
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA green process for recycling and synthesis of cathode materials LiMn2O4 from spent lithium-ion batteries using citric acid†
Junzhen Wang ,
Kui Huang*,
Haili Dong,
Yuanhuan Lu,
Kunjie Liu,
Zhangqing Chen,
Xinke Shan,
Guoliang Huang and
Lin Wei
,
Kui Huang*,
Haili Dong,
Yuanhuan Lu,
Kunjie Liu,
Zhangqing Chen,
Xinke Shan,
Guoliang Huang and
Lin Wei
School of Resources, Environment and Materials, Guangxi University, 100 Daxue Road, Nanning 530004, China. E-mail: khuang@gxu.edu.cn
First published on 19th August 2022
Abstract
In view of the reducing reagent consumption and secondary pollution caused by recycling spent lithium-ion batteries (LIBs), a relatively green process has been proposed, because the complex process to separate metals and the use of a large number of environmentally unfriendly chemical reagents are not involved. This process combines acid leaching with the resynthesis of the cathode material to recycle LiMn2O4 (LMO) from spent LIBs. The leaching efficiencies of Li and Mn exceeded 94% under the conditions of 1.0 M citric acid concentration, solid–liquid ratio of 60 g L−1, and 60 min leaching time. After the leaching process, spinel LMO was successfully resynthesized by the sol–gel process using leachate. The sample calcined at 700 °C has the best electrochemical performances, and the initial discharge capacity at a 2C rate and capacity retention after 100 cycles were 87.85 mA h g−1 and 93.63%, respectively. The resynthesized cathode material possessed excellent cycling performance, which may result from Al doping. Furthermore, the mechanism of overall reaction and the formation process of complex Mn(C6H6O7)·H2O in the leaching process were explored. This study indicates that citric acid is an effective reagent for recycling cathode materials and the process is feasible.
1. Introduction
The application of lithium-ion batteries (LIBs) has expanded from mobile electronic devices to electric vehicles due to their high voltage platform, high energy density, low self-discharge, and good cycling performance.1 In the near future, LIBs will also become a key driving force in the field of drones and the Internet of Things.2 The development trend of LIBs shows that a huge rise in their market share is immeasurable. LiMn2O4 (LMO) with spinel structure is one of the most promising cathode materials (the others including LiFePO4, LiNixCoyMnzO2, et al.) and has been successfully commercialized in much large-scale energy storage equipment because it has the advantages of rich raw material resources, high safety, and good charge–discharge performance.3,4LIBs will flow into the waste stream after about 1000 charge–discharge cycles (3–5 years).5 Considering the adverse environmental impact of inappropriate disposal, it is urgent to treat these spent batteries with sustainable methods. Hydrometallurgical technology is considered to be more sustainable and excellent in current technologies for recycling spent LIBs because of its features of high recovery efficiency and low energy consumption.6 Generally, the hydrometallurgical processes involve the leaching process to dissolve metals in cathode materials using acid or alkali solution, and then separating and purifying different metals by solvent extraction, chemical precipitation, and electro-deposition.7 However, the emission of waste liquid as a major defect appears due to the use of acid/alkali solution and various chemical reagents. In the traditional leaching processes, studies emphasize the treatment of cathode materials in spent LIBs using inorganic acids such as HNO3,8 HCl,9,10 H2SO4.11,12 Although high leaching efficiency could be provided by inorganic acid, a series of problems involving strong corrosion, toxic gas emissions (Cl2, SOx and NOx), and difficult treatment of waste liquid were caused by its excessive use. Thus, more interest is turned into biodegradable organic acid leaching to avoid secondary pollution. Citric acid,13,14 malic acid,15,16 ascorbic acid17,18 and so on have been used to dissolve spent cathode materials. The leaching of three different organic acids (citric acid, malic acid, aspartic acid) on LiCoO2 was compared in the presence of H2O2 and the results showed that high leaching efficiency was achieved when the leaching agent is citric acid.19 In addition, energy consumption and gas emission are minimal in the whole production of citric acid.19 Musariri et al.20 studied the leaching kinetic behavior of citric and malic acid where a faster kinetic was observed for citric acid (95% for Co, 97% for Li, 99% for Ni within 30 min).
Some organic acids can act as both leaching and reducing agent to simplify leaching processes due to the presence of reductive groups in their chemical structure. Ascorbic acid with alkene glycol group was used to recycle Co and Li as both leaching and reducing agents.17 Citrus fruit juices containing citric acid, malic acid, and ascorbic acid were used by D. Pant and T. Dolker to leach Li, Ni, Co and Mn from spent LIBs.21 The leaching efficiencies of as high as 94–100% can be achieved.
Separation and extraction of valuable metals in leachate after leaching processes is also an indispensable part,22 but complex processes and large reagent consumption often imply some risks of secondary pollution. Resynthesis of cathode materials is thus a good alternative in many recycling methods for spent LIBs to avoid these cumbersome operations. Spent cathode materials were leached and then synthesized by sol–gel process using organic acid as a chelating agent, and the results indicated that micro-morphology and electrochemical properties of resynthesized materials were as good as that of new synthetic materials.4,13 Another method of resynthesis is the coprecipitation method, that is, the metal ions in the leachate were coprecipitated to form a precursor. Subsequently, a cathode material with good electrochemical performances can be prepared by calcining lithium salt and precursor together.23–25 However, the coprecipitation method is only applicable to the recovery of ternary power batteries with many kinds of metal ions.
Based on the above studies, multipurpose organic acids can be used simultaneously as leaching, reducing and chelating agents. In this study, citric acid in the absence of reducing agent was used to leach LiMn2O4 and the optimal leaching conditions of system were examined. Besides, the reaction mechanism between citric acid and LMO was explained from the perspective of the decomposition in solution for citric acid. LMO was resynthesized directly by a simple sol–gel process using the leachate as raw material after the leaching process, and the resynthesized samples have excellent cycling performance.
