DOI:
10.1039/D2RA04468D
(Paper)
RSC Adv., 2022,
12, 25054-25059
Zinc dendrite suppression by a novel additive combination for rechargeable aqueous zinc batteries†
Received
19th July 2022
, Accepted 24th August 2022
First published on 2nd September 2022
Abstract
With the advantages of low cost, good safety, and easy assembly, aqueous zinc batteries (AZBs) are expected to be a promising energy storage device. However, AZBs are compromised by Zn dendrites and the hydrogen evolution reaction. Herein, we use polyethylene glycol-200 (PEG-200) and benzylidene acetone (BDA) as additives in the electrolyte of AZBs in order to inhibit Zn dendrite growth and side reactions, thus improving the cycle performance of the Zn electrode. PEG-200 can be not only used as a co-solvent for BDA but also as a surfactant to achieve a uniform interfacial electric field. As a brightening agent, BDA forms a diffusion layer on the plating substrate, which increases the electrochemical polarization and nucleation overpotential, increases the number of active nucleation sites, and finally refines the grain size of the zinc deposit. The surface of the symmetric battery electrode with electrolyte containing PEG-200 additive is smooth after cycling, and dendrite formation is successfully suppressed. The Zn–Zn symmetric cell with additive-containing electrolyte has a higher nucleation overpotential and a cyclic stability for as long as 890 h (only 48 h for the unmodified symmetric cell). This is due to the adsorption of the additive on the negative electrode, which homogenizes the deposition interface and reduces the contact of the negative electrode with water.
1. Introduction
With the merits of abundant sources, low price, chemical stability, high theoretical capacity (820 mA h g−1, 5855 mA h L−1), low reduction potential (−0.76 V vs. SHE), and non-toxicity, metallic zinc is considered a good negative electrode material for aqueous-based batteries (AZBs) and is widely used in rechargeable zinc-ion, zinc-cobalt, and zinc-air batteries.1–3 However, similar to metallic lithium batteries, the application of metallic zinc batteries is also hindered by dendrite formation and side reactions.4–8 It is well known that during zinc deposition, the nucleation and growth processes of zinc are driven by the electric field on the negative surface. Zinc is preferentially deposited at parts of the surface where the electric field is strong, and at the uneven surface of the negative electrode, more zinc will be deposited on the elevated parts of the surface, eventually forming dendrites that pierce the separators and thus cause a short circuit. Therefore, a uniform interfacial electric field can ensure the nucleation and growth of zinc ions, which will greatly improve the cycling performance of aqueous zinc batteries.9–11
Many methods have been developed for the inhibition of dendrite growth and side reactions, such as electrolyte optimization,12–14 artificial SEI protective layers,15–22 epitaxial electrodeposition,23 and alloying.6,24–26 Optimized electrolytes (e.g., diethyl ether,27 methanol,13 ethylene glycol,28,29 acetonitrile,30,31 sodium dodecyl sulfate12) are an effective and promising modification method, and additives can adsorb on depressions of the negative surface to flatten the negative electrode, homogenize the interfacial current, optimize the Zn2+ flux, avoid the “tip effect”, and allow the uniform nucleation of zinc ions.17,29 For example, Xu et al. added 2 vol% diethyl ether (Et2O) to the aqueous electrolyte. Due to the preferential adsorption of ether at the high-potential dendrite tips, Zn2+ deposition occurred in other regions, which flattened the Zn negative surface, reduced the dendrite growth rate, and at the same time reduced the evolution of hydrogen. Finally, the Zn–MnO2 cell exhibited excellent cycling performance.27
In the conventional galvanizing industry, polyethylene glycol-200 (PEG-200) is usually classified as the primary additive that levels and smoothens the zinc coating while also acting as a co-solvent for benzylidene acetone (BDA)32 (PEG-200 is soluble in aqueous solution, while BDA is insoluble in aqueous solution, but soluble in PEG-200, so PEG-200 and BDA must be used together to make the plating solution efficient33). BDA is classified as a secondary additive agent and used as a brightener in zinc electrodeposition. Both PEG-200 and BDA do not form complexes with Zn2+ ions. DBA can adsorb to zinc protrusions via the aromatic ring, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, and/or via the carbonyl group.34 Morón et al.33 used BDA and PEG200 as additives to compare the effects of the presence of a CC double bond and a carbonyl group in the aliphatic chain on the electrodeposition mechanism, morphology, preferred orientation and corrosion resistance of the Zn deposits. The zinc coatings obtained in the presence of BDA–PEG200 associates were metallic gray, compact, adherent and smooth. Meanwhile, the adsorption capacity of BDA is higher than that of PEG-200. Due to the unevenness of the negative electrode before deposition, PEG-200 and BDA adsorbed to the electrode protrusions can block all the active sites and impede the transport of ions to the electrode surface. When the potential is high enough, PEG-200 and BDA are desorbed from the zinc surface. Subsequently, our expectation was reached and zinc was deposited uniformly on the negative surface.17,33,34
