
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel thiazole derivatives incorporating phenyl sulphonyl moiety as potent BRAFV600E kinase inhibitors targeting melanoma†
Afaf Y. Khormia,
Thoraya. A. Farghalybc,
Abrar Bayazeedc,
Youssef O. Al-Ghamdid,
Hanan Gaber Abdulwahab *e and
Mohamed R. Shaabanb
*e and
Mohamed R. Shaabanb
aDepartment of Chemistry, Faculty of Science, King Khalid University, Abha, Saudi Arabia
bDepartment of Chemistry, Faculty of Science, Cairo University, Giza 12613, Egypt
cDepartment of Chemistry, Faculty of Applied Science, Umm Al-Qura University, Makkah Almukarramah, Saudi Arabia
dDepartment of Chemistry, College of Science Al-zulfi, Majmaah University, Al-Majmaah 11952, Saudi Arabia
eDepartment of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, Egypt. E-mail: hanangaber@azhar.edu.eg
First published on 27th September 2022
Abstract
Novel thiazole derivatives possessing phenyl sulfonyl moiety were designed and synthesized as B-RAFV600E kinase inhibitors based on the clinically-approved anticancer drug, dabrafenib. All target compounds showed significant inhibition of B-RAFV600E kinase enzyme at nanomolar levels. Compounds 7b and 13a revealed excellent B-RAFV600E inhibitory activity, superior to that of dabrafenib with IC50 values of 36.3 ± 1.9, 23.1 ± 1.2, and 47.2 ± 2.5 nM, respectively. Moreover, the title compounds were much more selective toward B-RAFV600E kinase than B-RAF wild type. In addition, the most potent compounds were further evaluated for their anticancer activity against B-RAFV600E-mutated and wild type melanoma cells. A positive correlation between the cytotoxic activity and selectivity for B-RAF V600E over B-RAF wild type was clearly observed for compounds 7b, 11c, 13a, and 17. All the screened compounds potently inhibited the growth of WM266.4 melanoma cells with IC50 values in the range from 1.24 to 17.1 μM relative to dabrafenib (IC50 = 16.5 ± 0.91 μM). Compounds 7b, 11a and 11c, 13a, and 17 were much more potent than dabrafenib against B-RAFV600E-mutated WM266.4 melanoma cells. Furthermore, compound 7b suppressed the phosphorylation of downstream ERK1/2 from WM266.4 cells. Also, the docking study revealed the proper orientation and well-fitting of the title compounds into the ATP binding site of B-RAFV600E kinase.
1. Introduction
Ras/Raf/Mek/ERK kinase cascade, also known as MAPK pathway, plays a pivotal role in inter and intracellular communication, which controls the fundamental cellular processes such as cell growth, survival, differentiation, and proliferation.1,2 This pathway is frequently activated or overexpressed in various disease conditions, particularly cancer.3,4RAF kinases are the key components of Ras/Raf/Mek/ERK pathway. Since their discovery in 1983, RAF kinases have been associated with cancer. Three isoforms (A-RAF, B-RAF, and C-RAF) are known for the RAF kinase family, showing different degrees of biochemical potencies (B-RAF > C-RAF > A-RAF).5 Being the most frequently mutated isoform in human cancers, B-RAF is the major activating kinase for the MEK/ERK pathway. Numerous activating mutations of B-RAF have been identified by several research groups. Accounting for more than 90% of B-RAF mutations in cancer, B-RAFV600E point mutation in which valine is substituted by glutamic acid in codon 600, is the most common, particularly in melanoma.6 The overexpression of B-RAFV600E kinase is associated with the proliferation, aggressiveness, and poor prognosis of malignant tumors. In melanoma, B-RAFV600E mutation accounts for 500 times activation relative to the wild-type B-RAF kinase.6 Therefore, B-RAFV600E kinase is considered as a research hotspot for the discovery of new anticancer agents, and great efforts have been devoted to the development of B-RAFV600E kinase inhibitors.7,8 As a consequence of this effort, clinically approved B-RAFV600E kinase inhibitors (Fig. 1) have been introduced into the market, resulting in a dramatic change in the treatment of B-RAFV600E-driven cancers, especially melanoma.9,10 Given that the B-RAF enzyme is crucial for normal cell processes, extensive efforts are currently being made by scientists to obtain potent and safe medications that selectively inhibit B-RAFV600E without interfering with the B-RAF wild-type.11–13
 | ||
| Fig. 1 Clinically-approved B-RAFV600E inhibitors. | ||
On the other hand, the thiazole nucleus has attracted significant attention in medicinal chemistry for the discovery and development of biologically active compounds, particularly anticancer agents.14–16 Several thiazole-containing compounds have been reported as potent B-RAFV600E kinase inhibitors (Fig. 2).8,17–23 Moreover, dabrafenib (Fig. 1), a thiazole derivative developed by GlaxoSmithKline as a potent and selective B-RAFV600E inhibitor, was approved by FDA in 2013 for the treatment of B-RAFV600E-driven tumors.19
 | ||
| Fig. 2 Reported thiazoles as B-RAFV600E inhibitors. | ||
In the light of these facts and in continuation of our effort toward the discovery of potent anticancer agents,24–36 herein, novel thiazole derivatives were designed and synthesized as potent B-RAFV600E kinase inhibitors with potential anticancer activities, based on the clinically-approved B-RAFV600E inhibitor, dabrafenib. The design of our target compounds is illustrated in Fig. 3. In this work, while conserving the thiazole core of dabrafenib, different moieties were incorporated into the thiazole nucleus, namely, arylazo 7a–c, arylhydrazono 11a–c, and aryl groups 13a, b, 15, 19. Motivated by the reported anticancer activity of phenyl sulphonyl derivatives,30 the 2,6-difluorophenylsulphonamide group of dabrafenib was also replaced by the phenylsulphonylmethyl (PhSO2CH2) moiety. In addition, the fluorophenyl moiety in dabrafenib was replaced by the arylidenehydrazine moiety in the target compounds. Moreover, the substituent effect (R1–R4) on B-RAFV600E kinase inhibitory activity was also explored. On surveying the literature, it was found that bis vemurafenib compounds, reported by Grasso et al.,37 possessed potent B-RAFV600E inhibitory activity in both cell-free and cell-based assays. Also, the anticancer and kinase inhibitory activity of bisthiazole compounds is well established.38,39 Inspired by these findings, bisthiazole compound 15 was also synthesized to study the effect of dimerization on B-RAFV600E inhibition.
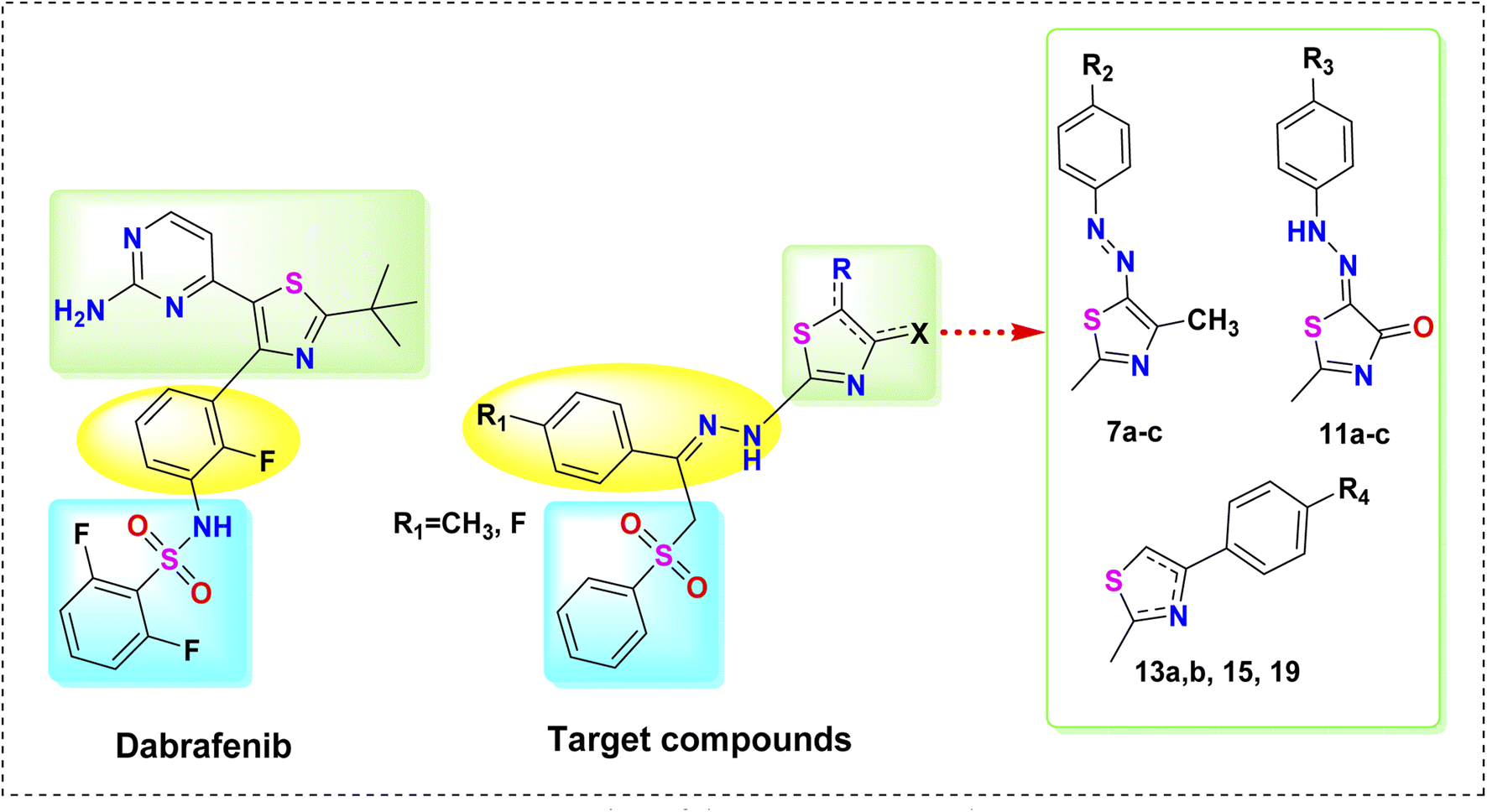 | ||
| Fig. 3 Design of the target compounds. | ||
In this work, all target compounds were screened in vitro for their ability to inhibit the kinase activity of B-RAFV600E. Furthermore, to evaluate the selectivity of the target compounds, the most active derivatives were tested in vitro against B-RAF wild type. The most potent compounds were also screened for their anticancer activity against B-RAFV600E-mutated and B-RAF wild type melanoma cells. Also, a cell-based assay was performed to measure the blocking effect of the title compounds on the phosphorylation of downstream ERK. Finally, a docking study of the most promising compounds was conducted to predict their binding modes within the active site of the B-RAFV600E kinase.