2. Experimental
2.1 Materials and reagents
The spent LIBs used in this work were purchased from a local small spent battery recycler. All reagents used in this work were of analytical grade, with citric acid and Li2CO3 having a purity of 99.5% and 99.0%, respectively. And all solutions were prepared with ultrapure water.2.2 Experimental procedure
The leaching efficiency χ can be calculated according to eqn (1):
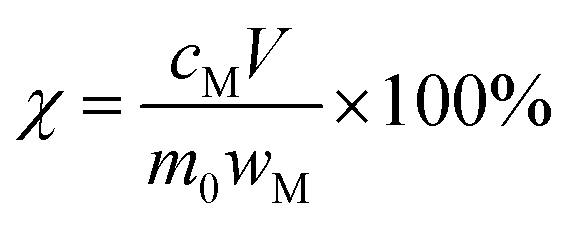 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn was adjusted to 1.05:2 by adding Li2CO3 as required and the pH was adjusted to 6.0 by adding a suitable amount of aqueous ammonia to leachate. And then leachate was heated and stirred at 75 °C in a magnetic stirrer with constant temperature until gel was formed. The gel was dried at 100 °C in over for 24 h to obtain precursor powder. The precursor was thermally treated in two-step in a furnace: it was precalcined at 450 °C for 4 h in the ambient atmosphere and then calcined at 600–800 °C for 16 h (labeled as LMO-600, LMO-700, LMO-800, respectively).
:Mn was adjusted to 1.05:2 by adding Li2CO3 as required and the pH was adjusted to 6.0 by adding a suitable amount of aqueous ammonia to leachate. And then leachate was heated and stirred at 75 °C in a magnetic stirrer with constant temperature until gel was formed. The gel was dried at 100 °C in over for 24 h to obtain precursor powder. The precursor was thermally treated in two-step in a furnace: it was precalcined at 450 °C for 4 h in the ambient atmosphere and then calcined at 600–800 °C for 16 h (labeled as LMO-600, LMO-700, LMO-800, respectively).2.3 Analytical method
The metal ions concentration in the leachate was analysed by inductively coupled plasma optical emission spectroscopy (ICP-OES; Optima 8000DV, PerkinElmer, USA). The phase of cathode materials, precipitates, and resynthesized LMO samples were determined by X-ray diffractometry (XRD; Rigaku, Japan) with Cu-Kα radiation. The surface micro-morphology of the precipitates and the resynthesized LMO samples were observed by scanning electron microscopy (SEM; 800-07334, Phenom, Holland) and field emission scanning electron microscope (FE-SEM; SU8020, Hitachi, Japan) attached with energy-dispersive X-ray spectroscopy (EDS), respectively. In addition, X-ray photoelectron spectroscopy (XPS; ESCALAB 250XI+, Thermo Fisher Scientific, USA) was utilized to examine the valence states of Mn and the chemical composition of the precipitates.Electrochemical tests were conducted by using a CR2032-type coin cell. The cathodes were fabricated by mixing the resynthesized LMO samples with polyvinylidene difluoride (PVDF) and acetylene black (8:1:1 by mass) in N-methyl-2-pyrrolidone to form electrode slurry, which was coated onto Al foil current collector and roll-pressed. The prepared cathode sheets were dried at 80 °C in a vacuum to evaporate solvent. And then coin cells were assembled in glove box filled with high-purity argon. In the coin cells, Li foil and Celgard 2500 membrane as the anode and the separator, respectively. The electrolyte was 1.0 M LiPF6 in a mixture of ethylene carbonate, diethyl carbonate, and dimethyl carbonate (1:1:1 by volume).
Electrochemical tests were performed using a Neware battery testing system (CT-4008-5V10mA-164, Neware, China) from 3.0 to 4.5 V. The assembled cells were tested for 100 cycles at 2C to analyse cycling performance. Various rates (1, 2, 3, 5, and 10C) were also tested to investigate the rate performance of the resynthesized LMO samples. Cyclic voltammogram (CV) tests and electrochemical impedance spectra (EIS) were recorded using an electrochemical workstation (CHI 660D, Chenghua, China).
3. Results and discussion
3.1 Pretreatment of the spent LIBs
The Al foil and the LiMn2O4 material were separated by calcining the cathode active materials at 550 °C in a muffle furnace to burn off the conducting media and binder. Fig. S1† shows XRD patterns before and after heating cathode active materials. The peak of graphite (the 2θ diffraction angle corresponding to graphite (002) crystal plane at about 2θ = 26°) disappeared and the peak intensities of LMO increased in the XRD pattern of cathode materials after heat treatment. Meanwhile, some weak peaks of Mn3O4 were observed. These analysis results indicated that the graphite carbon originating from PVDF and conductive substances has been removed and that the formation of Mn3O4 might be attributed to redox reaction of Mn(III)/Mn(IV) in LMO with carbon at high temperature as valence states of Mn element in Mn3O4 are +2 and +3.The chemical composition of cathode material LMO as determined by acid digestion is given in Table S1,† showing the presence of 3.78% Li and 64.75% Mn along with 0.9% Al.
3.2 Leaching process
The citric acid (C6H8O7) is an organic acid with several donor-active groups, containing three carboxyl groups (–COOH) and one hydroxyl group (–OH), which means that a single C6H8O7 molecule can coordinate with multiple metal ions simultaneously. All pKa values of citric acid had been reported: pK1 = 3.13, pK2 = 4.76, pK3 = 6.40 and pK(OH) ≈ 11.26 The ionization of C6H8O7 is three steps as follows:| H3Cit ⇌ H2Cit− + H+ | (2) |