C double bond, and/or via the carbonyl group.34 Morón et al.33 used BDA and PEG200 as additives to compare the effects of the presence of a CC double bond and a carbonyl group in the aliphatic chain on the electrodeposition mechanism, morphology, preferred orientation and corrosion resistance of the Zn deposits. The zinc coatings obtained in the presence of BDA–PEG200 associates were metallic gray, compact, adherent and smooth. Meanwhile, the adsorption capacity of BDA is higher than that of PEG-200. Due to the unevenness of the negative electrode before deposition, PEG-200 and BDA adsorbed to the electrode protrusions can block all the active sites and impede the transport of ions to the electrode surface. When the potential is high enough, PEG-200 and BDA are desorbed from the zinc surface. Subsequently, our expectation was reached and zinc was deposited uniformly on the negative surface.17,33,34
Inspired by this, we used PEG-200 with BDA as an additive to modify the electrochemical properties of AZBs. Compared with the conventional ZnSO4 electrolyte, the electrolyte containing PEG-200 and BDA additives exhibited a lower contact angle, higher corrosion potential, lower corrosion current density, and higher overpotential, and the cyclic stability of the symmetric cell reached up to 890 h, it was 18 times that of unmodified cell (unmodified cycle time is only 48 h).
2. Experimental section
2.1 Material preparation
ZnSO4·7H2O (99.5%) was purchased from Macklin (Shanghai, China), PEG-200 was purchased from Kelong (Chengdu, China), and BDA was purchased from Damas-beta (Shanghai, China). Zinc foil (thickness: 0.2 mm) was purchased from Aiweixin (Tianjin, China). All other reagents were of analytical grade and used directly without purification. Deionized water was used to prepare all aqueous electrolytes. ZnSO4 solution (1 mol L−1) was used as the electrolyte, which was modified by adding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm PEG-200 and 10–100 ppm BDA.
000 ppm PEG-200 and 10–100 ppm BDA.
1 mol L−1 ZnSO4 solution was used as the electrolyte, which was modified by adding 10000 ppm PEG-200 (ref. 32) and 10–100 ppm BDA. The zinc foil without further modification was cut into discs with the diameter of 1/2 inches, washed with acetone, and then vacuum dried. Glass fiber filters were used as a separator. A simple device was used to assemble the cells, which facilitated the disassembly of the electrodes, as shown in Fig. S1.† According to the cycle results of Zn–Zn symmetric batteries containing different amounts of BDA additive, 10000 ppm PEG-200 and 50 ppm BDA were the optimal additives, and three different electrolytes named A1, A2, and A3 were chosen for comparative studies, as shown in Table S1 and Fig. S1.†
The optimal amount of BDA was mainly used as a reference for the cycling performance of symmetric cells with different amounts, as shown in Fig. S1(a).† BDA addition was studied using an electrolyte of 1 M ZnSO4 + 10000 ppm PEG-200, since BDA is insoluble in water, but PEG-200 increases the solubility of BDA. Four gradients were also set for the addition of BDA, and the optimal cycle performance of the symmetric cell was achieved at 50 ppm for 890 h. Therefore, 1 M ZnSO4 + 10000 ppm PEG-200 + 50 ppm BDA was chosen as the electrolyte with optimal concentration. The impedance of cells containing the different electrolytes also increased with the added amount, which was due to the adsorption of PEG-200 and BDA on the electrode surface and resulted in increase in the resistance to ion transfer in the cell, as shown in Fig. S1(b).† So, the main study ratio was determined by Fig. S1† as A3 (1 M ZnSO4 + 10000 ppm PEG-200 + 50 ppm BDA).
2.2 Materials characterization
The wettability of the negative electrode with the electrolyte was tested with a contact angle tester (KRUSS, Germany). Scanning electron microscopy (SEM, EVO MA15 ZEISS) was used to characterize the morphology. The surface composition of the samples has been confirmed by X-ray diffraction (XRD, X Pert PRO MPD) with Cu Kα radiation (1.54 Å) in the scan range of 2θ = 10° to 90°.
