
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring room-temperature ferromagnetism in WXBC (X = W, Mn, Fe) monolayers†
Yusuf Zuntu Abdullahi‡
 *a,
Sohail Ahmad‡b and
Fatih Ersan‡c
*a,
Sohail Ahmad‡b and
Fatih Ersan‡c
aDepartment of Physics, Faculty of Science, Kaduna State University, P.M.B. 2339 Kaduna State, Nigeria. E-mail: yusufzuntu@gmail.com
bDepartment of Physics, College of Science, King Khalid University, P O Box 9004, Abha, Saudi Arabia
cDepartment of Physics, Aydin Adnan Menderes University, Aydin 09010, Turkey
First published on 5th October 2022
Abstract
Two-dimensional (2D) transition metal boron-carbide is a novel material that has unique properties suitable for advanced spintronics and storage applications. Through first-principles calculations based on density functional theory (DFT) calculations, we report a new class of stable 2D ceramic WXBC (X = W, Mn, Fe) monolayers. We find that all WXBC monolayers prefer a ferromagnetic ground state with metallic electronic property. DFT calculations proved that WXBC monolayers exhibit good energetic, mechanical, and dynamic stabilities. More importantly, these monolayers exhibit large magnetic anisotropy energy (MAE) of 1213 μeV, 247 μeV and 20 μeV per magnetic atom for W2BC, WMnBC, and WFeBC, respectively. An out-of-plane easy axis (EA) magnetization direction is found for W2BC whereas the EA for WMnBC and WFeBC are in-plane. By performing Monte Carlo (MC) simulations based on the 2D Heisenberg model, we predict Curie temperatures (TC) of 155 K for the W2BC monolayer. The Berezinskii–Kosterlitz–Thouless transition (BKT) temperature values of WMnBC and WFeBC are as high as 374.69 K and 417.39 K. For further investigations, the adsorption properties of Li, Na, and K atoms on WXBC (atm-WXBC) systems are examined. It is revealed that the initial ferromagnetic metallic properties of bare WXBC monolayers are maintained for all atm-WXBC systems. The obtained strong chemisorption energies are high enough to make adsorbed Li, Na, and K immobile on WXBC surfaces. All these findings demonstrate the unique potential of WXBC monolayers as multifunctional candidates for advanced magnetic device and storage applications.
1 Introduction
After the pioneering experimental synthesis of the ferromagnetic CrI3 and Cr2Ge2Te6 layers,1,2 a great deal of attention has been paid to two-dimensional (2D) magnetic materials. These CrI3 and Cr2Ge2Te6 layers were key to experimentally achieving room-temperature ferromagnetism in the 2D magnetic materials.3,4 The obtained results also raised hopes for the future development of quantum technology, which would allow information to be processed at ultra-high speeds by utilizing the electron spin degree of freedom. To further consolidate these prospects, new 2D stable ferromagnetic materials with a considerable magnetic moment, elevated Curie temperature (TC), and large magnetic anisotropy energy (MAE) should be explored. Recently, transition metal (TM) boride (popularly referred to as MBene) structures were theoretically proposed as a potential candidate for room-temperature TC with a sizable MAE.5,6 Because of the MBene layered nature, selective etching techniques would make it easier to reduce them to 2D form, from their bulk equivalent.7Besides theoretical success on MBene sheets for spintronics applications,5,6 however, there is still a need for designing sustainable 2D magnetic materials to satisfy the modern electronic devices. Taking advantage of the MBene structural and physical properties, it will be intriguing to expand the search to other new 2D materials similar to MBene. For example, the MnX (X = As, P)8 and Mn2BC9 sheets with half-metallic and metallic properties respectively are reported to exhibit above room-temperature TC values. The Ti2BN10 and X2BC (X = Mg, Ca, Sr, Ti, V, Mo)11 sheets were theoretically demonstrated for both Li+ and Na+ ion batteries with enhanced battery performance better than that reported for MBene.12–15 It's worth noting that these reported materials share similar MBene building blocks in monolayer and bulk forms.
Here, we aimed to explore tetragonal (t) ceramic WXBC (X = W, Fe, Mn) sheets as potential candidates for spintronic applications. According to our knowledge, WXBC sheets has not been theoretically characterised, especially for spin-related applications. We know that layered ceramic W2B2 sheets crystallize in an orthorhombic phase with the space group Cmcm and WFeB2 nanostructure exists as one of the component of NiCrBSi/WC–Co composite.16 Using high-power pulsed magnetron sputtering (HPPMS), a comparable ceramic Mo2BC bulk material was synthesized at a substrate temperature of 400 °C.17 Because of its exceptional mechanical and thermal properties, it can be used as an electrode in a nuclear reactor. As mentioned earlier, selective etching would be an appropriate method for the synthesis of WXBC sheets. A previous study has demonstrated that having at least two TM atoms in the MBene structure may promote easy synthesis process.7 It could also provide a favorable platform to produce strong spin-orbital coupling (SOC) interactions for the large MAE and large charge carriers for enhanced TC.
