
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBF3·Et2O-Mediated annulation of α-keto acids with aliphatic ketones for the synthesis of γ-hydroxy-butenolides and γ-alkylidene-butenolides†
Zhenfeng Cheng,
Qingyun Gu,
Yushan Xie,
Yanan Zhang and
Xiaobao Zeng *
*
School of Pharmacy, Nantong University, Nantong 226001, P. R. China. E-mail: zengxb@ntu.edu.cn
First published on 25th August 2022
Abstract
Annulation reaction of α-keto acids with cyclic or acyclic aliphatic ketones is reported herein to divergently access γ-hydroxy-butenolides and γ-alkylidene-butenolides depending on the amount of BF3·Et2O. This protocol features good functional tolerance and ease of operation, to open a route to access butenolides via an annulation and dehydration process.
Introduction
Butenolides are privileged structures widely existing in bioactive terpene natural products. Most of them are naturally-occurring structures and display various bio-activities.1 They are also widely used in traditional Chinese medicines to relieve cough and invigorate blood circulation.2 For example, bioactive eudesmanolides sesquiterpene atractylenolide I, II and III isolated from dried roots are used in diverse disorders like chronic asthenia, anorexia (Fig. 1).3 Styxlactone is a new lactone isolated from Myrmekioderma Styx.4 To this end, ever since the initial report of morpholine promoted cyclization of cyclohexanone with 2-chloro-2-methoxyethyl acetate by Baiocchi in 1979,5 the construction of butenolides has been of interest in the synthetic community.6 For example, Brønsted or Lewis acids promoted annulation reaction α-keto acids with linear aliphatic ketones for the preparation of butenolides have been separately developed by several groups,7 while the preparation of highly substituted fused butenolides from α-keto acids and aliphatic ketones has not been reported yet. Thus, developing novel approaches to divergently access γ-hydroxy-butenolides and γ-alkylidene-butenolides with commercially available starting materials and ease of operation is still desirable.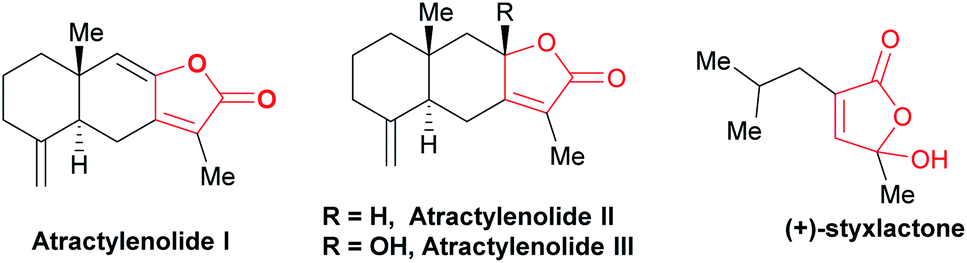 | ||
| Fig. 1 Representative bioactive eremophilanolides. | ||