2.3 Electrochemical tests
The symmetric cells were assembled in a simple device (Fig. S2†). Linear sweep voltammetry (LSV) and Tafel plots were measured on a three-electrode device. LSV was measured using a platinum electrode as the working electrode, zinc foil as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode at the scan rate of 1 mV s−1. For a better comparison, all potentials measured against SCE were converted to the reversible hydrogen electrode (RHE) scale in this work using E(RHE) = E(Hg/HgCl2) + 0.0591 × pH + 0.24, where pH values of electrolytes were determined by BANTE 90Q.35 Tafel plots were measured using metallic zinc as the working electrode, a platinum electrode as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. All electrochemical tests were performed at room temperature. Linear sweep voltammetry (LSV), Tafel plots, and electrochemical impedance spectroscopy (EIS) were measured using an electrochemical workstation (Reference 3000, Gamry, USA). The electrochemical stability windows of the different electrolytes were tested via LSV at the scan rate of 1 mV s−1. EIS data were recorded between 106 and 0.01 Hz at an amplitude of 10 mV. The corrosion potential and corrosion current were calculated by fitting the Tafel plots. Galvanostatic charge/discharge (GCD) and long-term stability were tested on a NEWARE multi-channel workstation (CT-4800T-5V10mA, China) at 30 °C.
3. Results and discussion
3.1 Characterization of the basic electrolyte properties
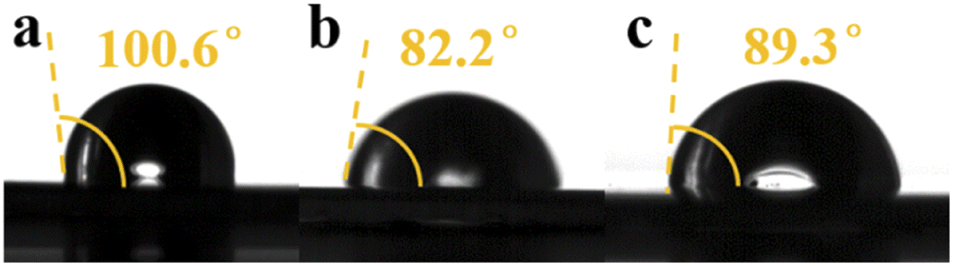
The wettability of the Zn electrode directly influences the energy barrier for Zn nucleation and H2 evolution.13 Therefore, we tested the contact angle between the electrolyte and metallic zinc. Compared with A1, the contact angle decreased from 100.6° to 82.2° with the addition of PEG-200 to the electrolyte, while the contact angle increased from 82.2° to 89.3° with the addition of BDA, as shown in Fig. 1. The addition of hydrophilic PEG-200 increased the overall hydrophilicity, and the addition of hydrophobic BDA increased the overall hydrophobicity. An increased hydrophilicity can accelerate the diffusion kinetics of Zn2+ ions and thus avoid the formation of dendrites. However, high hydrophilicity usually improves the contact between Zn and water, which may aggravate side reactions.7
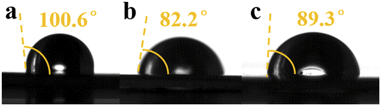 |
| | Fig. 1 Contact angle between metallic zinc and (a) A1, (b) A2, and (c) A3. | |
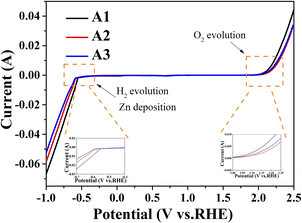
The electrochemical stability window of the electrolyte was measured by anodic and cathodic LSV scans at a scan rate of 10 mV s−1, as shown in Fig. 2. As shown below is the hydrogen evolution reaction (HER)7 in acidic solution:
and the oxygen evolution reaction (OER) in acidic solution is
| | |
2H2O → O2 + 4H+ + 4e−
| (2) |
 |