Aside from exploring the MBene structural diversity for room-temperature TC, one of the most recognized strategies to achieve enhanced TC and MAE involves the application of mechanical strain and electric field strength.18 For example, the TC of the Fe3P sheet remains above room temperature under applied biaxial strain as large as 10%.18 The fact that MAE arises due to spin–orbit coupling (SOC), it was shown that the MAE of CrFeBC can be tuned by the external electric field from in-plane to out-of-plane magnetization direction.19 In another report, an enhanced TC high above room temperature with a sizable MAE were reported for structures under mechanical strain.20,21
In this study, density functional theory (DFT) and mean-field approximation of the 2D classical Heisenberg model calculations are combined to investigate the suitability of WXBC monolayers for spin-based electronics. The free-standing WXBC sheets display good stability based on their mechanical, phonon and thermal properties. Ferromagnetic (FM) spin ordering corresponds to the ground state for WXBC monolayers. The W2BC shows a large MAE of 1213 μeV per W atom with an out-of-plane easy axis (EA), whereas WXBC (X = Fe, Mn) preferred an in-plane EA. The MAE value for WMnBC is 247 μeV per magnetic atom whereas that of WFeBC is 20 μeV per magnetic atom. Since we are only interested in FM with out-of-plane EA, the predicted MAE of the W2BC sheet can be further tuned by an apply electric field strength and mechanical strain. Based on the 2D Heisenberg MC simulations, we predict TC of 155 K for the W2BC monolayer. The BKT temperature values of WMnBC and WFeBC are as high as 374.69 K and 417.39 K.
We also explore the adsorption properties of Li, Na, and K ions on WXBC sheets. We found that, due to strong steric coulomb repulsion, the obtained strong chemisorption energies are high enough to make adsorbed Li, Na, and K immobile on WXBC surfaces.
2 Computational method
All the ground state properties calculations have been performed using the Vienna ab initio simulation package (VASP)22 which are based on density functional theory (DFT)23 method. The exchange–correlation have been treated with generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE)24 parametrization. The Hubbard U correction25 (PBE + U) has been used to described strongly localized d orbitals of W, Mn and Fe atoms. The estimated Ueff values for d orbitals of W, Mn and Fe atoms are Ueff = 3.01 eV, Ueff = 2.97 eV and Ueff = 3.04 eV, respectively. More information about our obtained Ueff values can be found in previous studies26,27 and the ESI.† The projected augmented wave (PAW) pseudopotentials method24 has been used to describe the core and valence electrons for all atoms in WXBC monolayers. For the dispersion correction, DFT-D2 correction of Grimme28 has been used. The irreducible Brillouin zone (BZ) in reciprocal space has been represented by Monkhorst–Pack set of k-points grid meshes.29 For optimizations and total density of state calculations we have used (8 × 8 × 1) and (12 × 12 × 1) grid meshes respectively. An energy cut-off of 500 eV was taken for plane wave basis set expansion. A vacuum space of 18 Å along the z-direction was set to eliminate interaction between the adjacent layers of WXBC. The criteria for convergence of the total energy and remaining force on each atom for two iterative steps were set smaller than 10−5 eV and 0.001 eV Å−1 respectively. The dynamic stability of WXBC monolayers have been examined with aid of phonon dispersions calculations. For the phonon calculations we have employed (2 × 2) supercell of WXBC monolayers to reduce the constraint of periodic boundary conditions. Then, we use supercell method of the PHONOPY code30 to obtain phonon band structures. To evaluate the thermal stability of the WXBC monolayers, we performed ab initio molecular dynamics (AIMD) simulations with a time step of 3.0 fs. The temperature was monitored with the aid of Nose–Hoover thermostat.31 To examine the ferromagnetic ordering at finite temperature, we performed MC simulations based on 2D Heisenberg model. Similar details of the MC calculations can be found in our previous published paper.93 Results and discussions