2. Experimental
2.1. Chemistry
The instruments utilized for recording the spectral data are illustrated in the ESI [see ESI† for more details]. The used hydrazonoyl chlorides were prepared by the same method cited in the literature reports.40,412.1.1.1. 2-Benzenesulfonyl-1-p-tolyl-ethanone thiosemicarbazone (3). Pale yellow solid (86% yield), mp 160–162 °C; IR (KBr) νmax 3417, 3248, 3163 (NH and NH2), 3055, 3001 (sp2 C–H), 2916 (sp3 C–H), 1612 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1496, 1458, 1427, 1303, 1141, 1080 cm−1; 1H NMR (DMSO-d6) δ 2.28 (s, 3H, CH3), 5.28 (s, 2H, CH2), 7.07 (d, J = 8.1 Hz, 2H, Ar–H), 7.55–7.85 (m, 5H, Ar–H), 7.86 (d, J = 8.1 Hz, 2H, Ar–H), 7.93 (s, 1H, NH), 8.38 (s, 1H, NH), 10.52 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 14.2, 51.8, 122.6, 127.9, 129.0, 129.2, 130.7, 134.1, 134.8, 135.3, 139.0, 179.0. MS m/z (%) 348 (M+ + 1, 0.36), 347 (M+, 1.3), 192 (9), 147 (7), 117 (100), 115 (40), 105 (17), 91 (35), 77 (60), 65 (21). Anal. calcd for C16H17N3O2S2 (347.46): C, 55.31; H, 4.93; N, 12.09. Found: C, 55.26; H, 4.81; N, 11.94%.
N), 1496, 1458, 1427, 1303, 1141, 1080 cm−1; 1H NMR (DMSO-d6) δ 2.28 (s, 3H, CH3), 5.28 (s, 2H, CH2), 7.07 (d, J = 8.1 Hz, 2H, Ar–H), 7.55–7.85 (m, 5H, Ar–H), 7.86 (d, J = 8.1 Hz, 2H, Ar–H), 7.93 (s, 1H, NH), 8.38 (s, 1H, NH), 10.52 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 14.2, 51.8, 122.6, 127.9, 129.0, 129.2, 130.7, 134.1, 134.8, 135.3, 139.0, 179.0. MS m/z (%) 348 (M+ + 1, 0.36), 347 (M+, 1.3), 192 (9), 147 (7), 117 (100), 115 (40), 105 (17), 91 (35), 77 (60), 65 (21). Anal. calcd for C16H17N3O2S2 (347.46): C, 55.31; H, 4.93; N, 12.09. Found: C, 55.26; H, 4.81; N, 11.94%.
2.1.1.2. 2-Benzenesulfonyl-1-(4-fluorophenyl)-ethanone thiosemicarbazone (17). White crystals (88% yield), mp 205–207 °C; IR (KBr) νmax 3387, 3317, 3263 (NH and NH2), 3062, (sp2 C–H), 2970, 2900 (sp3 C–H), 1604 (C
N), 1504, 1473, 1435, 1303, 1234, 1157, 1141, 1080 cm−1; 1H NMR (DMSO-d6) δ 5.33 (s, 2H, CH2), 7.05 (t, J = 9 Hz, 2H, Ar–H), 7.55–7.94 (m, 7H, Ar–H), 8.02 (s, 1H, NH), 8.41 (s, 1H, NH), 10.60 (s, 1H, NH); 13C NMR (DMSO-d6) δ 52.9 (CH2), 115.2, 115.4 (d, 2JCF, 20 Hz), 128.5, 129.7, 130.1, 130.2 (d, 3JCF, 8 Hz), 133.2, 134.7, 135.7, 139.7, 162.3, 164.0 (d, 1JCF, 246 Hz), 179.7 (CS). MS m/z (%) 353 (M+ + 2, 1.2), 352 (M+ + 1, 2.5), 351 (M+, 11), 210 (5), 196 (48), 151 (43), 135 (6), 121 (100), 109 (28), 101 (47), 95 (22), 77 (68). Anal. calcd for C15H14FN3O2S2 (351.42): C, 51.27; H, 4.02; N, 11.96. Found: C, 51.05; H, 3.94; N, 11.86%.
 | ||
| Scheme 1 Synthesis of the thiosemicarbazone–phenylsulfone derivative 3. | ||
 | ||
| Scheme 2 Reaction of thiosemicarbazone–phenylsulfone derivative 3 with hydrazonoyl chlorides 4a–c. | ||
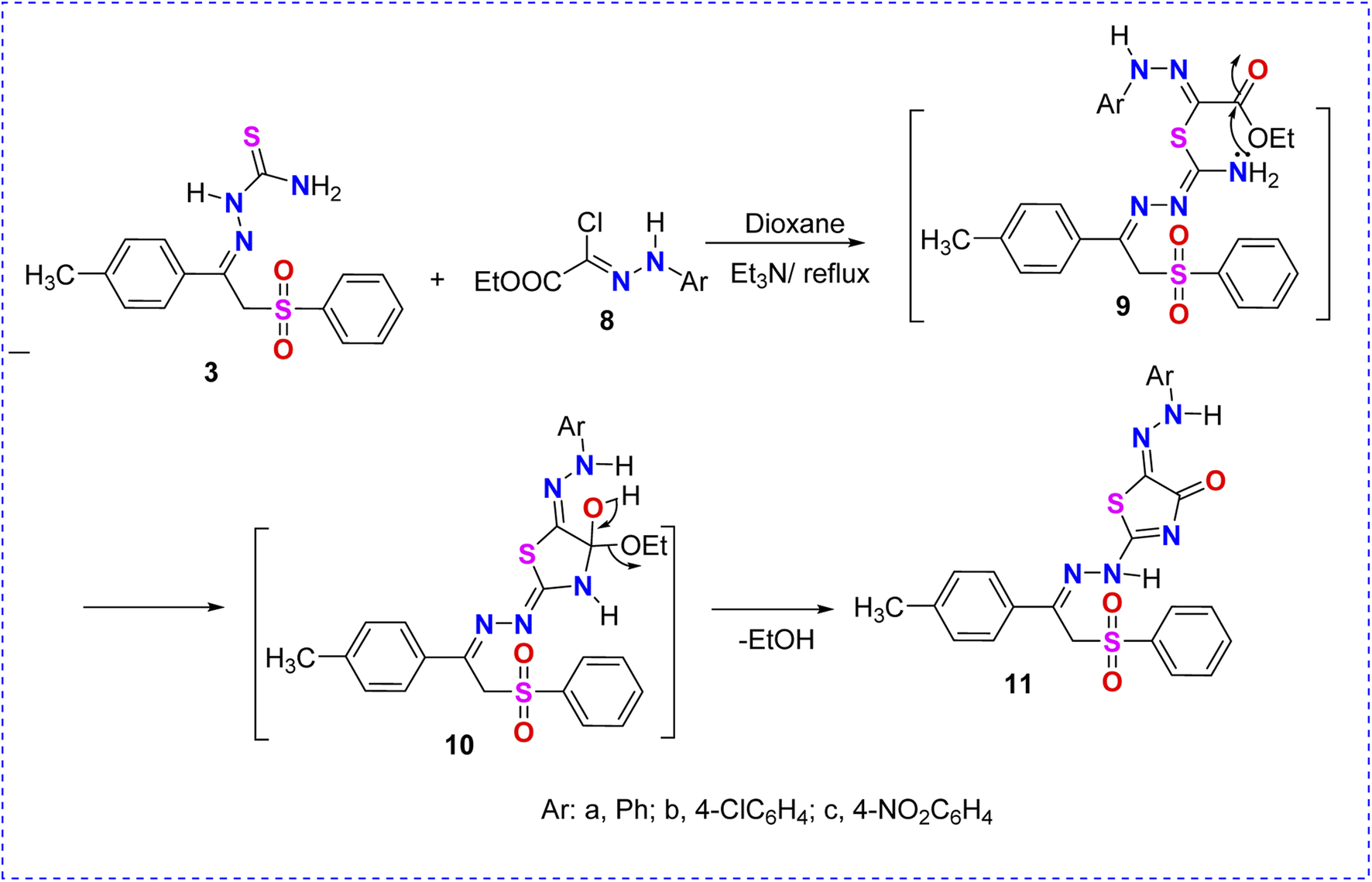 | ||
| Scheme 3 Reaction of thiosemicarbazone–phenylsulfone derivative 3 with hydrazonoyl chloride 8a–c. | ||
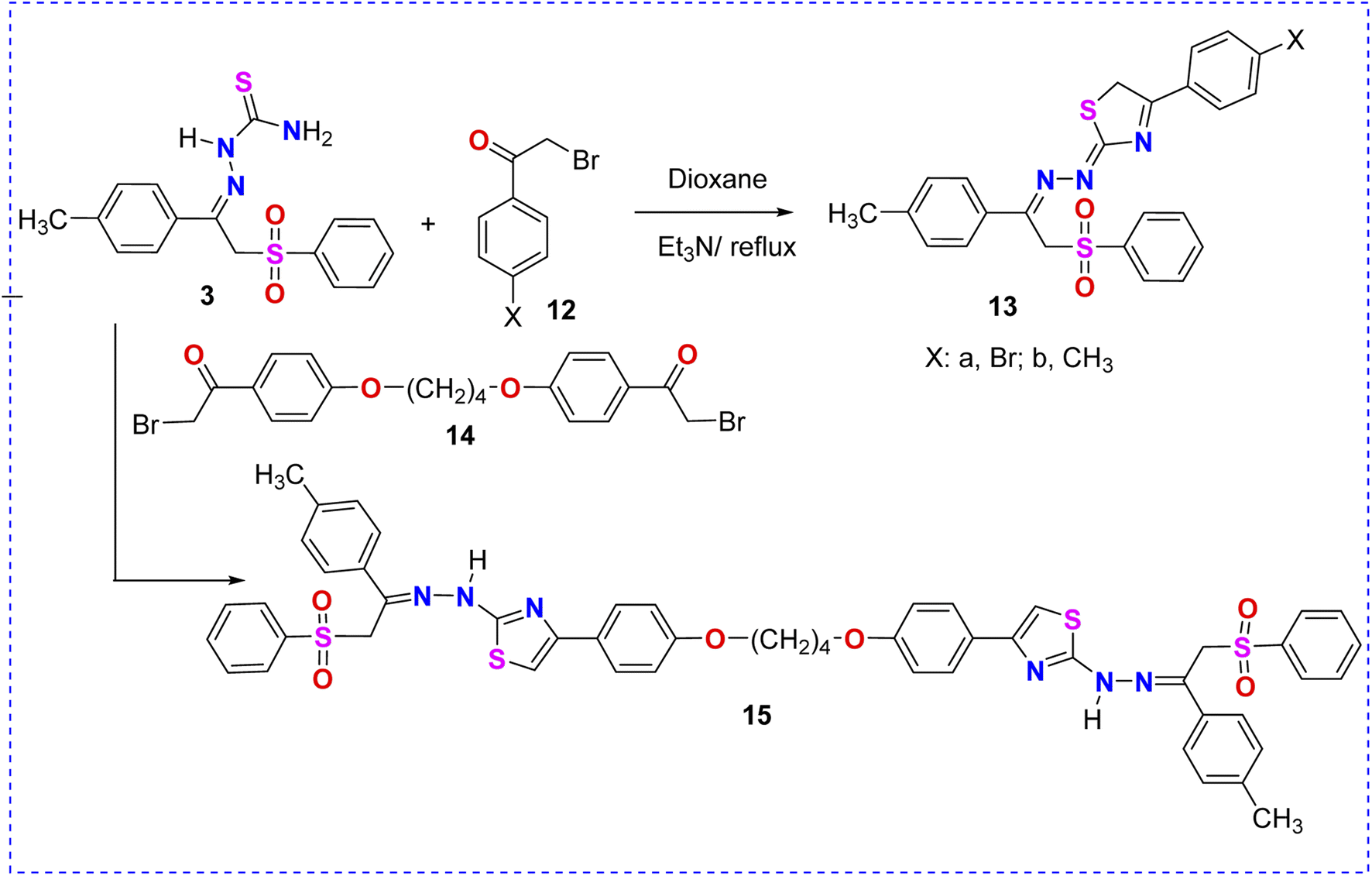 | ||
| Scheme 4 Reaction of thiosemicarbazone–phenylsulfone derivative 3 with phenacyl bromide derivatives 12a, 12b and 14. | ||
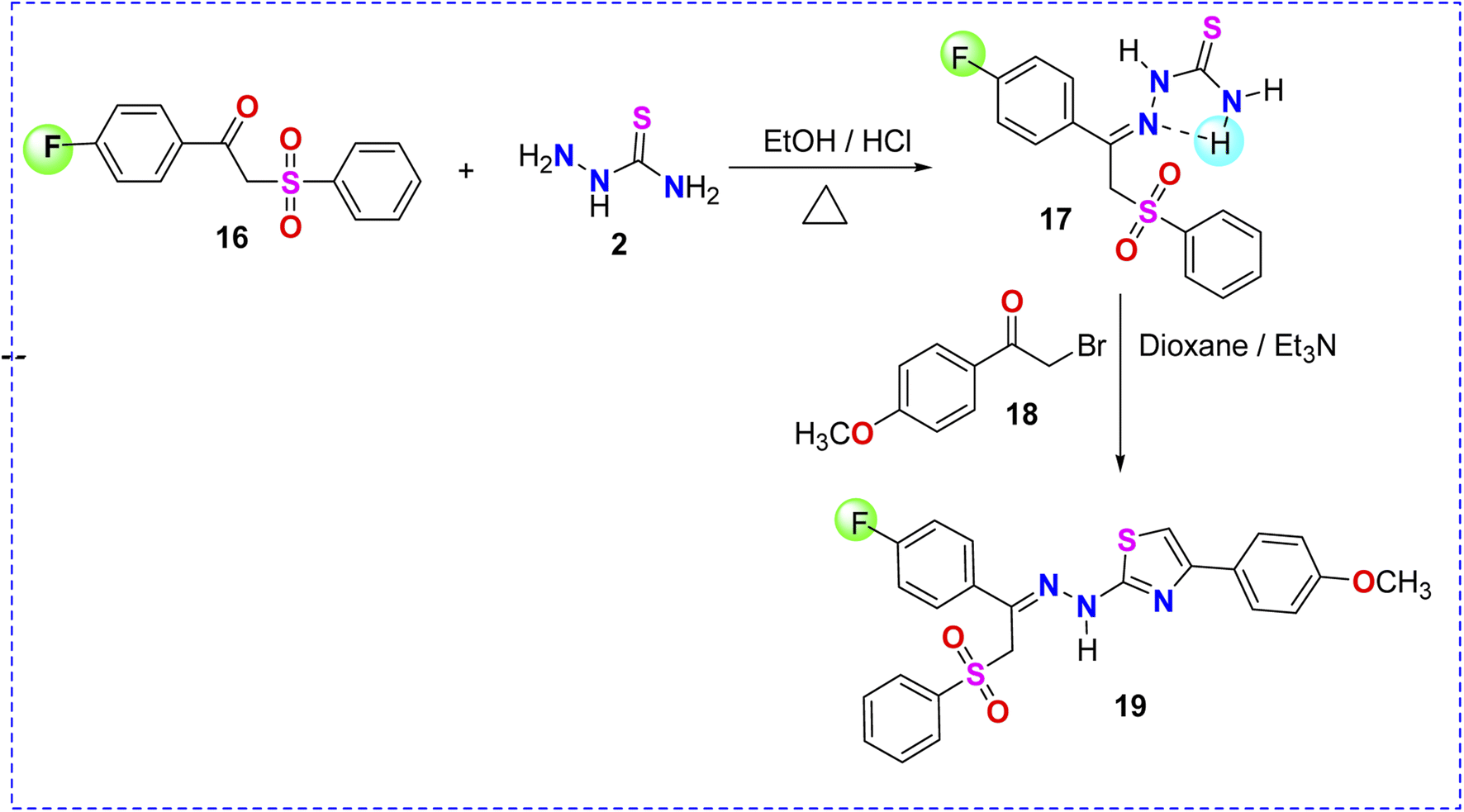 | ||
| Scheme 5 Synthesis of thiosemicarbazone–phenylsulfone derivative 17 and thiazole derivative 19. | ||
2.1.2.1. N-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-N′-(4-methyl-5-phenylazo-thiazol-2-yl)-hydrazine (7a). Dark red solid (82% yield), mp 173–174 °C; IR (KBr) νmax 3255 (NH), 3055, 3001 (sp2 C–H), 2924 (sp3 C–H), 1589 (C
N), 1535, 1489, 1411, 1373, 1311, 1242, 1157, 1072 cm−1; 1H NMR (DMSO-d6) δ 2.35 (s, 3H, CH3), 2.56 (s, 3H, CH3), 5.26 (s, 2H, CH2), 6.65–7.86 (m, 14H, Ar–H), 10.67 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 14.0, 32.1, 53.3, 120.4, 124.2, 127.9, 129.1, 129.5, 131.4, 131.5, 133.8, 134.6, 139.2, 141.9, 152.4, 152.5, 164.2, 166.7, 171.6. MS m/z (%) 490 (M+ + 1, 0.6), 489 (M+, 1.6), 347 (10), 231 (2.7), 143 (2), 117 (74), 105 (6), 92 (12), 91 (37), 77 (100). Anal. calcd for C25H23N5O2S2 (489.6): C, 61.3; H, 4.7; N, 14.3. Found: C, 61.2; H, 4.6; N, 14.2%.