| | Fig. 2 Electrochemical stability windows of three electrolytes. | |
All electrolytes tested in this work were acidic, and the pH values are shown in Table S2.† As shown in Fig. 2, the hydrogen and oxygen evolution potentials of the electrolyte were changed after the addition of additives, with the hydrogen evolution potential being more negative and the oxygen evolution potential being more positive. The hydrogen evolution potential of A3 is more negative than that of A2, indicating that BDA as a brightener can effectively reduce the occurrence of side reactions. And the oxygen evolution potential of A2 was more positive than that of A3, indicating that PEG-200 played a dominant role in the increase of oxygen evolution potential. The results showed that the additive makes the electrochemical stability window of the electrolyte larger and the electrolyte becomes more stable, and H2O was more difficult to decompose, which inhibits the occurrence of side reactions.7,12
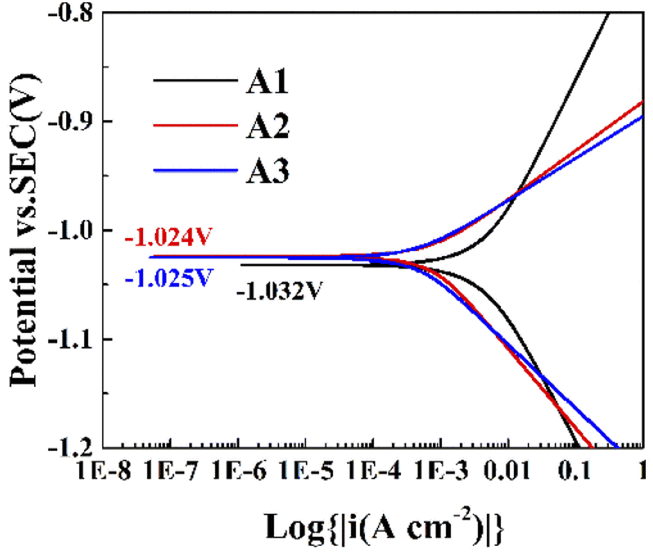
Fig. 3 shows the Tafel plots of the zinc negative in the three electrolytes, and the corrosion potentials and corrosion current densities of the three electrolytes can be obtained by fitting the curves. According to Table S3,† the corrosion potentials of A1, A2, and A3 were −1.032, −1.024, and −1.025 V, and corrosion current densities of A1, A2, and A3 were 4.45 × 10−3, 0.6 × 10−3, and 0.9 × 10−3 A cm−2. After the addition of PEG-200 to 1 M ZnSO4 solution, a more positive corrosion potential and smaller corrosion current density were obtained, showing that the corrosion reaction occurred with a smaller tendency and a lower rate.17,36,37 After the addition of BDA, the corrosion potential decreased, the corrosion current density decreased, and the electrode exhibited a slightly larger corrosion reaction trend and a lower corrosion rate. As a hydrophobic molecule, BDA decreased the wettability of the electrode with the electrolyte, which reduced the diffusion kinetics of zinc ions and decreased the corrosion rate of the electrode.
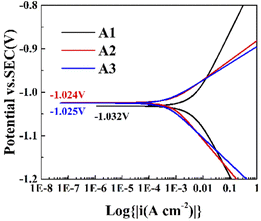 |
| | Fig. 3 Tafel plots of metallic zinc in three electrolytes. | |
To observe the by-products of the reaction between the metallic zinc and the electrolyte, we immersed the zinc flakes in the three electrolytes for seven days. The weakly acidic electrolyte showed a higher HER thermodynamic trend due to its higher H+ activity, as shown by the following reaction equations:1
| | |
2H2O(l) + 2e− → H2(g) + 2OH−(aq.)
| (3) |
| | |
4Zn2+(aq.) + 6OH−(aq.) + SO42−(aq.) → Zn4SO4(OH)6(s)
| (4) |
| | |
Zn2+(aq.) + 2OH−(aq.) → ZnO(s) + H2O(l)
| (5) |
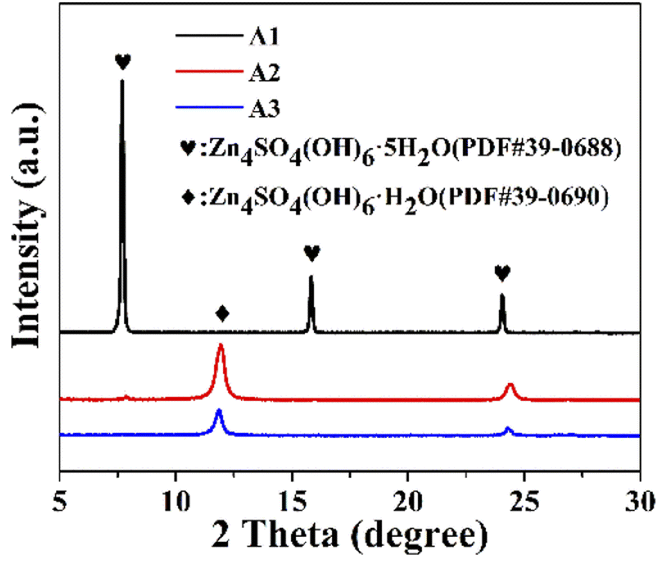
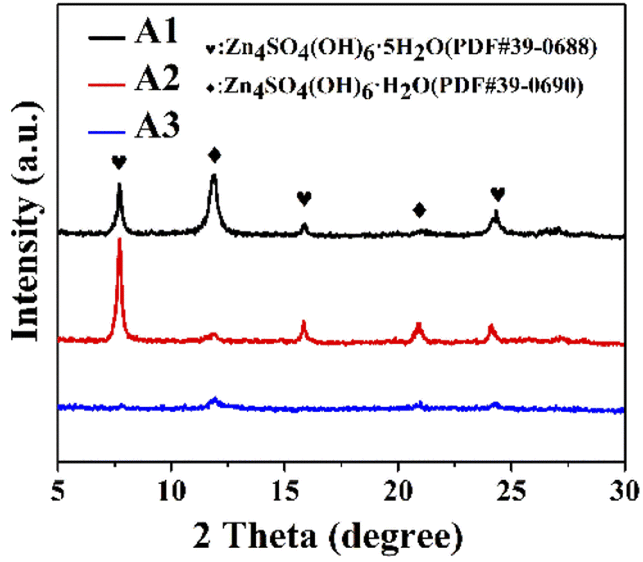
As the acidic electrolyte corroded on the metallic zinc surface, hydrogen was released, which resulted in a locally alkaline electrolyte, while solvation generated Zn4SO4(OH)6·5H2O, and the inert corrosion by-products affected the diffusion of electrons and ions at the interface.20 The by-product generated on the surface of the zinc flakes after immersion was Zn4SO4(OH)6·5H2O (PDF#39-0688), as shown in Fig. 4. After immersion in the ZnSO4 electrolyte, the peaks of Zn4SO4(OH)6·5H2O were mainly located at 7.7°, 15.83°, and 24.08°. In contrast, after the addition of PEG-200 or PEG-200 and BDA, the XRD characteristic peaks of the by-products at 11.9° and 24.32° were mainly attributed to Zn4SO4(OH)6·H2O (PDF#39-0690). This suggests that the addition of PEG-200 and BDA reduced the contact between water and metallic zinc, which may reduce the occurrence of side reactions.
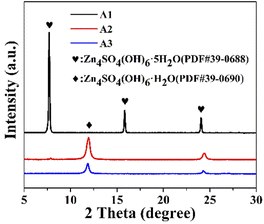 |
| | Fig. 4 XRD of Zn electrodes after immersion in A1, A2 and A3 for 7 days, respectively. | |
The SEM results showed that metallic zinc generated many hexagonal shapes on the surface after seven days of immersion, as shown in Fig. 5. It can be seen that metallic zinc immersed in A1, A2, and A3 generated hexagonal zinc at the surface, which is consistent with the literature.23,38 However, much more hexagonal zinc was generated by immersion in A1 than in A2 and A3. The size of the hexagonal zinc generated by immersion in A3 solution was much smaller than that generated by immersion in A1 and A2, as BDA in the additive can have the effect of refining the deposited particles as a brightening agent. The immersion results indicate that the additives can improve zinc deposition.
 |
| | Fig. 5 SEM images of Zn electrodes after immersion in A1 (a), A2 (b) and A3 (c) for 7 days, respectively. | |
3.2 The impact of electrolyte additives on Zn dendrites
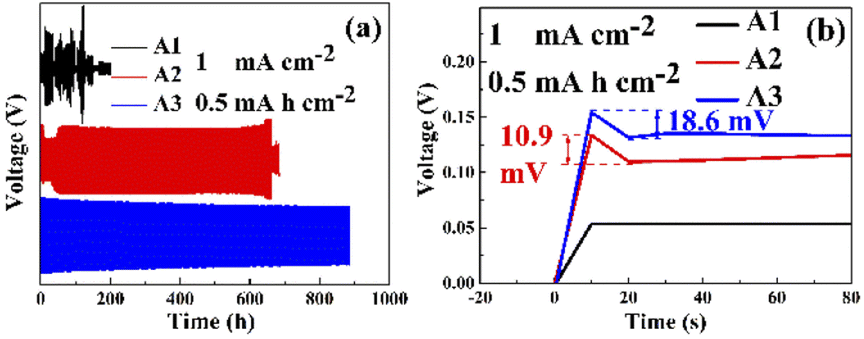
Fig. 6(a) shows the GCD of Zn–Zn symmetric cells with different electrolytes. Due to the irregularity of the zinc sheet surface, the zinc sheet of the cell with PEG-200 electrolyte reached uniformity after 48 cycles before the voltage reached the plateau. The symmetric cells assembled using A1 failed shortly after startup. However, after the addition of PEG-200 to the electrolyte, the symmetric cell exhibited a cyclic stability of 659 h, which increased to 890 h after the addition of PEG-200 and BDA. It showed that both PEG-200 and BDA could improve the cycling performance of AZBs. For A2 and A3, the adsorption of additives on the electrode surface increased the impedance of the cells and resulted in a larger onset voltage with increased electrochemical polarization and nucleation overpotential, while the number of active sites for nucleation was increased and the grain size of the zinc deposit was refined.17,33 Fig. 6(b) shows the nucleation overpotentials of A2 and A3 were 10.9 and 18.6 mV, respectively, while the nucleation overpotential of A1 was only 0.3 mV, as the adsorption of PEG-200 and BDA on the electrode surface blocks all the active sites on the electrode surface and hinders the transport of ions to the electrode surface. However, when the voltage was sufficiently high, a large amount of zinc was deposited on the negative surface after the desorption of PEG-200 and BDA from the electrode surface. This was also confirmed by EIS (Fig. 7), revealing charge-transfer resistances (Rct) for A1, A2, and A3 of 63, 517.2, and 722.4 Ω, and surface film (electrode/electrolyte interface) resistances (Rsf) for A2 and A3 of 316.3 and 436.9 Ω, respectively. This indicates that the additive was successfully adsorbed on the electrode surface.17
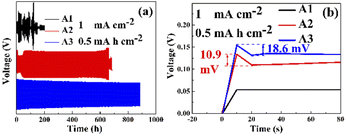 |
| | Fig. 6 (a) GCD performances of Zn–Zn symmetric cells with different electrolytes (with 1 mA h cm−2 at 0.5 mA cm−2), (b) the nucleation overpotentials of Zn–Zn symmetric cells with different electrolytes (with 1 mA h cm−2 at 0.5 mA cm−2). | |
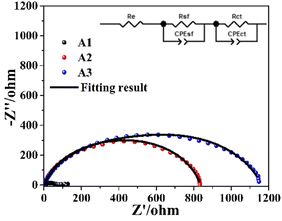 |
| | Fig. 7 EIS data for zinc negative in different electrolytes. | |
The negative electrode after 50 cycles was tested by XRD (Fig. 8), showing the characteristic peaks of the ZnSO4 electrolyte at 7.7°, 15.83°, and 24.08° for Zn4SO4(OH)6·5H2O and 11.9° for Zn4SO4(OH)6·H2O. However, after the addition of PEG-200, the characteristic peaks of Zn4SO4(OH)6·5H2O were observed at 7.7°, 15.83°, and 24.08°, and the characteristic peaks of Zn4SO4(OH)6·H2O were observed at 11.9° and 20.9°. In contrast, no characteristic peaks of by-products were observed after the addition of PEF-200 and BDA, indicating that the production of by-products was successfully suppressed.
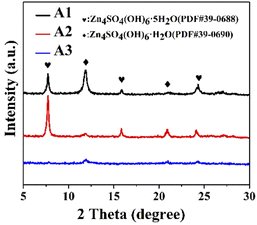 |
| | Fig. 8 XRD of Zn electrodes after 50 cycles with A1, A2 and A3, respectively. | |
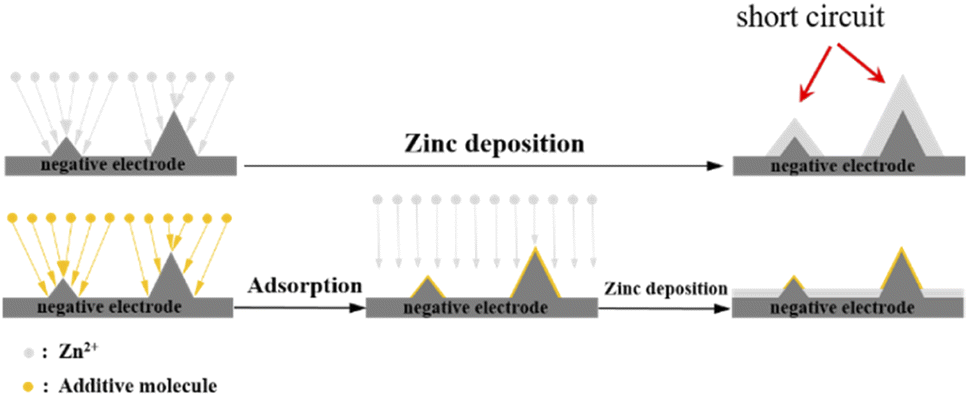
Fig. 9 shows the SEM images of the negative electrode surface of the symmetric cell after 50 cycles. Fig. 9(a) shows that when A1 was used as the electrolyte, significant dendrite formation occurred on the negative electrode surface after cycling. In contrast, when A2 and A3 were used as the electrolytes, the additives adsorbed on the negative electrode surface after cycling. This hindered the continuous growth of dendrites, indicating that the additives can inhibit the formation of dendrites during cycling. The adsorption of the additives PEG-200 and BDA on elevated regions of the negative electrode surface shielded the local high current and avoided the “tip effect”, allowing uniform zinc deposition on the negatively charged surface. As a brightening agent, BDA refined the deposited particles, resulting in a uniform zinc layer, as shown in Fig. 10. Therefore, the cycle performance of the symmetric battery can be significantly improved by the addition of PEG-200 and BDA into the electrolyte.
 |
| | Fig. 9 SEM images of Zn electrodes after 50 cycles with A1 (a), A2 (b) and A3 (c), respectively. | |
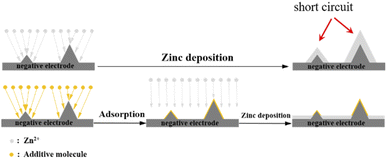 |
| | Fig. 10 Zinc deposition process with and without additives. | |
4. Conclusions
In this paper, we used PEG-200 and BDA to modify the ZnSO4 electrolyte of AZBs. PEG-200 and BDA adsorbed on the electrode surface can block the active sites and impede the transport of ions to the electrode surface. However, when the overpotential is high enough, PEG-200 and BDA desorbed from the zinc surface followed by zinc deposition on the negative electrode surface. The results show that the addition of PEG-200 and BDA to the ZnSO4 electrolyte improved the wettability of the metallic zinc with the electrolyte, increased the electrochemical window of the electrolyte, and H2O was more difficult to decompose. Since PEG-200 and BDA can adsorb on the negative surface, these additives reduced the corrosion tendency and corrosion rate of the negative electrode in the electrolyte and also facilitated the uniform deposition of zinc on the negative surface. With the additive combination in the electrolyte, the long-term cyclic stability of the Zn–Zn symmetric cell exceeded 890 h.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by National Key R&D Program of China (2021YFB4001502), National Natural Science Foundation of China (No. 21875056 and No. 22075231), and the Sichuan Science and Technology Program (No. 2020YFSY0026 and No. 2021YFSY0022). The authors also appreciate the financial support by special project for the central government to guide the development of local science and technology in Sichuan Province.
References
- W. C. Du, E. H. X. Ang, Y. Yang, Y. F. Zhang, M. H. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- B. Y. Tang, L. T. Shan, S. Q. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- L. B. Yuan, J. N. Hao, C. C. Kao, C. Wu, H. K. Liu, S. X. Dou and S. Z. Qiao, Energy Environ. Sci., 2021, 14, 5669–5689 RSC.
- T. S. Zhang, Y. Tang, S. Guo, X. X. Cao, A. Q. Pan, G. Z. Fang, J. Zhou and S. Q. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 RSC.
- F. Wang, O. Borodin, T. Gao, X. L. Fan, W. Sun, F. D. Han, A. Faraone, J. A. Dura, K. Xu and C. S. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- S. B. Wang, Q. Ran, R. Q. Yao, H. Shi, Z. Wen, M. Zhao, X. Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef PubMed.
- M. Li, Z. L. Li, X. P. Wang, J. S. Meng, X. Liu, B. K. Wu, C. H. Han and L. Q. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC.
- V. C. Ho, H. Lim, M. J. Kim and J. Mun, Chem.–Asian J., 2022, 17, e202200289 CrossRef CAS PubMed.
- Q. Yang, G. J. Bang, Y. Guo, Z. X. Liu, B. X. Yon, D. H. Wang, Z. D. Huang, X. L. Li, J. Fan and C. Y. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed.
- M. Zhou, Y. Chen, G. Fang and S. Liang, Energy Storage Mater., 2022, 45, 618–646 CrossRef.
- Y. Li, Z. H. Wang, Y. Cai, M. E. Pam, Y. K. Yang, D. H. Zhang, Y. Wang and S. Z. Huang, Energy Environ. Mater., 2022, 1–29 Search PubMed.
- Z. G. Hou, X. Q. Zhang, X. N. Li, Y. C. Zhu, J. W. Liang and Y. T. Qian, J. Mater. Chem. A, 2017, 5, 730–738 RSC.
- J. N. Hao, L. B. Yuan, C. Ye, D. L. Chao, K. Davey, Z. P. Guo and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- T. N. T. Tran, M. S. Zhao, S. J. Geng and D. G. Ivey, Batteries Supercaps, 2022, 5, e202100420 Search PubMed.
- Q. Q. Lu, C. C. Liu, Y. H. Du, X. Y. Wang, L. Ding, A. Omar and D. Mikhailova, ACS Appl. Mater. Interfaces, 2021, 13, 16869–16875 CrossRef CAS PubMed.
- X. Zeng, K. Xie, S. Liu, S. Zhang, J. Hao, J. Liu, W. K. Pang, J. Liu, P. Rao and Q. Wang, Energy Environ. Sci., 2021, 14, 5947–5957 RSC.
- Z. M. Zhao, J. W. Zhao, Z. L. Hu, J. D. Li, J. J. Li, Y. J. Zhang, C. Wang and G. L. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- L. S. Cao, D. Li, T. Deng, Q. Li and C. S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19292–19296 CrossRef CAS PubMed.
- M. W. Cui, Y. Xiao, L. T. Kang, W. Du, Y. F. Gao, X. Q. Sun, Y. L. Zhou, X. M. Li, H. F. Li, F. Y. Jiang and C. Y. Zhi, ACS Appl. Energy Mater., 2019, 2, 6490–6496 CrossRef CAS.
- X. T. Zhang, J. X. Li, D. Y. Liu, M. K. Liu, T. S. Zhou, K. W. Qi, L. Shi, Y. C. Zhu and Y. T. Qian, Energy Environ. Sci., 2021, 14, 3120–3129 RSC.
- L. Tan, C. Wei, Y. Zhang, Y. An, S. Xiong and J. Feng, Chem. Eng. J., 2022, 431, 134277 CrossRef CAS.
- R. Guo, X. Liu, F. Xia, Y. Jiang, H. Zhang, M. Huang, C. Niu, J. Wu, Y. Zhao, X. Wang, C. Han and L. Mai, Adv. Mater., 2022, 34, e2202188 CrossRef PubMed.
- J. X. Zheng, Q. Zhao, T. Tang, J. F. Yin, C. D. Quilty, G. D. Renderos, X. T. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. H. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- X. Y. Fan, H. Yang, X. X. Wang, J. X. Han, Y. Wu, L. Gou, D. L. Li and Y. L. Ding, Adv. Mater. Interfaces, 2021, 8, 2002184 CrossRef CAS.
- Z. Cai, Y. T. Ou, J. D. Wang, R. Xiao, L. Fu, Z. Yuan, R. M. Zhan and Y. M. Sun, Energy Storage Mater., 2020, 27, 205–211 CrossRef.
- P. Xiao, H. Li, J. Fu, C. Zeng, Y. Zhao, T. Zhai and H. Li, Energy Environ. Sci., 2022, 15, 1638–1646 RSC.
- W. N. Xu, K. N. Zhao, W. C. Huo, Y. Z. Wang, G. Yao, X. Gu, H. W. Cheng, L. Q. Mai, C. G. Hu and X. D. Wang, Nano Energy, 2019, 62, 275–281 CrossRef CAS.
- N. Wang, Y. Yang, X. Qiu, X. L. Dong, Y. G. Wang and Y. Y. Xia, ChemSusChem, 2020, 13, 5556–5564 CrossRef CAS PubMed.
- R. Z. Qin, Y. T. Wang, M. Z. Zhang, Y. Wang, S. X. Ding, A. Y. Song, H. C. Yi, L. Y. Yang, Y. L. Song, Y. H. Cui, J. Liu, Z. Q. Wang, S. N. Li, Q. H. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS.
- J. Q. Shi, K. X. Xia, L. J. Liu, C. Liu, Q. Zhang, L. Li, X. Z. Zhou, J. Liang and Z. L. Tao, Electrochim. Acta, 2020, 358, 136937 CrossRef CAS.
- Z. Hou, H. Tan, Y. Gao, M. H. Li, Z. H. Lu and B. Zhang, J. Mater. Chem. A, 2020, 8, 19367–19374 RSC.
- S. J. Banik and R. Akolkar, J. Electrochem. Soc., 2013, 160, D519–D523 CrossRef CAS.
- L. E. Moron, A. Mendez, F. Castaneda, J. G. Flores, L. Ortiz-Frade, Y. Meas and G. Trejo, Surf. Coat. Technol., 2011, 205, 4985–4992 CrossRef CAS.
- L. E. Moron, A. Mendez, J. C. Ballesteros, R. Antano-Lopez, G. Orozco, Y. Meas, R. Ortega-Borges and G. Trejo, J. Electrochem. Soc., 2011, 158, D435–D444 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- M. S. Abdel-All, Z. A. Ahmed and M. S. Hassan, J. Appl. Electrochem., 1992, 22, 1104–1109 CrossRef.
- M. Abdallah, Corros. Sci., 2003, 45, 2705–2716 CrossRef CAS.
- A. L. Xia, X. M. Pu, Y. Y. Tao, H. M. Liu and Y. G. Wang, Appl. Surf. Sci., 2019, 481, 852–859 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiaoliang Zhou
a,
Xiaoliang Zhou