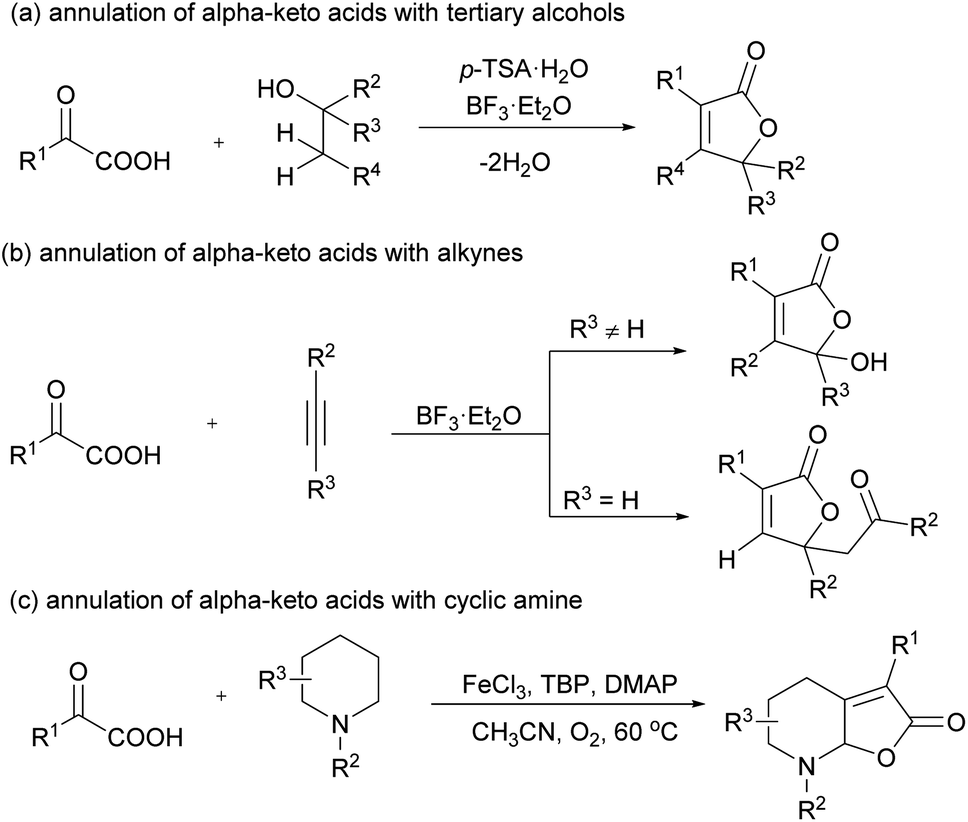
On the other hand, α-keto acids have long been serving as acylating agents to deliver a carbonyl group via metal or oxidant-promoted decarboxylative coupling reactions.8 However, the annulation reactions of α-keto acids are far less studied.9 In 2015, Zhu and coworkers10 innovatively reported a synergistic acid promoted annulation of α-keto acid with alkenes generated from tertiary alcohols to afford highly substituted butenolides (Scheme 1a). Following the preliminary report, the annulation of α-keto acid with internal11 or terminal alkynes12 to afford γ-hydroxybutenolides or multiply substituted butenolides was also reported by the same group (Scheme 1b). The strategy was then extended to the annulation reaction of α-keto acids with functionalized alkynes by Wang13 and Cui group.14 In 2018, Fan and coworkers15 reported an elegant synthesis of furan-2(5H)-one fused N,O-containing bicyclic compounds via the annulation reaction of N-aryl substituted saturated cyclic amines with α-keto acids (Scheme 1c). Despite the substantial progress achieved, the annulation with unsaturated bonds is far less than well developed.
 | ||
| Scheme 1 Previous reports in the annulation of α-keto acid. | ||
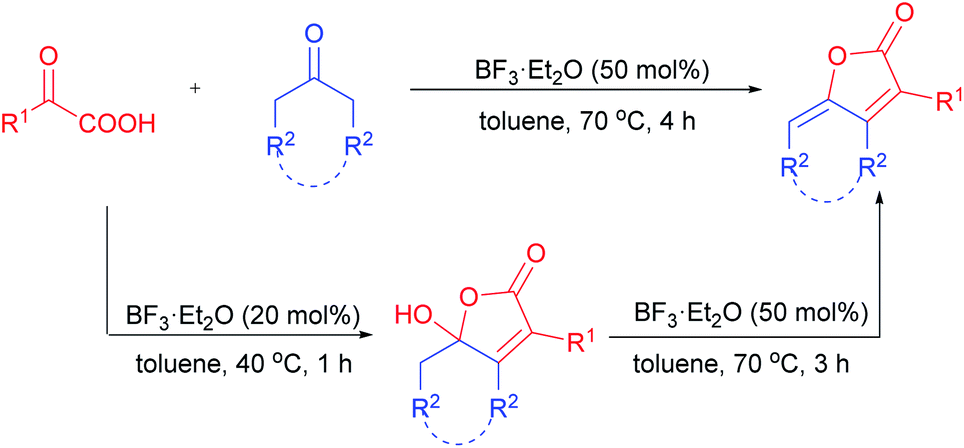
Meanwhile, ketones are among the most readily available building blocks in organic synthesis. Great efforts have been devoted to the direct α-functionalization16 and annulation reaction17 of ketones by in situ generated enols. However, to the best of our knowledge, the simple acid-promoted annulation reaction of α-keto acid with cyclic or linear aliphatic ketones has not been explored yet. As part of our continuous effort in radical decarboxylation and annulation reaction of α-keto acid,18 we reported herein a novel avenue to divergently access γ-hydroxy-butenolides and γ-alkylidene-butenolides via BF3·Et2O-catalyzed annulation of α-keto acids with aliphatic ketones (Scheme 2). Water was the only by-product generated in the transformation, thus making it a simple and efficient process for the construction of various butenolide.
