The reaction proceeds by the transfer of an amino group from aminomethylene derivatives of resorc[4]arene to the reaction product (2). The aminocoumarin derivatives of resorc[4]arene formed are conformationally labile and the population of conformers depends on the type and polarity of the solvent. For the derivative 2c, a search for the lowest energy conformers was performed using MTD sampling with the CREST and GFN2−xTB programmes. The calculations resulted in a final five conformations for which geometry optimisation was performed at the higher level of DFT theory by r2scan-3c in acetone. The population of conformers at 298 K calculated from the energies obtained agrees reasonably well with the conformers observed on the NMR spectrum of derivative 2c in acetone. The mechanism of the reaction of the morpholine derivative of resorc[4]arene with 4-hydroxycoumarin based on multiscale (ONIOM) calculations of the reaction energy profile using the NEB method is discussed. The transition state structure of this reaction was determined and the activation energy (ΔE≠TS = 39.48 kcal mol−1) and reaction energy (ΔEP–R = 10.77 kcal mol−1) – in chloroform were calculated using the M06-2X functional and the 6-311G** basis set.Conflicts of interest
The author declare no conflict of interest.Acknowledgements
Calculations was funded by Wrocław Centre for Networking and Supercomputing, Poland: grant number 27538605.References
- M.-Y. Yu, J. Yang, T.-T. Guo and J.-F. Ma, Efficient Catalytic Oxidative Desulfurization toward Thioether and Sulfur Mustard Stimulant by Polyoxomolybdate-Resorcin[4]arene-Based Metal-Organic Materials, Inorg. Chem., 2020, 59(7), 4985 CrossRef CAS PubMed.
- C. M. A. Gangemi, A. Pappalardo and G. T. Sfrazzetto, Applications of supramolecular capsules derived from resorcin[4]arenes, calix[n]arenes and metallo-ligands: from biology to catalysis, RSC Adv., 2015, 5, 51919 RSC.
- D. T. Payne, W. A. Webre, Y. Matsushita, N. Zhu, Z. Futera, J. Labuta, W. Jevasuwan, N. Fukata, J. S. Fossey, F. D'Souza, K. Ariga, W. Schmitt and J. P. Hill, Multimodal switching of a redox-active macrocycle, Nat. Commun., 2019, 1007, 1 Search PubMed.
- A. Szafraniec and W. Iwanek, Synthesis of a coumarin derivative of resorcin[4]arene with solvent-controlled chirality, RSC Adv., 2020, 10, 12747 RSC.
- Y. Matsushita and T. Matsui, Synthesis of aminomethylated calix[4]resorcinarenes, Tetrahedron Lett., 1993, 46, 7433 CrossRef.
- N. K. Beyeh and K. Rissanen, N-Alkyl Ammonium Resorcinarene Salts: A Versatile Family of Calixarene-Related Host Molecules, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer, 2016, pp. 255–284 Search PubMed.
- A. Szafraniec and W. Iwanek, Intramolecular Hydrogen Bond Driven Conformational Selectivity of Coumarin Derivatives of Resorcin[4] arene, Int. J. Mol. Sci., 2020, 21, 6160 CrossRef CAS.
- W. Iwanek, R. Froehlich, P. Schwab and V. Schurig, The synthesis and crystallographic structures of novel boraoxazino–oxazolidine derivatives of resorcarene, Chem. Commun., 2002, 2516 RSC.
- A. Karmen A, F. Wroblewski F and J. S. Ladue, Transaminase activity in human blood, J. Clin. Investig., 1955, 34(1), 126 CrossRef PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 11, e01493 Search PubMed.
- G. Henkelman and H. Jonsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113(22), 9978 CrossRef CAS.
- A. Olyaei and M. Sadeghpour, Recent advances in the synthesis and synthetic applications of Betti base (aminoalkylnaphthol) and bis-Betti base derivatives, Rsc. Adv., 2019, 9, 18467–18497 RSC.
- K. Stefańska, A. Szafraniec, M. P. Szymański, M. Wierzbicki, A. Szumna and W. Iwanek, Chiral chromane[4]arenes synthesised by cycloaddition reactions of o-quinomethine resorcin[4]arenes, New J. Chem., 2019, 43, 2687 RSC.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, r2SCAN-3c: A “Swiss army knife” composite electronic-structure method, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- Semiempirical extended tight-binding program package xtb version 6.50, https://github.com/grimme-lab/xtb, accessed 15 May 2022 Search PubMed.
- S. Grimme, Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 2019, 15(5), 2847 CrossRef CAS PubMed.
- S. Ehlert, M. Stahn and S. Grimme, Robust and Efficient Implicit Solvation Model for Fast Semiempirical Methods, J. Chem. Theory Comput., 2021, 17(7), 4250 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22, 7169 RSC.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Best Practice DFT Protocols for Basic Molecular Computational Chemistry, Angew. Chem., Int. Ed., 2022, e202205735 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS.
- T. Vreven and K. Morokuma, Chapter 3 Hybrid Methods: ONIOM(QM:MM) and QM/MM, Annual Reports in Computational Chemistry, 2006, vol. 2, p. 35 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04610e |
| This journal is © The Royal Society of Chemistry 2022 |