| H2Cit− ⇌ HCit2− + H+ | (3) |
| HCit2− ⇌ Cit3− + H+ | (4) |
In the structure of spinel LMO, approximately cubic closepacked array of oxides incorporates MnO6 octahedron sharing two opposing corners with tetrahedral LiO4, with Mn atoms residing on the octahedral 16d sites, O atoms on the 32e sites, and the Li atoms occupying the tetrahedral 8a sites.27 The three-dimensional network of octahedral (16c) and tetrahedral (8a) sites is the (111) channels of the spinel lattice during Li+ intercalation and extraction.2 The reaction between C6H8O7 and LiMn2O4 is a multiphase process involving liquid–solid phase. C6H8O7 in solution ionize to numerous H+ to attack and break the strong M–O bond (M is metal ion), and then citrate anions combine with metal ions through complexation. Fig. 1 clearly illustrates the reaction procedure of multiphase. C6H8O7 also played the role of reduction (E0 = −0.18 V) during the metal dissolution process due to the hydroxyl group.7,28,29
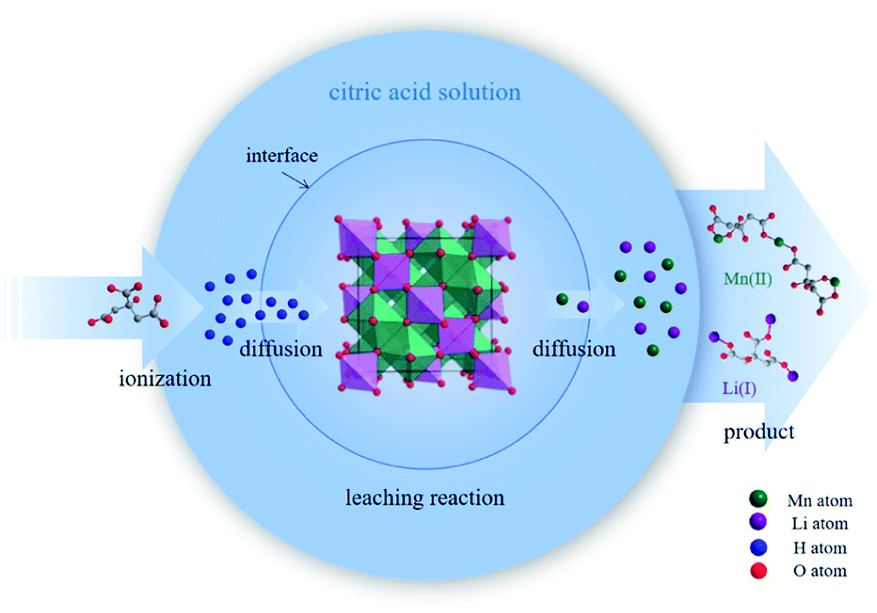 | ||
| Fig. 1 Multiphase reaction of C6H8O7 and LMO. | ||
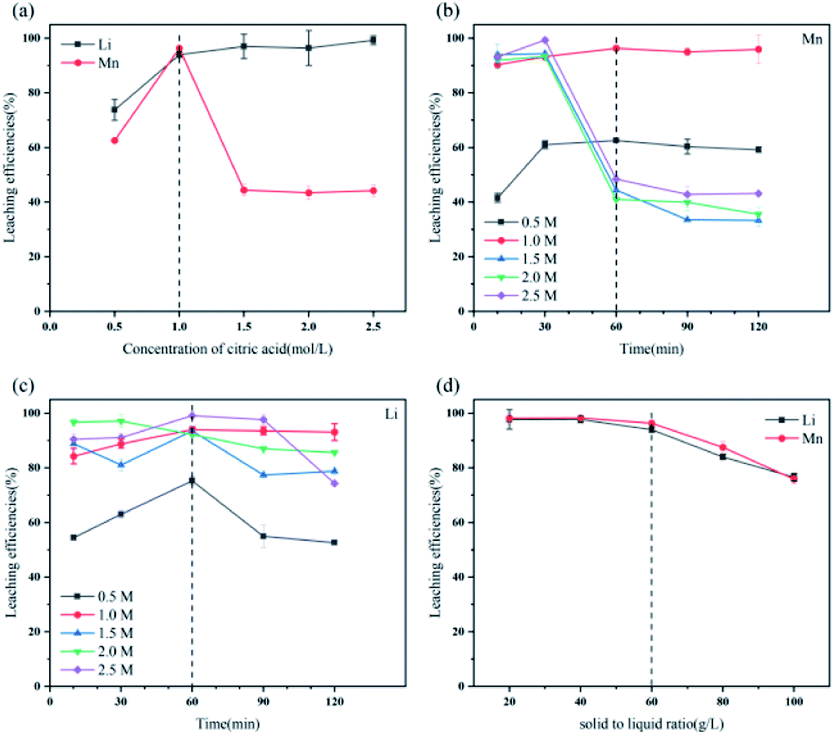 | ||
| Fig. 2 Effect of (a) C6H8O7 concentration, (b) and (c) time, (d) solid–liquid ratio on leaching efficiencies of Li and Mn. | ||
3.3 Analysis of the leached precipitates
The leaching efficiencies of citric acid on LMO were affected significantly due to the formation of white precipitate in the solution of high acid concentration. Fig. S2† shows the XRD pattern of white precipitate during leaching processes. According to the matching result in the DIFFRAC.EVA standard database, the substance in this work was identified as C6H8MnO8, namely monohydrate manganese citrate Mn(C6H6O7)·H2O, which are similar to other studies and belongs to orthorhombic crystal lattice, with space group P212121.30,31Fig. 3 shows the SEM images of white precipitate Mn(C6H6O7)·H2O in leaching processes. As shown in Fig. 3a, single Mn(C6H6O7)·H2O particles were observed in cuboid bar shape (the range from a few micrometers to dozens of micrometers in length), which is consistent with another previous study.30 However, it occurred in situation where particles aggregated with cluster morphology (Fig. 3b) and particle size of them was increasing (Fig. 3c and d) with acid concentration (1.5 to 2.5 M), which are not mentioned in the literature. These might be attributed to higher acid concentration in this work than that (0.3 M citric acid) in other work.28 Similarly, Nefedova et al.31 claimed that after reagents containing manganese ions were added to citric acid solution, small clusters of Mn(C6H6O7)·H2O at molecular level began forming in solution. These clusters then were the role of primary nuclei of particles on which larger aggregates were continuously formed. Mn(C6H6O7)·H2O obtained in leaching processes fitted well a mesokineitc model of solid formation.32 The formation of a solid substance in a supersaturated environment can be regarded as the totality of the processes of nucleation, growth, and aggregation of particles, since the aggregation not only leads to the formation of primary nuclei of particles, but also forms aggregates with a multilevel hierarchical structure.
 | ||
| Fig. 3 SEM images of white precipitate (a) single particles and (b) aggregates formed at 1.5 M acid concentration, and the comparison of aggregates (c) at 1.5 M acid concentration and (d) at 2.5 M acid concentration under the same magnification. | ||
XPS was performed to examine the chemical composition of the precipitates and the overall valence states of Mn element (Fig. 4). After a fine sweep of Mn 2p, Fig. 4a shows that the two broad shoulders with binding energies of 641.5 and 653.8 eV were consistent with the peak of Mn 2p3/2 and Mn 2p1/2, respectively. Based on the result of peak fitting for the Mn 2p3/2 spectrum, the two peaks with binding energies of 642.6 and 641.1 eV represented Mn(IV) and Mn(II), respectively.33 This suggested that the precipitates in leaching processes contained two valence states of Mn(IV) and Mn(II). However, the oxidation state of Mn in the Mn(C6H6O7)·H2O was bivalent, then Mn(IV) should be derived from unreacted LMO. Strong evidence was that the characteristic peak of Li 1s (binding energy between 48.7 and 57.0 eV) in XPS survey (Fig. 4c) was observed,34 which can be indicated that a small quantity of LMO particles were not involved in the leaching reaction. In addition, the 100% of leaching efficiency for Li in this study was unable, even at high acid concentrations of 1.5–2.5 M. It is possible that the precipitate MnC6H6O7·H2O formed after 60 min of leaching was present as a product layer on the surface of the unreacted LMO particles, thus inhibiting their further leaching. The reason that unreacted LMO was not detected by XRD and SEM might result from the low content of this fraction and the minute proportion in solid substance after leaching processes. Strictly speaking, the solid substance after the leaching processes should be a mixture of product Mn(C6H6O7)·H2O and leaching residue LMO.