The 2D WXBC monolayers can be theoretically created from the Mn2BC monolayer.32 Fig. 1 displays the relaxed structures of WXBC, which assume tetragonal symmetry with a space group of P4/mmm (no. 129). The black square line (top view) in Fig. 1 mapped out the unit-cell of the WXBC monolayers. For the W2BC, the unit consists of two W atoms and B/C atoms, while one of the W atoms is substituted with Mn and Fe atoms in the WMnBC and WFeBC cases, respectively. Each atom in the WXBC structures is chemically bonded by four neighboring atoms. To determine the chemical bonding nature of the WXBC monolayers, we display the electron localization functions (ELF) in Fig. 1. As is known, the ELF takes values in the range of 0 to 1 e−Å−3. In each side view, it is evident that the ionic bonding in the WXBC monolayers dominates due to the concentration of charge density around the B/C atoms, which is absent in the rest of the atoms. The charge localization around the B/C atoms validates their dominant electronegativity over the W, Mn, and Fe atoms. Bader charge analysis33 shows that the charge transfers of around 1.45 e−, 1.85 e−, and 2.91 e− occur from W2, WMn, and WFe atoms into BC compounds.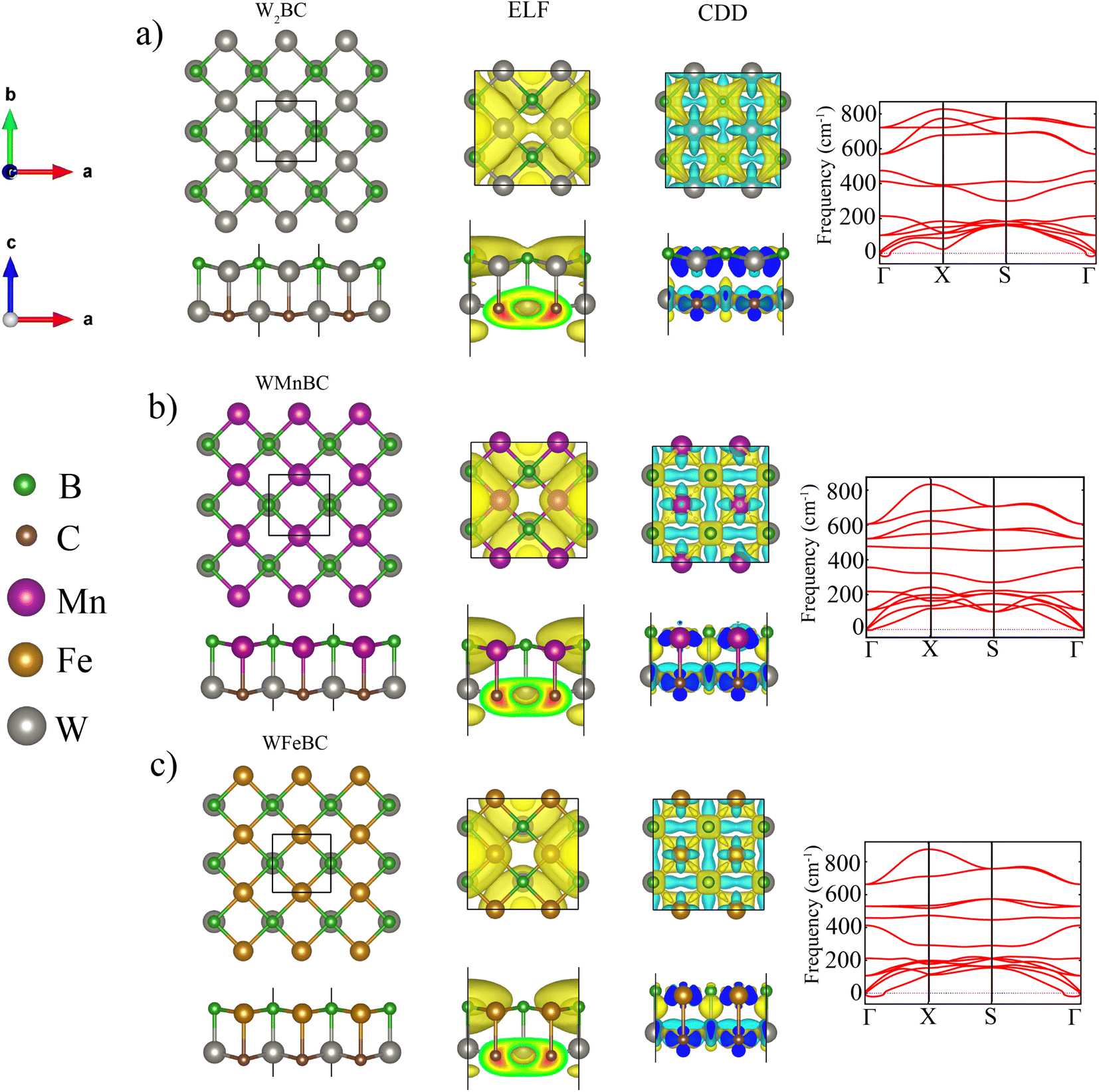 | ||
| Fig. 1 From left to right, the first three figures illustrate the top and side view of the optimized unit cell, the electron localization functions (ELF), and the charge density difference (CDD) in the 2 × 2 supercell. The ELF iso-surface values is approximately 0.280 e− Å−3 and that of charge density difference is 0.016 e− Å−3 for WXBC monolayers. The coloured balls on the left side of the figure denotes the atoms in the WXBC structures. The last figure on the right side represent the phonon dispersions curves for WXBC monolayers. | ||
According to the PBE (PBE + U) calculations, the lattice constant (a = b) of the W2BC, WMnBC, and WFeBC monolayers were found to be 3.00 Å (3.00 Å), 2.89 Å (2.91 Å), 2.87 Å (2.89 Å). It is noted that the PBE + U calculations for the WMnBC and WFeBC cases produces enhanced lattice constant. This shows that the Hubbard U correction yields low bonding stiffness in these structures as compared to that obtained in PBE calculations. It is anticipated that the inclusion of the Hubbard U will affect the magnetic properties of the WXBC monolayers. For PBE calculations, the estimated W–B (W–C), W–B (Mn–C) and W–B (Fe–C) bond lengths are 2.30 Å (2.18 Å), 2.22 Å (2.38 Å) and 2.20 Å (2.36 Å) respectively along the direction (interlayer height). The bond lengths in the plane are 2.15 Å (2.14 Å), 2.05 Å (2.09 Å) and 2.05 Å (2.07 Å) for W–B (W–C), W–C (Mn–B) and W–C (Fe–B) respectively. Overall, the estimated lattice constants and bond length values are either larger or consistent with those reported for TM2BC sheets.32 To assess the energetic stability of the WXBC monolayers, we examine both the cohesive energy (Ecoh) and formation energy (Ef) per atom. The following equation is used to calculate Ecoh:
| Ecoh = (EW + EX + EB + EC − EWXBC)/4 | (1) |
| Ef = (EWXBC − μW − μX − μB − μC)/4 | (2) |
As a further check on the stability of the WXBC monolayers, we compute the in-plane stiffness (Y) and Poisson's ratio (ν) using the strain-energy approach.35,36 The calculated Y (ν) along the x or y strain directions are summarized in Table S1.† The estimated Y (ν) values illustrate the good mechanical stability of WXBC monolayers. We expect a stiffer bonding network in the W2BC monolayer than the bonds in WMnBC and WFeBC monolayers. It is noted that all positive Y (ν) values are isotropic for W2BC and WMnBC monolayers and anisotropic for the WFeBC case. For the WFeBC case, the Y value in b direction is larger than that of a direction, indicating that this WFeBC would resists compression along the b-axis than the a-axis. However, all calculated Y values show that they satisfy the minimum Born criteria for elastic stability of tetragonal 2D materials.37 In comparison, the Y values for W2BC (210.63 N m−1) and WFeBC (208.93/216.09 N m−1 along a/b) are larger than those reported for Ti2BN (200.40 N m−1) sheet.10
To evaluate the dynamic stability of these monolayers, we have plotted phonon dispersion curves in Fig. 1. The phonon curves for WMnBC illustrate no imaginary phonon frequency along to whole high symmetry points in the Brillouin zone. The highest frequency of these monolayers reaches up to 800 cm−1, comparable to most of the well-known MBene sheets.38 It can be concluded that the WXBC monolayers are robust materials owing to their high optical vibration modes. To demonstrate the thermal stabilities of WXBC monolayers we carried out AIMD at 300 K using 4 × 4 × 1 supercell. As displayed in Fig. S2,† the structural integrity of WXBC are preserved at room temperature with no structural deformation. In general, the obtained stability results show that the WXBC monolayers could potentially be synthesized in the experiment. To establish an easy comparison with the previous reports, all the stability checks have been done using PBE calculations.
