
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, biological activity and toxicity to zebrafish of benzamides substituted with pyrazole-linked 1,2,4-oxadiazole†
Yingying Shaoa,
Minting Tua,
Sen Yanga,
Yingying Wanga,
Binlong Suna,
Jianjun Shic,
Chengxia Tan *a and
Xuedong Wang*b
*a and
Xuedong Wang*b
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: tanchengxia@zjut.edu.cn
bSchool of Environmental Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China. E-mail: zjuwxd@163.com
cSchool of Chemistry and Chemical Engineering, Huangshan University, Huangshan 245041, China
First published on 18th August 2022
Abstract
To find pesticidal lead compounds with high activity, a series of novel benzamides substituted with pyrazole-linked 1,2,4-oxadiazole was designed and synthesized by using the splicing principle of active substructures. The chemical structures of the target compounds were confirmed by 1H NMR, 13C NMR and HRMS. The preliminary bioassay showed that most compounds displayed good larvicidal activities against mosquito larvae at 10 mg L−1. In particular, compound 12g exhibited obvious activity; its lethal rate reached up to 100% (at 5 mg L−1) and 55% (at only 2 mg L−1). Furthermore, compound 12f (70.6%) and 12h (100%) showed good fungicidal activities against Pyricularia oryzae, with EC50 values of 8.28 and 5.49 μg mL−1, respectively, which were superior to that of the control drug bixafen (9.15 μg mL−1). Finally, the LC50 of compound 12h to zebrafish embryo was 0.39 mg L−1, so it was classified as a high-toxic compound. Thus, this compound may be used as a potential lead compound for further structural optimisation to develop new compounds with high activity and low toxicity.
1. Introduction
Nitrogenous heterocyclic compounds play an important role in the process of new pesticide development. Among them, possessing the advantages of variable structure, high-selectivity, high-efficiency, and low-toxicity and in line with the development concept of modern green pesticides, pyrazole derivatives have become the focus in the field of pesticides. In addition, pyrazole amide derivatives have good insecticidal,1–7 fungicidal8–16 and other biological activities.17 Developed in recent years, pyrazole amide derivatives, such as furametpyr, penthiopyrad, bixafen, sedaxane, penflufen, chlorantraniliprole, cyantraniliprole and tebufenpyrad (Fig. 1), have important shares in the pesticide market. However, the skeleton structure changes of them are mainly the replacement and modification of the pyrazole amide acid parent nucleus and aromatic amine, which is prone to cross-resistance.18–23 Therefore, through introducing a new skeleton, there is room for pyrazole amide derivatives to further improve their biological activities. | ||
| Fig. 1 Chemical structures of furametpyr, penthiopyrad, bixafen, sedaxane, penflufen, chlorantraniliprole, cyantraniliprole, tebufenpyrad. | ||
The 1,2,4-oxadiazole heterocycle, as a bio-isostere of the amide bond, not only has biological activities such as insecticidal,24,25 fungicidal26–30 and herbicidal,31,32 but also has anti-inflammatory,33–37 hypotensive38 and other physiological activities. Our previous work showed that benzamides substituted with 1,2,4-oxadiazole compounds have good insecticidal and fungicidal activities.39 Therefore, in this study, taking bixafen as lead compound, introducing benzoic acid substituted with 1,2,4-oxadiazole between the pyrazole ring and the amide bond, 18 novel benzamides substituted with pyrazole-linked 1,2,4-oxadiazole compounds were designed and synthesized (Fig. 2). The chemical structures of all the target compounds were confirmed by 1H NMR, 13C NMR and HRMS. Their insecticidal activities and fungicidal activities were studied and a toxicity test of zebrafish embryo was performed. The synthetic route of the target compounds is shown in Scheme 1.
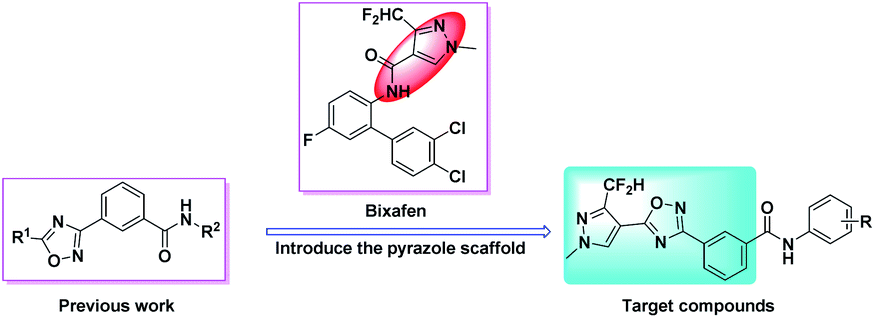 | ||
| Fig. 2 Design strategy of target compounds. | ||
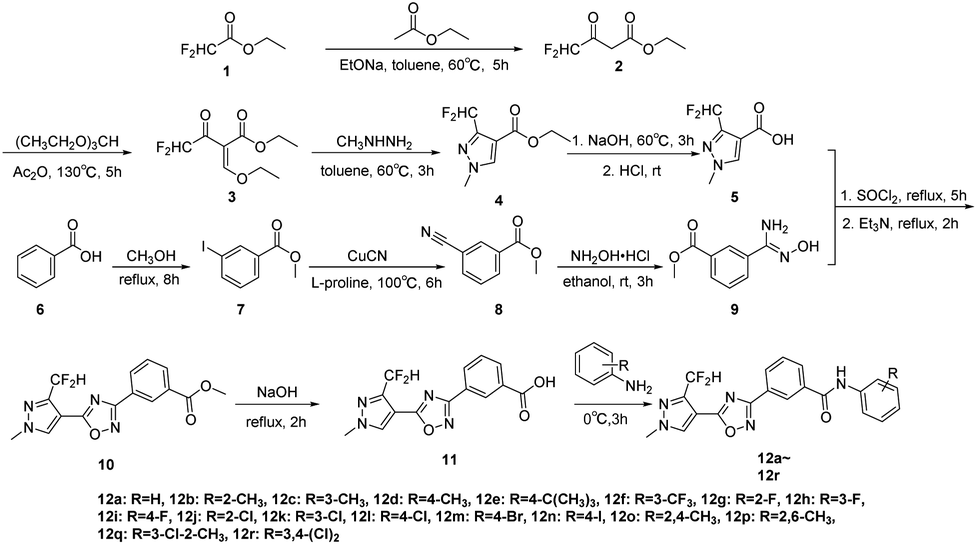 | ||
| Scheme 1 Synthetic route of target compounds 12a–12r. | ||
2. Results and discussion
2.1. Synthesis of target compounds
The target compounds 12a–12r were synthesized taking ethyl difluoroacetate 1 and 3-iodobenzoic acid 6 as starting materials. The compound 1 underwent Claisen condensation, addition–elimination, cyclization and hydrolysis reaction to give 3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylic acid 5. The compound 6 experienced esterification, cyanidation reaction, then reacted with hydroxylamine to give methyl (Z)-3-(N′-hydroxycarbamimidoyl) benzoate 9. Subsequently, the cyclization reaction of compound 5 and compound 9 yielded intermediate 10. Finally, the compound 10 was hydrolyzed under alkaline condition and acidified, then condensation reaction of intermediate 11 and substituted aniline obtained target compounds 12a–12r.The mechanism of step 3, cyclization reaction, is shown in Scheme 2. From this mechanism, it could be seen that the late stage of the reaction was a nucleophilic reaction of intermediate hydrazone, however, its nucleophilic activity was not strong enough to react at a high rate. Thus, to improve the reaction rate and yield, through increasing the temperature appropriately (from 0 to 80 °C), the effect of temperatures on reaction yield was studied and the results showed that its yield reached up to highest (85.3%) at 60 °C.
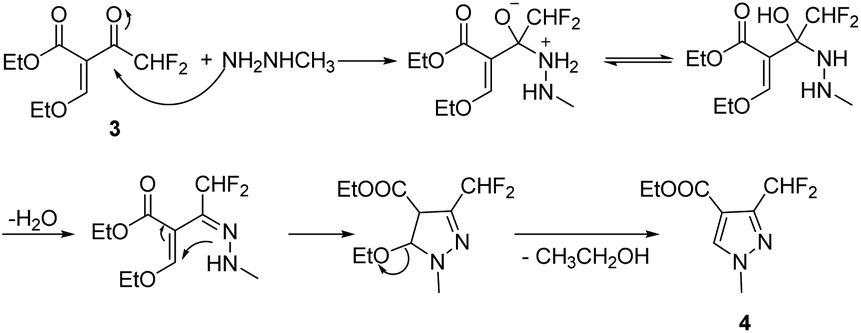 | ||
| Scheme 2 Synthetic mechanism of the intermediate 4. | ||
2.2. Biological activities of target compounds and SAR
The insecticidal activities of the target compounds against Mythimna separata, Pyrausta nubilalis, Helicoverpa armigera and Spodoptera frugiperda are listed in Table 1. And, the larvicidal activities of target compounds against Mosquito larvae are shown in Table 2. Etoxazole and broflanilide was used as the control drug. As shown in Table 1, most of the target compounds were found to exhibit certain insecticidal activities. Among them, compounds 12h (55%) and 12l (65%) exhibited moderate insecticidal activities against Mythimna separata at 500 mg L−1, which was inferior than that of etoxazole (100%) and broflanilide (100%). In addition, some of them showed good lethal activities against Mosquito larvae, and it was worth mentioning that compound 12g exhibited obvious activity, its lethal rate reached up to 100% (at 5 mg L−1) and even at 2 mg L−1, the lethal activity of compound 12g was 55%, which was definitely superior than etoxazole.| Compound | Insecticidal activities (death rates%, 500 mg L−1) | |||
|---|---|---|---|---|
| Mythimna separata | Pyrausta nubilalis | Helicoverpa armigera | Spodoptera frugiperda | |
| 12a | 40 | 0 | 15 | 0 |
| 12b | 10 | 0 | 0 | 20 |
| 12c | 0 | 20 | 0 | 0 |
| 12d | 25 | 10 | 15 | 10 |
| 12e | 10 | 0 | 0 | 0 |
| 12f | 15 | 0 | 5 | 0 |
| 12g | 45 | 25 | 35 | 0 |
| 12h | 55 | 0 | 10 | 0 |
| 12i | 20 | 0 | 15 | 0 |
| 12j | 30 | 0 | 0 | 0 |
| 12k | 30 | 10 | 25 | 10 |
| 12l | 65 | 30 | 35 | 0 |
| 12m | 0 | 10 | 5 | 15 |
| 12n | 0 | 0 | 0 | 0 |
| 12o | 0 | 5 | 10 | 0 |
| 12p | 10 | 10 | 0 | 0 |
| 12q | 20 | 0 | 10 | 0 |
| 12r | 5 | 30 | 5 | 10 |
| Etoxazole | 100 | 100 | 100 | 100 |
| Broflanilide | 100 | 100 | 100 | 100 |
| QCK | 0 | 0 | 0 | 0 |
| Compound | Larvicidal activities (death rates%) | |
|---|---|---|
| Concentration (mg L−1) | Mosquito larvae | |
| 12a | 10 | 0 |
| 12b | 10 | 0 |
| 12c | 10 | 100 |
| 5 | 50 | |
| 12d | 10 | 0 |
| 12e | 10 | 0 |
| 12f | 10 | 5 |
| 12g | 10 | 100 |
| 5 | 100 | |
| 2 | 55 | |
| 12h | 10 | 0 |
| 12i | 10 | 25 |
| 12j | 10 | 0 |
| 12k | 10 | 0 |
| 12l | 10 | 15 |
| 12m | 10 | 0 |
| 12n | 10 | 10 |
| 12o | 10 | 0 |
| 12p | 10 | 0 |
| 12q | 10 | 5 |
| 12r | 10 | 35 |
| Etoxazole | 10 | 100 |
| 5 | 35 | |
| QCK | 0 | 0 |
The fungicidal activity of target compounds 12a–12r against 9 tested funguses at 50 mg L−1 are listed in Table 3. Bixafen was used as the control drug. Overall, most of the target compounds were found to exhibit certain antifungal activities. Among them, the target compounds exhibited better fungicidal activities against Pyricularia oryzae and Sclerotinia sclerotiorum. For example, compounds 12f, 12h, 12k and 12r exhibited an inhibition rate of 52.9–100% against Pyricularia oryzae, especially compound 12h showed an inhibition rate of 100% against Pyricularia oryzae, which was comparable to that of bixafen (100%). For Sclerotinia sclerotiorum, compounds 12f (50.0%), 12h (50.0%), 12k (69.2%) and 12o (60.6%) also showed moderate fungicidal activities.
| Compound | Fungicidal activities (inhibition rate%, 50 mg L−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| AS | FG | CA | PO | SS | BC | TC | FO | PP | |
| a Alternaria solani (AS), Fusarium graminearum (FG), Cercospora arachidicola (CA), Pyricularia oryzae (PO), Sclerotinia sclerotiorum (SS), Botrytis cinerea (BC), Thanatephorus cucumeris (TC), Fusarium oxysporum (FO), Physalospora piricola (PP). | |||||||||
| 12a | 21.4 | 30.8 | 16.1 | 17.6 | 47.1 | 21.1 | 6.5 | 9.1 | 25.0 |
| 12b | 14.3 | 3.8 | 9.7 | 17.6 | 23.5 | 15.8 | 6.5 | 4.5 | 7.1 |
| 12c | 28.6 | 30.8 | 16.1 | 29.4 | 49.0 | 31.6 | 43.5 | 13.6 | 7.1 |
| 12d | 28.6 | 19.2 | 9.7 | 5.9 | 23.5 | 15.8 | 10.9 | 13.6 | 10.7 |
| 12e | 35.7 | 23.1 | 16.1 | 41.2 | 37.3 | 26.3 | 10.9 | 4.5 | 17.9 |
| 12f | 28.6 | 42.3 | 29.0 | 70.6 | 70.6 | 42.1 | 10.9 | 18.2 | 35.7 |
| 12g | 21.4 | 15.4 | 22.6 | 5.9 | 43.1 | 5.3 | 10.9 | 4.5 | 10.7 |
| 12h | 28.6 | 38.5 | 35.5 | 100.0 | 68.6 | 36.8 | 43.5 | 27.3 | 17.9 |
| 12i | 28.6 | 3.8 | 16.1 | 41.2 | 39.2 | 21.1 | 26.1 | 22.7 | 7.1 |
| 12j | 21.4 | 19.2 | 22.6 | 17.6 | 39.2 | 26.3 | 10.9 | 4.5 | 7.1 |
| 12k | 21.4 | 38.5 | 22.6 | 76.5 | 62.7 | 42.1 | 30.4 | 13.6 | 35.7 |
| 12l | 21.4 | 11.5 | 22.6 | 5.9 | 19.6 | 15.8 | 6.5 | 13.6 | 10.7 |
| 12m | 21.4 | 30.8 | 16.1 | 29.4 | 37.3 | 10.5 | 23.9 | 18.2 | 17.9 |
| 12n | 21.4 | 30.8 | 16.1 | 17.6 | 23.5 | 10.5 | 8.7 | 13.6 | 7.1 |
| 12o | 14.3 | 19.2 | 22.6 | 5.9 | 64.7 | 21.1 | 10.9 | 9.1 | 21.4 |
| 12p | 21.4 | 23.1 | 22.6 | 5.9 | 47.1 | 5.3 | 26.1 | 4.5 | 25.0 |
| 12q | 21.4 | 3.8 | 16.1 | 17.6 | 29.4 | 5.3 | 17.4 | 9.1 | 17.9 |
| 12r | 21.4 | 38.5 | 22.6 | 52.9 | 47.1 | 15.8 | 34.8 | 22.7 | 25.0 |
| Bixafen | 92.9 | 70.6 | 86.7 | 100.0 | 100.0 | 72.7 | 92.7 | 73.9 | 77.8 |
| QCK | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
On the basis of the preliminary fungicidal activity results, compounds 12f, 12h, 12k (>70% inhibitory) and bixafen (control drug) were selected for further EC50 bioassays, and the results are shown in Table 4. The results showed that compounds 12f and 12h possessed excellent fungicidal activities against Pyricularia oryzae with EC50 of 8.28 and 5.49 μg mL−1, respectively, and their EC50 values were significantly superior than that of bixafen (9.15 μg mL−1).
| Compound | Fungus | y = a + bx | r2 | EC50 (μg mL−1) |
|---|---|---|---|---|
| 12f | PO | y = 1.3031x + 3.8036 | 0.9846 | 8.2819 |
| 12f | SS | y = 1.1444x + 3.6775 | 0.9843 | 14.3106 |
| 12h | PO | y = 1.0839x + 4.1985 | 0.9809 | 5.4879 |
| 12k | PO | y = 1.2952x + 3.5555 | 0.9986 | 13.0409 |
| Bixafen | PO | y = 1.7973x + 3.2715 | 0.9766 | 9.1549 |
| SS | y = 1.3923x + 3.5714 | 0.9707 | 6.8965 |
From Table 3, it was concluded that substituent on the benzene ring can greatly influence the activity. Structure–activity relationship (SAR) results for these target compounds showed that the order of fungicidal activity of target compounds was F > CF3 > CH3 on the benzene ring, such as the high, middle and low active compounds 12h, 12f and 12c. Moreover, in the target compounds substituted with F (12g–12i), the meta substitution of benzene ring can increase the fungicidal activity. And, when the meta-directing of the benzene ring was electron-withdrawing group, its inhibitory activity against Pyricularia oryzae was better than others. Overall, compared compound 12q (3-Cl-2CH3) with 12b (2-CH3) and 12k (3-Cl), the insecticidal activities (against Mythimna separata, Pyrausta nubilalis, and Spodoptera frugiperda) and fungicidal activities showed that their biological activities were inhibited (3-Cl > 3-Cl-2CH3 > 2-CH3) when methyl group was introduced into the ortho position. Meanwhile, these experimental data provide ideas for further exploration and optimization of such compounds.
2.3. Toxicity to zebrafish (Danio rerio)
According to the test results of fungicidal activity, we selected compound 12h with higher fungicidal activity to further detect its zebrafish toxicity. After exposure to its gradient concentrations (0–0.80 mg L−1), the LC50 calibration curve was achieved by plotting the mortality percentages vs. the concentrations of analyte (Fig. 3). Obviously, the mortality rate increased dramatically with increases in the exposure concentrations (<0.60 mg L−1). However, over 0.60 mg L−1, the mortality rate reached as high as 100%. As a consequence, the LC50 value of compound 12h was calculated to be 0.39 mg L−1, which should be attributable to a highly toxic compound according to ISO 15088-2007.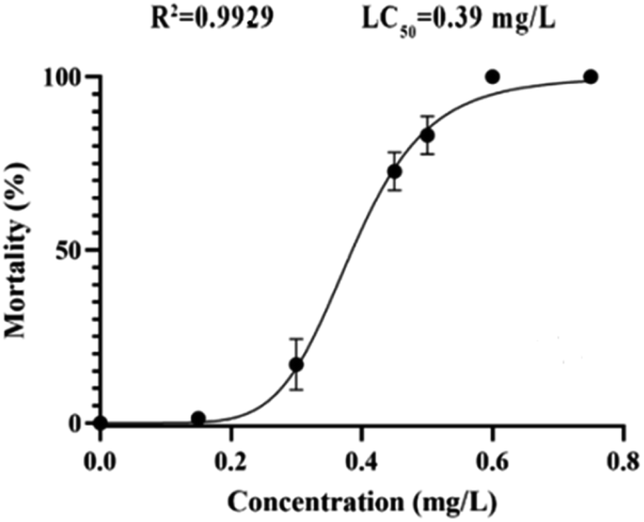 | ||
| Fig. 3 Zebrafish embryo mortality rates after exposure to compound 12h. | ||
From 24 to 96 hpf (hours post fertilization), the embryonic-larval zebrafish were acutely exposed to compound 12h across the concentration range of 0.049–0.390 mg L−1 (Fig. 4). The concentration selection of compound 12h was based on its LC50 value. In the 0.049 and 0.098 mg L−1 treatment groups, zebrafish body length significantly increased when compared to the control group, demonstrating the low-level exposure promoted larval growth and development. In sharp contrast, in the 0.195 and 0.390 mg L−1 treatments, a series of typical abnormalities were observed, including spinal curvature, head deformity, pericardial edema, yolk cyst, and melanin loss. Especially in the 0.390 mg L−1 treatment, typical yolk cyst and swim sac closure were observed for the 96 hpf larvae, reflecting in that their swim speed and activity were substantially decreased.
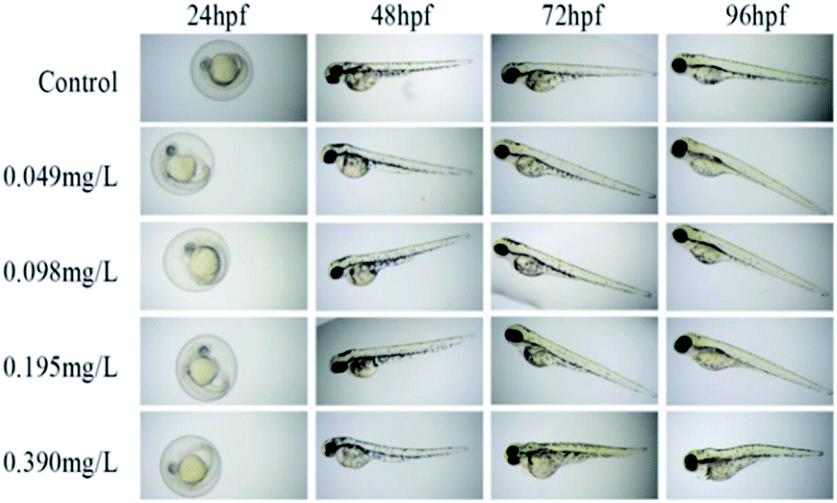 | ||
| Fig. 4 Zebrafish embryo malformation after exposure to compound 12h. | ||
3. Materials and methods
3.1. General information
1H NMR and 13C NMR spectra were measured on BRUKER Avance 500 MHz spectrometer (Bruker 500 MHz, Fällanden, Switzerland) using DMSO-d6 or CDCl3 as the solvent. High-resolution electrospray mass spectra (HR-ESI-MS) were determined using an UPLC H CLASS/QTOF G2 XS mass spectrometer (Waters, Milford, CT, USA). The HPLC analysis was conducted on Shimadzu LC-20AT. Melting points were determined using an M-565 melting point (Buchi, Switzerland). All the reagents were analytical grade or synthesized in our laboratory. The characterization data for all synthetic compounds are provided in the ESI.†Ethics statement: The Institutional Animal Care and Use Committee (IACUC) at Wenzhou Medical University (SYXK 2019-0009, April 4, 2019 to April 4, 2024) approved our study plan for proper use of zebrafish. All studies were carried out in strict accordance with the guidelines of the IACUC. All dissections were performed on ice, and all efforts were made to minimize suffering.
3.2. Synthesis
3.2.10.1 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-phenylbenzamide (12a). White solid, yield 63.8%, mp 217–219 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.51 (s, 1H), 8.86 (s, 1H), 8.61 (s, 1H), 8.23 (dd, J = 28.5, 7.5 Hz,2H), 7.82 (d, J = 8.0 Hz, 2H), 7.75 (t, J = 7.5 Hz, 1H), 7.58–7.28 (m, 3H), 7.13 (t, J = 7.5 Hz, 1H), 4.02 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.92, 167.53, 164.75, 143.62 (t, J = 26.0 Hz), 139.00, 135.95, 135.52, 130.70, 130.00 (d, J = 11.2 Hz), 129.48, 128.66, 126.45, 126.26, 123.91, 120.54, 110.77 (t, J = 237.1 Hz), 104.29, 39.55; HRMS calcd for C20H16F2N5O2 [M + H]+ 396.1267, found 396.1261.
3.2.10.2 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(2-tolyl)benzamide (12b). White solid, yield 61.4%, mp 211–214 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.87 (s, 1H), 8.65 (s, 1H), 8.25 (dd, J = 25.0, 8.0 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.56–7.32 (m, 2H), 7.30 (d, J = 7.0 Hz, 1H), 7.28–7.16 (m, 2H), 4.03 (s, 3H), 2.27 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.95, 167.57, 164.61, 143.55 (t, J = 26.0 Hz), 136.24, 135.56, 133.85, 130.68, 130.41, 130.08, 129.97 (d, J = 2.4 Hz), 129.65, 129.59, 129.55 (d, J = 2.7 Hz), 126.71, 126.27 (d, J = 9.4 Hz), 126.10, 110.83 (t, J = 246.5 Hz), 104.30, 39.57, 17.95; HRMS calcd for C21H18F2N5O2 [M + H]+ 410.1423, found 410.1420.