 | ||
| Scheme 2 BF3·Et2O-catalyzed annulation of α-keto acids and aliphatic ketones. | ||
Results and discussion
We commenced our study with 1a (0.5 mmol) and 2a (2.0 equiv.) as model substrates in the presence of BF3·Et2O (1.0 equiv.) as catalyst in toluene at 40 °C, 3aa was obtained in 70% yield (Table 1, entry 1). Inspired by the preliminary results, the effects of different catalysts were first investigated. The utilization of other Lewis acids such as FeCl3 and Bi(OTf)3 led to the decrease in the yield (entries 2 and 3), while product 3aa could not be detected with p-TSA as catalyst (entry 4). A survey of the solvents revealed that toluene was the optimal solvent, inferior yield was obtained with MeCN, DCE, or fluorobenzene (entries 1 and 5–7). By fine-tuning the amount of BF3·Et2O, 3aa was obtained in 86% yield (entries 8 and 9). The yield of 3aa was decreased to 78% by lowering the ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a from 1:2 to 1:1.5 (entry 10). Finally, the optimal reaction conditions were deemed as entry 8 to provide product 3aa in 86% yield after a comprehensive optimization of conditions.
a
:2a from 1:2 to 1:1.5 (entry 10). Finally, the optimal reaction conditions were deemed as entry 8 to provide product 3aa in 86% yield after a comprehensive optimization of conditions.
a
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (3aa) (%) |
| a Standard conditions: 1a (0.5 mmol), 2a (2.0 equiv), BF3·Et2O (1 equiv.), toluene (2.0 mL), 40 °C, 1 h.b Isolated yields. | ||
| 1 | None | 70 |
| 2 | FeCl3 instead of BF3·Et2O | 45 |
| 3 | Bi(OTf)3 instead of BF3·Et2O | 53 |
| 4 | p-TSA instead of BF3·Et2O | n.d. |
| 5 | MeCN instead of toluene | 35 |
| 6 | DCE instead of toluene | 46 |
| 7 | Fluorobenzene instead of toluene | 69 |
| 8 | BF3·Et2O (0.2 equiv.) | 86 |
| 9 | BF3·Et2O (0.1 equiv.) | 76 |
| 10 | 1a:2a = 1:1.5 |
78 |
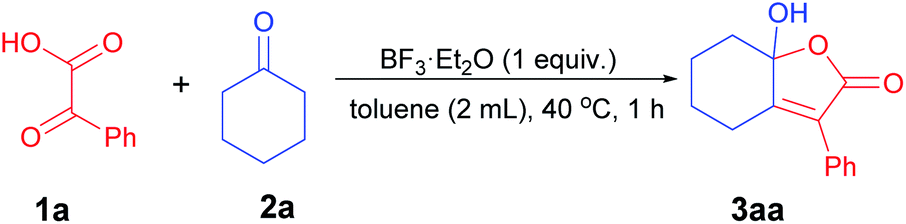
Interestingly, it was found that 4aa could be obtained in 82% yield at elevated reaction temperature by prolonging the reaction time to 4 h during the optimization process for the preparation of 3aa (Table 2, entry 1). Then we proceeded to evaluate the effects of temperature on the reaction. The reaction was significantly affected by the temperature. A dramatic erosion in the yield of γ-alkylidene-butenolides (4aa) was observed when the reaction was performed at 40 °C (entry 2). Continuous increasing the temperature to 90 °C didn't improve the yield (entry 3). Then the loading of catalyst was further evaluated. A comparable yield was obtained when the loading of BF3·Et2O was decreased to 0.5 equiv. (entry 4), however, a sharp decrease in yield was observed by lowering the loading of BF3·Et2O to 0.2 equiv., indicating that BF3·Et2O is crucial for the dehydration process of 3aa to form desired product 4aa (entry 5). Furthermore, switching to other Lewis acid provided 4aa in decreasing yield (entries 6 and 7). The replacement of toluene with other solvents failed to improve the yield (entries 8 and 9). Additionally, 4aa was afforded in a similar yield when increasing the ratio of 1a:2a to 1:3 (entry 10). Then the optimal condition for 4aa was identified as entry 4.