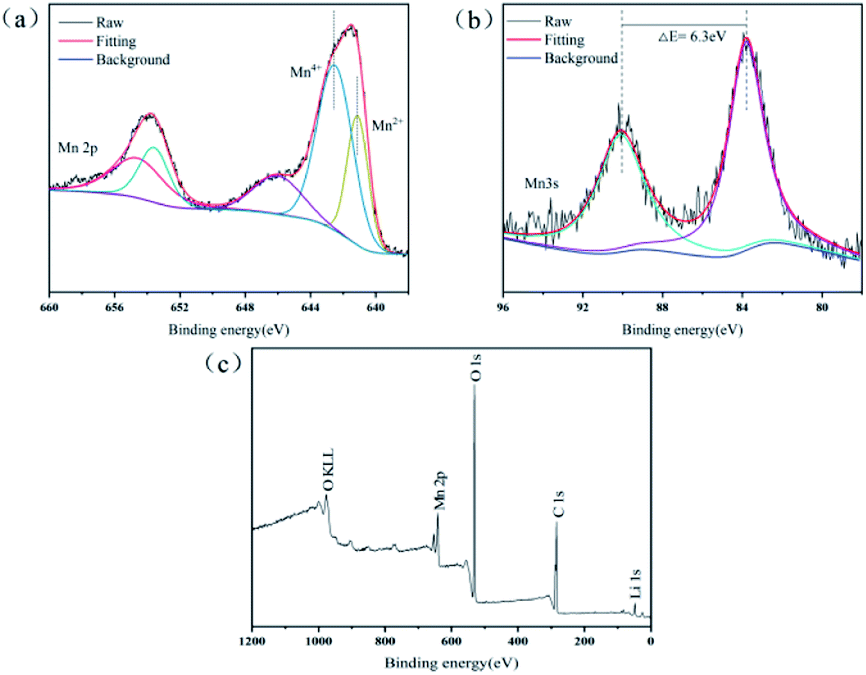 | ||
| Fig. 4 XPS spectra of the Mn(C6H6O7)·H2O. (a) Mn 2p, (b) Mn 3s, (c) XPS Survey. | ||
3.4 Leaching process between citric acid and Li, Mn
Fig. S3† illustrates the ionization of C6H8O7 at different pH values. When the leaching reaction between C6H8O7 and LMO was taken place, four protons of C6H8O7 could be liberated depending on pH values in solution.35 Therefore, at different pH-value conditions, the actual ionization of carboxyl group (–COOH) in the acid solution has some differences, and the form of citrate anion (H2Cit−, HCit2−, Cit3−) varies accordingly. The pH values of leachate after leaching processes under different citric acid concentrations (setting leaching time of 60 min) thus were accurately measured, as shown in Fig. S4.† The pH values of leachate ranged from 4.0 to 7.0 at the condition of 0.5 and 1.0 M, and now the trivalent citrate anion Cit3− (eqn (4)) is predominant in leachate, sufficiently complexing with Li and Mn, which are stable in solution as the structure of Li3Cit and Mn3(Cit)2 respectively. These results are also consistent with the final leaching product of most studies.7,14,36 Similarly, C6H8O7 in the leachate mainly was secondary ionization (eqn (3)) at 1.5–2.5 M. The citrate dianion HCit2− forms Li2HCit and Mn(HCit) (i.e. MnC6H6O7·H2O) with Li and Mn respectively, and MnC6H6O7·H2O is a chelate with poorly solubility.37 Fig. 5a describes the reaction mechanism involved in this leaching process.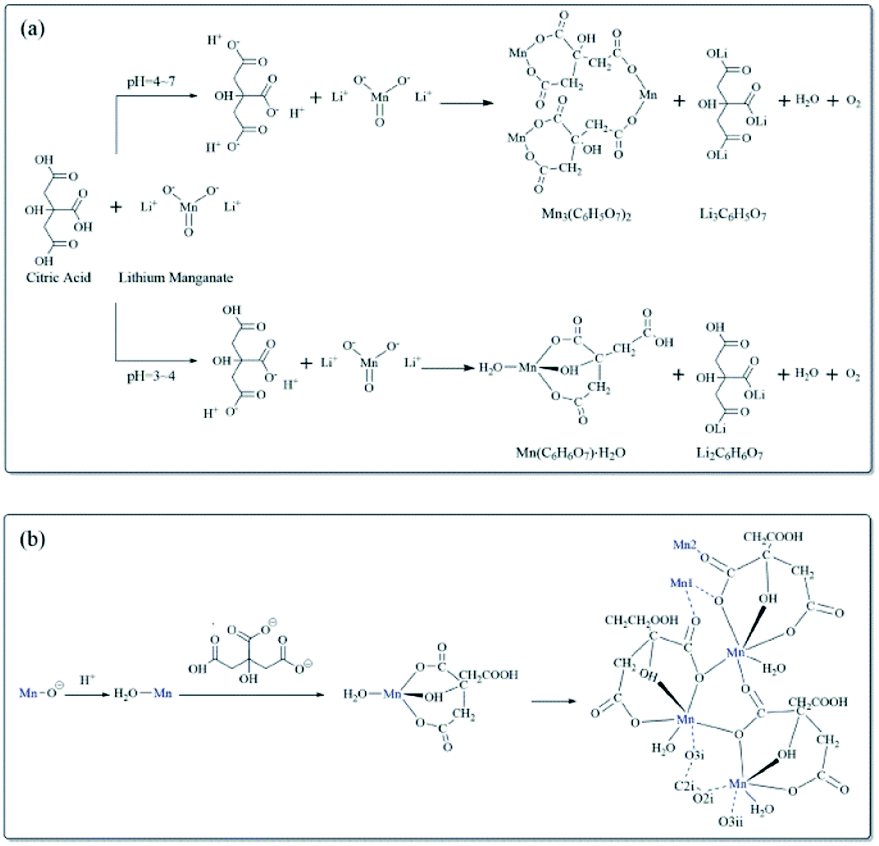 | ||
| Fig. 5 (a) Leaching reaction mechanism of C6H8O7 and LMO. (b) Processes of the Mn(C6H6O7)·H2O nucleation, growth and aggregation. | ||
The leaching reaction of C6H8O7 and metal ions consists of two steps: (1) breaking of M–O bond at the interface of liquid and solid under protons; (2) complexation of citrate anion and metal ions. The leaching of metal Li and Mn in solution depends on not only concentration of H+ but also anionic forms of selected organic acids.38 At high acid concentration, the ionization of citrate anion was inhibited, which then affects the complexation of citrate anion and metal ions, due to the presence of numerous H+ in leachate after leaching processes. Appropriate acid concentration leading to increased pH value of leachate is helpful to complete ionization of C6H8O7 to provide system with more carboxyl groups to complex the metal ions.
Based on a structural study of Mn(C6H6O7)·H2O in other reports, as for the formation of Mn(C6H6O7)·H2O in this study, it can be speculated that hydrogen ions combine with O atoms to form Mn–H2O structure containing a water molecule through the formation of electron pair, when excess hydrogen ions in 1.5 to 2.5 M citric acid solution are provided to disturb Mn–O bonds, and then the citrate dianion chelates to the Mn atom through the α-hydroxyl, the α-carboxyl and one β-carboxyl O atom, leading to tridentate ligand Mn(C6H6O7)·H2O with a free β-carboxylic acid group. The bonded O atom of each α-carboxyl in Mn(C6H6O7)·H2O units further coordinates with an adjacent Mn atom, while the other O atom of this carboxyl group is strongly bonded to another Mn atom. Finally, a helical chain is formed at the molar ratio of Mn/H3Cit = 1:1 due to α-carboxyl as a bridging ligand.39 The tight crystal structure is achieved by hydrogen bonds between neighboring chains. Fig. 5b describes the above coordination and consolidation for a series of atoms, indicating all processes of nucleation, growth, and aggregation of particles were completed in a supersaturated environment, which corresponds to cluster morphology in SEM images shown in Fig. 3b–d.
3.5 Characterization and electrochemical testing of the resynthesized LiMn2O4
The XRD patterns of the resynthesized LMO samples calcined at 600–800 °C were shown in Fig. 6. All the samples corresponding XRD patterns revealed the typical diffraction peaks of the spinel LMO with a space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, and without any impurity peaks. Among all the samples, the LMO700 sample possessed the strongest and sharpest diffraction peaks, indicating the crystallization of samples calcined at 700 °C was relatively good. The lattice parameters of LMO600-800 obtained from Rietveld analysis of these XRD are listed in Table S2† and were increased with the increase of calcined temperature. The (111) peak of sample LMO800 showed a small shift to the left, which corresponded to an increase in lattice parameter from 8.20 to 8.23 Å and an increase in lattice volume from 551.9 to 557.75 Å3. The crystal parameters and cell volume of LMO700-800 sample were significantly greater than those of the LMO600 sample, which may supply faster Li+ movement in three-dimensional channel and better electrochemical performance.4,40
m, and without any impurity peaks. Among all the samples, the LMO700 sample possessed the strongest and sharpest diffraction peaks, indicating the crystallization of samples calcined at 700 °C was relatively good. The lattice parameters of LMO600-800 obtained from Rietveld analysis of these XRD are listed in Table S2† and were increased with the increase of calcined temperature. The (111) peak of sample LMO800 showed a small shift to the left, which corresponded to an increase in lattice parameter from 8.20 to 8.23 Å and an increase in lattice volume from 551.9 to 557.75 Å3. The crystal parameters and cell volume of LMO700-800 sample were significantly greater than those of the LMO600 sample, which may supply faster Li+ movement in three-dimensional channel and better electrochemical performance.4,40
 | ||
| Fig. 6 XRD patterns of LMO sample calcined at 600 °C, 700 °C, 800 °C. | ||
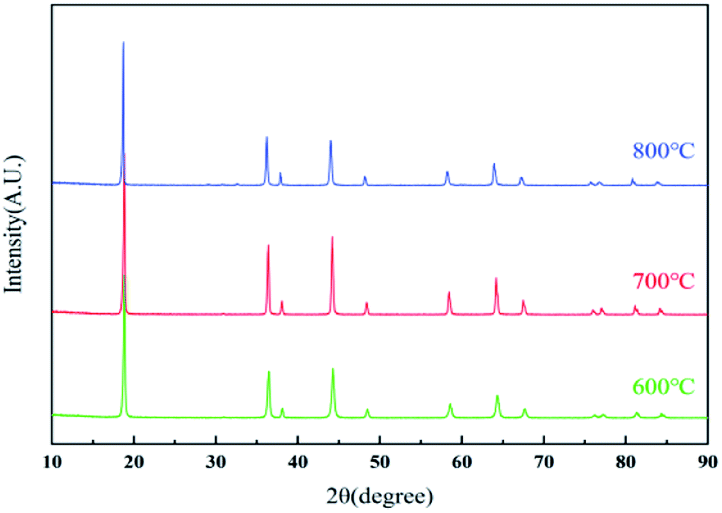
The effect of calcined temperatures on the micro-morphologies of resynthesized LMO was observed by SEM. As shown in Fig. 7a, only a few particles in LMO600 sample had regular spinel morphology and the agglomeration was serious, indicating the crystallinity of sample is poor. Fig. 7b and c show the SEM images of LMO700-800 sample, which possessed more spinel morphologies compared with LMO600 sample, facilitating to improve electrochemical performance. However, a certain degree of agglomeration was observed in the samples calcined at 800 °C, making the non-uniform dispersion of particles. Samples calcined at all three temperatures suffered from a non-uniform particle size distribution, with particles ranging from micrometer to nanometer scale. Studies have shown that nanostructured cathode materials can increase the contact area between the active cathode material and the electrolyte, providing superior electrochemical properties over ordinary materials.41–43 Moreover, partial amorphous particles of small sizes were observed, which is attributed to inadequate time during the calcination process. It is noted that trace Al was detected and uniformly distributed like Mn element and O element when using EDS to verify the element distribution in the samples, as shown in Fig. 8. The presence of trace Al may result from incomplete separation of cathode active materials and Al foil as a current collector, as previously shown (Table S1†), the LMO obtained pretreatment still contained 0.9 wt% of Al impurities.
 | ||
| Fig. 7 SEM images of the resynthesized LMO samples calcined at (a) 600 °C, (b) 700 °C, (c) 800 °C and (d) the single spinel LMO700 particle. | ||
 | ||
| Fig. 8 EDS images of resynthesized LMO700 sample. | ||
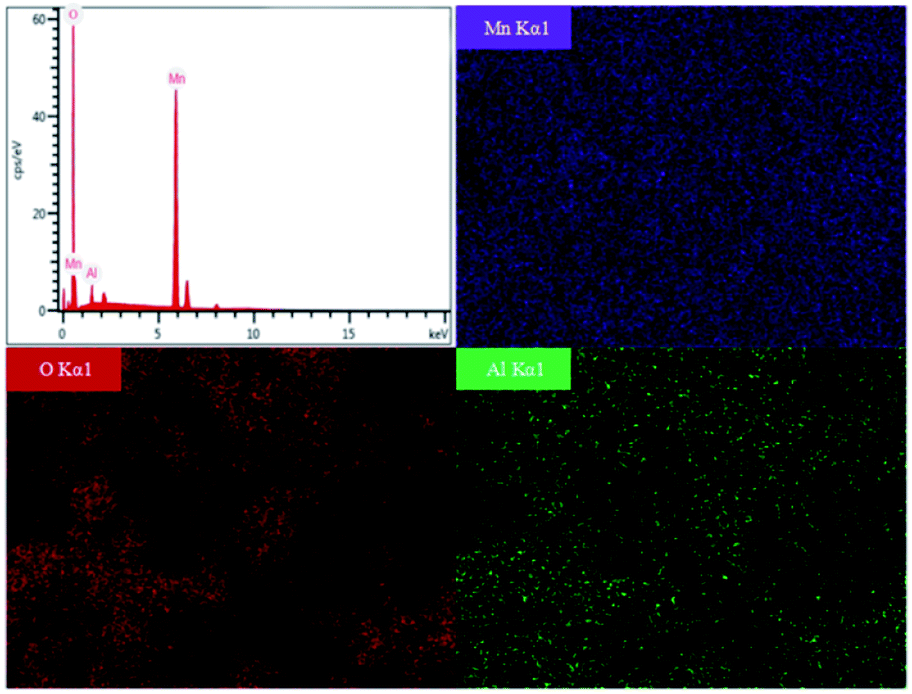
To systematically evaluate the electrochemical performance of resynthesized LMO sample, a series of electrochemical tests were performed from 3.0 to 4.5 V (Fig. 9). Fig. 9a shows that the initial discharge capacities of LMO600-800 sample at 2C rate were 58.11, 87.85, 84.33 mA h g−1 respectively, and the capacity retentions of that after 100 cycles reached 73.98%, 93.63%, 89.64% respectively. The LMO700 and 800 samples possessed excellent spinel structure and thus exhibited better cycling performances than LMO600 sample. The calcined samples at 600 °C lacked regular spinel structure, leading to an obstruction of Li+ movement to exhibit low discharge capacity and fast capacity fading, and these results are consistent with the analysis of XRD and SEM. The rate performance of LMO700 sample was studied (Fig. 9b) to further analyse its electrochemical performance. The reason why discharge capacity decreased with the increase of current density is that electrochemical polarization is enhanced under high current density, causing blocked diffusion of Li+ and incomplete electrochemical reactions.4,13,44 The LMO700 sample was charged and discharged at 1, 2, 3, 5 and 10C for cycling and then back at 1C, and found that the capacity has no decay, showing excellent cycling performance.
 | ||
| Fig. 9 Electrochemical performances of resynthesized LMO sample. (a) Discharge capacities of LMO600-800 sample at 2C, (b) rate test, (c) initial charge–discharge curves (d) CV curves of LMO700 sample. | ||
Fig. 9c and d were initial charge–discharge and CV curves of LMO700 sample, respectively. There are two plateaus in charge–discharge process, corresponding to the realization of two-phase reaction (eqn (5) and (6)) of Li+ intercalation and extraction in electrochemistry.44,45 Two pairs of redox peaks were observed in CV curves of LMO700 sample, showing intrinsic reduction peaks at 4.14 and 4.25 V and oxidation peaks at 3.89 and 4.05 V. These peaks further demonstrate the two-phase reaction of Li+ intercalation and extraction.46
| Mn2O4 + xLi+ + xe− ⇌ LixMn2O4 | (5) |
| LixMn2O4 + (1 − x)Li+ + (1 − x)e− ⇌ LiMn2O4 | (6) |
The impedance plot of resynthesized LMO sample (Fig. 10) exhibited typical Nyquist characteristics, a semicircle at middle and high frequency representing Li+ charge transfer resistance (Rct) at the interface, an inclined line in the lower frequency region representing the Warburg impedance (W), corresponding to the diffusion of Li+ in LMO particles.13,45 The large resistance value means that the movement and diffusion of Li+ were affected by trace Al. When cathode material LMO is doped with Al component, Mn atoms are replaced by Al atoms to occupy the center of octahedron; and strong Al–O binding (Al–O bind: 512 kJ mol−1; Mn–O bind: 402 kJ mol−1) can reduce the collapse of spinel structure due to the Jahn–Taller effect,47 improving cycling stability of spinel LMO.45,48,49 However, if the doped Al component is excessive, Al atoms replace Li atoms in the tetrahedral sites instead of all entering the octahedral sites where the Mn atoms are located.50 Thus, in this situation, Al atoms hinder the channel of Li+ movement and suppress electrochemical reactions in charge–discharge process.45 In a word, the Al element affected the electrochemical performances of LMO sample in our work to a certain extent.
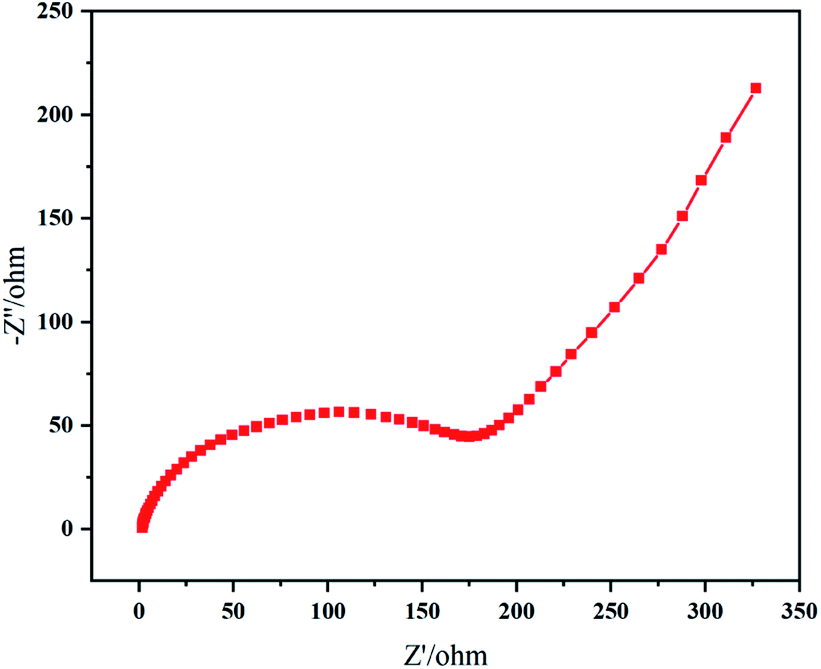 | ||
| Fig. 10 EIS plot of LMO700 sample. | ||
4. Conclusions
The secondary pollution caused by use of reagents (especially inorganic acid) in the process of hydrometallurgical treatment of spent LIBs cannot be ignored. The use of organic acid like citric acid may be a promising method in future industrial applications because it is environmentally friendly while ensuring satisfactory leaching efficiencies.(1) The optimal conditions of citric acid leaching LMO were citric acid concentration of 1.0 M, leaching time of 60 min, and solid–liquid ratio of 60 g L−1, and more than 94% of metal Li and Mn could be leached.
(2) Based on XRD, SEM, XPS analysis results, at relatively high acid concentrations of 1.5–2.5 M, the complex formed by complexation process of citrate anion and Mn atoms is monohydrate manganese citrate Mn(C6H6O7)·H2O, which is poorly soluble.
(3) By analysing the reaction mechanism of C6H8O7 and LMO and the formation of Mn(C6H6O7)·H2O in leaching process, it is further confirmed that the leaching of metal ions in organic acids was related to the type of anion ionized from the selected acid in the solution.
(4) The initial discharge capacities of LMO600-800 sample at 2C rate were 58.11, 87.85, 84.33 mA h g−1, and the capacity retention after 100 cycles were 73.98%, 93.63%, 89.64%, respectively. Rate performance test also showed that LMO700 sample has good cycling performance. The electrochemical performances of resynthesized LMO samples are affected by trace Al in leachate, which is attributed to the fact that in the process of Li+ movement, an appropriate amount of Al inhibits the Jahn–Taller distortion of spinel structure.
(5) Two routes for recycling waste LMO can be proposed in this study. The first route is to leach and chelate cathode materials with citric acid from spent LIBs, and then new cathode materials can be synthesized by sol–gel process. The appropriate control of impurities and the particle size distribution of resynthesized cathode materials contribute to improved electrochemical performances. In addition, based on the formation of precipitate Mn(C6H6O7)·H2O, the following scheme can also be considered. LMO is leached using citric acid (as leaching and precipitant agent) with high concentration to separate Mn, as in the case of oxalic acid to recycle LiCoO2, metal Li in solution is then extracted by other methods.
Author contributions
Kui Huang: Conceptualization, Writing-Review & Editing. Junzhen Wang: Investigation, Writing-original draft. Haili Dong: Methodology, Writing-Review & Editing. Yuanhuan Lu: Writing-Review & Editing. Kunjie Liu: Writing-Review &Editing. Zhangqing Chen: Writing-Review & Editing. Xinke Shan: Writing-Review & Editing. Guoliang Huang: Writing-Review & Editing. Lin Wei: Writing-Review & Editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21767003), Guangxi Natural Science Foundation under Grant No. 2018GXNSFAA281261, the Scientific Research Foundation of Guangxi University (Grant No. XBZ110928) and Young Teachers Innovation Cultivation Program (BRP180247) from Guangxi Bossco Environmental Protection Technology Co., Ltd. Authors are grateful to the reviewers who help us improve the paper by many pertinent comments and suggestions.Notes and references
- J. H. Fang, Z. P. Ding, Y. Ling, J. P. Li, X. Q. Zhuge, Z. H. Luo, Y. R. Ren and K. Luo, Chem. Eng. J., 2022, 440, 135880 CrossRef CAS.
- J. P. Wang, Arch. Metall. Mater., 2021, 66, 745–750 CAS.
- M. Y. Chen, X. T. Ma, B. Chen, R. Arsenault, P. Karlson, N. Simon and Y. Wang, Joule, 2019, 3, 2622–2646 CrossRef CAS.
- L. Yao, Y. B. Xi, H. J. Han, W. W. Li, C. J. Wang and Y. Feng, J. Alloys Compd., 2021, 868, 159222 CrossRef CAS.
- X. P. Chen, H. R. Ma, C. B. Luo and T. Zhou, J. Hazard. Mater., 2017, 326, 77–86 CrossRef CAS PubMed.
- X. X. Zhang, L. Li, E. Fan, Q. Xue, Y. F. Bian, F. Wu and R. J. Chen, Chem. Soc. Rev., 2018, 47, 7239–7302 RSC.
- Y. J. Shih, S. K. Chien, S. R. Jhang and Y. C. Lin, J. Taiwan Inst. Chem. Eng., 2019, 100, 151–159 CrossRef CAS.
- H. Chen, S. Gu, Y. X. Guo, X. Dai, L. D. Zeng, K. T. Wang, C. L. He, G. Dodbiba, Y. Z. Wei and T. Fujita, Hydrometallurgy, 2021, 205, 105746 CrossRef CAS.
- S. P. Barik, G. Prabaharan and L. Kumar, J. Clean. Prod., 2017, 147, 37–43 CrossRef CAS.
- Y. Guo, F. Li, H. C. Zhu, G. M. Li, J. W. Huang and W. Z. He, Waste Manag., 2016, 51, 227–233 CrossRef CAS PubMed.
- F. Wang, R. Sun, J. Xu, Z. Chen and M. Kang, RSC Adv., 2016, 6, 85303–85311 RSC.
- H. Li, S. Z. Xing, Y. Liu, F. J. Li, H. Guo and G. Kuang, ACS Sustainable Chem. Eng., 2017, 5, 8017–8024 CrossRef CAS.
- L. Yao, Y. Feng and G. X. Xi, RSC Adv., 2015, 5, 44107–44114 RSC.
- F. Meng, Q. C. Liu, R. Kim, J. X. Wang, G. Liu and A. Ghahreman, Hydrometallurgy, 2020, 191, 105160 CrossRef CAS.