3.2.10.3 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(3-tolyl)benzamide (12c). White solid, yield 69.3%, mp 170–173 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.87 (s, 1H), 8.61 (s, 1H), 8.22 (dd, J = 36.5, 7.5 Hz, 2H), 7.76 (t, J = 7.5 Hz, 1H), 7.68–7.57 (m, 2H), 7.43 (t, J = 53.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 6.95 (d, J = 7.0 Hz, 1H), 4.03 (s, 3H), 2.33 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.95, 167.56, 164.69, 143.75 (t, J = 25.8 Hz), 138.91, 137.86, 136.01, 135.56, 130.69 (d, J = 11.0 Hz), 129.99 (d, J = 14.8 Hz), 129.51, 128.53, 126.43, 126.26, 124.64, 121.07, 117.73, 109.93 (t, J = 233.1 Hz), 104.30, 39.56, 21.24; HRMS calcd for C21H18F2N5O2 [M + H]+ 410.1423, found 410.1418.
3.2.10.4 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(4-tolyl)benzamide (12d). White solid, yield 61.4%, mp 225–229 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.86 (s, 1H), 8.59 (s, 1H), 8.21 (dd, J = 37.5, 8.0 Hz, 2H), 7.74 (t, J = 7.5 Hz, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 53.0 Hz, 1H), 7.17 (d, J = 8.5 Hz, 2H), 4.01 (s, 3H), 2.28 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.95, 167.56, 164.55, 143.64, 136.45, 136.04, 135.57 (d, J = 2.5 Hz), 132.93, 130.66 (d, J = 11.9 Hz), 129.89, 129.51 (d, J = 2.3 Hz), 129.08 (d, J = 3.7 Hz), 129.04, 126.24, 120.56 (d, J = 4.2 Hz), 109.86 (t, J = 212.94 Hz), 104.29, 39.56, 20.54; HRMS calcd for C21H18F2N5O2 [M + H]+ 410.1423, found 410.1418.
3.2.10.5 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(4-(tert-butyl)phenyl)benzamide (12e). White solid, yield 69.2%, mp 146–149 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.85 (s, 1H), 8.62 (s, 1H), 8.22 (dd, J = 26.5, 8.0 Hz, 2H), 7.79–7.66 (m, 3H), 7.58–7.23 (m, 3H), 4.02 (s, 3H), 1.28 (s, 9H); 13C NMR (126 MHz, DMSO-d6) δ 169.91, 167.54, 164.52, 146.26, 143.63 (t, J = 25.9 Hz), 136.42, 135.97, 135.49 (d, J = 2.8 Hz), 130.61 (d, J = 12.8 Hz), 129.91 (d, J = 13.2 Hz), 129.39, 126.43, 126.25, 125.24, 120.32, 109.84 (t, J = 225.54 Hz), 104.31, 39.54, 34.06, 31.19; HRMS calcd for C24H24F2N5O2 [M + H]+ 452.2893, found 452.1891.
3.2.10.6 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(3-(trifluorome-thyl)phenyl)benzamide (12f). White solid, yield 76.8%, mp 191–194 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.86 (s, 1H), 8.63 (s, 1H), 8.27 (s, 2H), 8.15 (dd, J = 61.0, 7.5 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.56–7.29 (m, 2H), 4.02 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.94, 167.46, 165.08, 143.62 (t, J = 26.1 Hz), 139.79, 135.50, 135.38, 130.76, 130.32, 129.88, 129.55, 129.29, 126.43, 126.34, 124.16 (q, J = 272.2 Hz), 123.92, 120.18, 116.53 (d, J = 3.8 Hz), 109.85 (t, J = 234.9 Hz), 104.27, 39.53; HRMS calcd for C21H15F5N5O2 [M + H]+ 464.1140, found 464.1136.
3.2.10.7 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(2-fluorophenyl)benzamide (12g). White solid, yield 59.7%, mp 221–224 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.88 (s, 1H), 8.64 (s, 1H), 8.25 (dd, J = 36.0, 7.5 Hz, 2H), 7.78 (t, J = 8.0 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.44 (t, J = 53.5 Hz, 1H), 7.34–7.21 (m, 3H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.96, 167.51, 164.73, 156.85, 154.88, 143.63, 135.55, 134.94, 130.80, 130.29, 129.62, 127.19 (d, J = 8.0 Hz), 126.57, 126.31, 125.53 (d, J = 12.6 Hz), 124.37 (d, J = 3.2 Hz), 115.91 (d, J = 19.8 Hz), 109.87 (t, J = 234.6 Hz), 104.28 (d, J = 2.1 Hz), 39.55; HRMS calcd for C20H15F3N5O2 [M + H]+ 414.1172, found 414.1172.
3.2.10.8 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(3-fluorophenyl)benzamide (12h). White solid, yield 62.5%, mp 192–195 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.87 (s, 1H), 8.60 (s, 1H), 8.23 (dd, J = 42.5, 8.0 Hz, 2H), 7.86–7.70 (m, 2H), 7.60 (d, J = 8.0 Hz, 1H), 7.55–7.30 (m, 2H), 6.96 (t, J = 8.5 Hz, 1H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 170.37, 167.91, 165.42, 163.48, 161.56, 144.06 (t, J = 26.0 Hz), 141.18 (d, J = 11.0 Hz), 135.97 (d, J = 11.3 Hz), 131.13, 130.72, 130.64, 129.93, 126.88, 126.76, 116.57 (d, J = 2.4 Hz), 110.77 (d, J = 20.16 Hz), 110.29 (t, J = 234.36 Hz), 107.61 (d, J = 26.2 Hz), 104.73, 39.98; HRMS calcd for C20H15F3N5O2 [M + H]+ 414.1172, found 414.1167.
3.2.10.9 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(4-fluorophenyl)benzamide (12i). White solid, yield 61.6%, mp 241–245 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.55 (s, 1H), 8.86 (s, 1H), 8.60 (s, 1H), 8.22 (dd, J = 36.5, 7.5 Hz, 2H), 7.82 (dd, J = 9.0, 5.0 Hz, 2H), 7.75 (t, J = 7.5 Hz, 1H), 7.43 (t, J = 53.0 Hz, 1H), 7.22 (t, J = 9.0 Hz, 2H), 4.02 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 170.36, 167.94, 165.08, 159.84, 157.93, 144.05 (t, J = 26.0 Hz), 136.19, 135.95, 135.75 (d, J = 2.6 Hz), 131.08, 130.49, 129.92, 126.77 (d, J = 14.6 Hz), 122.81 (d, J = 7.9 Hz), 115.67 (d, J = 22.2 Hz), 110.30 (t, J = 234.7 Hz), 104.72, 39.97; HRMS calcd for C20H15F3N5O2 [M + H]+ 414.1172, found 414.1170.
3.2.10.10 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(2-chlorophenyl)benzamide (12J). White solid, yield 62.8%, mp 208–211 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.88 (s, 1H), 8.66 (s, 1H), 8.27 (d, J = 28.0 Hz, 2H), 7.88–7.21 (m, 6H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 170.39, 167.93, 165.16, 144.07 (t, J = 25.9 Hz), 135.98, 135.43, 135.35, 131.13, 130.73, 130.16, 130.08, 129.07, 128.18, 128.00, 126.98, 126.80, 110.31 (t, J = 235.0 Hz), 111.29, 104.72, 39.99; HRMS calcd for C20H15ClF2N5O2 [M + H]+ 430.0877, found 430.0880.
3.2.10.11 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(3-chlorophenyl)benzamide (12k). White solid, yield 65.0%, mp 194–197 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.88 (s, 1H), 8.61 (s, 1H), 8.23 (dd, J = 45.5, 7.5 Hz, 2H), 7.99 (s, 1H), 7.82–7.71 (m, 2H), 7.58–7.29 (m, 2H), 7.19 (d, J = 7.0 Hz, 1H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.97, 167.49, 165.01, 143.64 (t, J = 26.2 Hz), 140.47, 135.56, 133.01, 130.78, 130.39, 130.28, 129.60, 126.45, 126.33, 123.62, 119.90, 118.80, 110.81 (t, J = 234.6 Hz), 104.27, 99.55, 39.56; HRMS calcd for C20H15ClF2N5O2 [M + H]+ 430.0877, found 430.0872.
3.2.10.12 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(4-chlorophenyl)benzamide (12l). White solid, yield 65.4%, mp 228–230 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.87 (s, 1H), 8.60 (s, 1H), 8.23 (dd, J = 42.0, 8.0 Hz, 2H), 7.85 (d, J = 8.5 Hz, 2H), 7.76 (t, J = 8.0 Hz, 1H), 7.64–7.26 (m, 3H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.94, 167.50, 164.83, 143.63 (t, J = 26.2 Hz), 137.95, 135.67, 135.54, 130.72, 130.16, 129.53, 128.58, 127.55, 126.43, 126.29, 122.01, 109.86 (t, J = 234.7 Hz), 104.27, 39.55; HRMS calcd for C20H15ClF2N5O2 [M + H]+ 430.0877, found 430.0880.
3.2.10.13 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(4-bromophenyl)benzamide (12m). White solid, yield 67.3%, mp 218–223 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.88 (s, 1H), 8.60 (s, 1H), 8.23 (dd, J = 44.5, 7.5 Hz, 2H), 7.79 (m, 3H), 7.57 (d, J = 8.5 Hz, 2H), 7.44 (t, J = 53.5, 1H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.95, 167.49, 164.84, 143.73 (t), 138.37, 135.68, 135.55, 131.50, 130.74, 130.17, 129.56, 126.42, 126.29, 122.38, 115.63, 110.80 (t, J = 234.8 Hz), 104.26, 39.55; HRMS calcd for C20H15BrF2N5O2 [M + H]+ 474.0372, found 474.0369.
3.2.10.14 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(4-iodophenyl)benzamide (12n). White solid, yield 68.8%, mp 241–245 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.87 (s, 1H), 8.59 (s, 1H), 8.22 (dd, J = 43.5, 7.5 Hz, 2H), 7.81–7.58 (m, 5H), 7.43 (t, J = 53.0 Hz, 1H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.94, 167.49, 164.82, 143.63 (t, J = 26.0 Hz), 138.86, 137.34, 135.69, 135.54, 130.73, 130.16, 129.53, 126.43, 126.29, 122.63, 110.80 (t, J = 234.6 Hz), 104.27, 87.65, 39.56; HRMS calcd for C20H15IF2N5O2 [M + H]+ 522.0233, found 522.0233.
3.2.10.15 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(2,4-dimethylphenyl)benzamide (12o). White solid, yield 65.0%, mp 218–222 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.86 (s, 1H), 8.65 (s, 1H), 8.24 (dd, J = 22.0, 8.0 Hz, 2H), 7.75 (t, J = 7.5 Hz, 1H), 7.44 (t, J = 53.5 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.10 (s, 1H), 7.04 (d, J = 7.8 Hz, 1H), 4.02 (s, 3H), 2.29 (s, 3H), 2.23 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.92, 167.56, 164.58, 143.63 (t, J = 26.0 Hz), 135.60, 135.51, 135.32, 133.63, 133.62, 130.90, 130.56, 129.92, 129.50, 126.61, 126.58, 126.49, 126.28, 109.87 (t, J = 234.8 Hz), 104.30, 39.54, 20.56, 17.84; HRMS calcd for C22H20F2N5O2 [M + H]+ 424.1580, found 424.1576.
3.2.10.16 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(2,6-dimethylphenyl)benzamide (12p). White solid, yield 65.4%, mp 259–264 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.87 (s, 1H), 8.66 (s, 1H), 8.26 (dd, J = 17.0, 8.0 Hz, 2H), 7.78 (t, J = 8.0 Hz, 1H), 7.43 (t, J = 53.5 Hz, 1H), 7.15 (s, 3H), 4.02 (s, 3H), 2.22 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 170.38, 168.01, 167.91, 167.80, 164.78, 143.98 (t, J = 23.2 Hz), 136.04, 135.97, 135.84, 135.52, 130.86, 130.42, 130.06, 128.24, 127.28, 126.83, 111.25 (t, J = 235.0 Hz), 104.73, 39.99, 18.50; HRMS calcd for C22H20F2N5O2 [M + H]+ 424.1580, found 424.1577.
3.2.10.17 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(3-chloro-2-methylphenyl)benzamide (12q). White solid, yield 67.3%, mp 214–217 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.87 (s, 1H), 8.65 (s, 1H), 8.26 (dd, J = 28.0, 7.5 Hz, 2H), 7.77 (t, J = 8.0 Hz, 1H), 7.56–7.31 (m, 3H), 7.31–7.25 (m, 1H), 4.03 (s, 3H), 2.28 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.93, 167.50, 164.79, 143.62 (t, J = 26.1 Hz), 137.88, 135.51, 135.13, 133.86, 132.29, 130.68, 130.18, 129.58, 126.95, 126.93, 126.52, 126.34, 125.92, 109.86 (t, J = 234.8 Hz), 104.27, 39.54, 15.37; HRMS calcd for C21H17ClF2N5O2 [M + H]+ 444.1033, found 444.1028.
3.2.10.18 3-(5-(3-(Difluoromethyl)-1-methyl-1H-pyrazol-4-yl)-1,2,4-oxadiazol-3-yl)-N-(3,4-dichlorophenyl)benzamide(12r). White solid, yield 68.8%, mp 204–208 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.87 (s, 1H), 8.60 (s, 1H), 8.28 (m, 3H), 7.79–7.76 (m, 2H), 7.63 (d, J = 8.5 Hz, 1H), 7.43 (t, J = 53.0 Hz, 1H), 4.03 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.96, 167.45, 165.01, 143.74 (t, J = 26.4 Hz), 139.12, 135.54, 135.29, 130.92, 130.78, 130.60, 130.39, 129.62, 126.42, 126.35, 125.39, 121.60, 120.38, 109.86 (t, J = 234.6 Hz), 104.26, 39.56; HRMS calcd for C20H14Cl2F2N5O2 [M + H]+ 464.0487, found 464.0481.
3.3. Biological activity and toxicity determination
The insecticidal and fungicidal activities were investigated in the National Pesticide Engineering Research Centre, Nankai University, according to ref. 40 and 41, and the results of the activity test are shown in Tables 1–3. Through acute exposure, we assessed the toxicity of compounds 12h on zebrafish embryo. According to the preliminary exposure experiments, a series of gradient concentrations of compounds 12h was set on the basis of mortality rates in the range of 10–95%. LC50 values for zebrafish embryos exposed to compound 12h from 24 to 96 hpf: control (0 mg L−1 of 12h), 0.049, 0.098, 0.195, 0.390 mg L−1 of 12h. The LC50 (median lethal concentration) value was computed by the Boltzmann equation.42,43 The observational indexes included mortality rate and teratogenic effects. Full experimental details are available in the ESI.†4. Conclusions
In conclusion, taking ethyl difluoroacetate and 3-iodobenzoic acid as starting materials, a total of 18 unreported pyrazole-linked 1,2,4-oxadiazole substituted benzamides were synthesized by a multi-step reaction based on the splicing principle of active substructures. The structures of the target compounds were confirmed by 1H NMR, 13C NMR and HRMS. The preliminary insecticidal activity results showed that compound 12g had excellent lethal activity against mosquito larvae (100% inhibition at 5 mg L−1). Furthermore, the fungicidal activity results showed that compounds 12f (70.6%) and 12h (100%) exhibited obvious activities against Pyricularia oryzaes, with EC50 of 8.28 and 5.49 μg mL−1, respectively, which were lower than bixafen (9.15 μg mL−1). Although, the resulting LC50 value for compound 12h was 0.39 mg L−1, which was classified as a high-toxic compound, it may be used as a potential leading compound for further structural optimisation to develop new compounds with high activity and low toxicity.Author contributions
Y. S., M. T., S. Y., Y. W., B. S. and J. S. carried out experimental work; Y. S. prepared the manuscript; C. T. designed the material and supervised the project; And C. T. revised the paper. X. W. acquired toxic data and revised the section of zebrafish toxicity. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge Hui-Li Wang for support with the toxicity determination. This work was supported by the Wynca Group (grant number KYY-HX-20180412), Siga Co. Ltd. R & D Program (grant number KYY-HX-20180737) and the Natural Science Foundation of Department of Education of Anhui Province (grant number KJHS2017B08).References
- H. Dai, K. Liang, Q. Zhou, Y. Ni, C. Qian, H. Qian, L. Li, Y. Shi, Z. Liang, J. Shi, L. Gao and X. Wu, Chin. J. Org. Chem., 2020, 40, 4350–4356 CrossRef CAS
.
- W. L. Dong, J. Y. Xu, L. X. Xiong and Z. M. Li, Molecules, 2012, 17, 10414–10428 CrossRef CAS PubMed
- G. P. Lahm, T. M. Stevenson, T. P. Selby, J. H. Freudenberger, D. Cordova, L. Flexner, C. A. Bellin, C. M. Dubas, B. K. Smith, K. A. Hughes, J. G. Hollingshaus, C. E. Clark and E. A. Benner, Bioorg. Med. Chem. Lett., 2007, 17, 6274–6279 CrossRef CAS PubMed
- G. P. Lahm, T. P. Selby, J. H. Freudenberger, T. M. Stevenson, B. J. Myers, G. Seburyamo, B. K. Smith, L. Flexner, C. E. Clark and D. Cordova, Bioorg. Med. Chem. Lett., 2005, 15, 4898–4906 CrossRef CAS PubMed
- J. Cui and L. Xu, Agrochemicals, 2017, 56, 797–800 Search PubMed
- A. Numata, K. Maeda, F. Saito and K. Ando, Jpn. Pat., JP2014034540-A, 2014 Search PubMed
- H. J. Song, Y. X. Liu, L. X. Xiong, Y. Q. Li, N. Yang and Q. M. Wang, J. Agric. Food Chem., 2012, 60, 1470–1479 CrossRef CAS PubMed
- S. Sun, L. Chen, J. Huo, Y. Wang, S. Kou, S. Yuan, Y. Fu and J. Zhang, J. Agric. Food Chem., 2022, 70, 3447–3457 CrossRef CAS PubMed
- P. Desbordes, B. Essigmann, S. Gary, O. Gutbrod, M. Maue and H. G. Schwarz, Pest Manage. Sci., 2020, 76, 3340–3347 CrossRef CAS PubMed
- X. W. Hua, W. R. Liu, Y. Chen, J. Ru, S. J. Guo, X. B. Yu, Y. H. Cui, X. H. Liu, Y. C. Gu, C. M. Xue, Y. Liu, J. K. Sui and G. Q. Wang, J. Agric. Food Chem., 2021, 69, 11470–11484 CrossRef CAS PubMed
- X. Wang, X. Y. Wang, B. H. Zhou, J. F. Long and P. Li, J. Heterocycl. Chem., 2021, 58, 2109–2116 CrossRef CAS
- G. Yang, and H. Li, WO Pat., 2019120218-A1, 2019 Search PubMed
- J. Wu, J. Wang, D. Y. Hu, M. He, L. H. Jin and B. A. Song, Chem. Cent. J., 2012, 6, 51 CrossRef CAS PubMed
- T. T. Liu, Y. Ni, L. K. Zhong, H. Y. Huang, W. Q. Hu, T. M. Xu and C. X. Tan, Chin. J. Org. Chem., 2015, 35, 422–427 CrossRef CAS
- W. Wang, J. H. Wang, F. R. Wu, H. Zhou, D. Xu and G. Xu, J. Agric. Food Chem., 2021, 69, 5746–5754 CrossRef CAS PubMed
- Y. Xie, H. Y. Gong, X. B. Wang, X. H. Ruan, J. P. Zhang, Q. Li, W. Xiao and W. Xue, Agrochemicals, 2016, 55, 872–876 Search PubMed
- J. Q. Huo, B. Zhao, Z. Zhang, J. H. Xing, J. L. Zhang, J. G. Dong and Z. J. Fan, Molecules, 2018, 23, 12 Search PubMed
- J. J. Xiao, M. Liao, M. J. Chu, Z. L. Ren, X. Zhang, X. H. Lv and H. Q. Cao, Molecules, 2015, 20, 807–821 CrossRef PubMed
- X. L. Chen, Y. X. Xiao, G. L. Wang, Z. Li and X. Y. Xu, Res. Chem. Intermed., 2016, 42, 5495–5508 CrossRef CAS
- Y. Y. Zhao, H. G. Li, P. W. Sun, L. Gao, J. B. Liu, S. Zhou, L. X. Xiong, N. Yang, Y. X. Li and Z. M. Li, Bioorg. Med. Chem. Lett., 2020, 30, 127535 CrossRef CAS PubMed
- K. Liang, Q. Zhou, X. Xun, Y. D. Ni, J. F. Li, Y. J. Shi, H. Y. Zhou, X. X. Wu, J. Shi, L. Gao and H. Dai, Chin. J. Org. Chem., 2020, 40, 1665–1672 CrossRef CAS
- B. X. Geng, W. Y. Chen and Z. L. Xiu, Agrochemicals, 2014, 53, 242–244 CAS
- B. L. Wang, H. X. Wang, H. Liu, L. X. Xiong, N. Yang, Y. Zhang and Z. M. Li, Chin. Chem. Lett., 2020, 31, 739–745 CrossRef CAS
- Y. H. Li, H. J. Zhu, K. Chen, R. Liu, A. Khallaf, X. N. Zhang and J. P. Ni, Org. Biomol. Chem., 2013, 11, 3979–3988 RSC
- M. M. Zhang, J. X. Sun, L. Z. Xiu and M. H. Wang, Mod. Agrochem., 2019, 18, 17–20 Search PubMed
- W. Grammenos, L. Brahm, T. Grote, B. Mueller, I. R. Craig, C. H. Winter, B. J. Merget, G. C. Rudolf, J. K. Lohmann, T. A. Stoesser, M. Seet, M. Fehr, and A. Koch, WO Pat., 2021175669-A1, 2021 Search PubMed
- W. Chen, Q. Chen, Q. Y. Wu and G. F. Yang, Chin. J. Org. Chem., 2005, 25, 1477–1481 CAS
- V. Terteryan-Seiser, M. A. Quintero Palomar, W. Grammenos, C. Wiebe, D. Strobel, P. S. J. D. Santos, J. Barnes, J. S. Barnes, M. A. Q. Palomar, P. M. A. Quintero, WO Pat., 2018202428-A1, 2018 Search PubMed
- J. N. Sangshetti, R. R. Nagawade and D. B. Shinde, Bioorg. Med. Chem. Lett., 2009, 19, 3564–3567 CrossRef CAS PubMed
- C. Krishna, M. V. Bhargavi, C. P. Rao and G. L. D. Krupadanam, Med. Chem. Res., 2015, 24, 3743–3751 CrossRef CAS
- T. H. Huang, H. Chen, J. Chen and A. D. Zhang, Chin. J. Struct. Chem., 2014, 33, 1455–1459 CAS
- T. H. Huang, H. Y. Tu, M. Liu, C. J. Hou and A. D. Zhang, Chin. J. Org. Chem., 2011, 31, 891–896 CAS
- M. Touaibia, D. C. Faye, J. A. Doiron, A. I. Chiasson, S. Blanchard, P. P. Roy and M. E. Surette, J. Nat. Prod., 2022, 85, 225–236 CrossRef CAS PubMed
- M. F. A. Mohamed, A. A. Marzouk, A. Nafady, D. A. El-Gamal, R. M. Allam, G. E. A. Abuo-Rahma, H. I. El Subbagh and A. H. Moustafa, Bioorg. Chem., 2020, 105, 104439 CrossRef CAS PubMed
- S. Yatam, R. Gundla, S. S. Jadav, N. R. Pedavenkatagari, J. Chimakurthy, B. N. Rani and T. Kedam, J. Mol. Struct., 2018, 1159, 193–204 CrossRef CAS
- A. Pace, S. Buscemi, A. P. Piccionello and I. Pibiri, in Advances in Heterocyclic Chemistry, eds. E. F. V. Scriven and C. A. Ramsden, Elsevier Academic Press Inc, San Diego, 2015, vol. 116, pp. 85–136 Search PubMed
- M. Gobec, T. Tomasic, T. Markovic, I. Mlinaric-Rascan, M. S. Dolenc and Z. Jakopin, Chem.-Biol. Interact., 2015, 240, 200–207 CrossRef CAS PubMed
- E. Meyer, A. C. Joussef, H. Gallardo and A. J. Bortoluzzi, J. Mol. Struct., 2003, 655, 361–368 CrossRef CAS
- S. Yang, C. L. Ren, T. Y. Ma, W. Q. Zou, L. Dai, X. Y. Tian, X. H. Liu and C. X. Tan, Int. J. Mol. Sci., 2021, 22, 2367 CrossRef CAS PubMed
- Y. Zhang, H. W. Zhu, J. F. Shang, B. L. Wang and Z. M. Li, Chin. J. Org. Chem., 2019, 39, 861–866 CrossRef CAS
- J. X. Mu, Y. X. Shi, M. Y. Yang, Z. H. Sun, X. H. Liu, B. J. Li and N. B. Sun, Molecules, 2016, 21, 11 CrossRef PubMed
- Y. N. Zhang, X. D. Wang, X. H. Yin, M. R. Shi, R. A. Dahlgren and H. L. Wang, Environ. Toxicol., 2016, 31, 736–750 CrossRef CAS PubMed
- Y. H. Zhang, M. Liu, J. F. Liu, X. D. Wang, C. H. Wang, W. M. Ai, S. B. Chen and H. L. Wang, Environ. Toxicol. Pharmacol., 2018, 57, 9–18 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04327k |
| This journal is © The Royal Society of Chemistry 2022 |