a
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (4aa) (%) |
| a Standard conditions: 1a (0.5 mmol), 2a (2.0 equiv.), BF3·Et2O (1 equiv.), toluene (2.0 mL), 70 °C, 4 h.b Isolated yields. | ||
| 1 | None | 82 |
| 2 | 40 °C instead of 70 °C | 13 |
| 3 | 90 °C instead of 70 °C | 62 |
| 4 | BF3·Et2O (0.5 equiv.) | 82 |
| 5 | BF3·Et2O (0.2 equiv.) | 63 |
| 6 | Sc(OTf)3 instead of BF3·Et2O | 61 |
| 7 | AlCl3 instead of BF3·Et2O | 55 |
| 8 | Xylene instead of toluene | 73 |
| 9 | Chlorobenzene instead of toluene | 71 |
| 10 | 1a:2a = 1:3 |
79 |
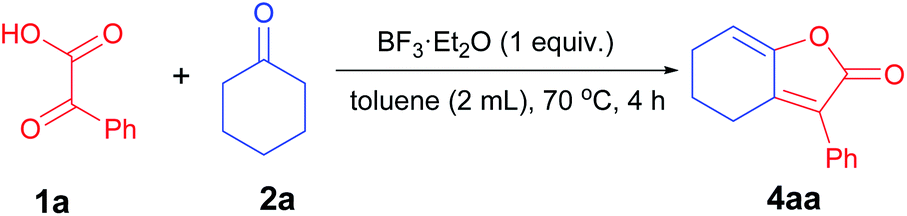
With the optimal conditions in hand, we set out to investigate the scope for the synthesis of γ-hydroxy-butenolides (3aa–p) (Table 3). The reaction was slightly affected by the steric hindrance of cyclohexanone, to furnish 3aa and 3ac with relatively higher yields than 3ab with ortho-methyl substituents. Tetrahydro-4-pyrone was a suitable substrate for the reaction, to afford 3ad with moderate yield. Additionally, seven-membered cycloheptanone proceeded well to give 3ae in 79% yield. The reaction also tolerated 1,3-cyclohexanedione, delivering the corresponding product 3af and 3ag in synthetically useful yields. The structure of 3af was unambiguously assigned by the single crystal X-ray crystallography. It was worth noting that acetone was well accommodated to afford the corresponding 3ah in 59% yield. Encouraged by the broad generality of aliphatic ketones, we then examined the scope of α-keto acids. The reaction proceeded well when naphthenic α-keto acid was employed as the