Having established the stability of the WXBC monolayers, we determined the favorable magnetic ground state for WXBC monolayers. As illustrated in Fig. S3,† a ferromagnetic (FM) and two different antiferromagnetic (AFM) states in the 2 × 2 × 1 supercell has been considered. The estimated values of the exchange energies for these magnetic states are listed in Table S2.† It is clear that the magnetic ground state for WXBC monolayers corresponds to the FM state. Table S1† lists the magnetic moments obtained at PBE levels for WXBC monolayers, and the W2BC is found to be non-magnetic. However, the magnetic moment for W2BC appeared when the Hubbard U correction was included. This implies that the Hubbard U correction in W2BC structure yields a polarized spin in the 5d orbitals of the W atom after orbital reorientation.
For PBE + U calculations, there is a magnetic moment of 1.6 μB, 2 μB, 3 μB per atom for W2BC, WMnBC, and WFeBC monolayers, respectively. Table S1† summarises the magnetic moment of each magnetic ion. The magnetic moment is mainly from Mn (2 μB), Fe (2 μB) magnetic ions for the WMnBC and WFeBC cases. The remaining atoms have their magnetic moment reduced. It should be noted that with respect to Hund's rules, an isolated Mn and Fe atoms would have about 5 μB and 4 μB, respectively. However, due to the bonding influence of surrounding atoms in the W2BC, WMnBC and WFeBC monolayers, the total magnetic moment becomes decreased. The charge density difference (CDD) plot, evidently show that the concentration of magnetic moment around magnetic ions, namely Mn, Fe and W atoms in the WMnBC and WFeBC monolayers (see Fig. 1).
Based on the FM ground-state of WXBC structures, the electronic property is then examined. To correct the self-interaction errors which can be seen in these kinds of WXBC monolayers (involving strongly correlated atoms), the PBE + U calculations have been considered. This is the fact that the electronic properties of strongly correlated systems can be improved by using the PBE + U method.25 The band structures and their corresponding projected density of state (PDOS) for the FM WXBC monolayers are displayed in Fig. 2. It is clear that all structures exhibit a metallic electronic character. There is clear evidence of asymmetric spin states around the Fermi level and beyond for all cases, confirming the presence of magnetic moments. From the PDOS plots, the contributions of the states around the Fermi level are mainly dominated by d orbital of W, Mn, and Fe atoms in both spin channels.
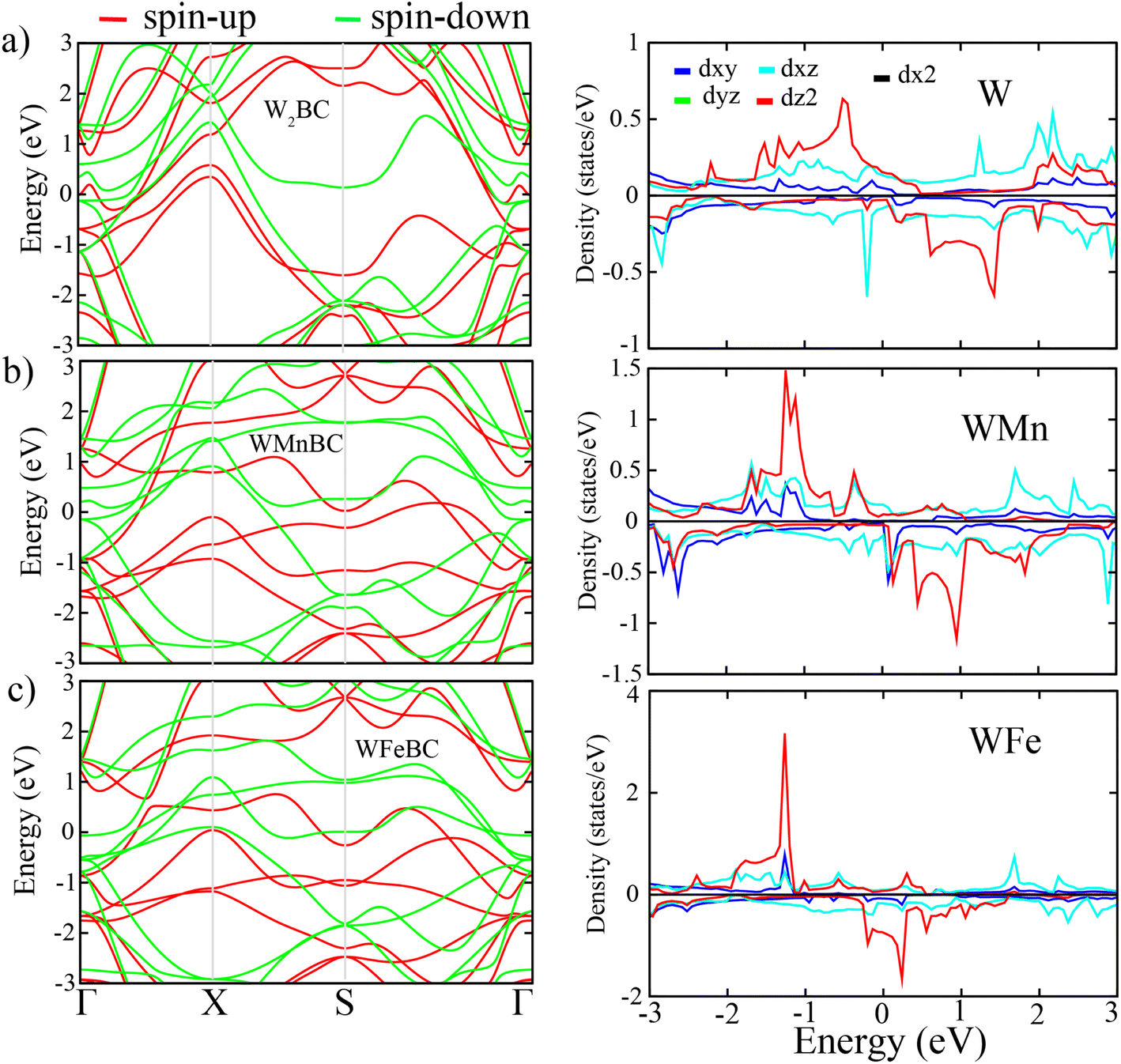 | ||
| Fig. 2 From left to right, the figure shows the band structure and partial density of states (PDOS) of WXBC monolayers. | ||