- L. Yao, H. S. Yao, G. X. Xi and Y. Feng, RSC Adv., 2016, 6, 17947–17954 RSC.
- C. H. Sun, L. P. Xu, X. P. Chen, T. Y. Qiu and T. Zhou, Waste Manag. Res., 2018, 36, 113–120 CrossRef CAS PubMed.
- L. Li, J. Lu, Y. Ren, X. X. Zhang, R. J. Chen, F. Wu and K. Amine, J. Power Sources, 2012, 218, 21–27 CrossRef CAS.
- D. D. Chen, S. Rao, D. X. Wang, H. Y. Cao, W. M. Xie and Z. Q. Liu, Chem. Eng. J., 2020, 388, 124321 CrossRef CAS.
- L. Li, J. B. Dunn, X. X. Zhang, L. Gaines, R. J. Chen, F. Wu and K. Amine, J. Power Sources, 2013, 233, 180–189 CrossRef CAS.
- B. Musariri, G. Akdogan, C. Dorfling and S. Bradshaw, Miner. Eng., 2019, 137, 108–117 CrossRef CAS.
- D. Pant and T. Dolker, Waste Manag., 2017, 60, 689–695 CrossRef CAS PubMed.
- K. Huang, H. Xiong, H. L. Dong, Y. L. Liu, Y. H. Lu, K. J. Liu and J. Z. Wang, Process Saf. Environ., 2022, 165, 278–285 CrossRef CAS.
- X. P. Chen, J. Z. Li, D. Z. Kang, T. Zhou and H. R. Ma, Green Chem., 2019, 21, 6342–6352 RSC.
- X. Yang, P. Dong, T. Hao, Y. J. Zhang, Q. Meng, Q. X. Li and S. Y. Zhou, JOM, 2020, 72, 3843–3852 CrossRef CAS.
- B. W. Zhu, Y. J. Zhang, Y. L. Zou, Z. L. Yang, B. Zhang, Y. Zhao, M. Y. Zhang, Q. Meng and P. Dong, J. Environ. Manage., 2021, 300, 113710 CrossRef CAS PubMed.
- J. L. Pierre and I. Gautier-Luneau, BioMetals, 2000, 13, 91–96 CrossRef CAS PubMed.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 124–147 CrossRef.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- A. Van Hoonacker and P. Englebienne, Curr. Nanosci., 2006, 2, 359–371 CrossRef CAS.
- Q. X. Zheng, K. Shibazaki, T. Ogawa and M. Watanabe, ACS Sustainable Chem. Eng., 2021, 9, 10970–10976 CrossRef CAS.
- K. V. Nefedova, V. D. Zhuravlev, S. M. Khaliullin, A. P. Tyutyunnik and L. Y. Buldakova, Theor. Found. Chem. Eng., 2021, 55, 117–122 CrossRef CAS.
- I. V. Melikhov and E. F. Simonov, Theor. Found. Chem. Eng., 2011, 45, 581–588 CrossRef CAS.
- V. R. Galakhov, M. Demeter, S. Bartkowski, M. Neumann, N. A. Ovechkina, E. Z. Kurmaev, N. I. Lobachevskaya, Y. M. Mukovskii, J. Mitchell and D. L. Ederer, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 113102 CrossRef.
- K. N. Wood and G. Teeter, ACS Appl. Energy Mater., 2018, 1, 4493–4504 CrossRef CAS.
- A. Topolski, Chem. Pap., 2011, 65, 389–392 CAS.
- X. P. Chen, B. L. Fan, L. P. Xu, T. Zhou and J. R. Kong, J. Clean. Prod., 2016, 112, 3562–3570 CrossRef CAS.
- M. S. Kachhawaha and A. K. Bhattacharya, Z. Anorg. Allg. Chem., 1962, 315, 104–109 CrossRef.
- R. Golmohammadzadeh, F. Rashchi and E. Vahidi, Waste Manag., 2017, 64, 244–254 CrossRef CAS PubMed.
- Y. F. Deng and Z. H. Zhou, J. Coord. Chem., 2009, 62, 778–788 CrossRef CAS.
- H. P. Gao, Q. Z. Yan, P. P. Xu, H. D. Liu, M. Q. Li, P. Liu, J. Luo and Z. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 51546–51554 CrossRef CAS PubMed.
- S. Al Khateeb and T. D. Sparks, J. Mater. Res., 2019, 34, 2456–2471 CrossRef CAS.
- R. Santos-Ortiz, T. Rojhirunsakool, J. K. Jha, S. Al Khateeb, R. Banerjee, K. S. Jones and N. D. Shepherd, Solid State Ionics, 2017, 303, 103–112 CrossRef CAS.
- K. S. Chen, R. Xu, N. S. Luu, E. B. Secor, K. Hamamoto, Q. Q. Li, S. Kim, V. K. Sangwan, I. Balla, L. M. Guiney, J. W. T. Seo, X. K. Yu, W. W. Liu, J. S. Wu, C. Wolverton, V. P. Dravid, S. A. Barnett, J. Lu, K. Amine and M. C. Hersam, Nano Lett., 2017, 17, 2539–2546 CrossRef CAS PubMed.
- Y. L. Ding, J. Xie, G. S. Cao, T. J. Zhu, H. M. Yu and X. B. Zhao, J. Phys. Chem. C, 2011, 115, 9821–9825 CrossRef CAS.
- X. Yi, X. Y. Wang, B. W. Ju, Q. L. Wei, X. K. Yang, G. S. Zou, H. B. Shu and L. Hu, J. Alloys Compd., 2014, 604, 50–56 CrossRef CAS.
- X. F. Zhang, H. H. Zheng, V. Battaglia and R. L. Axelbaum, J. Power Sources, 2011, 196, 3640–3645 CrossRef CAS.
- Y. K. Sun, C. S. Yoon, C. K. Kim, S. G. Youn, Y. S. Lee, M. Yoshio and I. H. Oh, J. Mater. Chem., 2001, 11, 2519–2522 RSC.
- S. T. Myung, N. Kumagai, S. Komaba and H. T. Chung, Solid State Ionics, 2001, 139, 47–56 CrossRef CAS.
- H. B. Wang, R. Gao, Z. Y. Li, L. M. Sun, Z. B. Hu and X. F. Liu, Inorg. Chem., 2018, 57, 5249–5257 CrossRef CAS PubMed.
- A. D. Robertson, S. H. Lu, W. F. Averill and W. F. Howard, J. Electrochem. Soc., 1997, 144, 3500–3505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04391b |
| This journal is © The Royal Society of Chemistry 2022 |