substrate to afford product 3ai in 85% yield. Both electron-donating and withdrawing aryl substituents of α-keto acids were tolerated to give 3aj–n in moderate to good yields. Moreover, thienyl α-keto acid reacted smoothly to deliver 3ao in 72% yield. Notably, pyruvic acid was also compatible in this reaction, giving 3ap in 71% yield. Disappointedly, the free hydroxyl group (2h) and alkenyl moiety (2i) could not be tolerated in the reaction.
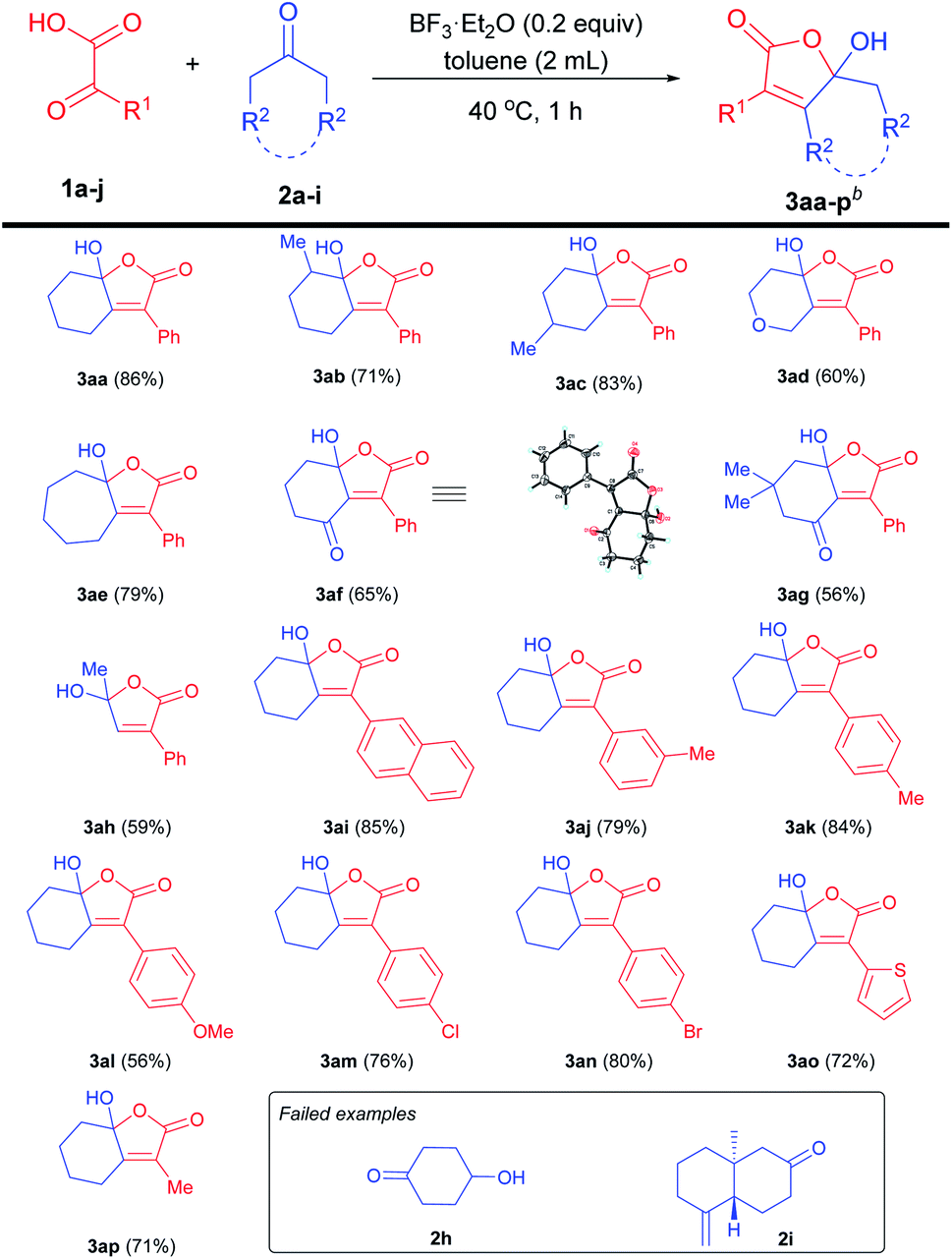
Subsequently, the substrate scope of γ-alkylidene-butenolides 4aa–i was examined (Table 4). The reaction worked well with aryl α-keto acids bearing methyl and chloride substituents to give 4ab–d in good yields, indicating that the electronic properties of the phenyl ring have no obvious influence on the reaction efficiency. Significantly, α-keto acid with thienyl group was a viable substrate for the reaction to furnish 4ae in 61% yield. In addition, both 1,3-cyclohexandione and five-membered cyclopentanone were suitable substrates to afford 4af and 4ag in moderate yields. It was worth mentioning that acyclic ketones such as acetone and 3-pentanone were well tolerated in the reaction to give rise to the corresponding 4ah and 4ai in a relatively lower yield of 45% and 39%, respectively.
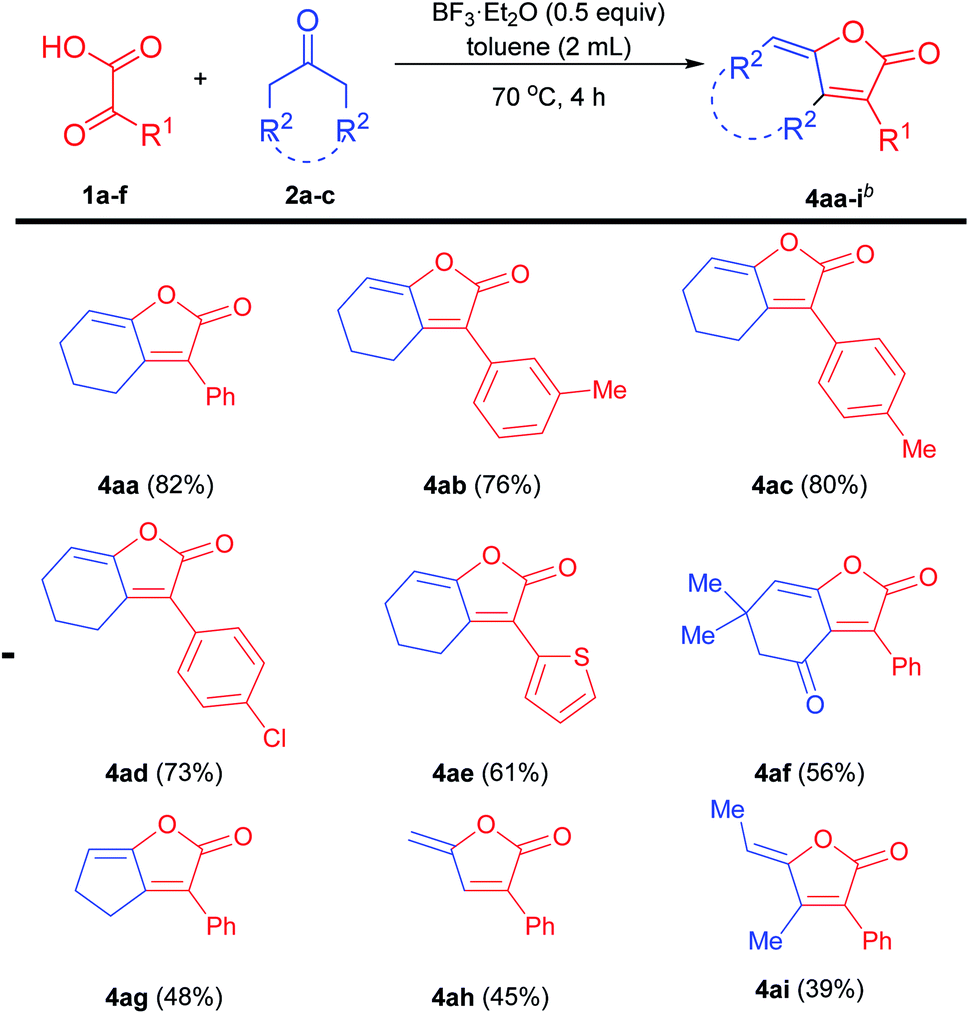
To demonstrate the practicality of this protocol, a gram-scale reaction was performed (Scheme 3). Gratifyingly, the substrates were divergently converted to 3aa and 4aa under standard conditions in good yields, further implying this approach's potential utility.
 | ||
| Scheme 3 Gram-scale reactions. | ||
To delineate the mechanism, control experiment was conducted with 3aa as substrate in the presence of excess amount of BF3·Et2O at 70 °C, γ-alkylidene-butenolide (4aa) was obtained in 92% yield, to further imply that BF3·Et2O was crucial for the dehydration process in this transformation (Scheme 4).
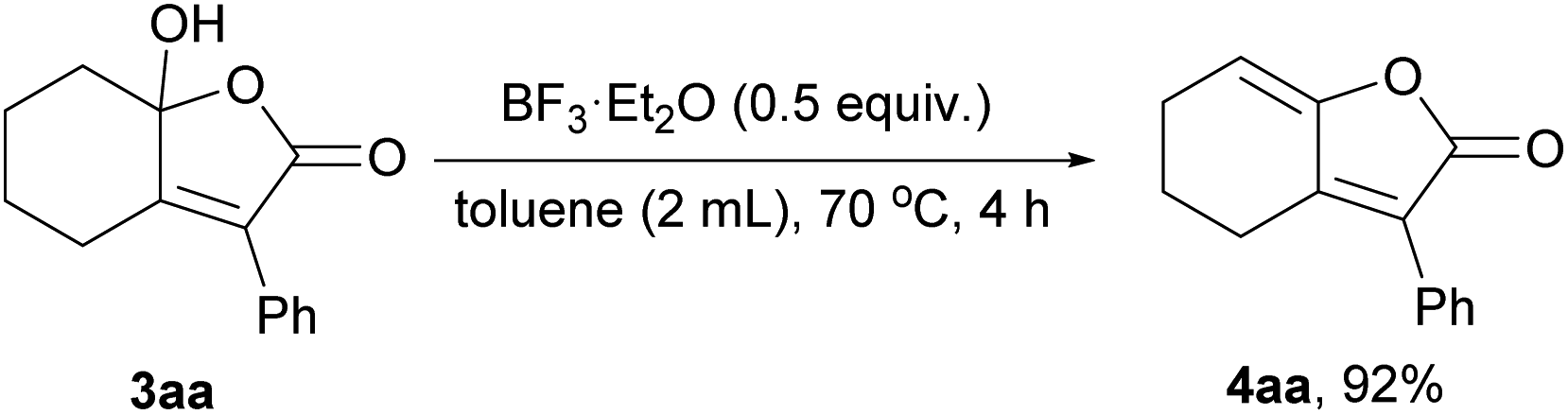 | ||
| Scheme 4 Control experiment. | ||
Based on the results and previous reports,10–15 we proposed a plausible pathway accounting for the formation of 3aa and 4aa (Scheme 5). Initially, the enolized cyclohexanone A from 2a underwent nucleophilic attack with α-keto acid (1a) with the assistance of Lewis acid to afford intermediate B. The intermediate B was then cyclized intramolecularly to give intermediate C, followed by dehydration to give 3aa. With increased amount of catalyst at elevated reaction temperature, 3aa was able to transformed to produce 4aa after dehydration.
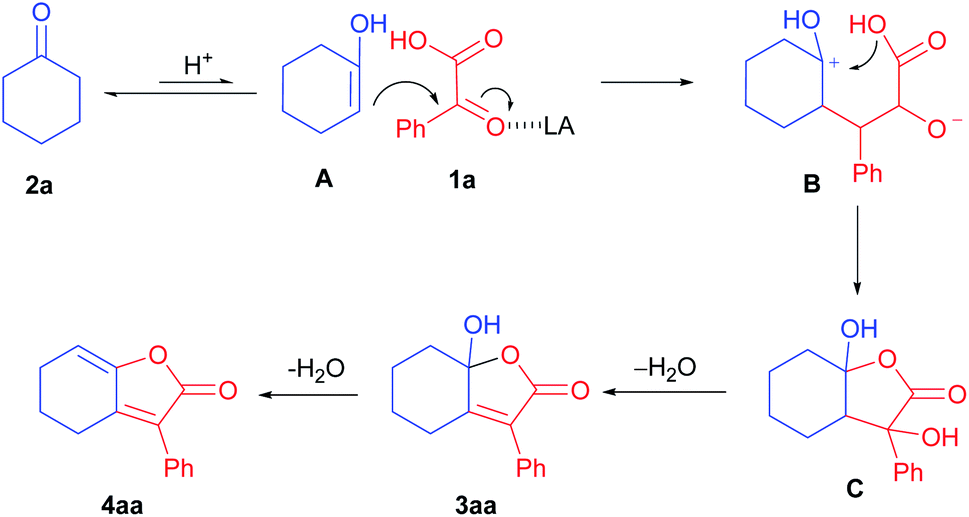 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
In summary, we have developed an efficient and convenient BF3·Et2O-mediated annulation reaction of α-keto acids with aliphatic ketones to simultaneously synthesize γ-hydroxy-butenolides and γ-alkylidene-butenolides. The method features good functional group tolerance and moderate to good yields, thus providing a facial and practical approach for the diversification of the library of butenolides. The application of this method for the synthesis of natural products is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 21KJB150014). The Large Instruments Open Foundation of Nantong University (No. KFJN2269). The Jiangsu College Students Innovation and Entrepreneurship Training Program (No. 202210304157Y).Notes and references
- (a) L. Wu, Z. Liao, C. Liu, H. Jia and J. Sun, Chem. Biodiversity, 2016, 13, 645–671 CrossRef CAS PubMed; (b) I. Muhammad, S. Shams ul Hassan, S. Cheung, X. Li, R. Wang, W.-D. Zhang, S.-K. Yan, Y. Zhang and H.-Z. Jin, Fitoterapia, 2021, 148, 104800 CrossRef CAS PubMed; (c) L. Li-Bin, J. Xiao, Q. Zhang, R. Han, B. Xu, S.-X. Yang, W.-B. Han, J.-J. Tang and J.-M. Gao, J. Agric. Food Chem., 2021, 69, 11878–11889 CrossRef PubMed; (d) M. Zhou, F. Duan, Y. Gao, X. Peng, X. Meng and H. Ruan, Bioorg. Chem., 2021, 115, 105247 CrossRef CAS PubMed; (e) Z. Meng and B. Liu, Org. Biomol. Chem., 2018, 16, 957–962 RSC.
- (a) W.-J. Zhang, X.-H. Li and Y.-P. Shi, J. Nat. Prod., 2010, 73, 143–146 CrossRef CAS PubMed; (b) B. M. Fraga, Nat. Prod. Rep., 2010, 27, 1681–1708 RSC; (c) Q. Liu, J. H. Ahn, S. B. Kim, C. Lee, B. Y. Hwang and M. K. Lee, Phytochemistry, 2013, 87, 112–118 CrossRef CAS PubMed.
- (a) H. Hikino, Y. Hikino and I. Yosioka, Chem. Pharm. Bull., 1962, 10, 641–642 CrossRef CAS PubMed; (b) S. Ramesh and G. Mehta, Tetrahedron Lett., 2015, 56, 5545–5548 CrossRef CAS.
- J. Peng, S. G. Franzblau, F. Zhang and M. T. Hamann, Tetrahedron Lett., 2002, 43, 9699–9702 CrossRef CAS.
- L. Baiocchi, M. Bonanomi, M. Giannangeli and G. Picconi, Synthesis, 1979, 434–436 CrossRef CAS.
- (a) S. K. Bagal, R. M. Adlington, J. E. Baldwin, R. Marquez and A. Cowley, Org. Lett., 2003, 5, 3049–3052 CrossRef CAS PubMed; (b) S. K. Bagal, R. M. Adlington, J. E. Baldwin and R. Marquez, J. Org. Chem., 2004, 69, 9100–9108 CrossRef CAS PubMed; (c) P. Srinivas, D. S. Reddy, K. S. Kumar, P. K. Dubey, J. Iqbal and P. Das, Tetrahedron Lett., 2008, 49, 6084–6086 CrossRef CAS; (d) S. Chatterjee, R. Sahoo and S. Nanda, Org. Biomol. Chem., 2021, 19, 7298–7332 RSC; (e) S. V. Fedoseev and M. Y. Belikov, Chem. Heterocycl. Compd., 2018, 54, 759–761 CrossRef CAS.
- (a) H. Wyss, L. Révész and R. Scheffold, Helv. Chim. Acta, 1981, 64, 2272–2278 CrossRef CAS; (b) Y.-H. Yang and M. Shi, Org. Lett., 2006, 8, 1709–1712 CrossRef CAS PubMed; (c) S. D. Townsend and G. A. Sulikowski, Org. Lett., 2013, 15, 5096–5098 CrossRef CAS PubMed; (d) A. S. Mwakaboko and B. Zwanenburg, Eur. J. Org. Chem., 2016, 21, 3495–3499 CrossRef PubMed; (e) B. Almohaywi, T. T. Yu, G. Iskander, D. S. H. Chan, K. K. K. Ho, S. Rice, D. S. Black, R. Griffith and N. Kumar, Bioorg. Med. Chem. Lett., 2019, 29, 1054–1059 CrossRef CAS PubMed.
- (a) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380–7391 RSC; (b) F. Penteado, E. F. Lopes, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, Chem. Rev., 2019, 119, 7113–7278 CrossRef CAS PubMed.
- (a) Z. He, F. Fang, J. Lv and J. Zhang, Tetrahedron Lett., 2017, 58, 1034–1036 CrossRef CAS; (b) R. Yabe, Y. Ebe and T. Nishimura, Chem. Commun., 2021, 57, 5917–5920 RSC.
- W. Mao and C. Zhu, Org. Lett., 2015, 17, 5710–5713 CrossRef CAS PubMed.
- W. Mao and C. Zhu, Org. Chem. Front., 2017, 4, 1029–1033 RSC.
- W. Mao and C. Zhu, Chem. Commun., 2016, 52, 5269–5272 RSC.
- B. Zhao, Z. Zhang, P. Li, T. Miao and L. Wang, Org. Lett., 2021, 23, 5698–5702 CrossRef CAS PubMed.
- X. Bao, L. Zeng, J. Jin and S. Cui, J. Org. Chem., 2022, 87, 2821–2830 CrossRef PubMed.
- X. Shi, Y. He, X. Zhang and X. Fan, Adv. Synth. Catal., 2018, 360, 261–266 CrossRef CAS.
- (a) G. A. Shevchenko, B. Oppelaar and B. List, Angew. Chem., Int. Ed., 2018, 57, 10756–10759 CrossRef CAS PubMed; (b) I. Felker, G. Pupo, P. Kraft and B. List, Angew. Chem., Int. Ed., 2015, 54, 1960–1964 CrossRef CAS PubMed; (c) E. A. Merritt and B. Olofsson, Synthesis, 2011, 2011, 517–538 CrossRef; (d) S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS PubMed; (e) S. Liang, K. Xu, C.-C. Zeng, H.-Y. Tian and B.-G. Sun, Adv. Synth. Catal., 2018, 360, 4266–4292 CrossRef CAS; (f) J. Li, Z. Yang, T. Yang, J. Yi and C. Zhou, New J. Chem., 2018, 42, 1581–1584 RSC.
- (a) J.-F. Marcoux, E. G. Corley, K. Rossen, P. Pye, J. Wu, M. A. Robbins, I. W. Davies, R. D. Larsen and P. J. Reider, Org. Lett., 2000, 2, 2339–2341 CrossRef CAS PubMed; (b) J.-F. Marcoux, F.-A. Marcotte, J. Wu, P. G. Dormer, I. W. Davies, D. Hughes and P. J. Reider, J. Org. Chem., 2001, 66, 4194–4199 CrossRef CAS PubMed; (c) S. Agasti, T. Pal, T. K. Achar, S. Maiti, D. Pal, S. Mandal, K. Daud, G. K. Lahiri and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 11039–11043 CrossRef CAS PubMed; (d) F. Benfatti, F. de Nanteuil and J. Waser, Chem.–Eur. J., 2012, 18, 4844–4849 CrossRef CAS PubMed; (e) M. N. Palange, R. G. Gonnade and R. Kontham, Org. Biomol. Chem., 2019, 17, 5749–5759 RSC; (f) Y. Tanabe, K. Mitarai, T. Higashi, T. Misaki and Y. Nishii, Chem. Commun., 2002, 2542–2543 RSC; (g) X. Li, Y. Wang, K. Fu, Z. Hu, Z. Li, W. Ma, M.-M. Xun and C. Yuan, J. Heterocycl. Chem., 2020, 57, 2056–2062 CrossRef CAS.
- (a) X. Zeng, C. Liu, W. Yang, Y. Weng, X. Wang and Y. Hu, J. Org. Chem., 2019, 84, 3656–3661 CrossRef CAS PubMed; (b) X. Zeng, C. Liu, W. Yang, X. Wang, X. Wang and Y. Hu, Chem. Commun., 2018, 54, 9517–9520 RSC; (c) X. Zeng, C. Liu, X. Wang, J. Zhang, X. Wang and Y. Hu, Org. Biomol. Chem., 2017, 15, 8929–8935 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2182801. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04546j |
| This journal is © The Royal Society of Chemistry 2022 |