To evaluate the suitability of WXBC monolayers in practical spintronic devices, we estimate their MAE and TC parameters. The MAE and TC are well-known parameters used to measure the preservation of FM long-range ordering against thermal fluctuations. For MAE, an out-of-plane [001] (along the z-direction) and four in-plane magnetization directions ([100], [010], [110], [111]) were considered. The relative energy values for these magnetization directions are summarized in Table S3(a)–(c).† The W2BC monolayer magnetization direction is out-of-plane [001] (see Table S3(a)†), whereas the remaining monolayer magnetization direction is in-plane [100]. It is noted that, the magnetization easy axis (EA) switches from out-of-plane to EA in-plane when one of the W atoms are substituted with either an Mn or Fe atom. This is due to the dominant feature of the orbital moment (Lz) of the W in both spin channels around Fermi level. As a result, the total energy of the sheet is reduced, favoring the out-of-plane direction magnetization axis, as shown in Table S3(a).† For the remaining WMnBC and WFeBC cases, sharp peaks in the minority spin around the Fermi level, which resulted in an in-plane MAE magnetization axis. The estimated out-of-plane MAE value of the W2BC monolayer is approximately 1213 μeV per W atom. The in-plane MAE values of WMnBC and WFeBC monolayers are 247 μeV and 20 μeV per magnetic atom, respectively. It is also noted that the MAE decreases for both WMnBC and WFeBC cases. This means that substitutional doping of the W atom with the Fe atom corresponds to the low spin-orbital coupling (SOC) interaction in the WFeBC structure, which reduces the MAE. Our calculated MAE values are larger than those reported for MBene and related structures.8,38
We also investigate the effects of electric field strength and mechanical strain on MAE. Since in-plane MAE is not our target, henceforth, we shall focus on the W2BC monolayer. Table S4 (a)† summarizes the dependence of electric field on MAE values in the range of 0.2–1.0 eV Å−1. The computed MAE value under the electric field strength decreased as compared with the one obtained for the bare W2BC monolayer. However, despite the reduction in MAE under electric field strength, a sizable MAE value is still maintained and is comparable to or larger than most of MBene's reported MAE values. However, the MAE values show an increasing pattern as larger electric field strength is applied until it reaches the value of 0.8 eV Å−1 and then abruptly decreases at 1.0 ev Å−1. It should be noted that we did not observe any change in the EA up to a maximum of 1.0 eV Å−1 electric field strength. Concerning the effects of mechanical strain on the MAE, we applied biaxial strain in the range of −3% to 6% using a 3% step size. The obtained results of MAE vs. strain are listed in Table S4(b).† It is noted that the mechanical compression @ −3% and stretching @ 6% favor enhancement of MAE. It can be deduced that these strain points correspond to larger SOC as a result of increased W–W interactions.