2.1.2.2. N-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-N′-(4-methyl-5-m-tolylazo-thiazol-2-yl)-hydrazine (7b). Dark red solid (89% yield), mp 148–150 °C; IR (KBr) νmax 3425 (NH), 3032 (sp2 C–H), 2916 (sp3 C–H), 1589 (C
N), 1535, 1489, 1373, 1311, 1265, 1188, 1072 cm−1; 1H NMR (DMSO-d6) δ 2.27 (s, 3H, CH3), 2.34 (s, 3H, CH3), 2.54 (s, 3H, CH3), 5.25 (s, 2H, CH2), 6.78–7.86 (m, 13H, Ar–H), 10.64 (s, 1H, NH); MS m/z (%) 504 (M+ + 1, 1.3), 503 (M+, 4), 362 (12), 161 (2), 142 (11), 132 (10), 117 (64), 106 (31), 91 (100), 77 (89). Anal. calcd for C26H25N5O2S2 (503.6): C, 62.0; H, 5.0; N, 13.9. Found: C, 61.9; H, 4.9; N, 13.8%.
2.1.2.3. N-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-N′-(4-methyl-5-p-tolylazo-thiazol-2-yl)-hydrazine (7c). Red solid (90% yield), mp 180–182 °C; IR (KBr) νmax 3441 (NH), 3032 (sp2 C–H), 2978, 2924 (sp3 C–H), 1589 (C
N), 1535, 1481, 1411, 1381, 1311, 1265, 1188, 1072 cm−1; 1H NMR (DMSO-d6) δ 2.28 (s, 3H, CH3), 2.34 (s, 3H, CH3), 2.55 (s, 3H, CH3), 5.25 (s, 2H, CH2), 6.79–7.85 (m, 13H, Ar–H), 10.65 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 8.98 (CH3), 21.56 (CH3), 21.79 (CH3), 45.99, 112.33, 115.35, 116.02, 122.89, 128.40, 128.65, 129.54, 129.63, 130.18, 133.01, 134.28, 139.19, 139.81, 143.87, 155.07. MS m/z (%) 504 (M+ + 1, 1.9), 503 (M+, 5.5), 362 (18), 161 (3), 142 (8), 132 (8), 117 (65), 106 (28), 91 (100), 77 (88). Anal. calcd for C26H25N5O2S2 (503.6): C, 62.0; H, 5.0; N, 13.9. Found: C, 62.0; H, 5.1; N, 13.9%.
2.1.2.4. 2-[N′-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-hydrazino]-5-(phenyl-hydrazono)-thiazol-4-one (11a). Yellow solid (72% yield), mp 90–92 °C; IR (KBr) νmax 3425, 3263 (2NH), 3032 (sp2 C–H), 2985 (sp3 C–H), 1705 (C
O), 1612 (CN), 1543, 1496, 1311, 1226, 1157, 1072 cm−1; 1H NMR (DMSO-d6) δ 2.33 (s, 3H, CH3), 5.21 (s, 2H, CH2), 6.95–7.83 (m, 14H, Ar–H), 10.46 (s, 1H, NH), 12.40 (s, 1H, NH); MS m/z (%) 491 (M+, 10), 350 (8), 120 (14), 117 (91), 105 (13), 92 (39), 91 (55), 77 (100). Anal. calcd for C24H21N5O3S2 (491.6): C, 58.6; H, 4.3; N, 14.3. Found: C, 58.5; H, 4.3; N, 14.1%.
2.1.2.5. 2-[N′-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-hydrazino]-5-[(4-chloro-phenyl)-hydrazono]-thiazol-4-one (11b). Yellow solid (71% yield), mp 100–102 °C; IR (KBr) νmax 3417, 3248 (2NH), 2978, 2924 (sp3 C–H), 1705 (C
O), 1612 (CN), 1489, 1458, 1311, 1234, 1157, 1080 cm−1; 1H NMR (DMSO-d6) δ 2.34 (s, 3H, CH3), 5.28 (s, 2H, CH2), 7.06–7.88 (m, 13H, Ar–H), 10.56 (s, 1H, NH), 12.40 (s, 1H, NH); MS m/z (%) 528 (M+ + 2, 1), 527 (M+ + 1, 3), 526 (M+, 10), 142 (11), 129 (13), 117 (91), 115 (41), 111 (25), 91 (44), 83 (24), 77 (100). Anal. calcd for C24H20ClN5O3S2 (526.0): C, 54.8; H, 3.8; N, 13.3. Found: C, 54.7; H, 3.7; N, 13.2%.
2.1.2.6. 2-[N′-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-hydrazino]-5-[(4-nitro-phenyl)-hydrazono]-thiazol-4-one (11c). Orange solid (65% yield), mp 180–181 °C; IR (KBr) νmax 3417, 3248 (2NH), 3062 (sp2 C–H), 2993, 2931 (sp3 C–H), 1705 (C
O), 1612 (CN), 1496, 1450, 1327, 1311, 1234, 1149, 1111, 1072 cm−1; 1H NMR (DMSO-d6) δ 2.29 (s, 3H, CH3), 5.27 (s, 2H, CH2), 7.07–8.40 (m, 13H, Ar–H), 10.52 (s, 1H, NH), 11.18 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 21.3 (CH3), 46.1 (CH2), 124.5, 126.3, 127.8, 128.0, 128.2, 128.5, 128.5, 128.6, 129.1, 129.5, 129.7, 133.9, 134.6, 136.8, 139.8, 180.0. MS m/z (%) 536 (M+, 2.36), 423 (3), 306 (3), 130 (6), 117 (100), 105 (11), 91 (45), 84 (13), 77 (82). Anal. calcd for C24H20N6O5S2 (536.58): C, 53.7; H, 3.8; N, 15.7. Found: C, 53.6; H, 3.5; N, 15.5%.
