
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP-doped porous carbon derived from walnut shell for zinc ion hybrid capacitors†
Haibin Sun *a,
Congcong Liua,
Dongfang Guob,
Shuangshuang Lianga,
Wenhe Xiea,
Shenghong Liua and
Zijiong Lic
*a,
Congcong Liua,
Dongfang Guob,
Shuangshuang Lianga,
Wenhe Xiea,
Shenghong Liua and
Zijiong Lic
aSchool of Physics and Electronic Engineering, Xinyang Normal University, Xinyang, 464000, People's Republic of China. E-mail: sunhaibin@xynu.edu.cn
bSchool of Physics and Microelectronics, Zhengzhou University, Zhengzhou, 450001, People's Republic of China
cSchool of Physics & Electronic Engineering, Zhengzhou University of Light Industry, Zhengzhou, 450002, Peoples' Republic of China
First published on 31st August 2022
Abstract
Zinc ion hybrid capacitors (ZHCs) are expected to be candidates for large-scale energy storage products due to their high power density and large energy density. Due to their low cost and stability, carbon materials are generally the first choice for the cathode of ZHCs, but they face a challenge in the serious self-discharge behavior. Herein, zinc ion hybrid capacitors with high-performance are successfully assembled using a porous carbon cathode derived from low-cost p-doped waste biomass and a commercial zinc foil anode. The p-doped walnut shell ZHCs delivered a specific capacity of 158.9 mA h g−1 with an energy density of 127.1 W h kg−1 at a low current density. More importantly, the device had outstanding anti-self-discharge characteristics (retaining 77.98% of its specific capacity after a 72 h natural self-discharge test) and long-term cycle stability (retaining 88.2% of its initial specific capacity after 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 7.5 A g−1). This work presents guidance and support for the design and optimization of electrode materials for zinc ion supercapacitors and next-generation aqueous zinc ion energy storage performance.
000 cycles at 7.5 A g−1). This work presents guidance and support for the design and optimization of electrode materials for zinc ion supercapacitors and next-generation aqueous zinc ion energy storage performance.
Introduction
With the rapid requirements of portable electronic products and electric vehicles, developing novel energy storage devices with a high energy density, large output power, long service life, safety, and efficiency has been the primary focus of energy storage research.1–11 Supercapacitors, due to their high specific power, safety, environmental friendliness, low cost, and competitive cycle life, have shown promising application prospects.12–14 However, their low specific energy limits their development. Lithium-ion batteries have achieved great success since their commercialization and are widely used in various electronic products.15–17 Lithium ion batteries have a wide application prospect, attributed to their high energy density and wide working potential window.18 They have many shortcomings, such as a low power density, limited lithium resources, environmental pollution, poor cycle stability, and safety.19,20 Although the sodium/potassium-ion batteries that have emerged in recent years also show excellent energy storage performance, they suffer from a low power density.21–26 Due to the high conductivity and energy provided by multivalent cations, pure metals can be directly used as electrode materials.27–30 Theoretically, the sensitive induction between electrons and the redox reaction between metal cations and electrodes provide a steady electrochemical foundation for energy storage devices assembled based on metal electrodes with a large energy density and outstanding cycling stability.31,32Zinc ion hybrid capacitors (ZHCs), which display the potentials of both supercapacitors and batteries, have received extensive addressing in the energy storage field.33 Compared with the common lithium metal, zinc metal has more abundant resources, better economic benefits, lower cost and higher theoretical volumetric specific capacity (5855 mA h cm−3 vs. Zn/Zn2+), thus serving as a promising lithium substitute.27 As an emerging branch of supercapacitors, ZHCs can significantly increase the energy density without sacrificing power density.34 For conventional ZHCs, commercial zinc foil and carbon materials are used as the battery anodes and capacitive cathodes in energy storage systems, respectively. They rely on the reversible oxidation/reduction reaction on the surface of the zinc anode (stripping/deposition of zinc ions) and the efficient physical ion adsorption/desorption in the porous cathode to complete the sustainable electrochemical energy storage process. Comparative results show that dendrite growth on zinc anodes and the formation of a zinc oxide/zinc hydroxide insulating layer are significantly inhibited in neutral electrolytes such as ZnSO4 aqueous solution.35,36 This provides a stable environment for the efficient operation of aqueous ZHCs.37,38
Finding suitable cathode materials is the key to assembling high-performance ZHCs.39 Commercial activated carbon is a popular cathode for ZHCs. However, an excessively high degree of carbonization results in a lower content of heteroatom functional groups in commercial activated carbon, which is not conducive to high-speed ion transport or charge transfer during electrochemical energy storage. Ordinary commercial activated carbon cathodes are difficult to adapt to the quick performance and long cycle lifetimes of energy storage devices at a high current density.40 Biomass has abundant resources, low costs, and is a sustainable resource.41 Porous carbon materials rich with functional groups, large specific surface area, and adjustable pores can be obtained through a simple carbonization and activation process, which provides a good choice for the synthesis of high-performance porous carbon cathodes.42 Heteroatom doping is an interesting means to enhance the electrochemical performance of carbon materials. Taking phosphorus doping as an example, the P–C bond is longer than that of C–C bond due to the large atomic size of phosphorus, which distorts the carbon skeleton and generates defects.43 In addition, the lower electronegativity is also conducive to the redistribution of charges between P and C atoms, which improves the electrochemical energy storage.44 More importantly, the functional oxygen-containing group, PCO3, in carbon lattice can enhance wettability of the electrode and accelerate ion diffusion and charge transfer. Based on this, Lee et al. proposed phosphorus (P) and boron (B) co-doped activated carbon cathodes for zinc ion capacitors.45 The device showed a superior specific capacity of 169.4 mA h g−1 at 0.5 A g−1, and excellent capacity retention (88% after 30000 cycles at 10 A g−1). Huang et al. directly carbonized a polyaniline/sodium alginate hydrogel to synthesize nitrogen (N) and sulfur (S) co-doped carbon (NSC) with a hierarchical porous structure.46 Due to its unique nanopore structure and the synergistic effect of heteroatom doping, NSC exhibited excellent specific capacitance (309 F g−1 at 0.5 A g−1). The assembled symmetrical supercapacitor also showed excellent energy density (15.9 W h L−1) and power density (426 W h L−1). This strategy improved rate performance and service life defects of conventional activated carbon cathodes caused by low functional group contents.
In this work, we synthesized a P-doped porous carbon cathode material from inexpensive biomass waste (walnut shell) and assembled it with zinc foil to form typical ZHCs. The hybrid device showed excellent energy storage characteristics in 2 M ZnSO4 electrolyte. ZHC-WAPC-4/P showed an excellent specific capacity of 158.9 mA h g−1 at 0.5 A g−1. Even then at 20 A g−1, the specific capacity of the device remained at 70.1 mA h g−1. The calculation results showed that the device had an ultra-high energy density close to 127.1 W h kg−1 with a maximum power density of 16000 W kg−1. In addition, the device had outstanding anti-self-discharge characteristics (retaining 77.98% of its specific capacity after a 72 h natural self-discharge test) and long-life cycle stability (retaining 88.2% of its initial specific capacity after 15000 cycles at a high current density). This work demonstrates the optimization of electrode materials and the design of high-performance aqueous zinc ion energy storage devices.
Materials and methods
The walnut shell particles were washed in acetone, ethanol, and deionized water for 30 min with 3–5 times. The washed walnut shells were dried under vacuum at 80 °C for 12 h. The obtained dried walnut shell particles were calcined at 600 °C for 2 h under N2 at a heating rate of 3 °C min−1. Then, the calcined products were ground into powder, mixed with deionized water, and then dried under vacuum at 80 °C for 12–14 h to obtain the original walnut shell-derived carbon sample (WAC). After that, 2 g of WAC powder was mixed uniformly with different mass ratios of KOH (MKOH:MWAC = 2:1, 3:1, 4:1, or 5:1, respectively) and further pyrolyzed at 800 °C for 2 h under the protection atmosphere of N2. The residual KOH was removed using 0.5 M HCl, and washed with deionized water. The black powders were heated at 80 °C under vacuum for 12 h to acquire walnut shell-derived porous carbon WAPC-x (x is the ratio of MKOH:MWAC). Finally, 1 g WAPC-4 and 2 g red phosphorus powders were mixed in 10 ml deionized water and thoroughly stirred for 1 h, and subsequently put in a hot blast-drying oven at 90 °C for 12 h. Next, it was heated in a tube furnace at 800 °C for 2 h under the protection atmosphere of Ar. It was washed with deionized water and vacuum dried at 80 °C for 12 h to achieve the final P-doped porous carbon WAPC-4/P, as illustrated in Fig. 1a.
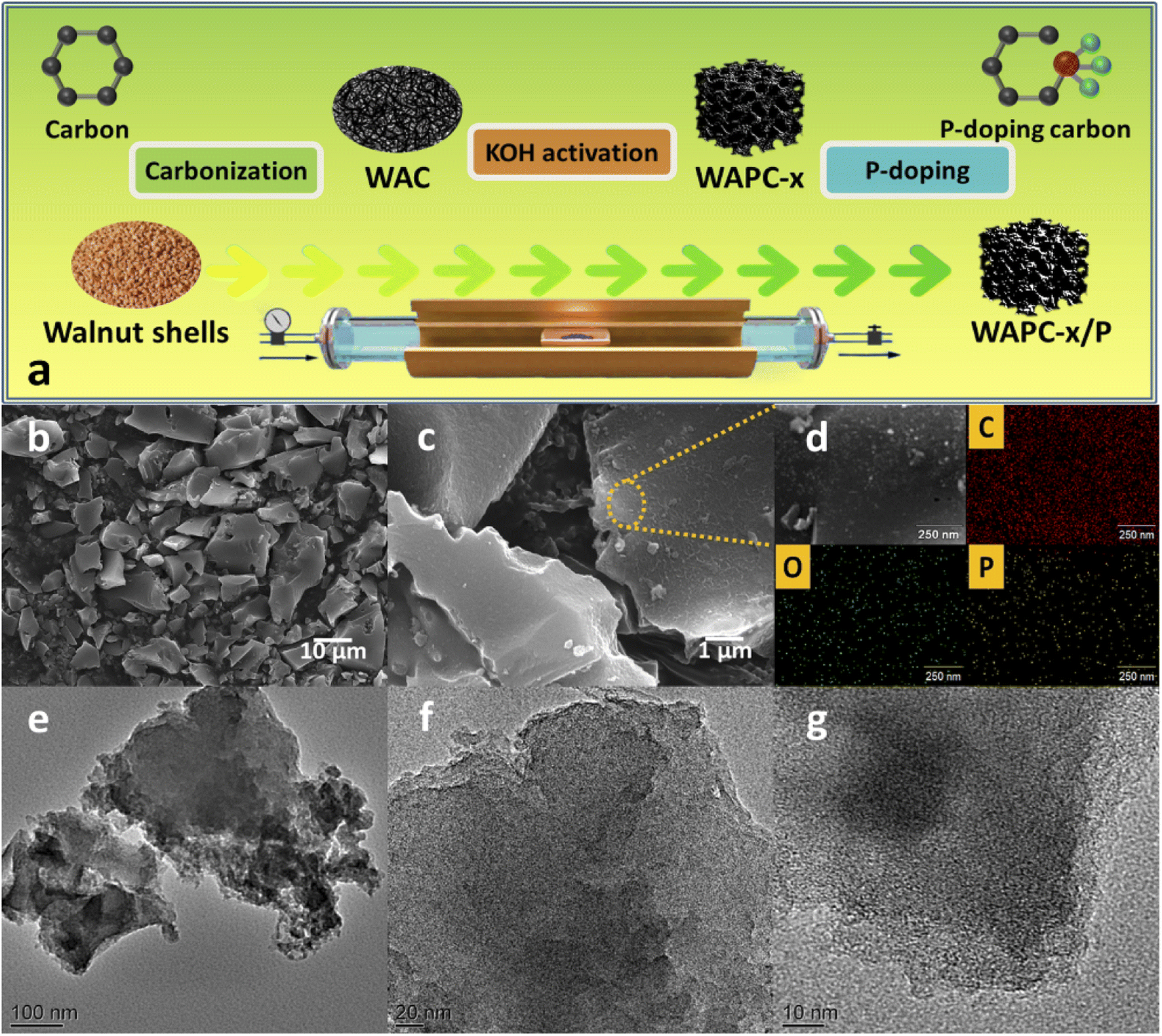 | ||
| Fig. 1 (a) Schematic of preparation of high-performance WAPC-x cathode, (b and c) SEM images, (d) EDX analysis, and (e–g) TEM images of WAPC-4/P. | ||
Zinc foil (thickness of 100 μm) was purchased from Kluder Group Chemical Reagent Co., Ltd. First, acetone and deionized water were used to wash and dry the nickel foam for electrode preparation. During cathode preparation, the carbon powder, carbon black, and polytetrafluoroethylene (PTFE) were mixed uniformly in ethanol at a mass ratio of 8:1:1. The obtained slurry was coated on the surface of a nickel foam, and subsequently dried at 60 °C for 12 h under vacuum. The mass of the porous carbon on a single positive electrode was 1.2–1.5 mg cm−2. The electrode was then used directly to assemble ZHCs after being pressed. Porous carbon cathodes and commercial zinc foil anodes were used to assemble ZHCs in a 2 M ZnSO4 aqueous electrolyte (the zinc foil surface was not oxidized before use). The electrochemical performance of the WAPC-x//Zn2+(aq)//Zn hybrid device was measured in a potential window of 0.3–1.9 V, which can be analyzed by cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) curves. The electrochemical kinetics was evaluated by electrochemical impedance spectroscopy (EIS). All measurements were performed on a CHI 660E electrochemical workstation. Since zinc foils served as active materials and current collectors at the same time in ZHCs, their mass was in excess.
Based on the mass of the WAPC-x cathode, the specific capacity of ZHCs was calculated by the following equation:
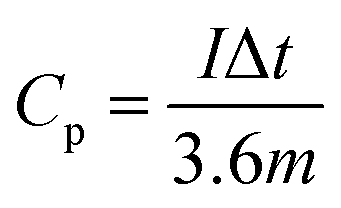 | (1) |
The specific power (P, W kg−1) and specific energy (E, W h kg−1) of the capacitors depicted in the Ragone plot were measured by the equations:
 | (2) |
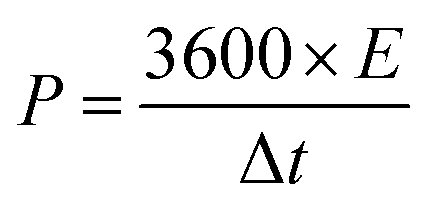 | (3) |
The morphology and structure of the as-prepared materials was represented by scanning electron microscopy (SEM, JSM-7001F) and transmission electron microscopy (TEM, JSM-2100). The structural compositions of the samples were characterized by X-ray powder diffraction (XRD, D8 ADVANCE) and Raman spectroscopy (Renishaw in Via). The elemental characteristics were determined by energy-dispersive X-ray spectroscopy (EDX). The porous structures were analyzed by N2 ad/de-sorption measurements (BELSORP-max). The pore size distributions were measured by the NLDFT method. The chemical states and element contents were determined by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi).
Results and discussion
The microstructure and morphology of porous carbon WAPC-x were preliminarily characterized by lots of SEM images. The SEM results (Fig. 1b and c, S1 and S2†) showed that all products had an irregular block structure, and the surface of the material was relatively rough. The product did not change significantly after activation and P doping. The EDX results of WAPC-4/P (Fig. 1d) show the uniform element distribution on the porous carbon surface, demonstrating successful P element doping. The TEM image showed that the sample has a well-developed microporous structure. This was ascribed to the effective etching effect of carbon atoms by KOH (6KOH + 2C → 2K + 3H2 + 2K2CO3).47 The insertion of potassium destroyed the crystal structure and formed many pores under the chemical etching process.48The crystal phase and structural defects of porous carbon WAPC-x were further analyzed by XRD and Raman spectroscopy. The XRD patterns (Fig. 2a) revealed the structure and changes of layered porous carbon.49 All samples displayed two broad characteristic peaks near 2θ = 22.6° and 43.8°, which matched the (002) and (101) crystal planes of hexagonal graphitic carbon, respectively. The low-angle (002) peak and the low-intensity (101) peak indicated that the sample was amorphous or had a small crystalline region.50 The (002) peak of the activated product WAPC-x gradually shifted to a lower diffraction angle, and its peak intensity decreased, indicating that the degree of defects of the porous carbon increased after activation. According to Bragg's law, activation processes increase the interlayer distance of a material.51 The typical D peak (∼1338 cm−1) and G peak (∼1590 cm−1) of the Raman spectra (Fig. 2b) correspond to the defects in the graphitic lattice and the tensile vibration of the crystal plane inside the graphitic carbon, respectively. The 2D peak at 2698 cm−1 was caused by two-phonon resonance scattering, while the peak at 2922 cm−1 was contributed by the (D + G) peak. The weak peak near 3120 cm−1 further confirmed the existence of defects.52 In addition, the intensity ratio of the D to G peak (ID/IG) corresponds to the defect and disorder degree of carbon materials, and a larger ID/IG value indicates more structural defects in a material.53,54 The calculated results show that the ID/IG values of WAC, WAPC-2, WAPC-3, WAPC-4, WAPC-5 and WAPC-4/P are 1.18, 1.78, 2.02, 2.26, 2.43 and 2.29, respectively. The results show that WAPC-5 has the most defects, which should be caused by adding more activator.
 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra, and (c) N2 adsorption–desorption isotherms of WAPC-x. (d) Contact angle of the electrolyte at the interface of WAPC-4 and WAPC-4/P. | ||
The N2 adsorption–desorption isotherms (Fig. 2c and S3a†) were used to acquire the pore structure of the material. All samples showed a typical type I adsorption isotherm,55 indicating that porous carbon was mainly composed of micropores and mesopores. Based on the Branauer–Emmett–Teller (BET) model, porosity of the material was calculated (Table S1†). The specific surface areas of WAPC-2, WAPC-3, WAPC-4, WAPC-4/P and WAPC-5 were 1416.3, 2158.8, 2542.6, 2518.4 and 2856.6 m2 g−1, respectively. WAPC-5 has the most developed pore structure, which is consistent with the Raman results. But the micropore contribution of WAPC-5 decreased further. The micropore ratio of WAPC-4/P obtained a small increase after phosphorus doping, which indicated that the defects induced by phosphorus doping into the carbon lattice promoted the formation of more micropores. It is worth noting that WAPC-x has a superior hierarchical porous structure. In porous carbon, the existence of mesopores can promote the deep penetration of the electrolyte, which is conducive to full contact interface between the electrode and electrolyte, and increase the conductivity of the electrode. Micropores can improve active sites, which is the key to improving the capacitance of a carbon electrode. This hierarchical porous structure composed of both micropores and mesopores is favorable to the rapid transfer of ions/charges for the charging–discharging process. As shown in Fig. 2d, contact angle analysis was used to study the wettability between the electrolyte and electrode. The contact angles of the electrolyte at the interface of WAPC-4 and WAPC-4/P were 116.4° and 78.5°, respectively. A lower contact angle indicated better wettability. The introduction of phosphorus atoms promoted the formation of functional more oxygen-containing groups (PCO3) in the carbon material, suggesting the improved wettability between the electrolyte and electrode surface.45
The surface element chemical states of porous carbon WAPC-4 and WAPC-4/P was analyzed by XPS (Fig. 3 and S3b,† Table 1). The analysis confirmed the elemental coexistences of C, O, and P, and the successful doping of P atoms in WAPC-4/P. The C 1s peaks of WAPC-4 and WAPC-4/P in Fig. 3a and d, which could be decomposed into four peaks at 283.83, 284.32, 286.31, and 288.66 eV (C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and O–CO, respectively).56 The O 1s spectra (Fig. 3b and e) showed three peaks at 531.11, 532.08, and 533.08 eV (C–O or P–O, –OH, and O–CO, respectively).57 The P 2p spectra are shown in Fig. 3c and f. WAPC-4 did not show a similar peak because it was not doped with P. In contrast, P-doped WAPC-4/P displayed strong peaks at 134.43 eV and 135.53 eV (P–C and P–O groups, respectively). The presence of the oxygen functional group PCO3 in the carbon lattice enhanced the wettability of the material and accelerated charge transfer, which improved the ion diffusion efficiency and optimized the cycle stability of the electrode.58–60
O, and O–CO, respectively).56 The O 1s spectra (Fig. 3b and e) showed three peaks at 531.11, 532.08, and 533.08 eV (C–O or P–O, –OH, and O–CO, respectively).57 The P 2p spectra are shown in Fig. 3c and f. WAPC-4 did not show a similar peak because it was not doped with P. In contrast, P-doped WAPC-4/P displayed strong peaks at 134.43 eV and 135.53 eV (P–C and P–O groups, respectively). The presence of the oxygen functional group PCO3 in the carbon lattice enhanced the wettability of the material and accelerated charge transfer, which improved the ion diffusion efficiency and optimized the cycle stability of the electrode.58–60
| Samples | Element (%) | ||
|---|---|---|---|
| C | O | P | |
| WAPC-4 | 82.7 | 17.3 | — |
| WAPC-4/P | 74.4 | 23.5 | 2.1 |
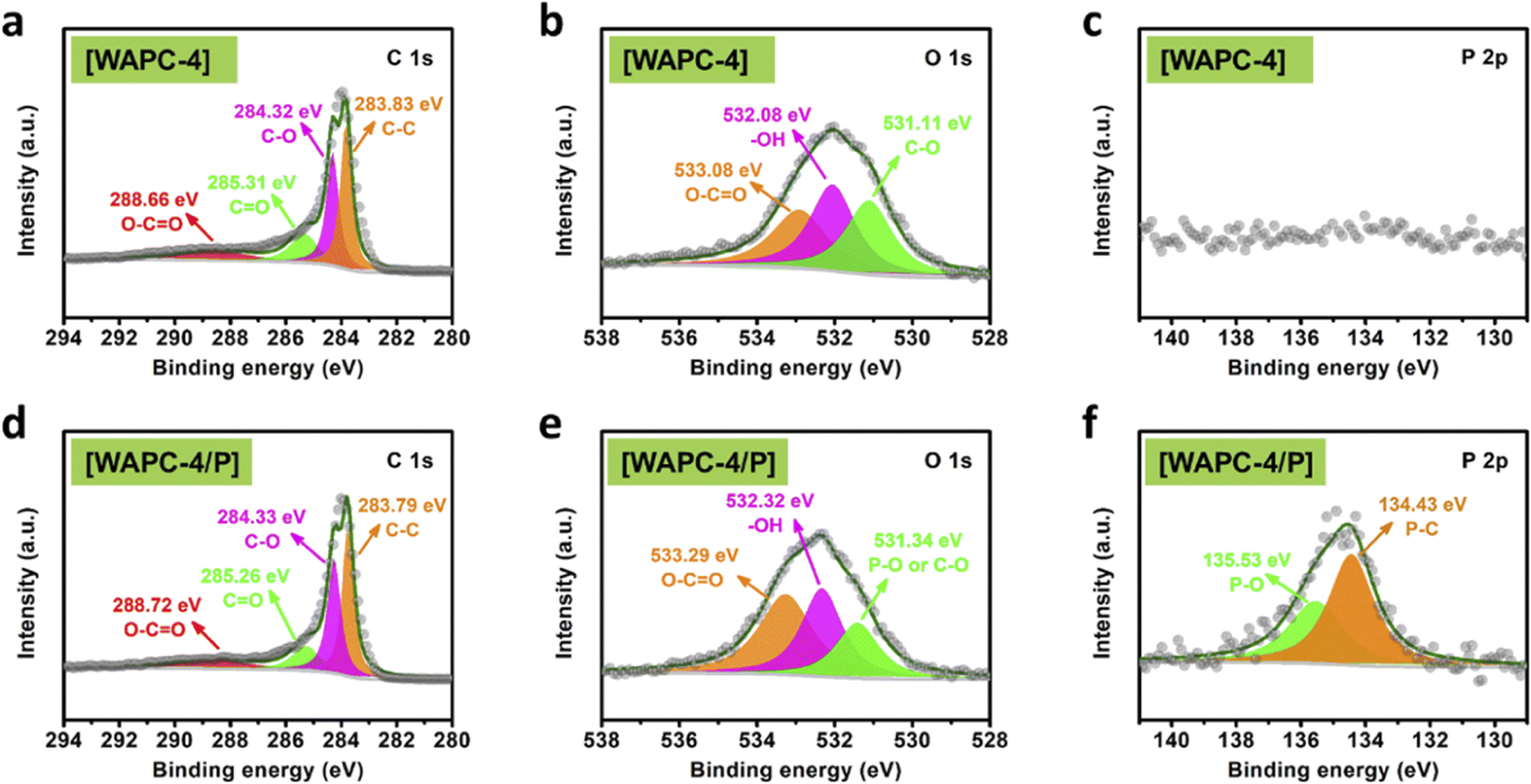 | ||
| Fig. 3 HR XPS spectra of WAPC-4 and WAPC-4/P, (a and d) C 1s, (b and e) O 1s, and (c and f) P 2p. | ||
To confirm that obtaining porous carbon derived from biomass materials is feasible, the performance of WAPC-x was evaluated in ZHCs. Here, the working voltage of the device was evaluated by the CV curve at a low scan rate (Fig. S4†). When the voltage was lower than 0.3 V or higher than 1.9 V, the CV curve displayed an obvious jumping phenomenon, so the optimal working voltage range of the device was 0.3–1.9 V. The obvious redox peak in the curve corresponds to the deposition of zinc ions and the stripping of the zinc anode (Zn ↔ 2e + Zn2+).
The CV curves of ZHC-WAPC-4/P (Fig. 4a) were described in the scan range of 5–100 mV s−1. As the scanning rate increased, the CV curves maintained a linear change, indicating not significant deformation, demonstrating its excellent charging capability and rapid dynamic characteristics. The GCD tests were evaluated in the different current density of 0.5–20 A g−1 (Fig. 4b), which showed the similar quasi-triangular shape, indicating the highly reversible reaction process. Preliminary results confirmed that the hierarchical porous carbon WAPC-4/P had good electrochemical reaction kinetic adaptability and reaction reversibility.61 The specific capacity of ZHC-WAPC-x was deduced from the loaded mass of the porous carbon cathode. ZHC-WAPC-4/P had a specific capacity of 158.9 mA h g−1 at 0.5 A g−1. The capacity of ZHC-WAPC-4 was very close to this result (158 mA h g−1) and better than the test results of ZHC-WAPC-2 (119.6 mA h g−1), ZHC-WAPC-3 (138.5 mA h g−1), and ZHC-WAPC-5 (149.8 mA h g−1) (Fig. 4d). At 20 A g−1, the capacity of the ZHC-WAPC-4/P electrode (70.1 mA h g−1) was significantly superior to the ZHC-WAPC-4 (61.7 mA h g−1), which is better than WAPC-2 (36.1 mA h g−1), WAPC-3 (44.3 mA h g−1) and WAPC-5 (52.5 mA h g−1). Although WAPC-5 has the most developed pore structure, WAPC-4 exhibits the best overall performance among the undoped P samples. The better rate performance may be attributed to a more reasonable pore distribution and protection from overactive activation, which is the reason why WAPC-4 was selected for further phosphating. As expected, the rate capability of ZHC-WAPC-4/P at high current densities is further enhanced due to the introduction of oxygen-containing functional groups by P doping.
 | ||
| Fig. 4 (a) CV curve and (b) GCD curve of ZHC-WAPC-4/P. (c) Nyquist plot of the electrodes (the inset is corresponding equivalent circuit model). (d) Specific capacitance as a function of current density in the range of 0.5–20 A g−1. | ||
To better understand the electrochemical performance of the electrodes, EIS were used to analyze the charge kinetic process. Fig. 4c showed the Nyquist plots of ZHC-WAPC-4 and ZHC-WAPC-4/P measured in the variable frequency of 0.01–105 Hz. The plots were mainly composed of a semicircle arc in the high/medium-frequency area and a slope line in the low-frequency area. The intercept of the arc and the real axis corresponds to solution resistance (Rs) caused by the electrolyte between the electrodes. ZHC-WAPC-4 (7.47 Ω) and ZHC-WAPC-4/P (7.33 Ω) have similar Rs values (Table S2†). The diameter of the semicircle in the high/medium-frequency area describes the charge transfer resistance (Rct). Compared with ZHC-WAPC-4 (140.6 Ω), ZHC-WAPC-4/P had a significantly lower resistance of 96.59 Ω. The slope of the low-frequency area of the straight line was related to interfacial ion diffusion (Zw, Warburg impedance) between the electrode and electrolyte. ZHC-WAPC-4/P had the best ion diffusion performance.
The existence of the oxygen-containing functional group, PCO3, caused by P-doping enhanced the wettability of the material, promoted charge transfer, and increased the ion diffusion rate. P atoms with a larger atomic radius caused more defects after entering the carbon lattice.62 The lower electronegativity also caused the redistribution of charges between P and C atoms, which was beneficial for energy storage applications. Wang et al. introduced P atoms into carbon to induce more edges and topological defects, thereby accelerating the diffusion of metal ions and achieving excellent energy storage performance.63 The benefits of P atom doping have been proved in many works.64,65
The electrochemical kinetic behavior of ZHCs can be preliminarily analyzed by analyzing the peak current response of the CV curve with the rising scan rates. The relationship between the peak current (I) and the scan rate (V) is described as the following equation (Fig. 5a and b):
| I = aVb | (4) |
|
log(I) = blog(V) + log(a)
| (5) |
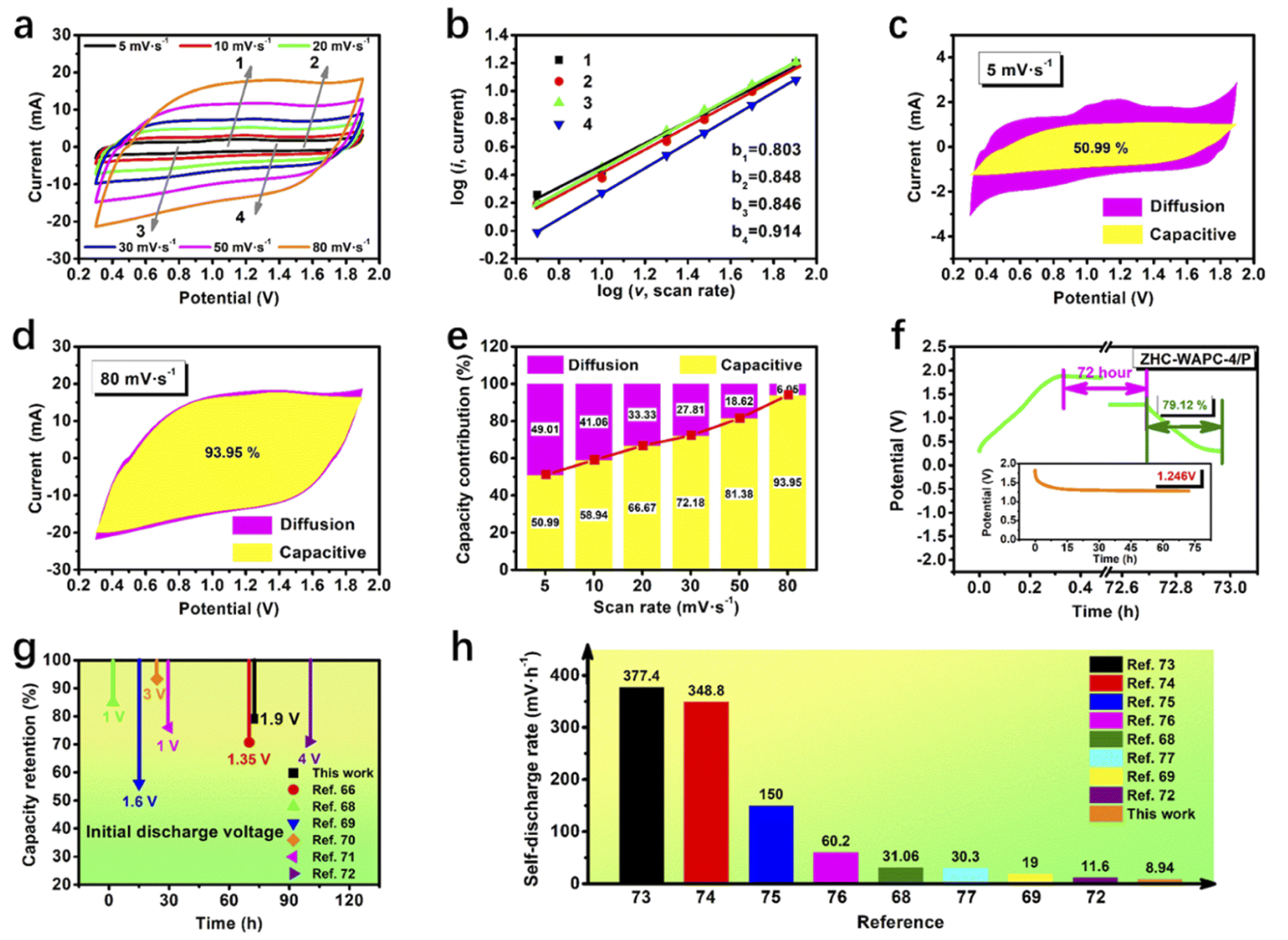 | ||
| Fig. 5 Electrochemical kinetic studies of ZHC-WAPC-4/P. (a) CV curves of ZHC-WAPC-4/P at different scan rates. (b) log(i) versus log(v) plots at specific peak currents. (c) Capacitance contribution at 5 mV s−1. (d) Capacitance contribution at 80 mV s−1. (e) Capacitance contribution as a function of scan rate. (f) Self-discharge examination combining the GCD test. (g) The capacitance retention and initial discharge voltage comparison of different capacitors and ion hybrid energy storage systems. (h) The self-discharge rate compared with other reported energy storage devices. | ||
The behavior of an energy storage system can be preliminarily judged based on the parameter b. When b is 0.5, it is controlled by ion diffusion. b is 1, meaning that the capacitance contribution is dominant during energy storage system.66 The fitting values show that the b values were 0.803, 0.848, 0.846, and 0.914, respectively, indicating that the electrochemical behavior of the ZHCs was caused by both ion diffusion and capacitance. The larger proportion of the capacitance contribution may be related to the reversible redox reaction (deposition/exfoliation of Zn2+/Zn).
To reveal the electrochemical behavior of ZHCs more accurately, Dunn's method was used to show the energy storage mechanism and electrochemical kinetics of hybrid devices at different scan rates. The formulas that could further quantify the changes in the cathode capacitance contribution were shown as follows:
| I = Icap + Idiff = k1V + k2V1/2 | (6) |
| I/V1/2 = k1V1/2 + k2 | (7) |
As shown in Fig. 5c–e, the capacitance contribution (yellow shaded area) accounted for 50.99% of the total electrochemical energy storage behavior at 5 mV s−1, and the capacitance contribution was close to 93.95% at 80 mV s−1. As the rising scan rate, the capacitance contribution ratio gradually increased. Diffusion occurred more easily at lower scan rates and provided more capacitance. The capacitance contribution increased with the scan rate increasing. This energy storage mechanism was profitable to an quick charging/discharging of the Zn2+ hybrid in aqueous solution.67 ZHC-WAPC-4 had a capacitance contribution of 46.04–90.35% in the range of 5–80 mV s−1 (Fig. S4d–f†), which is slightly lower than that of ZHC-WAPC-4/P. The capacitance contribution corresponds to the rapid electrochemistry of the energy storage device, and a higher capacitance contribution indicates a better rate performance. In summary, the larger specific capacity and higher capacitance contribution during energy storage indicate that P doping improved the rate performance of the porous carbon electrode material. This also explains the better ion diffusion performance of ZHS-WAPC-4/P.
Self-discharge has become an important indicator for evaluating energy storage devices. In this experiment, we discussed the anti-self-discharge ability of ZHC-WAPC-4 and ZHC-WAPC-4/P. Specifically, the energy storage device was charged to the highest working potential at 0.5 A g−1, and then the decay of the open-circuit voltage was monitored under long-term static conditions. The self-discharge curve showed that the open-circuit voltage of the hybrid power device dropped sharply in the initial stage of self-discharge. The voltage drop speed gradually decreased and tended to stabilize after a period of time. The potential of ZHC-WAPC-4/P dropped from 1.90 V to 1.25 V after 72 h (Fig. 5f), while the potential of ZHC-WAPC-4 dropped from 1.90 V to 1.28 V after 72 h (Fig. S5†). The low zinc anode potential (∼0.75 V vs. SHE in 2 M ZnSO4) helped stabilize the electric field inside the ZHCs, thereby inhibiting anion desorption from the surface of cathode and self-discharge of the system.68 Zn2+ was reduced to Zn and then firmly fixed on the surface of anode. Electrically neutral zinc atoms were difficult to transfer to the cathode in the open state due to the electrostatic adsorption of SO42−. In short, the introduction of zinc atoms made it difficult for the ions adsorbed on the anode to self-diffuse, and the possibility of spontaneous conversion of zinc atoms into zinc ions was very low. Finally, the zinc atoms were tightly anchored onto the anode surface and ultimately formed excellent discharge resistance.
The valence transition of Zn2+ on the anode side played a key role in the anti-self-discharge function. Moreover, the migration of ions in the hierarchical pores of the porous carbon surface was relatively easy, resulting in better anti-self-discharge characteristics. According to the continuous discharge curves, the corresponding capacity retention rates of ZHC-WAPC-4 and ZHC-WAPC-4/P after long-term self-discharge were calculated as 79.12% and 77.98%, respectively. The fewer functional oxygen-containing groups on the WAPC-4 surface also helped improve the self-discharge resistance of the ZHC-WAPC-4 cathode because oxygen-containing functional groups usually weaken electrostatic forces between the carbon electrode and electrolyte ions.69 This result (Fig. 5g) was better than many other related works.66,68–72 The calculated self-discharge rate (Fig. 5h) was also better than in many other reports.68,69,72–77 The ZHCs assembled from low-cost biomass-derived porous carbon and commercial zinc foil had good anti-self-discharge characteristics and cost-effectiveness.
To evaluate the energy density and power density, the zinc foil anode and WAPC-x cathode were assembled into a zinc ion hybrid capacitor device ZHC-WAPC-x in 2 M ZnSO4 (Fig. 6a). According to the mass of the porous carbon cathode, the values of energy density and power density of the ZHCs were deduced. ZHC-WAPC-4/P had the best energy density of 127.1 W h kg−1 at 400 W kg−1. The energy density of the device remained at 56.08 W h kg−1 at a high power density of 16000 W kg−1. Fig. 6b compares the Ragone diagram with other types of devices under similar conditions. In summary, the energy storage device using the ZHCs exhibited not only a high energy density used as a battery and but also a high power density used as a supercapacitor. Under a large current density, the structure of the active material was easily destroyed by high-speed charge–discharge, which led to the rapid capacity loss of the electrode (Fig. S6†). However, high-current charging and discharging are essential in practical applications. Therefore, the long-term charge–discharge cycle stability of devices under a high current is of key importance. Due to the stable structure of the carbon cathode, the hybrid device ZHC-WAPC-4/P exhibited an initial capacitance retention rate of 88.2% after being subjected to 15000 GCD cycles at 7.5 A g−1 (Fig. 6c), which is better than the cycle stability of ZHC-WAPC-4 (84.7%). The existence of the oxygen-containing functional group PCO3 induced by P doping significantly optimized the ion diffusion rate and cycle stability during the energy storage process of the ZHCs. The electrode did not change significantly before and after the cycling, and no foreign matter was generated, as shown by the SEM images and XRD patterns (Fig. 6d and e) of the WAPC-4/P electrode after long-term charge/discharge cycles. These results indicate its stable structure and chemical properties.
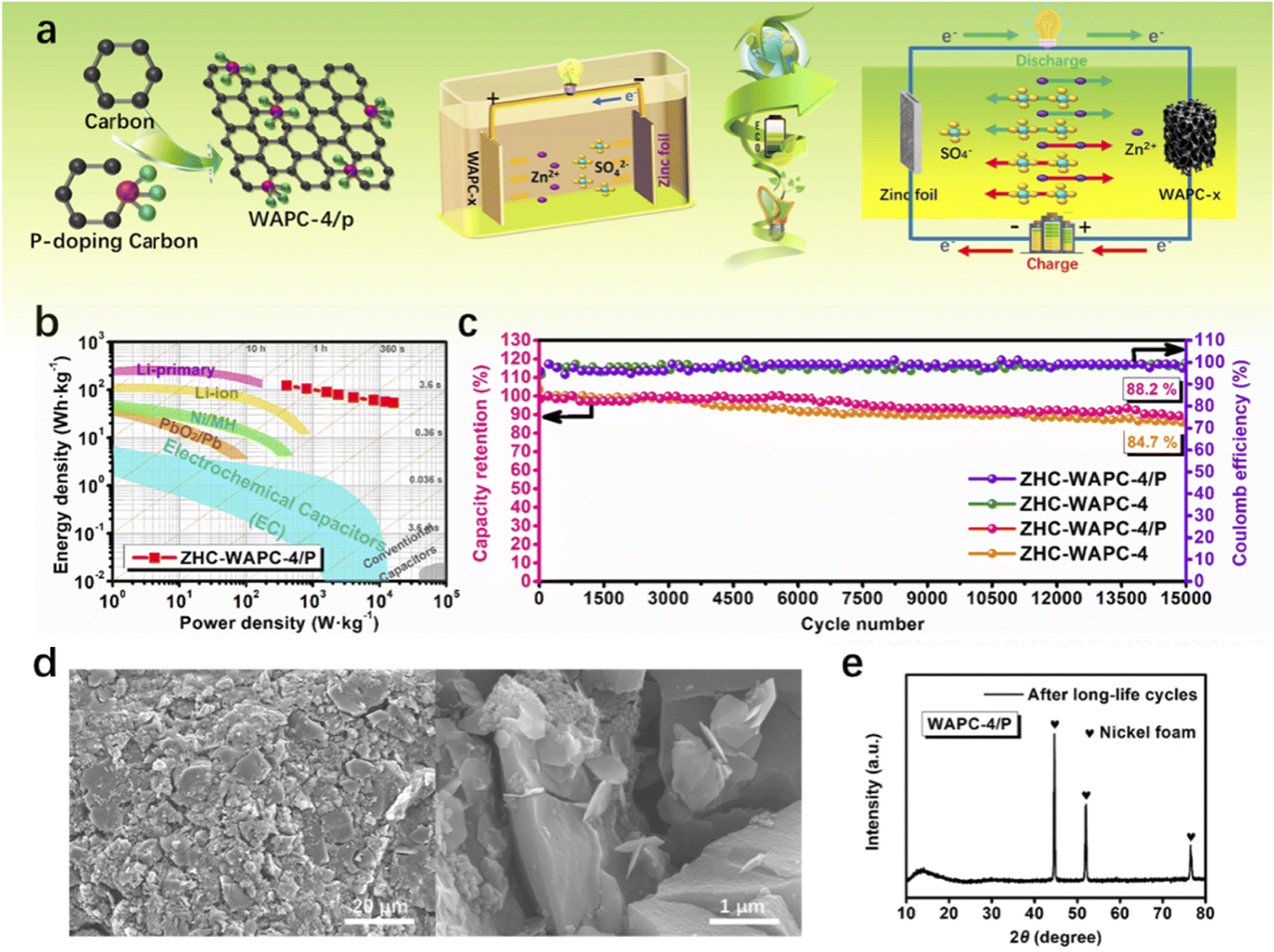 | ||
| Fig. 6 (a) Schematic diagram of the assembly and energy storage mechanism of ZHC-WAPC-x, (b) Ragone plots of ZHC-WAPC-4/P and different energy storage devices in terms of energy density and power density. (c) The cycling stability after in-depth 15000 GCD cycles procedure of ZHCs at 7.5 A g−1. (d) SEM image and (e) XRD pattern of WAPC-4/P cathode after long-term charge/discharge cycles. | ||
Conclusions
Choosing a suitable cathode material is critical when assembling high-performance ZHCs. In this paper, zinc ion hybrid capacitors were assembled through low-cost heteroatom-doped biomass-derived porous carbon cathodes and commercial zinc foil anodes. The porous carbons that passed the preliminary screening were further phosphated to introduce a large number of phosphooxy functional groups on the premise of maintaining the basic structure. The microstructure and redox activity of the final hierarchical porous carbon are further optimized and lead to the final excellent electrochemical performance. The assembled ZHC-WAPC-4/P exhibited an excellent specific capacity of 158.9 mA h g−1 and an ultra-high energy density of 127.1 W h kg−1 at a low current density. More importantly, the device had outstanding anti-self-discharge characteristics (retaining 77.98% of its specific capacity after 72 h natural self-discharge test) and long-term cycle stability (retaining 88.2% of its initial specific capacity after 15000 cycles at 7.5 A g−1). This work demonstrates an effective strategy for the optimization of electrode materials and the design of next-generation aqueous zinc ion energy storage devices.
Author contributions
Research design, H. S. and C. L.; experiments and data analysis, H. S., C. L., D. G., S. L., W. X.; S. L. and Z. L.; paper writing, H. S.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for support from the National Natural Science Foundation of China (11874317), Key Scientific Research Projects of Colleges and Universities in Henan Province (22A140009), Excellent Research Project for Overseas Students in Henan Province and Nanhu Scholars Program for Young Scholar of XYNU. This work made use of instruments in analysis testing centre of XYNU.References
- L. Dong, C. Xu, L. Yang, Z. H. Huang, F. Kang, Q. H. Yang and Z. Xin, J. Mater. Chem. A, 2016, 4, 4659–4685 RSC.
- Y. Cao, T. Zhou, K. Wu, Z. Yong and Y. Zhang, RSC Adv., 2021, 11, 6628–6643 RSC.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li and P. Bhattacharya, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- E. Adhamash, R. Pathak, Q. Qiao, Y. Zhou and R. McTaggart, RSC Adv., 2018, 13, 96–102 Search PubMed.
- P. Liu, Y. Gao, Y. Tan, W. Liu, Y. Huang, J. Yan and K. Liu, Nano Res., 2019, 12, 2835–2841 CrossRef CAS.
- J.-T. Cao, J.-L. Lv, Y.-X. Dong, X.-J. Liao, S.-W. Ren and Y.-M. Liu, J. Electroanal. Chem., 2021, 895, 115468 CrossRef CAS.
- H. Wang, M. Wang and Y. Tang, Energy Storage Mater., 2018, 13, 1–7 CrossRef CAS.
- Z. Li, D. Guo, Y. Liu, H. Wang and L. Wang, Chem. Eng. J., 2020, 397, 125418 CrossRef CAS.
- J. G. Wang, Z. Zhang, X. Zhang, X. Yin, X. Li, X. Liu, F. Kang and B. Wei, Nano Energy, 2017, 39, 647–653 CrossRef CAS.
- S. S. Haider, S. Zakar, M. Z. Iqbal and S. Dad, J. Electroanal. Chem., 2022, 904, 115881 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- M. Gheytanzadeh, A. Baghban, S. Habibzadeh, A. Mohaddespour and O. Abida, Res Adv., 2021, 11, 5479–5486 CAS.
- S. Wu, Y. Chen, T. Jiao, J. Zhou, J. Cheng, B. Liu, S. Yang, K. Zhang and W. Zhang, Adv. Energy Mater., 2019, 9, 1902915 CrossRef CAS.
- M. H. Alfaruqi, S. Islam, J. Gim, J. Song, S. Kim, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Chem. Phys. Lett., 2016, 650, 64–68 CrossRef CAS.
- R. Mukkabla, M. Deepa and A. K. Srivastave, ChemPhysChem, 2015, 16, 3242–3253 CrossRef CAS PubMed.
- X. Tong, F. Zhang, B. Ji, M. Sheng and Y. Tang, Adv. Mater., 2017, 28, 9979–9985 CrossRef.
- N. Shin, M. Kim, J. Ha, Y.-T. Kim and J. Choi, J. Electroanal. Chem., 2022, 914, 116296 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- S. Liu, W. Zheng, W. Xie, H. Cui, Y. Li, C. Zhang, Z. Ji, F. Liu, R. Chen, H. Sun and J. Xu, Carbon, 2022, 192, 162–169 CrossRef CAS.
- B. Li, J. Zheng, H. Zhang, L. Jin, D. Yang, H. Lv, C. Shen, A. Shellikeri, Y. Zheng and R. Gong, Adv. Mater., 2018, 30, 1705670 CrossRef PubMed.
- M. Sajjad, F. Cheng and W. Lu, RSC Adv., 2021, 11, 25450–25460 RSC.
- Z.-G. Liu, R. Du, X.-X. He, J.-C. Wang, Y. Qiao, L. Li and S.-L. Chou, ChemSusChem, 2021, 14, 3724–3743 CrossRef CAS PubMed.
- D. Su, A. Mcdonagh, S. Z. Qiao and G. Wang, Adv. Mater., 2017, 29, 1604007 CrossRef PubMed.
- A. Bouibes, N. Takenaka, K. Kubota, S. Komaba and M. Nagaoka, RSC Adv., 2022, 12, 971–984 RSC.
- Z. Jian, Z. Xing, C. Bommier, Z. Li and X. Ji, Adv. Energy Mater., 2016, 6, 1501874 CrossRef.
- K. Cao, H. Liu, W. Li, C. Xu, Q. Han, Z. Zhang and L. Jiao, J. Electroanal. Chem., 2019, 841, 51–55 CrossRef CAS.
- G. H. An, J. Hong, S. Pak, Y. Cho, S. Lee, B. Hou and S. Cha, Adv. Energy Mater., 2020, 10, 1902981 CrossRef CAS.
- M. S. Balogun, W. Qiu, W. Wang, P. Fang, X. Lu and Y. Tong, J. Mater. Chem. A, 2015, 3, 1364–1387 RSC.
- R. F. Ashour, D. H. Kelley, A. Salas, M. Starace, N. Weber and T. Weier, J. Power Sources, 2018, 378, 301–310 CrossRef CAS.
- Y. Sun, X. Liu, Y. Jiang, J. Li, J. Ding, W. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 18183–18208 RSC.
- X. Cao, L. Wang, J. Chen and J. Zheng, ChemElectroChem, 2018, 5, 2789–2794 CrossRef CAS.
- K. Li, Y. Shao, S. Liu, Q. Zhang, H. Wang, Y. Li and R. B. Kaner, Small, 2017, 13, 1700380 CrossRef.
- E. Bassey, L. Yang, M. Cao, Y. Feng and J. Yao, J. Electroanal. Chem., 2021, 881, 114972 CrossRef CAS.
- B. Li, F. Dai, Q. Xiao, L. Yang, J. Shen, C. Zhang and M. Cai, Energy Environ. Sci., 2016, 9, 102–106 RSC.
- Z. Li, D. Guo, D. Wang, M. Sun and H. Sun, J. Power Sources, 2021, 506, 230197 CrossRef CAS.
- F. R. McLarnon and E. J. Cairns, J. Electrochem. Soc., 1991, 138, 645 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- Y. Jiang, J. Li, Z. Jiang, M. Shi, R. Sheng, Z. Liu, S. Zhang, Y. Cao, T. Wei and Z. Fan, Carbon, 2021, 175, 281–288 CrossRef CAS.
- S. Yang, S. Wang, X. Liu and L. Li, Carbon, 2019, 147, 540–549 CrossRef CAS.
- T. Chen and L. Dai, Mater. Today, 2013, 16, 272–280 CrossRef CAS.
- H. Wang, Y. Shao, S. Mei, Y. Lu, M. Zhang, J. K. Sun, K. Matyjaszewski, M. Antonietti and J. Yuan, Chem. Rev., 2020, 120, 9363–9419 CrossRef CAS.
- H. Zhang, X. Li, D. Zhang, L. Zhang, M. Kapilashrami, T. Sun, P. A. Glans, J. Zhu, J. Zhong, Z. Hu, J. Guo and X. Sun, Carbon, 2016, 103, 480–487 CrossRef CAS.
- Y. G. Lee and G. H. An, ACS Appl. Mater. Interfaces, 2020, 12, 41342–41349 CrossRef CAS.
- J. Huang, W. Zhang, H. Huang, Y. Liu, Q. Yang and L. Li, ACS Appl. Mater. Interfaces, 2019, 7, 16710–16719 CAS.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- K. L. Van, T. L. T. Thu, H. N. T. Thu and H. V. Hoang, Russ. J. Electrochem., 2019, 55, 900–907 CrossRef.
- S. Liu, W. Zheng, M. Huang, Y. Xu, W. Xie, H. Sun and Y. Zhao, Nanotechnology, 2022, 33, 135401 CrossRef PubMed.
- J. Sun, J. Niu, M. Liu, J. Ji, M. Dou and F. Wang, Appl. Surf. Sci., 2018, 427, 807–813 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS.
- H. Sun, J. Xu, C. Wang, G. Ge, Y. Jia, J. Liu, F. Song and J. Wan, Carbon, 2016, 108, 356–362 CrossRef CAS.
- H. Sun, C. Fu, Y. Gao, P. Guo, C. Wang, W. Yang, Q. Wang, C. Zhang, J. Wang and J. Xu, Nanotechnology, 2018, 29, 305601 CrossRef.
- D. Wang, J. Nai, L. Xu and T. Sun, ACS Appl. Mater. Interfaces, 2019, 7, 18901–18911 CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- F. Sun, Z. Qu, J. Gao, H. B. Wu, F. Liu, R. Han, L. Wang, T. Pei, G. Zhao and Y. Lu, Adv. Funct. Mater., 2018, 28, 1804190 CrossRef.
- H. Jia, J. Sun, X. Xie, K. Yin and L. Sun, Carbon, 2019, 143, 309–317 CrossRef CAS.
- J. Wang, Z. Yang, F. Pan, X. Zhong, X. Liu, L. Gu and Y. Yu, RSC Adv., 2015, 5, 55136–55142 RSC.
- X. Zhao, Q. Zhang, C.-M. Chen, B. Zhang, S. Reiche, A. Wang, T. Zhang, R. Schlögl and D. Su, Nano Energy, 2012, 1, 624–630 CrossRef CAS.
- S. Chen, J. Wang, L. Fan, R. Ma, E. Zhang, Q. Liu and B. Lu, Adv. Energy Mater., 2018, 8, 1800140 CrossRef.
- H. M. Wang, H. X. Wang, Y. Chen, Y. J. Liu, J. X. Zhao, Q. H. Cai and X. Z. Wang, Appl. Surf. Sci., 2013, 273, 302–309 CrossRef CAS.
- J. Wang, Z. Yang, F. Pan, X. Zhong, X. Liu, L. Gu and Y. Yu, RSC Adv., 2015, 5, 55136–55142 RSC.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257–3261 CrossRef.
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- H. Li, J. Lang, S. Lei, J. Chen, K. Wang, L. Liu, T. Zhang, W. Liu and X. Yan, Adv. Funct. Mater., 2018, 28, 1800757 CrossRef.
- L. Dong, W. Yang, W. Yang, C. Wang, Y. Li, C. Xu, S. Wan, F. He, F. Kang and G. Wang, Nano-Micro Lett., 2019, 11, 94 CrossRef CAS PubMed.
- Q. Yang, Z. Huang, X. Li, Z. Liu, H. Li, G. Liang, D. Wang, Q. Huang, S. Zhang, S. Chen and C. Zhi, ACS Nano, 2019, 13, 8275–8283 CrossRef CAS PubMed.
- Y. Li, W. Yang, W. Yang, Z. Wang, J. Rong, G. Wang, C. Xu, F. Kang and L. Dong, Nano-Micro Lett., 2021, 13, 95 CrossRef CAS PubMed.
- A. M. Oicle, J. Tom and H. A. Andreas, Carbon, 2016, 110, 232–242 CrossRef.
- L. García-Cruz, P. Ratajczak, J. Iniesta, V. Montiel and F. Béguin, Electrochim. Acta, 2016, 202, 66–72 CrossRef.
- J. Q. Huang, T. Z. Zhuang, Q. Zhang, H. J. Peng, C. M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS PubMed.
- J. Menzel, E. Frackowiak and K. Fic, Electrochim. Acta, 2020, 332, 125435 CrossRef.
- R. Fei, H. Wang, Q. Wang, R. Qiu, S. Tang, R. Wang, B. He, Y. Gong and H. J. Fan, Adv. Energy Mater., 2020, 10, 2002741 CrossRef CAS.
- S. Dong, L. Wu, J. Wang, P. Nie, H. Dou and X. Zhang, J. Mater. Chem. A, 2017, 5, 5806–5812 RSC.
- Y. E. Zhu, L. Yang, J. Sheng, Y. Chen, H. Gu, J. Wei and Z. Zhou, Adv. Energy Mater., 2017, 7, 1701222 CrossRef.
- S. Li, J. Chen, X. Gong, J. Wang and P. S. Lee, Small, 2018, 14, 1804035 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04277k |
| This journal is © The Royal Society of Chemistry 2022 |