Next, we examine the angular dependence of MAE over a range of magnetization angle (θ) along the xz (MAExz), or yz (MAEyz) planes for W2BC monolayer. Firstly, we performed DFT calculations of MAExz and MAEyz as function of θ. The obtained DFT data is then fitted to an equation expressed as:
MAE(θ) = K1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ2 + K2sinθ4 sinθ2 + K2sinθ4
| (3) |
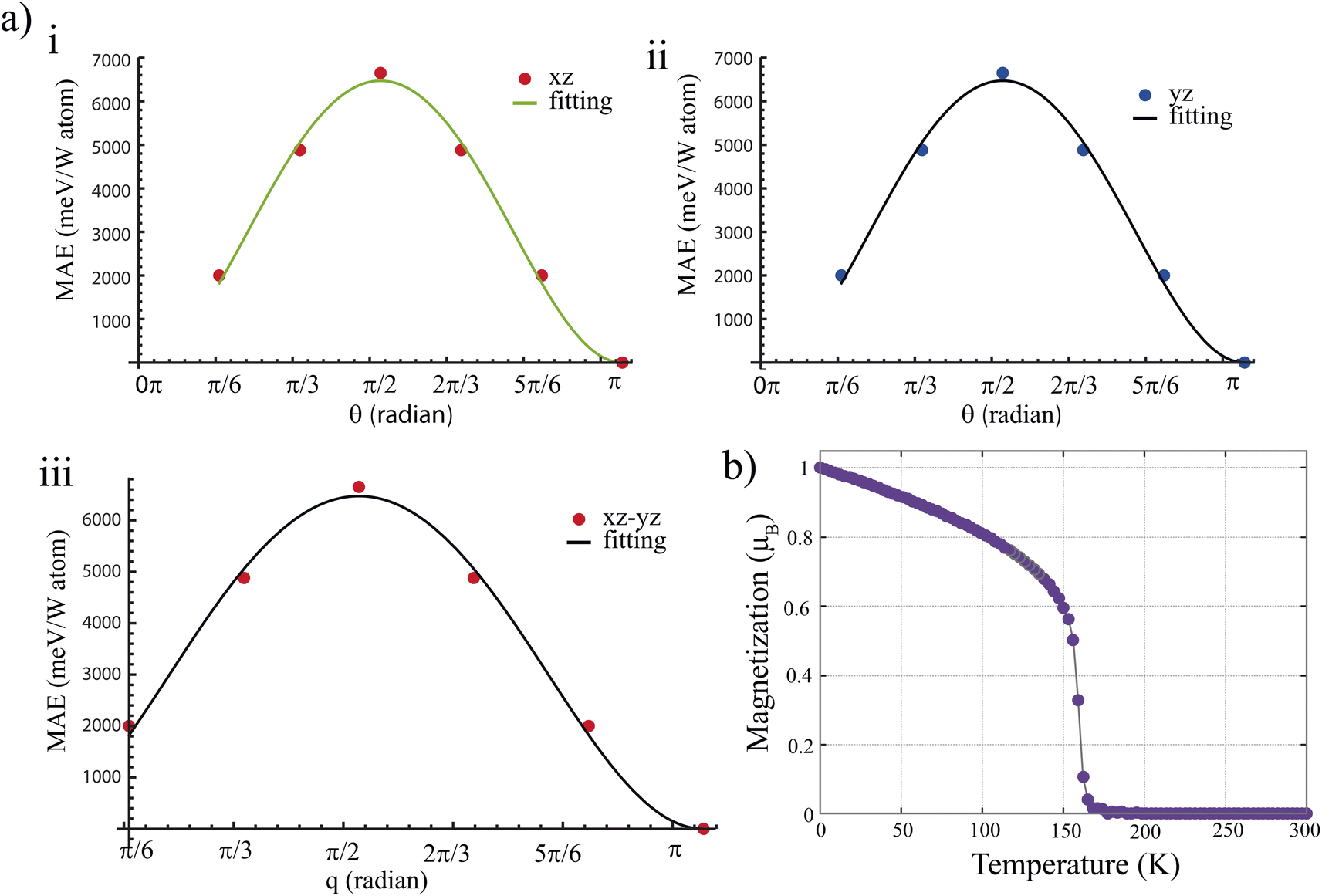 | ||
| Fig. 3 (a) The figure displays the MAE as function of magnetization angle of W2BC monolayer with the direction of magnetization lying on (i) xz, (ii) yz planes and the two (iii) xz, and yz planes respectively. (b) Total magnetization as a function of temperature for the W2BC monolayer. | ||
To investigate the magnetic phase transition temperatures of WXBC monolayers, 2 × 2 × 1 supercell and three different magnetic orientation are used. The analysis of the three magnetic states FM, AFM1 and AFM2 can be derived as follows:
| E(FM) = E0 − 8J1μ2 − 9J2μ2 | (4) |
| E(AFM1) = E0 + 8J1μ2 + J2μ2 | (5) |
| E(AFM2) = E0 − 8J1μ2 + 9J2μ2 | (6) |
| J1 = −(4E(FM) − 9E(AFM1) + 5E(AFM2))/(144μ2) | (7) |
| J2 = −(E(FM) − E(AFM2))/(18μ2) | (8) |
for W2BC, WMnBC, and WFeBC, respectively. However, using calculated magnetic exchange energies provided in Table S2,† we estimate the critical temperatures of these WXBC monolayers. The critical temperature, which is known as the Curie temperature, corresponds to the transition from the ferromagnetic to the paramagnetic phase. To maintain a robust long-range FM spin ordering, the candidate materials should have at least room temperature TC. We plot the total magnetization as a function of temperature for the W2BC monolayer in Fig. 3(b) based on MC results. We also provide the magnetic susceptibility and specific heat vs. temperature in Fig. S5.† The W2BC monolayer retains its ferromagnetic ordering at low temperatures, as shown in Fig. 3(b). The TC value appears at a temperature of 155 K. This means the phase transition from the FM to paramagnetic phase occurs at a low temperature due to the low value of exchange interaction constants of the W2BC monolayer. As a case of comparison, we also use the mean-filed approximation to estimate the TC of W2BC which is defined as| TC = 2ER/(3kBN) | (9) |
It should be emphasized that WXBC (X = Mn, Fe) monolayers exhibit an in-plane MAE. This means that the magnetic state of the WXBC (X = Mn, Fe) monolayers corresponds to XY model, which is a limiting case of the Heisenberg model. This phenomenon has been observed in our previous studies, that WXBC (X = Mn, Fe) monolayers exhibit a very unique Berezinskii–Kosterlitz–Thouless (BKT) transition39 at low temperature. This is because Mermin–Wagner theorem emphasizes that at a finite temperature, monolayer crystals e.g. WXBC cannot have long-range order.40 Thus, according to the XY model,41 the BKT can be evaluated as
| TC = 0.89(EAFM − EFM)/8kB. | (10) |
The EAFM − EFM has been listed in the Table S2.† The estimated BKT temperatures are 374.69 K and 417.39 K for WMnBC and WFeBC, respectively. The obtained BKT temperature values are above room temperature (300 K).