2.1.2.7. N-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-N′-[4-(4-bromo-phenyl)-5H-thiazol-2-ylidene]-hydrazine (13a). Pale yellow solid (92% yield), mp 120–121 °C; IR (KBr) νmax 3032 (sp2 C–H), 2954, 2916 (sp3 C–H), 1604 (C
N), 1489, 1442, 1365, 1311, 1149, 1111, 1080 cm−1; 1H NMR (DMSO-d6) δ 2.31 (s, 3H, CH3), 5.16 (s, 2H, CH2), 5.18 (s, 2H, CH2), 7.13 (d, J = 7.8 Hz, 2H, Ar–H), 7.54–7.71 (m, 9H, Ar–H), 7.80 (d, J = 7.8 Hz, 2H, Ar–H); MS m/z (%) 527 (M+ + 1, 0.22), 385 (11), 124 (15), 117 (100), 91 (32), 77 (77). Anal. calcd for C24H20BrN3O2S2 (526.5): C, 54.8; H, 3.8; N, 8.0. Found: C, 54.7; H, 3.7; N, 7.8%.
2.1.2.8. N-(2-Benzenesulfonyl-1-p-tolyl-ethylidene)-N′-(4-p-tolyl-5H-thiazol-2-ylidene)-hydrazine (13b). Pale orange solid (90% yield), mp 178–180 °C; IR (KBr) νmax 3032 (sp2 C–H), 2954, 2916 (sp3 C–H), 1604 (C
N), 1504, 1442, 1365, 1311, 1149, 1118, 1080 cm−1; 1H NMR (DMSO-d6) δ 2.31 (s, 3H, CH3), 2.36 (s, 3H, CH3), 5.16 (s, 2H, CH2), 5.19 (s, 2H, CH2), 7.13 (d, J = 8 Hz, 2H, Ar–H), 7.15 (d, J = 8 Hz, 2H, Ar–H), 7.29 (d, J = 8 Hz, 2H, Ar–H), 7.52–7.65 (m, 5H, Ar–H), 7.73 (d, J = 8 Hz, 2H, Ar–H); MS m/z (%) 462 (M+ + 1, 0.7), 461 (M+, 2.2), 320 (14), 176 (12), 147 (8), 142 (17), 117 (70), 105 (3), 94 (16), 91 (27), 77 (100). Anal. calcd for C25H23N3O2S2 (461.6): C, 65.1; H, 5.0; N, 9.1. Found: C, 65.0; H, 5.0; N, 9.0%.
2.1.2.9. 1,1′-[1,4-Butanediylbis(oxy)]bis[4-(2-{[2-(benzenesulfonyl)-1-(p-tolyl)ethylidene]-hydrazino}thiazol-4-yl)benzene] (15). Buff solid (78% yield), mp 185–187 °C; IR (KBr) νmax 3420 (NH), 2939 (sp3 C–H), 1604 (C
N), 1550, 1512, 1303, 1242, 1157, 1049 cm−1; 1H NMR (DMSO-d6) δ 1.91 (br.s, 4H, 2CH2), 2.30 (s, 6H, CH3), 4.10 (br.s, 4H, 2CH2), 5.18 (s, 4H, CH2), 6.99 (d, J = 8 Hz, 4H, Ar–H), 7.13 (d, J = 8 Hz, 4H, Ar–H), 7.61 (s, 2H, 2thiazole-H), 7.54–7.89 (m, 18H, Ar–H), 11.60 (br.s, 2H, 2NH); MS m/z (%) 982 (M+ + 1, 0.52), 981 (M+, 1), 256 (2), 139 (5), 117 (21), 110 (21), 98 (22), 91 (20), 84 (42), 77 (31). Anal. calcd for C52H48N6O6S4 (981.2): C, 63.7; H, 4.9; N, 8.6. Found: C, 63.4; H, 4.9; N, 8.4%.
2.1.2.10. N-[2-Benzenesulfonyl-1-(4-fluorophenyl)-ethylidene]-N′-[4-(4-methoxyphenyl)thiazol-2-yl]-hydrazine (19). Green solid (86% yield), mp 145–147 °C; IR (KBr) νmax 3271 (NH), 3062 (sp2 C–H), 2939 (sp3 C–H), 1604 (C
N), 1558, 1512, 1442, 1311, 1242, 1157, 1080 cm−1; 1H NMR (DMSO-d6) δ 3.79 (s, 3H, OCH3), 5.23 (s, 2H, CH2), 6.98–7.88 (m, 14H, Ar–H and thiazole-H), 11.75 (s, 1H, NH); MS m/z (%) 483 (M+ + 2, 0.36), 481 (M+, 1), 339 (14), 310 (10), 142 (10), 121 (100), 101 (44), 95 (19), 77 (82). Anal. calcd for C24H20FN3O3S2 (481.6): C, 59.9; H, 4.2; N, 8.7. Found: C, 59.7; H, 4.1; N, 8.7%.
2.2. Biological activity
3. Results and discussion
3.1. Chemistry
The synthesis of the unreported thiosemicarbazone 3 was achieved by the acid-catalyzed condensation of the phenacyl phenyl sulfone derivative 1 with thiosemicarbazide 2 in acidic ethanol, as shown in Scheme 1. The synthesized phenyl sulfone–thiosemicarbazone derivative 3 was further analyzed using spectroscopic techniques. The standard NMR techniques were utilized for assigning all the protons as well as carbons in its structural frame (Scheme 1).The 1H NMR spectrum for phenyl sulfone–thiosemicarbazone 3 in dimethylsulfoxide-d6 is described in Fig. 4. The 1H NMR spectrum of thiosemicarbazone 3 revealed the presence of the open-chain thioamide-tautomer with the NH2 protons being partially affected due to the outspread conjugation as well as the presence of the phenylsulfonylmethylene moiety. It was observed that the two protons of the amino group (NH2) were magnetically non-equivalent, displaying two different chemical shift values at δ = 7.9 and 8.4 ppm. This observation could be explained in terms of intramolecular hydrogen bond that restricted or slowed down rotation around the N–C bond.46,47 It is worth mentioning that the chemical shift values observed for all the NH protons in the presence of phenylsulfonylmethylene group showed a downfield shift of approximately +0.40 to +1.3 ppm compared to the previously reported analog lacking phenylsulfonylmethylene group.48 Also, the 13C NMR spectrum of the carbothioamide derivative 3 showed a signal at δ = 179 ppm assigned to the CS group, in addition to another signal at δ = 139 ppm corresponding to CN.
 | ||
| Fig. 4 1H NMR spectrum and intramolecular hydrogen-bonding of compound 3. | ||
Thiosemicarbazone 3 was reacted with hydrazonoyl halides 4a–c as cyclization reagents in dioxane/Et3N to afford the corresponding substituted phenylsulfone–thiazoles 7a–c pendant to the arylazo substituents (Scheme 2). The IR spectra of derivatives 7a–c revealed the demise of the NH2 absorption bands of the starting thiosemicarbazone as well as the carbonyl (CO) group of the reacted hydrazonoyl chlorides. Compared to the 1H NMR spectrum of the starting phenylsulfone–thiosemicarbazone 3, thiazole derivative 7a (taken as an example) revealed the disappearance of the magnetically-nonequivalent protons of NH2. Also, the 1H NMR spectrum of arylazothiazole derivative 7a displayed remarkable four singlet signals at δ = 2.38, 2.59, 5.28, and 10.86 corresponding to two CH3, CH2, and NH protons, respectively, in addition to fourteen aromatic protons in the δ range of 7.27–7.87 ppm (Scheme 2 and Fig. 5).
 | ||
| Fig. 5 Comparison of the downfield region of the 1H NMR spectra of the reactant thiosemicarbazone 3 and its product 7a. | ||
In a similar manner, the reactivity of thiosemicarbazone 3 toward C-1-(ethoxycarbonyl)-N-4-arylhydrazonoyl chlorides 8a–c was investigated. Thus, the reaction of (3) with halogenated reagents 8a–c in dioxane afforded the corresponding arylazothiazolone derivatives 11a–c, as shown in Scheme 3. The suggested structures of compounds 11a–c were confirmed based on the data extracted from their spectral analyses (Scheme 3).
The reaction of 2-(2-(phenylsulfonyl)-1-(p-tolyl)ethylidene)hydrazine-1-carbothioamide (3) with α-bromocarbonyl derivatives 12a and 12b in dioxane/Et3N furnished the corresponding phenylsulfone–thiazoles 13a and 13b, as illustrated in Scheme 4. The 1H NMR spectrum of derivative 13a showed three singlet signals at δ 2.31, 5.16, and 5.18 ppm assigned to the methyl and two methylene protons, respectively. Moreover, multiplet signals of the thirteen aromatic protons appeared in the region of δ 7.13–7.80 ppm. Molecular weights (m/z) of phenylsulfone–thiazole derivatives 13a and 13b, extracted from their mass spectra, were identical with the calculated values (Scheme 4).
The treatment of 2-(2-(phenylsulfonyl)-1-(p-tolyl)ethylidene)hydrazine-1-carbothioamide (3) with the interesting bis-α-bromocarbonyl derivative 14 in dioxane/Et3N afforded the corresponding bis-phenylsulfone–thiazole derivative 15, as depicted in Scheme 4. The 1H NMR spectrum of 15 revealed two singlet signals at δ 2.30 and 5.18 ppm assigned to the methyl and methylene protons, respectively. The protons of the four methylene (CH2) groups of the aliphatic ether spacer were confirmed at δ = 1.90 and 4.10 ppm. In addition, the aromatic protons as well as the thiazole-5H signals were detected in the region of δ 6.99–7.89 and 7.61 ppm, respectively.
Prompted by the role of fluorine atom in enhancing the biological activity of previously reported fluorinated compounds,49,50 p-fluorophenacyl sulfone 16 was reacted with thiosemicarbazide 2 in ethanolic solution containing HCl to obtain the corresponding thiosemicarbazone derivative 17 (Scheme 5). The reaction of the latter with p-methoxyphenacyl bromide 18 afforded the corresponding fluorinated thiazole product 19, as depicted in Scheme 5. Based on the spectroscopic data (experimental part), the structure of compound 19 was confirmed to be the N-[2-Benzenesulfonyl-1-(4-fluorophenyl)-ethylidene]-N′-[4-(4-methoxyphenyl)thiazol-2-yl]-hydrazine 19 (Scheme 5).
3.2. Biological activity
As presented in Table 1 and Fig. 6, all the tested compounds effectively inhibited the kinase activity of the B-RAFV600E enzyme with IC50 values at the nanomolar level in the range from 23.1 to 205 nM, which is equated to the reference drug dabrafenib (IC50 = 47.2 ± 2.5 nM). Among the screened compounds, derivatives 7b and 13a revealed excellent B-RAFV600E inhibitory activity, superior to that of dabrafenib with IC50 values of 36.3 ± 1.9, 23.1 ± 1.2, and 47.2 ± 2.5 nM, respectively. Furthermore, as expected, the bisthiazole derivative 15 showed potent B-RAFV600E inhibition (IC50 = 51.9 ± 2.7 nM), nearly equipotent to that of dabrafenib. In addition, 70% of the activity of dabrafenib toward B-rafV600E was observed for compound 17 (IC50 = 68.2 ± 3.5 nM). Meanwhile, compounds 3, 7c, 11a and 11c, and 19 displayed almost half the potency of dabrafenib with the IC50 values ranging from 84.9 ± 4.4 to 116.3 ± 8 nM. In this investigation, derivatives 7a, 11b, and 13b were the least potent against B-rafV600E kinase, exhibiting IC50 values of about 200 nM.
| Compound no. | B-RAF (V600E) (nM) | B-RAF (wild type) (nM) | WM266.4a (μM) | Sk-mel-23b (μM) |
|---|---|---|---|---|
| a B-RAFV600E-mutated WM266.4 melanoma cells.b B-RAF wild type Sk-mel-23 melanoma cells.c Not determined. | ||||
| 3 | 115.3 ± 6 | __c | __ | __ |
| 7a | 198.6 ± 10 | __ | __ | __ |
| 7b | 36.3 ± 1.9 | 123.5 ± 6.6 | 3.18 ± 0.17 | 7.87 ± 0.40 |
| 7c | 108.4 ± 5.6 | __ | __ | __ |
| 11a | 84.9 ± 4.4 | 361.1 ± 19.4 | 6.18 ± 0.34 | 0.43 ± 0.02 |
| 11b | 185.2 ± 9.6 | __ | __ | __ |
| 11c | 95.4 ± 5 | 229 ± 12.3 | 1.53 ± 0.08 | 22.3 ± 1.14 |
| 13a | 23.1 ± 1.2 | 97.9 ± 5.2 | 4.52 ± 0.25 | 50.2 ± 2.56 |
| 13b | 205.6 ± 11 | __ | __ | __ |
| 15 | 51.9 ± 2.7 | 251.2 ± 13.5 | 17.1 ± 0.94 | 17.6 ± 0.89 |
| 17 | 68.2 ± 3.5 | 153 ± 8.2 | 1.24 ± 0.07 | 12.4 ± 0.63 |
| 19 | 116.3 ± 8 | __ | __ | __ |
| Dabrafenib | 47.2 ± 2.5 | 231.2 ± 12.4 | 16.5 ± 0.91 | 9.03 ± 0.46 |
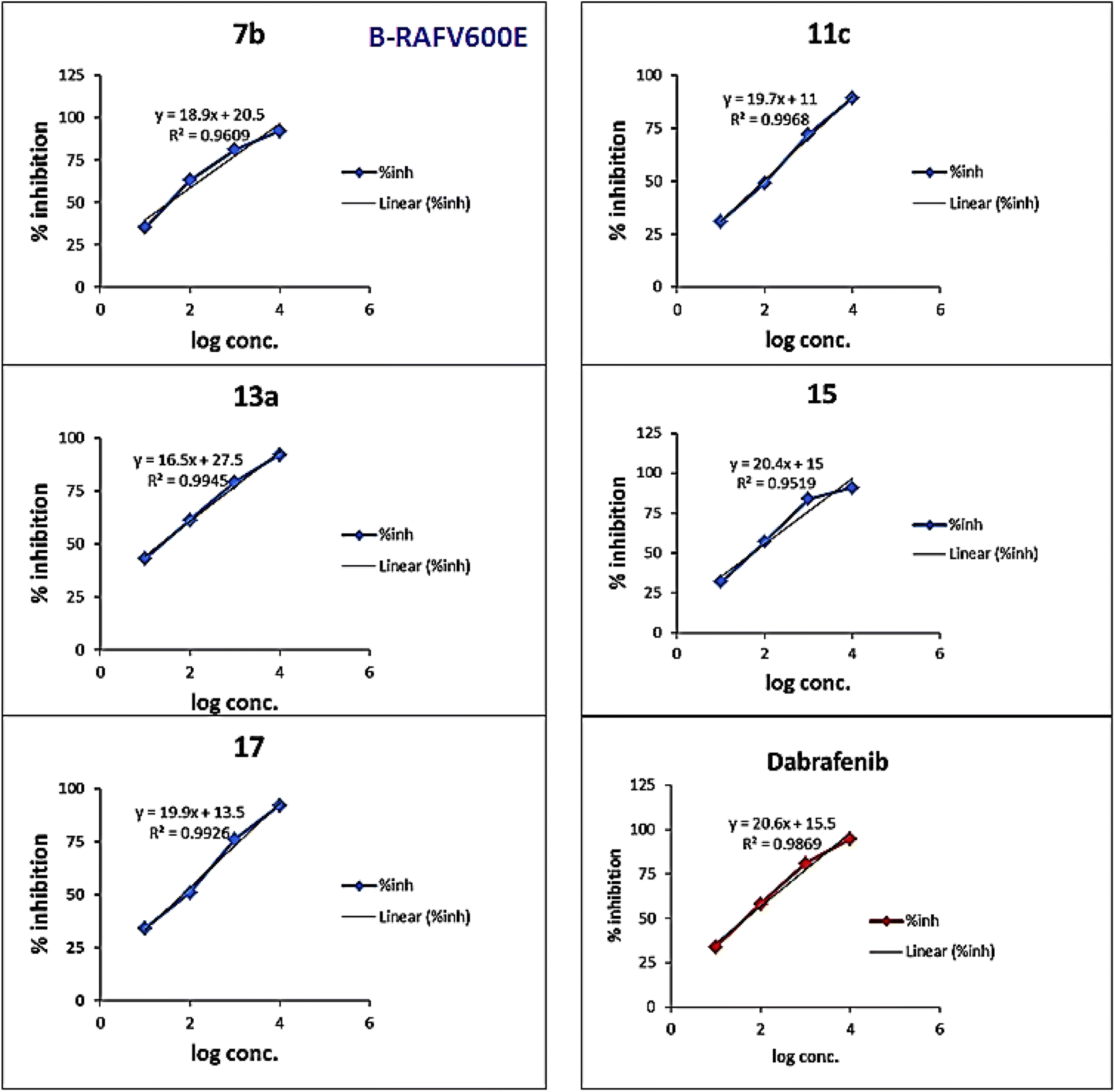 | ||
| Fig. 6 In vitro B-RAFV600E assay curves of compounds 7b, 11c, 13a, 15, 17, and dabrafenib. | ||
A brief investigation of the structure activity relationships revealed that, in general terms, arylazo thiazoles 7a–c showed higher B-RAFV600E kinase inhibitory activity than aryl hydrazono thiazole derivatives 11a–c. In addition, the tolyl derivatives 7b and 7c were much more potent than their unsubstituted phenyl analogue 7a (IC50 = 198.6 ± 10 nM). Besides, exceeding the activity of dabrafenib, the 3-tolyl analogue 7b (IC50 = 36.3 ± 1.9 nM) was 3-fold more active than its corresponding 4-tolyl derivative 7c (IC50 = 108.4 ± 5.6 nM). On the other hand, the unsubstituted phenyl 11a and 4-nitrophenyl 11c thiazolone derivatives possessed equipotent activity against B-RAFV600E kinase that was 2-fold higher than that elicited by their 4-chlorophenyl analogue 11b.
Regarding compounds 13a and 13b, bearing the aryl moiety at the 4-position of thiazole nucleus, interestingly, the 4-bromophenyl analogue 13a (IC50 = 23.1 ± 1.2 nM) was the most potent among the tested compounds, displaying double the activity of dabrafenib. Conversely, the replacement of bromine atom in 13a by a methyl group in derivative 13b resulted in a dramatic decrease in the activity with compound 13b (IC50 = 205.6 ± 11 nM), being the least potent among the screened compounds.
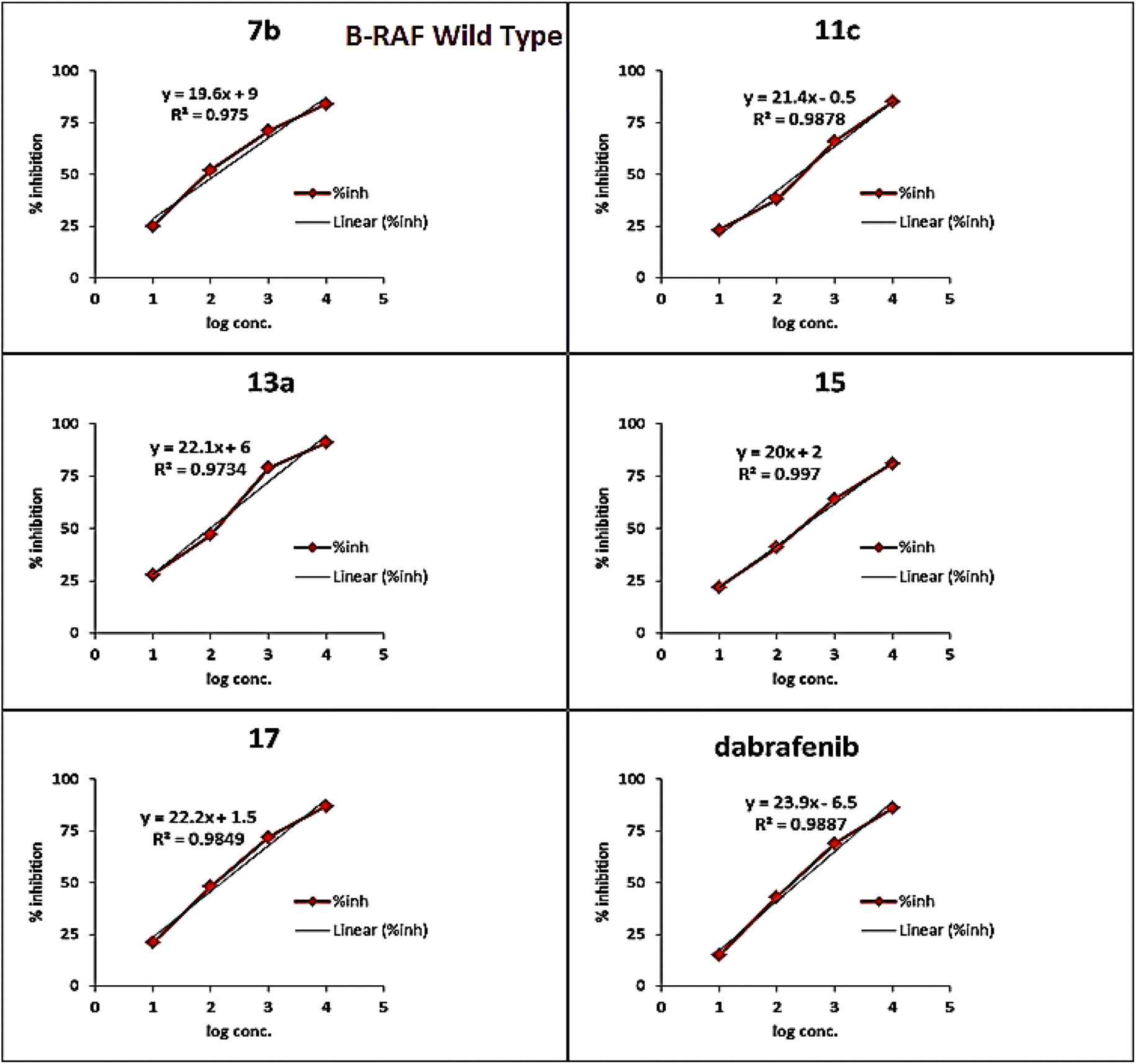 | ||
| Fig. 7 In vitro B-RAF wild type assay curves of compounds 7b, 11c, 13a, 15, 17, and dabrafenib. | ||
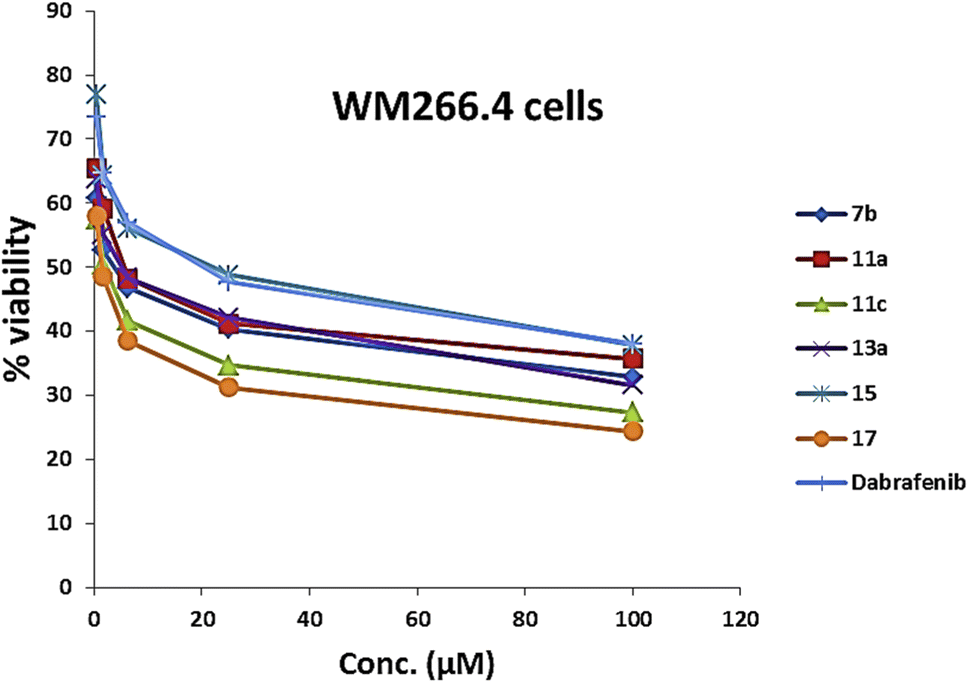 | ||
| Fig. 8 Cytotoxicity assay curve of compounds 7b, 11a and 11c, 13a, 15, 17, and dabrafenib in B-RAFV600E-mutated WM266.4 melanoma cells. | ||
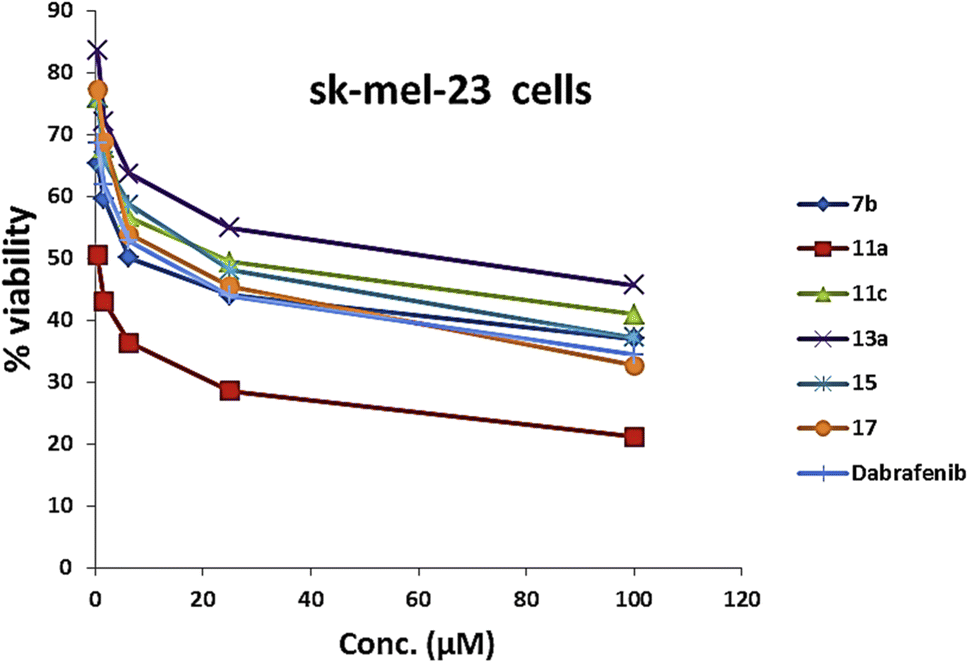 | ||
| Fig. 9 Cytotoxicity assay curve of compounds 7b, 11a and 11c, 13a, 15, 17, and dabrafenib in B-RAF wild type Sk-mel-23 melanoma cells. | ||
 | ||
| Fig. 10 The effect of compound 7b on the phosphorylation of ERK in the WM266.4 cell line. | ||
3.3. Docking study
To explain the potent B-RAFV600E kinase inhibitory activity elicited by the title compounds on a molecular basis, a docking study of the most promising compounds in this work was performed using MOE (2014.0901). A high-resolution B-RAFV600E kinase enzyme co-crystallized with dabrafenib was retrieved form protein data bank (PDB code: 4XV2) to be used in the docking study.Examining the docking results (Fig. 11–13) revealed good fitting and proper orientation of the docked compounds 7b, 11a and 11c, 13a, 15, and 17 into the ATP binding site of B-RAFV600E kinase enzyme with docking energy scores of −8.99, −8.74, −8.80, −7.88, −8.68, and −5.01 kcal mol−1, respectively, compared to the co-crystallized ligand dabrafenib (−9.95 kcal mol−1). Despite showing more potent inhibitory activity against the B-RAF V600E kinase, the docking score of compound 13a was relatively higher than that of compounds 7b, 11a, 11c, and 15. It is worth mentioning that inconsistencies between the inhibitory potency (experimental IC50 values) and calculated docking scores have been reported.51–53 Noticeably, all the analyzed compounds were involved in interactions with Cys532, a key residue in the hinge region of the enzyme active site. In addition, extensive hydrophobic interactions were observed between the docked compounds and Val471, Lys483, Leu505, Leu514, Phe583, and Phe595 residues, suggesting that the introduction of the hydrophobic groups (e.g., aryl and arylazo) into the thiazole core enhanced the affinity of the target compounds to the ATP binding site of the B-RAFV600E enzyme. The previous molecular docking results combined with the data of the enzyme assay confirm that the title phenylsulfone–thiazole derivatives are potential inhibitors of BRAFV600E kinase (see the ESI† for more details).
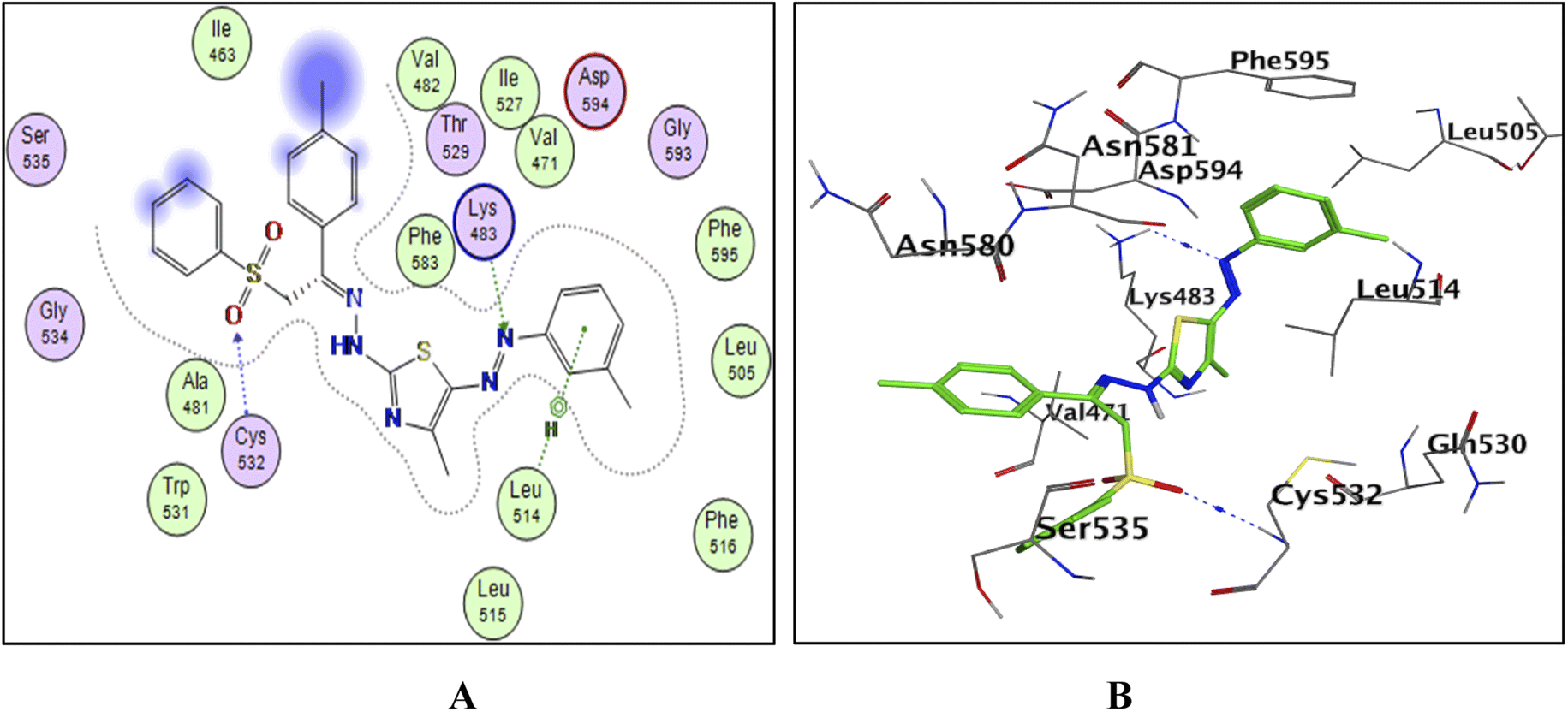 | ||
| Fig. 11 The docked model of compound 7b into the active site of the B-RAFV600E kinase; (A) 2D, (B) 3D. | ||
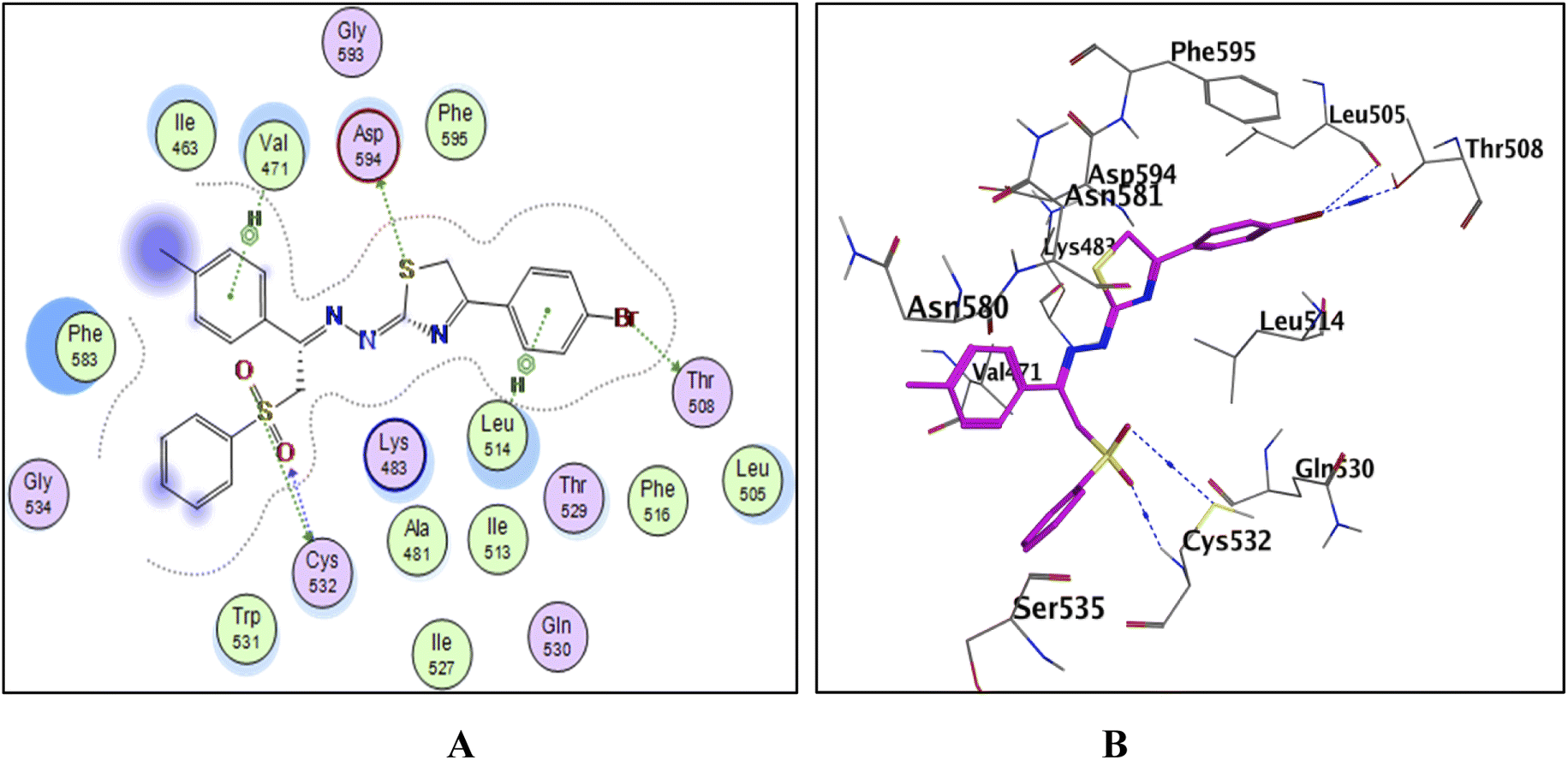 | ||
| Fig. 12 The docked model of compound 13a into the active site of the B-RAFV600E kinase; (A) 2D, (B) 3D. | ||
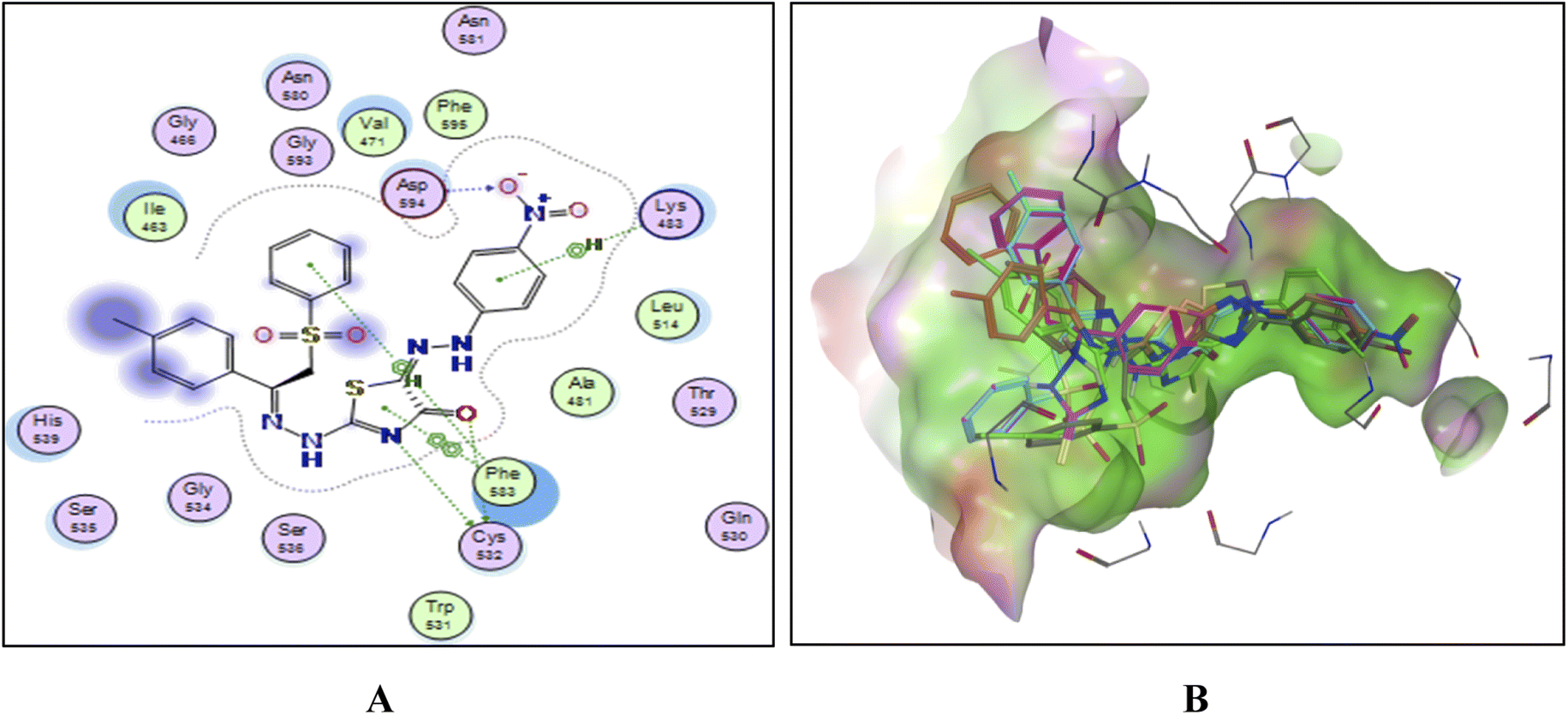 | ||
| Fig. 13 (A) The 2D docked model of compound 11c into the active site of the B-RAF V600E kinase, (B); overlay of the docked compounds into the active site of the B-RAF V600E kinase. | ||
4. Conclusion
To sum up, novel thiazole derivatives incorporating phenyl sulfonyl moiety were synthesized as the B-RAFV600E kinase inhibitors. All the target compounds inhibited the kinase catalytic activity of B-RAFV600E at nanomolar concentrations. Compounds 7b, 13a, and 15 revealed excellent B-RAFV600E inhibitory activities, superior or equipotent to that of the reference drug dabrafenib. Furthermore, the title compounds were remarkably more selective toward B-RAFV600E kinase than the B-RAF wild-type. In addition, the newly synthesized compounds potently inhibited the growth of B-RAFV600E-mutated melanoma cells, showing more selectivity toward B-RAF V600E-mutated over B-RAF WT melanoma cells. A docking study also displayed hydrogen bonding and hydrophobic interactions of the title compounds with key residues within the active site of the B-RAFV600E kinase.Conflicts of interest