To evaluate if the WXBC monolayers will favorably store Li-, Na- and K-ions, we estimate the adsorption energies (Eads) of these Li-, Na- and K-ions on the WXBC (atm@WXBC) surfaces. The Eads is defined as
| Eads = (EWXBC + Eatm) − Eatm@WXBC | (11) |
To assess the magnetic ground state, an AFM and FM calculations have been performed for all atm@WXBC systems. According to the obtained relative energy (see Table S5†) values, FM corresponds to magnetic ground state for all atm@WXBC systems. All the structural, energetic, electronic, and magnetic parameters for the energetically preferred atm@WXBC systems are listed in Table S4(b).† As is known, the W2BC monolayer is non-magnetic using the PBE method, however, an FM order with weak magnetic moment per unit cell is found when atm are adsorbed on the W2BC surface. This shows that there is an orbital reorientation in the atm@WXBC which produces polarized spins in the atm@WXBC system. The large magnetic moment per unit cell can be seen for atm@WXBC (X = Mn, Fe) cases. This indicates a parallel coupling (FM order) between the spins of the adsorbed atm with those of W, Mn, and Fe atoms electrons spin in this atm@WXBC (X = Mn, Fe) monolayers. Fig. S6† provides the CDD for atm@WXBC (X = Mn, Fe) systems. The CDD plots validate the magnetic moment distribution that show the concentration of magnetic moment between adatoms and nearest-neighbor atoms in the atm@WXBC (X = W, Mn, Fe) systems.
From eqn (4), we calculated Eads for atm at WXBC as listed in Table S6.† It is clear that all Eads values are large and positive, which guarantees the chemical adsorption of Li, Na, and K atoms on the WXBC monolayers without the tendency for Li, Na, and K cluster formation. However, the Eads' values are desperate. This is mainly due to electron transfer between positively charged Li+, Na+, and K+ ions and WXBC monolayers. According to Bader's charge analysis, all adsorbed Li, Na, and K transfer electrons to the WXBC monolayers, which corresponds well with the electronegativity of the surrounding atoms. The adsorption height (h) between the Li, Na, K atoms and WXBC surfaces is also provided in Table S6.† It is also evident that the h values obtained vary relatively according to Eads values. For example, the h (2.29 Å) is lower for the Li@WMnBC system with Eads (6.71 eV), and h (2.65 Å) is higher for the Na@WFeBC case with Eads (6.60 eV). We also provide the spin-polarized TDOS of each atm@WXBC systems in Fig. S7.† The metallic properties of bare WXBC monolayers are maintained for atm@WXBC cases. The majority and minority spin states of TDOS plots for atm@WXBC systems are asymmetric, which further validates the presence of magnetic moments.
4 Summary
We investigated the spin-related and adsorption properties of a new class of stable 2D WXBC (X = W, Mn, Fe) monolayers. DFT calculations confirmed that WXBC monolayers exhibit good energetic, mechanical, and dynamic stabilities. We find that all WXBC monolayers prefer a ferromagnetic ground state with metallic electronic property. Moreover, we investigate the potential of WXBC for spin-related applications. Based on the 2D Heisenberg MC simulations, we predict Curie temperatures (TC) of 155 K for the W2BC monolayer. The BKT temperature values of WMnBC and WFeBC are as high as 374.69 K and 417.39 K. More importantly, these monolayers exhibit large magnetic anisotropy energy (MAE) of 1213 μeV, 247 μeV and 20 μeV per atom for W2BC, WMnBC, and WFeBC, respectively. An out-of-plane easy axis (EA) magnetization direction is found for W2BC whereas the EA for WMnBC and WFeBC are in-plane. For W2BC monolayer, the MAE can be enlarged under biaxial strain in the range of −3% to 6%. For further investigations, the adsorption properties of Li, Na, and atoms on WXBC (atm-WXBC) systems are examined. It is revealed that the initial ferromagnetic metallic properties of bare WXBC monolayers are maintained for all atm-WXBC systems. The obtained strong chemisorption energies are high enough to make adsorbed Li, Na, and K immobile on WXBC surfaces. Our results suggest a promising route to design WXBC monolayers for spin-related and storage applications, which will arouse the further experimental interests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sohail Ahmad extends their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Large Groups Project under grant number (R. G. P. 2/139/43). This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) under Project No. 121F270. The computational resources are provided by TUBITAK ULAKBIM, High Performance, and Grid Computing Center (TR-Grid e-Infrastructure). Yusuf Zuntu Abdullahi would like to acknowledge Assoc. Prof. Dr Tiem Leong Yoon from the School of Physics, Universiti Sains Malaysia for providing the computing resources to carry out part of the calculations done in this paper. We would like to acknowledge Assoc. Prof. Dr Erol Vatansever from the Department of Physics Dokuz Eylul University, Izmir, Turkey for his valuable discussion on MC results. Figures showing atomic model, PDOS and band structure plots are generated using the VESTA program and XMGRACE, respectively.Notes and references
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire and D. H. Cobden, et al., Nature, 2017, 546, 270–273 CrossRef CAS.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang and Y. Wang, et al., Nature, 2017, 546, 265–269 CrossRef CAS.