There are no conflicts of interest to declare.References
- A. Plotnikov, E. Zehorai, S. Procaccia and R. Seger, The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation, Biochim. Biophys. Acta, 2011, 1813, 1619–1633 CrossRef CAS PubMed.
- Y. Sun, W.-Z. Liu, T. Liu, X. Feng, N. Yang and H.-F. Zhou, Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis, J. Recept. Signal Transduction Res., 2015, 35, 600–604 CrossRef CAS.
- T. Regad, Targeting RTK signaling pathways in cancer, Cancers, 2015, 7, 1758–1784 CrossRef CAS.
- U. Degirmenci, M. Wang and J. Hu, Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy, Cells, 2020, 9, 198 CrossRef CAS PubMed.
- T. Leicht, V. Balan, A. Kaplun, V. Singh-Gupta, L. Kaplun, M. Dobson and G. Tzivion, Raf kinases: function, regulation and role in human cancer, Biochim. Biophys. Acta, 2007, 1773, 1196–1212 CrossRef PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett and W. Bottomley, Mutations of the BRAF gene in human cancer, Nature, 2002, 417, 949–954 CrossRef CAS PubMed.
- P. Rusconi, E. Caiola and M. Broggini, RAS/RAF/MEK inhibitors in oncology, Curr. Med. Chem., 2012, 19, 1164–1176 CrossRef CAS.
- H. B. El-Nassan, Recent progress in the identification of BRAF inhibitors as anti-cancer agents, Eur. J. Med. Chem., 2014, 72, 170–205 CrossRef CAS.
- J. J. Luke, K. T. Flaherty, A. Ribas and G. V. Long, Targeted agents and immunotherapies: optimizing outcomes in melanoma, Nat. Rev. Clin. Oncol., 2017, 14, 463–482 CrossRef CAS PubMed.
- P. Koelblinger, O. Thuerigen and R. Dummer, Development of encorafenib for BRAF-mutated advanced melanoma, Curr. Opin. Oncol., 2018, 30, 125–133 CrossRef CAS PubMed.
- B. Agianian and E. Gavathiotis, Current insights of BRAF inhibitors in cancer: miniperspective, J. Med. Chem., 2018, 61, 5775–5793 CrossRef CAS PubMed.
- H.-L. Li, M.-M. Su, Y.-J. Xu, C. Xu, Y.-S. Yang and H.-L. Zhu, Design and biological evaluation of novel triaryl pyrazoline derivatives with dioxane moiety for selective BRAFV600E inhibition, Eur. J. Med. Chem., 2018, 155, 725–735 CrossRef CAS PubMed.
- R. Buck, S. Saleh, M. I. Uddin and H. C. Manning, Rapid, microwave-assisted organic synthesis of selective V600EBRAF inhibitors for preclinical cancer research, Tetrahedron Lett., 2012, 53, 4161–4165 CrossRef PubMed.
- A. Ayati, S. Emami, A. Asadipour, A. Shafiee and A. Foroumadi, Recent applications of 1, 3-thiazole core structure in the identification of new lead compounds and drug discovery, Eur. J. Med. Chem., 2015, 97, 699–718 CrossRef CAS PubMed.
- A. Ayati, S. Emami, S. Moghimi and A. Foroumadi, Thiazole in the targeted anticancer drug discovery, Future Med. Chem., 2019, 11, 1929–1952 CrossRef CAS PubMed.
- K. Mahmoud, T. A. Farghaly, H. G. Abdulwahab, N. T. Al-Qurashi and M. R. Shaaban, Novel 2-indolinone thiazole hybrids as sunitinib analogues: design, synthesis, and potent VEGFR-2 inhibition with potential anti-renal cancer activity, Eur. J. Med. Chem., 2020, 208, 112752 CrossRef PubMed.
- M.-Y. Zhao, Y. Yin, X.-W. Yu, C. B. Sangani, S.-F. Wang, A.-M. Lu, L.-F. Yang, P.-C. Lv, M.-G. Jiang and H.-L. Zhu, Synthesis, biological evaluation and 3D-QSAR study of novel 4, 5-dihydro-1H-pyrazole thiazole derivatives as BRAFV600E inhibitors, Bioorg. Med. Chem., 2015, 23, 46–54 CrossRef CAS.
- M. Ammar, M. S. Abdel-Maksoud and C.-H. Oh, Recent advances of RAF (rapidly accelerated fibrosarcoma) inhibitors as anti-cancer agents, Eur. J. Med. Chem., 2018, 158, 144–166 CrossRef PubMed.
- T. R. Rheault, J. C. Stellwagen, G. M. Adjabeng, K. R. Hornberger, K. G. Petrov, A. G. Waterson, S. H. Dickerson, R. A. Mook Jr, S. G. Laquerre and A. J. King, Discovery of dabrafenib: a selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors, ACS Med. Chem. Lett., 2013, 4, 358–362 CrossRef CAS PubMed.
- I. M. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, M. A. M. Gad-Elkareem, S. S. Abd El-Karim and E. S. Nossier, Synthesis and biological evaluation of new derivatives of thieno-thiazole and dihydrothiazolo-thiazole scaffolds integrated with a pyrazoline nucleus as anticancer and multi-targeting kinase inhibitors, RSC Adv., 2021, 12, 561–577, 10.1039/d1ra08055e.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Thiazole-containing compounds as therapeutic targets for cancer therapy, Eur. J. Med. Chem., 2020, 188, 112016, DOI:10.1016/j.ejmech.2019.112016.
- H. S. Anbar, M. I. El-Gamal, H. Tarazi, B. S. Lee, H. R. Jeon, D. Kwon and C. H. Oh, Imidazothiazole-based potent inhibitors of V600E-B-RAF kinase with promising anti-melanoma activity: biological and computational studies, J. Enzyme Inhib. Med. Chem., 2020, 35, 1712–1726, DOI:10.1080/14756366.2020.1819260.
- U. M. Ammar, M. S. Abdel-Maksoud, E. Ali, K. I. Mersal, K. Ho Yoo and C. H. Oh, Structural optimization of imidazothiazole derivatives affords a new promising series as B-Raf V600E inhibitors; synthesis, in vitro assay and in silico screening, Bioorg. Chem., 2020, 100, 103967, DOI:10.1016/j.bioorg.2020.103967.
- T. A. Farghaly and M. M. Abdalla, Synthesis, tautomerism, antimicrobial, anti-HCV, anti-SSPE, antioxidant and antitumor activities of arylazobenzosuberones, Bioorg. Med. Chem., 2009, 17, 8012–8019 CrossRef CAS PubMed.
- T. A. Farghaly, H. G. Abdulwahab, H. Y. Medrasi, M. A. Al-Sheikh, D. F. Katowah and A. Alsaedi, Novel 6,7,8-trihydrobenzo[6',7']cyclohepta[2',1'-e]pyrazolo[2,3-a]pyrimidine derivatives as Topo IIα inhibitors with potential cytotoxic activity, Bioorg. Chem., 2022, 128, 106043, DOI:10.1016/j.bioorg.2022.106043.
- A. A. Gaber, M. Sobhy, A. Turky, H. G. Abdulwahab, A. A. Al-Karmalawy, M. A. Elhendawy, M. M. Radwan, E. B. Elkaeed, I. M. Ibrahim, H. Elzahabi and I. H. Eissa, Discovery of new 1H-pyrazolo[3,4-d]pyrimidine derivatives as anticancer agents targeting EGFRWT and EGFRT790M, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2283–2303, DOI:10.1080/14756366.2022.2112575.
- R. E. Mansour, H. G. Abdulwahab and H. El-Sehrawi, Novel mannich bases derived from 2-substituted benzimidazole and (thio) hydantoin moieties as potent histone deacetylase 6 (hdac6) inhibitors, Azhar International Journal of Pharmaceutical and Medical Sciences, 2021, 1(3), 66–74, DOI:10.21608/aijpms.2021.78337.1077.
- S. A. Al-Hussain, T. A. Farghaly, M. E. Zaki, H. G. Abdulwahab, N. T. Al-Qurashi and Z. A. Muhammad, Discovery of novel indolyl-1, 2, 4-triazole hybrids as potent vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors with potential anti-renal cancer activity, Bioorg. Chem., 2020, 105, 104330 CrossRef CAS PubMed.
- Z. A. Muhammad, M. A. Radwan, T. A. Farghaly, H. M. Gaber and M. M. Elaasser, Synthesis and antitumor activity of novel [1, 2, 4, 5]-tetrazepino [6, 7-b] indole derivatives: marine natural product Hyrtioreticuline C and D analogues, Mini-Rev. Med. Chem., 2019, 19, 79–86 CrossRef CAS PubMed.
- A. M. Gouda, H. A. El-Ghamry, T. M. Bawazeer, T. A. Farghaly, A. N. Abdalla and A. Aslam, Antitumor activity of pyrrolizines and their Cu (II) complexes: Design, synthesis and cytotoxic screening with potential apoptosis-inducing activity, Eur. J. Med. Chem., 2018, 145, 350–359 CrossRef CAS PubMed.
- A. M. Al-Soliemy, T. A. Farghaly, E. M. Abbas, M. R. Shaaban and M. E. Zayed, Synthesis of thiazolyl-N-phenylmorpholine derivatives and their biological activities, Med. Chem., 2021, 17, 790–805 CrossRef CAS PubMed.
- A. M. A. Alnaja, T. A. Farghaly, H. S. El-Zahabi and M. R. Shaaban, Cytotoxicity, docking study of new fluorinated fused pyrimidine scaffold: Thermal and microwave irradiation synthesis, Med. Chem., 2021, 17, 501–518 CrossRef CAS PubMed.
- I. Althagafi, N. M. El-Metwaly, M. G. Elghalban, T. A. Farghaly and A. M. Khedr, Synthesis of Pyrazolone Derivatives and Their Nanometer Ag (I) Complexes and Physicochemical, DNA Binding, Antitumor, and Theoretical Implementations, Bioorg. Chem., 2018, 2018, 1–15 Search PubMed.
- D. H. Dawood, E. M. Abbas, T. A. Farghaly, M. M. Ali and M. F. Ibrahim, ZnO nanoparticles catalyst in the synthesis of bioactive fused pyrimidines as anti-breast cancer agents targeting VEGFR-2, Med. Chem., 2019, 15, 277–286 CrossRef CAS PubMed.
- T. Farghaly, I. Abbas, W. Hassan, M. Lotfy, N. Al-Qurashi and T. B. Hadda, Structure Determination and Quantum Chemical Analysis of 1, 3-Dipolar Cycloaddition of Nitrile Imines and New Dipolarophiles and POM Analyses of the Products as Potential Breast Cancer Inhibitors, Russ. J. Org. Chem., 2020, 56, 1258–1271 CrossRef CAS.
- M. R. Shaaban, T. A. Farghaly and A. M. Alsaedi, Synthesis, Antimicrobial and Anticancer Evaluations of Novel Thiazoles Incorporated Diphenyl Sulfone Moiety, Polycyclic Aromat. Compd., 2020, 1–17 Search PubMed.
- M. Grasso, M. A. Estrada, C. Ventocilla, M. Samanta, J. Maksimoska, J. Villanueva, J. D. Winkler and R. Marmorstein, Chemically Linked Vemurafenib Inhibitors Promote an Inactive BRAFV600E Conformation, ACS Chem. Biol., 2016, 11(10), 2876–2888, DOI:10.1021/acschembio.6b00529.
- X.-H. Gu, X.-Z. Wan and B. Jiang, Syntheses and biological activities of bis(3-indolyl)thiazoles, analogues of marine bis(indole)alkaloid nortopsentins, Bioorg. Med. Chem. Lett., 1999, 9, 569–572, DOI:10.1016/s0960-894x(99)00037-2.
- I. T. Radwan, A. H. M. Elwahy, A. F. Darweesh, M. Sharaky, N. Bagato, H. F. Khater and M. E. Salem, Design, synthesis, docking study, and anticancer evaluation of novel bis-thiazole derivatives linked to benzofuran or benzothiazole moieties as PI3k inhibitors and apoptosis inducers, J. Mol. Struct., 2022, 1265, 133454 CrossRef CAS.
- M. A. N. Mosselhi, M. A. Abdallah, Y. F. Mohamed and A. S. Shawali, Synthesis and Tautomeric Structure of 7-Arylhydrazono-7H-[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazines, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 487–498 CrossRef CAS.
- N. F. Eweiss and A. O. Osman, Synthesis of Heterocycles. Part II. New Routes to Acetylthiadiazolines and Alkylazothiazoles, J. Heterocycl. Chem., 1980, 17, 1713–1717 CrossRef CAS.
- M. A. Omar, G. S. Masaret, E. M. Abbas, M. M. Abdel-Aziz, M. F. Harras and T. A. Farghaly, Novel anti-tubercular and antibacterial based benzosuberone-thiazole moieties: Synthesis, molecular docking analysis, DNA gyrase supercoiling and ATPase activity, Bioorg. Chem., 2020, 104, 104316 CrossRef CAS.
- E. R. Cantwell-Dorris, J. J. O'Leary and O. M. Sheils, BRAFV600E: implications for carcinogenesis and molecular therapy, Mol. Cancer Ther., 2011, 10, 385–394 CrossRef CAS PubMed.
- N. M. Obaid, K. Bedard and W.-Y. Huang, Strategies for overcoming resistance in tumours harboring BRAF mutations, Int. J. Mol. Sci., 2017, 18, 585 CrossRef PubMed.
- T. Mosdam, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxic assay, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- M. M. Alsharekh, I. I. Althagafi, M. R. Shaaban and T. A. Farghaly, Microwave-assisted and thermal synthesis of nanosized thiazolyl-phenothiazine derivatives and their biological activities, Res. Chem. Intermed., 2019, 45, 127–154 CrossRef CAS.
- I. I. Althagafi, A. S. Abouzied, T. A. Farghaly, N. T. Al-Qurashi, M. Y. Alfaifi, M. R. Shaaban and M. R. Abdel Aziz, Novel Nano-sized bis-indoline Derivatives as Antitumor Agents, J. Heterocycl. Chem., 2019, 56, 391–399 CrossRef CAS.
- B. Gastaca, H. R. Sánchez, F. Menestrina, M. Caputo, M. de las Mercedes Schiavoni and J. J. P. Furlong, Thiosemicarbazones Synthesized from Acetophenones: Tautomerism, Spectrometric Data, Reactivity and Theoretical Calculations, Int. J. Anal. Mass Spectrom. Chromatogr., 2019, 7, 19–34 CrossRef CAS.
- D. Clemenceau, J. Cousseau, V. Martin, H. Molines, C. Wakselman, R. Mornet, F. Nogue and M. Laloue, Synthesis and cytokinin activity of two fluoro derivatives of N 6-isopentenyladenine, J. Agric. Food Chem., 1996, 44, 320–323 CrossRef CAS.
- O. V. El’kin, A. N. Bushuev, I. V. Tolstobrov, S. V. Fomin, E. S. Shirokova, A. V. Sazanov, V. A. Kozvonin and D. A. Kozulin, Organofluorine compounds in artificial blood circulation systems, Fascinating Fluoropolym. Their Appl., 2020, 401–424 Search PubMed.
- S.-Y. Huang and X. Zou, Advances and Challenges in Protein-Ligand Docking, Int. J. Mol. Sci., 2010, 11(8), 3016–3034, DOI:10.3390/ijms11083016.
- Y. C. Chen, Beware of docking!, Trends Pharmacol. Sci., 2015 Feb, 36(2), 78–95, DOI:10.1016/j.tips.2014.12.001.
- C. Mikra, G. Rossos, S. K. Hadjikakou and N. Kourkoumelis, Molecular Docking and Structure Activity Relationship Studies of NSAIDs. What do they Reveal about IC50?, Lett. Drug Des. Discovery, 2017, 14(8), 949–958, DOI:10.2174/1570180814666161207143231.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03624j |
| This journal is © The Royal Society of Chemistry 2022 |