- S. Fu, K. Kang, K. Shayan, A. Yoshimura, S. Dadras, X. Wang, L. Zhang, S. Chen, N. Liu and A. Jindal, et al., Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- L. Meng, Z. Zhou, M. Xu, S. Yang, K. Si, L. Liu, X. Wang, H. Jiang, B. Li and P. Qin, et al., Nat. Commun., 2021, 12, 1–8 CrossRef.
- Y. Z. Abdullahi, Z. D. Vatansever, E. Aktürk, Ü. Akıncı and O. Ü. Aktürk, Phys. Chem. Chem. Phys., 2020, 22, 10893–10899 RSC.
- M. Dou, H. Li, Q. Yao, J. Wang, Y. Liu and F. Wu, Phys. Chem. Chem. Phys., 2021, 23, 10615–10620 RSC.
- M. Khazaei, J. Wang, M. Estili, A. Ranjbar, S. Suehara, M. Arai, K. Esfarjani and S. Yunoki, Nanoscale, 2019, 11, 11305–11314 RSC.
- B. Wang, Y. Zhang, L. Ma, Q. Wu, Y. Guo, X. Zhang and J. Wang, Nanoscale, 2019, 11, 4204–4209 RSC.
- Y. Z. Abdullahi, Z. D. Vatansever, F. Ersan, U. Akinci, O. U. Akturk and E. Akturk, Phys. Chem. Chem. Phys., 2021, 23, 6107–6115 RSC.
- Y.-Y. Wu, T. Bo, X. Zhu, Z. Wang, J. Wu, Y. Li and B.-T. Wang, Appl. Surf. Sci., 2020, 513, 145821 CrossRef CAS.
- Y. Z. Abdullahi, F. Ersan, E. Akturk and O. U. Akturk, Phys. Rev. Appl., 2021, 16, 024031 CrossRef CAS.
- Z. Guo, J. Zhou and Z. Sun, J. Mater. Chem. A, 2017, 5, 23530–23535 RSC.
- T. Bo, P.-F. Liu, J. Zhang, F. Wang and B.-T. Wang, Phys. Chem. Chem. Phys., 2019, 21, 5178–5188 RSC.
- T. Bo, P.-F. Liu, J. Xu, J. Zhang, Y. Chen, O. Eriksson, F. Wang and B.-T. Wang, Phys. Chem. Chem. Phys., 2018, 20, 22168–22178 RSC.
- G. Yuan, T. Bo, X. Qi, P.-F. Liu, Z. Huang and B.-T. Wang, Appl. Surf. Sci., 2019, 480, 448–453 CrossRef CAS.
- H. Guo, B. Li, C. Lu, Q. Zhou and J. Jia, J. Alloys Compd., 2019, 789, 966–975 CrossRef CAS.
- H. Bolvardi, J. Emmerlich, S. Mráz, M. Arndt, H. Rudigier and J. M. Schneider, Thin Solid Films, 2013, 542, 5–7 CrossRef CAS.
- S. Zheng, C. Huang, T. Yu, M. Xu, S. Zhang, H. Xu, Y. Liu, E. Kan, Y. Wang and G. Yang, J. Phys. Chem. Lett., 2019, 10, 2733–2738 CrossRef CAS PubMed.
- Y. Z. Abdullahi, Z. D. Vatansever, E. Aktürk, Ü. Akıncı and O. Ü. Aktürk, Comput. Mater. Sci., 2022, 202, 110964 CrossRef.
- Z. Guan and S. Ni, ACS Appl. Electron. Mater., 2021, 3, 3147–3157 CrossRef CAS.
- T. Gorkan, E. Vatansever, U. Akıncı, G. Gokoglu, E. Akturk and S. Ciraci, J. Phys. Chem. C, 2020, 124, 12816–12823 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Cococcioni and S. De Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- Y. Z. Abdullahi, Z. D. Vatansever, E. Aktürk, Ü. Akıncı and O. Ü. Aktürk, Phys. Status Solidi B, 2021, 258, 2000396 CrossRef CAS.
- Y. Z. Abdullahi, Z. D. Vatansever, E. Aktürk, Ü. Akıncı and O. Ü. Aktürk, Phys. Chem. Chem. Phys., 2020, 22, 10893–10899 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355 CrossRef CAS.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- Y. Z. Abdullahi, Z. D. Vatansever, F. Ersan, U. Akinci, O. U. Akturk and E. Akturk, Phys. Chem. Chem. Phys., 2021, 23, 6107–6115 RSC.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- D. Fan, S. Lu, Y. Guo and X. Hu, J. Phys. Chem. C, 2018, 122, 15118–15124 CrossRef CAS.
- M. Topsakal, S. Cahangirov and S. Ciraci, Appl. Phys. Lett., 2010, 96, 091912 CrossRef.
- H. Şahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155453 CrossRef.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- Y. Z. Abdullahi, Z. D. Vatansever, F. Ersan, U. Akinci, O. U. Akturk and E. Akturk, Phys. Chem. Chem. Phys., 2021, 23, 6107–6115 RSC.
- J. M. Kosterlitz and D. J. Thouless, J. Phys. C: Solid State Phys., 1973, 6, 1181 CrossRef CAS.
- N. D. Mermin and H. Wagner, Phys. Rev. Lett., 1966, 17, 1133 CrossRef CAS.
- J. F. Fernández, M. F. Ferreira and J. Stankiewicz, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 292 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Linear response approach for Hubbard U parameter calculation, snapshots of the molecular dynamic simulations, magnetic orientations, total density of states for WXBC monolayers, charge density difference for alkali atoms on WXBC monolayers, total density of states for alkali adsorbed WXBC monolayers, tables (structural parameters, energies). See https://doi.org/10.1039/d2ra04488a |
| ‡ All authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |