
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of iron pincer complexes in hydrosilylation reactions
Rasheed Nihala
a,
Kalathingal Nasreen Hisana
b,
C. M. A. Afsina
b and
Gopinathan Anilkumar
 *abc
*abc
aInstitute for Integrated Programmes and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Priyadarsini Hills P.O., Kottayam, Kerala 686560, India, +91-481-2731036. E-mail: anilgi1@yahoo.com; anil@mgu.ac.in
bSchool of Chemical Sciences, Mahatma Gandhi University, Priyadarsini Hills P.O., Kottayam, Kerala 686560, India
cAdvanced Molecular Materials Research Centre (AMMRC), Mahatma Gandhi University, Priyadarsini Hills P.O., Kottayam, Kerala 686560, India
First published on 26th August 2022
Abstract
Due to its abundance, low cost and low toxicity, the first-row transition metal, iron is widely preferred as a catalyst in organic synthesis. The only drawback of lower selectivity due to high reactivity and low stability of the metal centre is tuned by using pincer ligands of different types. The different iron pincer complexes thus prepared are extensively used in catalyzing different types of organic reactions with great selectivity and functional group tolerance under moderate reaction conditions. In this review, we focus on the applications of iron pincer complexes in hydrosilylation reactions, especially the hydrosilylation of carbonyl derivatives and alkene/alkynes.
 Rasheed Nihala | Rasheed Nihala was born in Kerala, India in 1997. Currently she is in the final year of the Five Year Integrated Interdisciplinary MSc Programme at the Institute for Integrated Programmes and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Kerala. Her research interests include transition metal catalysis and organic synthesis. |
 Kalathingal Nasreen Hisana | Kalathingal Nasreen Hisana was born in Kerala, India in 1997. She obtained her BSc. Degree from St. Josephs College, Devagiri, Kozhikode, India (2018) and her MSc. degree from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam, India (2020). Her research interests mainly focus on developing new catalytic pathways in synthetic organic chemistry and applying them productively in various areas of human need. |
 C. M. A. Afsina | C. M. A. Afsina was born in Kerala, India, in 1993. She obtained her B.Sc. degree from Kannur University (Government Brennen College, Thalassery) in 2014 and her M.Sc. degree from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam, Kerala, in 2016. She has qualified for the CSIR-UGC National Eligibility Test 2019 with a research fellowship and is currently pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his Ph.D. in 1996 from the Regional Research Laboratory (renamed as the National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at the University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 170 papers in peer-reviewed journals, 7 patents, 7 book chapters and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, 2021). He has received the Dr S Vasudev Award from Govt. of Kerala, India for best research (2016) and the Evonik research proposal competition award (second prize 2016). He is a fellow of the Royal Society of Chemistry. |
Introduction
Hydrosilylation refers to the addition of silicon hydrides (organic/inorganic) across C–C, C-heteroatom multiple bonds. Sommer in 1947 reported the first hydrosilylation reaction between 1-octene and trichlorosilane.1 Later, Speier's discovery of hexachloroplatinic acid2 in 1957 paved the way for Karstedt's platinum catalyst3 which offered better selectivity in reactions. Hydrosilylation reaction is applied in industry for the production of silicone polymers4 and silane coupling reagents. The organosilicon reagents obtained as a result of hydrosilylation are utilized in synthesis for cross-coupling reactions,5,6 stereospecific oxidation,7,8 etc. The significance of organic silicon compounds for stereochemical control of organic synthesis has been reviewed already.9 Surveys on hydrosilylation have resulted in books10 and reviews11–13 of significance.Amongst the first-row transition metals, iron has gained importance in catalysis as it is inexpensive, earth-abundant, and easily available and the aspect has been reviewed.14 Reviews on hydrosilylation of alkenes using iron catalysis are known.15–18 Similarly, Lu et al. reviewed works on iron-catalyzed asymmetric hydrosilylation of alkenes in 2018.19 Although iron is abundant and inexpensive, the difficulty to control the reactivity of the Fe centre was a challenge. This could be overcome by using “pincer” ligands in iron complexes. Tridentate ligands that show mer-coordination to the metal centre are generally said to be pincer-type ligands. The general structure of the ligand consists of a 1,3-disubstituted benzene ring with 3 donor atoms. The substitution on 1 and 3 positions acts as the chelating arms. The third coordination is from the C or N of the aromatic ring and in some cases P (Fig. 1). Based on the donor atoms of the ligand, these are classified into various types-PCP, PNP, NNN, POCOP, ECE, ENE etc.20 The name “pincer” was coined as the ligand tunes the metal centre and stabilizes unusual oxidation states.21,22
 | ||
| Fig. 1 Pincer ligand framework. | ||
The first pincer-type ligands were reported in 1976 by Shaw.23 Afterwards, several types of pincer complexes of transition metals have been synthesized and used as efficient catalysts in various reactions. Amongst the transition metal pincer complexes, iron pincers have received notable recognition. Several iron pincer complexes have been used as excellent catalysts in hydrogenation, reductive cyclization, etc. and have been reviewed.24 Some catalysts reviewed here are regio-, stereo- and chemoselective for hydrosilylation. Most of the iron pincer complexes are highly enantioselective with high catalytic activity. Certain catalytic procedures are attractive by their product selectivity which can be monitored by choosing the appropriate reaction conditions. Mechanistic pathways of various hydrosilylation reactions involving different types of iron pincer complexes are proposed by comparing their experimental data and corresponding DFT calculations.
Excellent iron-based pincer complex catalytic systems were formed utilizing MACHO type ligands (PR2–CH2–CH2–NR′–CH2–CH2–PR2) for hydrogenating polar bonds under mild reaction conditions. A similar ruthenium-based catalyst Ru-MACHO by Takasago is well known for industrial application in the large scale hydrogenation of (R)-lactate.25
Due to the weak coordination and high bond enthalpy, Csp3–H activation is more difficult than Csp2–H activation. Precious metals like Ru, Rh, Pd, Ir, etc. are employed for attaining this mostly. But cheaper and benign iron makes it a better metal for C–H activation. The dual chelation offered by pincer ligands allows easier Csp3–H activation possible by double cyclometalation.26–30 Thus, transition metal pincer hydrides could be obtained.
This review comprehends the advances in the hydrosilylation reactions catalyzed by iron pincer complexes. To the best of our knowledge, this is the first review focusing on the applications of iron pincer catalytic systems for the hydrosilylation reaction of carbonyls, alkenes and alkynes. For simplicity and better understanding, the review has been classified based on the type of pincer ligand coordinated to the iron complex. A map of ligands and catalytic complexes discussed in this review is presented in Table 1.
| Complex | Ligands | References |
|---|---|---|
| NNN pincer iron complexes | ||
 |
[(2,6-ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me))2C5H3N] (Ar = substituted aryl group), 2N2 C(Me))2C5H3N] (Ar = substituted aryl group), 2N2 |
30 |
 |
31 | |
 |
Iminobipyridine (BPI) ligands | 32 |
 |
 |
33 |
 |
Pyrazinediimine ligand (PPzDI), CO ligands | 35 |
 |
36 | |
 |
 |
37 |
 |
Iminopyridine oxazoline ligands | 38 |
 |
Chiral bis(oxazolinylphenyl)amines (Bopa) | 39 |
 |
 |
42 |
 |
 |
45 |
 |
 |
46 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| PNN pincer iron complexes | ||
 |
47 | |
 |
 |
50 |
 |
 |
53 |
|
||
| ENE pincer iron complexes | ||
 |
21 | |
|
||
| PNP and PONOP pincer iron complexes | ||
 |
 |
57 |
|
||
| POCOP pincer iron complexes | ||
 |
 |
58 |
 |
61 | |
 |
 |
62 |
|
||
| PPP pincer iron complexes | ||
 |
 |
69 |
|
||
| PCP pincer iron complexes | ||
 |
 |
70 |
 |
 |
72 |
|
||
| NCN pincer iron complexes | ||
 |
 |
73 |
 |
 |
74 |
|
||
| PSiP pincer iron complexes | ||
 |
 |
76 |
 |
77 | |
|
||
| Miscellaneous | ||
 |
Chiral bpi ligands | 81 |
 |
Chiral tridentate boxmi ligand | 82 |
 |
85 | |
Hydrosilylation catalyzed by iron pincer complexes
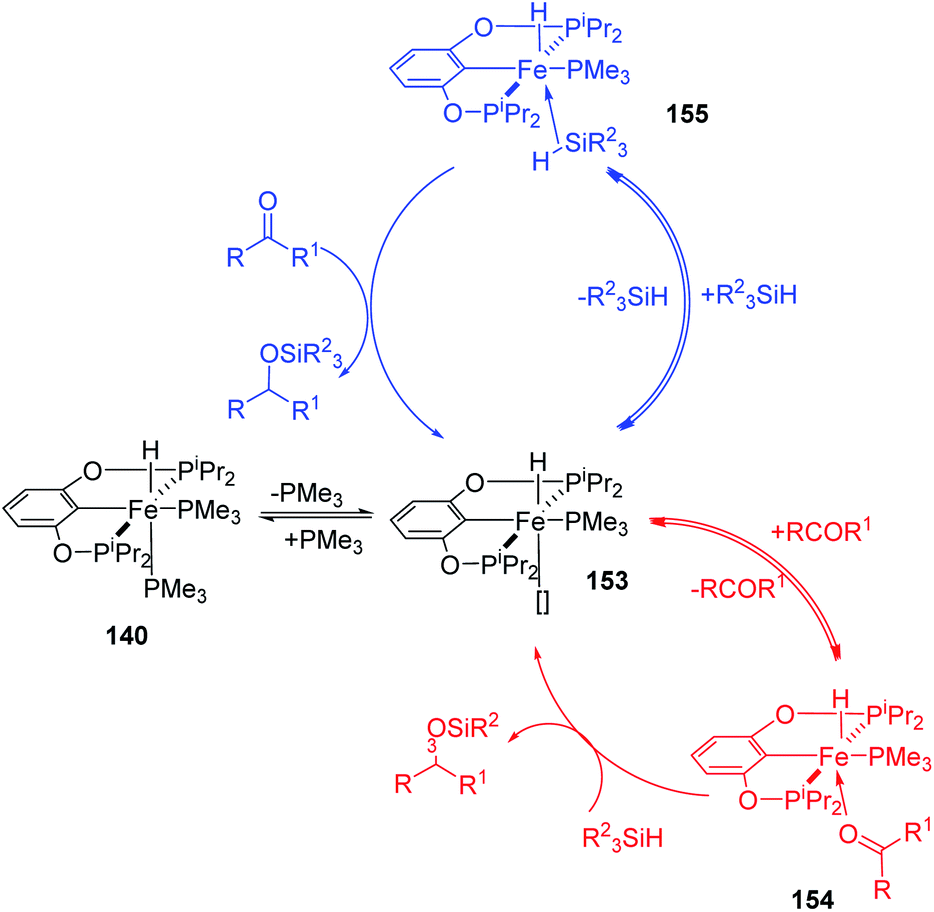
Iron pincer complexes are excellent catalyst for hydrosilylation reaction with high productivity and selectivity. Different types of chelating pincer ligand form catalytic complex with Fe to show varying catalytic activity. They are classified here based on structure of the chelating pincer ligands.NNN pincer iron complexes
In 2004, Chirik and co-workers reported the synthesis of bis(dinitrogen) iron(0) complex 3 which serves as a pre-catalyst in selective hydrosilylation reaction of alkenes and alkynes.31 On reducing 2,6-diisopropylphenyl iron(II) dihalides 1 or 2 with 0.5% sodium amalgam at 1 atm N2 for 48 h, 63% yield of bis(dinitrogen) iron(0) complex was obtained (Scheme 1). The complex catalyzed the hydrosilylation reaction without any activator and obtained TOF up to 364 h−1.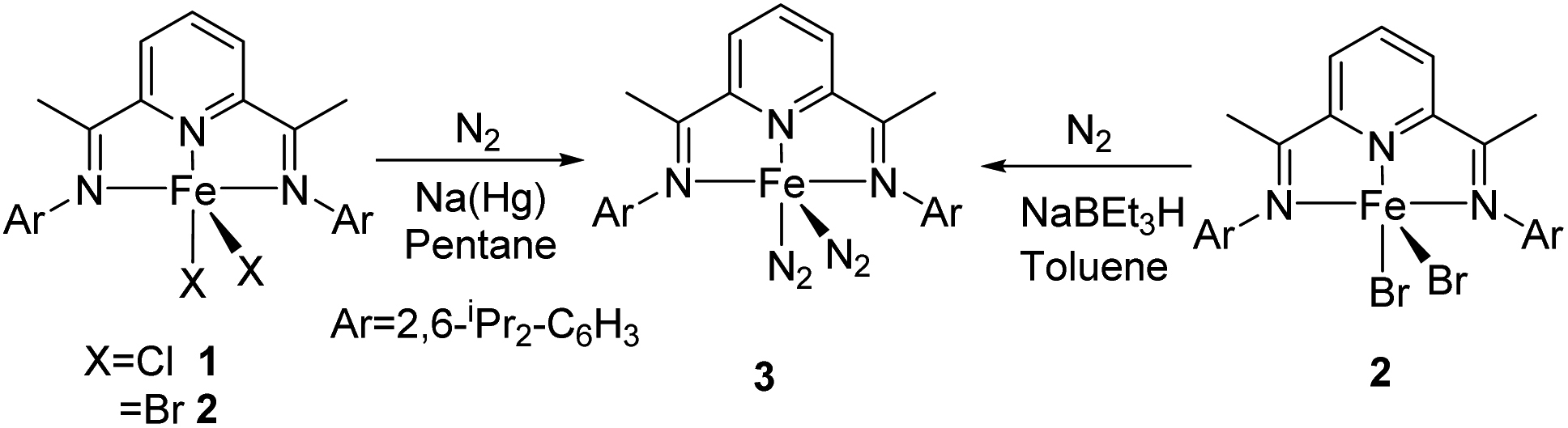 | ||
| Scheme 1 Synthesis of the bis(dinitrogen) iron(0) complex. | ||
Hydrosilylation of 1-hexene with Ph2SiH2 (or PhSiH3) catalyzed by 0.3 mol% 3 gave hydrosilylated anti-Markonikov product exclusively at 22 °C in 1 h. It was observed that reaction with PhSiH3 was rapid than that with Ph2SiH2. On analysing the substrate scope using different alkenes, it was found that terminal alkenes and styrene show rapid reaction followed by internal alkenes and gem-disubstituted alkenes. Also, terminal silanes predominated over internal silanes when trans-2-hexene was used as the substrate and dehydrogenative hydrosilylation products were not observed.
The iron dialkyl 5 (Fig. 2) was prepared in 83% yield by the reaction of bis-(imino)pyridine containing cyclohexyl substitution with (py)2Fe(CH2SiMe3)2 by Chirik et al. in 2008. On examining the iron dialkyls 4 and 5 as catalysts in carbonyl hydrosilylation, it was observed that a 1 mol% 4 or 0.1 mol% 5 and 2 equiv. of Ph2SiH2 at 23 °C followed by NaOH for 3 h in toluene gave the hydrosilylated product 7 in good to excellent yields32 (Scheme 2). The complexes exhibited good tolerance to functional groups also. Para-substituted acetophenones reacted rapidly whereas alkyl ketones were very sluggish. α,β-Unsaturated ketones like cyclohexenone and benzylidene acetone afforded 1,2-hydrosilylated products predominantly.
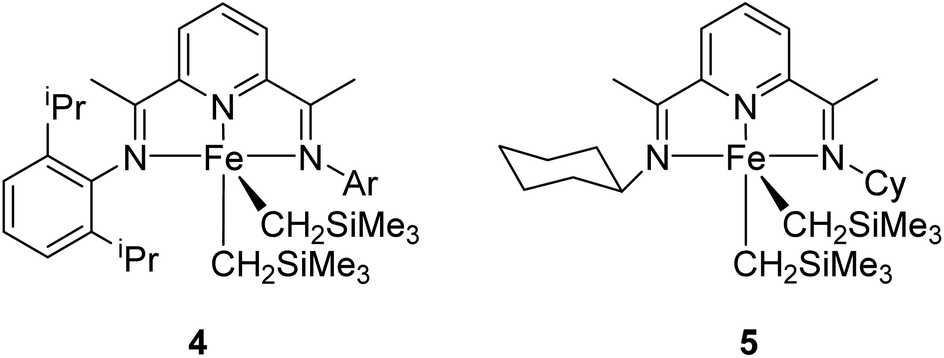 | ||
| Fig. 2 Iron dialkyl complexes. | ||
 | ||
| Scheme 2 Hydrosilylation of aldehydes and ketones catalyzed by bis(imino)pyridine iron dialkyl complexes. | ||
Nakazawa et al. in 2017 prepared a group of NNN type iron complexes using iminobipyridine (BPI) ligands of ketimine-type in the presence of anhyd. FeBr2 in THF at room temperature affording 56 to 99% yields (Scheme 3). Treatment of iminobipyridine derivatives 8 to 12 with FeBr2 in THF provided the iron complexes 13 to 17 ((HBPIAr,R)FeBr2) respectively. These complexes when activated with NaBHEt3 showed excellent catalytic activities in the hydrosilylation reaction of terminal alkenes when primary, secondary, and tertiary silanes were used.33 Examination of catalytic activity using different substituents on the imino carbon proved that reaction of imino nitrogen substituents and the imino carbon under appropriate conditions gave complexes with high catalytic activity with a turnover number up to 42000.
 | ||
| Scheme 3 Synthesis of iron complexes with BPI ligands. | ||
The room-temperature reaction of 1-octene and silane in 2:1 molar ratio with 0.01 mol% of (HBPIAr,R)FeBr2 catalyst and 0.2 mol% NaBHEt3 under N2 atmosphere room temperature resulted in the anti-Markovnikov product 1-octyl-silane. They also observed that primary and secondary silanes produced both monoalkylated and dialkylated silanes in the presence of the iron catalysts 13 and 15 and mostly monoalkylated products with 14 (Scheme 4). Thus, the olefin hydrosilylation was predicted to occur in two steps in which first the monoalkylated product is formed and then the dialkylated product. The selective synthesis of monoalkylated and dialkylated products could be attained by controlling the rate of each reaction which in turn could be done by altering the amount of catalyst used.
 | ||
| Scheme 4 Hydrosilylation of 1-octene using (BPI)FeBr2 complex catalysis. | ||
Lee and co-workers synthesized and determined the structure and catalytic power of the low-coordinated complex of iron (CztBu(PztBu)2)Fe[N(SiMe3)2] 27, containing NNN-pincer ligand 25 with dative pyrazole substitutions that are hemi labile and bis(trimethylsilyl)amide which offer steric hindrance (Scheme 5).34 At high temperature, they observed activation of intramolecular C–H bond at the 5-position of pyrazole group which furnished the pyrazolide-bridged complex of iron 27. Thus, the complex (CztBu(PztBu)2)Fe[N(SiMe3)2], could be obtained by reaction of Fe[N(SiMe3)2]2 26 and HCztBu(PztBu)2 25.35
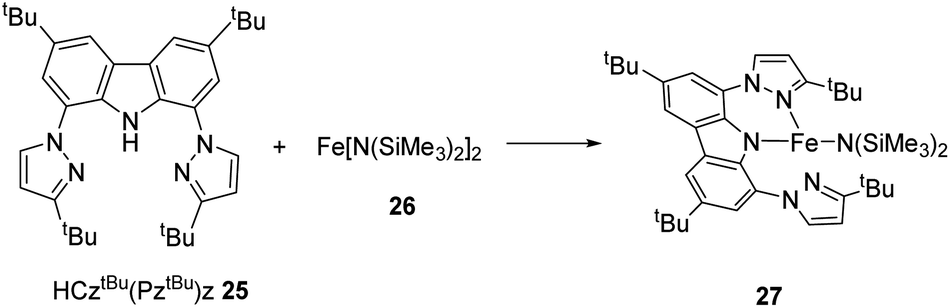 | ||
| Scheme 5 Reaction of HCztBu(PztBu)2 and Fe[N(SiMe3)2]2 to give (CztBu(PztBu)2)Fe[N(SiMe3)2]. | ||
The catalytic power of complex 27 was explored and it was found to be a chemoselective pre-catalyst in the hydrosilylation reaction of carbonyl compounds. Reaction of carbonyl compounds with PhSiH3 catalysed by 1 mol% 27 in C6D6 at mild temperature rapidly resulted in excellent yields of hydrosilylated products with TOF up to 55 m−1. But the reaction was slow for benzophenone. The products formed was mostly a concoction of PhSi[OCH(R1)R2], PhSiH[OCH(R1)R2]2 and PhSiH2[OCH(R1)R2]. By modifying the PhSiH3 to carbonyl ratio to 1:3 they got better TOFs and TONs as phenyl silane has three hydrides available for hydrosilylation. But increasing the catalyst loading to 10 mol% did not increase the catalytic performance. Although the catalytic mechanism is not correctly known, studies show the formation of an unstable paramagnetic species which is assumed to be the catalytically active iron hydride, HCztBu(PztBu)2 28, suggested to being formed by disproportionation of the pre-catalyst (Scheme 6).
 | ||
| Scheme 6 Disproportionation of the pre-catalyst. | ||
A novel NHC with redox activity, which acts as excellent π-acceptors and σ-donors was used to form a catalytic Fe/Rh complex by Roşca and co-workers.36 Simultaneous ligation of two metal centres is possible for the NHC. The catalyst 30 was obtained in 90% yield from the reaction of unstable NHC intermediate 29 using 0.5 equiv. of [Rh(COD)Cl]2 in THF for 2 h (Scheme 7). In order to study the electronic effects of the complex 29, they synthesized the Fe/Rh complex 30.
 | ||
| Scheme 7 Synthesis of COD substituted [FeRh] pre-catalyst. | ||
They studied the hydrosilylation of 4,4′-difluorobenzophenone 31 using Ph2SiH2 catalyzed by 2 mol% of the neutral and oxidized analogue of 30 in THF at 20 °C. It was observed that under similar reaction conditions, 31 was completely converted to products within 12 h and 2.5 h with catalysts 30 and 30+ respectively (Scheme 8). Thus, the oxidized form 30+ catalyses the reaction faster by ten times. Electronic structure of the NHC carbene and the rhodium complex were optimized through DFT calculations. It is cleared that reversible change in electronic properties of the hetero-bimetallic Fe/Rh system could be employed in redox-switch catalysis.
 | ||
| Scheme 8 Hydrosilylation of 4,4′-difluorobenzophenone by neutral or in situ-oxidised analogue of the catalyst. | ||
Lu in 2020 developed synthesis of chiral allyl silanes from racemic vinylcyclopropanes (VCP) 33 by 1,5-selective asymmetric hydrosilylation (Scheme 9) using NNN-FeCl2 35–39.37 This stereospecific cleavage of C–C bond not only furnished the allylic silanes 34 in high enantioselectivity but also recovered vinyl cyclopropanes 33 with very good enantioselectivity. The optimum condition for hydrosilylation of VCP with 1.5 equiv. PhSiEt3 at room temperature included 5 mol% NNN-FeCl2 35–39 catalyst with 15 mol% NaBHEt3 in dioxane. The alkyl or aryl groups bearing the VCP group follow a kinetic resolution pathway to form chiral allylic silanes with promising yields, stereoselectivity and enantioselectivity. Simultaneously, chiral VCPs are recovered with moderate to excellent yields and enantioselectivity here.
 | ||
| Scheme 9 Iron-catalyzed synthesis of chiral allyl silanes from racemic vinyl cyclopropanes. | ||
The authors also proposed a possible mechanism that proceeds via an iron-silyl complex (Scheme 10). From FeCl2 and silanes, iron-silyl species 40 is formed in the presence of NaBHEt3 and 3-chlorophenol. (S,S)-1-Phenyl-2-(1′-phenyl)vinylcyclopropane 33 undergo 1,2-insertion into the Fe–Si bond to form the tertiary alkyl iron species 41. The primary alkyl iron species 41 is formed through β-carbon elimination which further undergoes δ-bond metathesis with hydrosilane to obtain the desired product 34. The iron-silyl species is now ready for a new catalytic cycle.
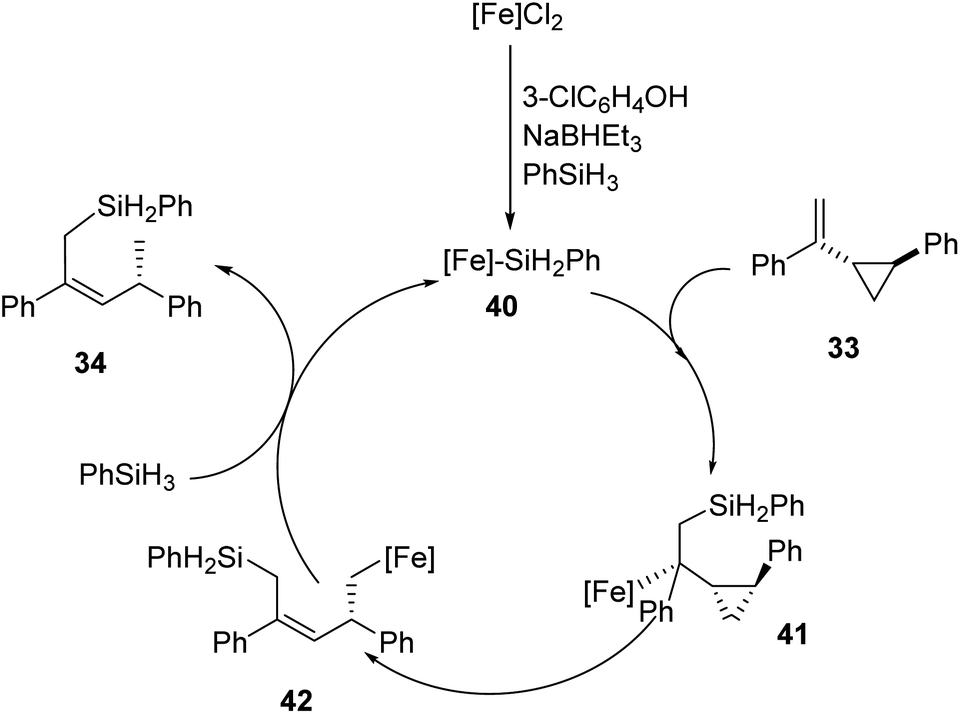 | ||
| Scheme 10 Possible mechanism that proceeds via iron-silyl complex. Reprinted from ref. 37, with permission from Elsevier. | ||
In 2016, Nakazawa et al. synthesized a series of iron complexes bearing an iminobipyridine ligand which were excellent catalysts for hydrosilylation of 1-octene along with primary, secondary, and tertiary silanes.38 NaBHEt3 activated the reaction to produce mono-, di- and tri-alkylated silanes. The catalyst TON of 12038 was obtained for the reaction between 1-octene and Ph2SiH2, and the catalyst was active at 100 °C.
Treatment of iminobipyridine derivatives 43 to 47 with FeBr2 in THF provided the iron complexes 48 to 52 ((HBPIAr,R)FeBr2) respectively (Scheme 11). Desired hydrosilylation products can be selectively synthesized by choosing suitable reaction conditions. A series of reaction solvents including hexane, THF, toluene, and diethyl ether facilitated the process with varying reaction temperatures and time. TONs were increasing with a decrease in pre-catalyst concentrations.
 | ||
| Scheme 11 Synthesis of iron complexes with BPI ligands. | ||
The team proposed a plausible mechanism for the hydrosilylation reaction of 1-octene with hydrosilane seems to follow Chalk–Harrod mechanism (Scheme 12). (BPI)FeBr2 53 reacted with NaBHEt3 to form (BPI)FeH2 54 which undergoes dehydrogenation to form a 14e species 55. 56 is formed through oxidative addition of 55 with a Si–H bond of silane where Fe and the 3 N donors make a plane perpendicular to the plane in which Si, Fe, and H are present. Olefin coordinate with Fe in 56 The olefin goes towards the Fe from the side close to the hydride ligand to form 57, which is followed by the insertion of olefin into the Fe–H bond to form 58 and finally, the reductive elimination of the alkyl and the silyl ligands forms the alkylsilane. Otherwise, the olefin may coordinate from the side close to the silyl ligand to form 59, which is followed by the insertion of olefin into the Fe–Si bond to form 60. From here, 60 takes two different paths which are reductive elimination to give alkylsilane and β-hydride elimination to give 61 which releases vinylsilane.
 | ||
| Scheme 12 Plausible mechanism for hydrosilylation of 1-octene catalyzed by iminobipyridene iron complexes. | ||
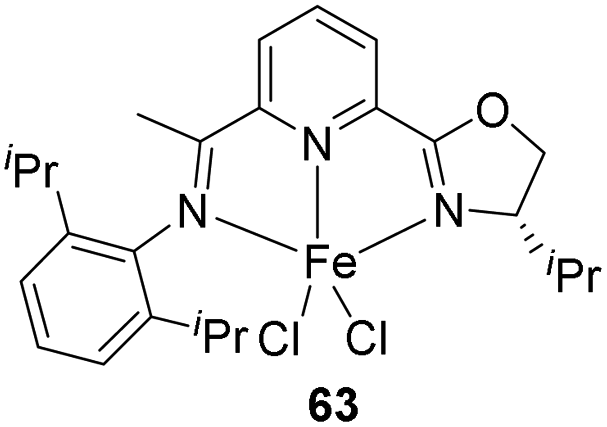
In 2015, Lu et al. introduced the highly regio- and enantioselective iron-catalysis for the anti-Markonikov asymmetric hydrosilylation of 1,1-disubstituted aryl alkenes 64 to form chiral organosilanes.39 The catalysis was enhanced with iminopyridine oxazoline ligands which forms a complex iron precatalyst 63 (Fig. 3). Easy preparation of a series of chiral organosilanes as well as organosilanols can be achieved from simple alkenes without the presence of a directing group.
 | ||
| Fig. 3 Iminopyridine-oxazoline iron complex. | ||
For hydrosilylation reaction, under optimized condition (alkene 64 (1.2 mmol), Ph2SiH2 (1.0 mmol), 63 (1–5 mol%), NaBHEt3 (3–15 mol%), neat/toluene (1 mL), Ar, RT, 12 h), chiral organo silanes with promising yields and excellent ee values were obtained for most of them (Scheme 13).
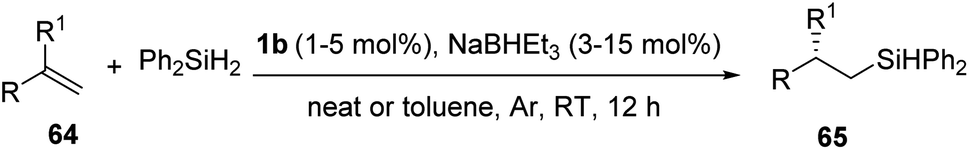 | ||
| Scheme 13 Hydrosilylation of 1,1-disubstituted aryl alkenes catalyzed by iminopyridine-oxazoline iron complex. | ||
On evaluating the substrate scope, electron-rich styrenes having ortho-, meta-, and para-substitutions afforded hydrosilylated products with good yields and excellent values of ee. Aldehyde and ketone protected by acetal group, amino, and imine groups well tolerated with the protocol to give products with 78–92% yields and 79–93% ee. Unfortunately, product wasn't forming when 4-(prop-1-en-2-yl)benzonitrile and 2-(prop-1-en-2-yl)benzonitrile were subjected to the reaction. Enantioselectivity of electron-deficient styrenes were affected by the position of substituent. Product yields were good but enantioselectivity was lower for meta-substituted styrenes. Ortho-substituted styrenes gave 66–84% ee while 1-(propen-2-yl)naphthalene and 2-(propen-2-yl)naphthalene afforded excellent yields and excellent ee values. Cyclic hydrosilylation products were excellent in yield and ee values. Even though aliphatic 1,1-disubstituted olefins and 1,2-disubstituted olefins afforded good yields, they were compromising in enantioselectivity.
Chiral bis(oxazolinylphenyl)amines (Bopa) were utilized as an excellent auxiliary ligand for iron catalyst for the asymmetric hydrosilylation ketones by Nishiyama and co-workers in 2010.40 Bopa was used as an excellent chiral ligand for various asymmetric catalysis previously.41,42 A mixture of 2 mol% Fe(OAc)2 and 3 mol% of Bopa-dpm were heated at 65 °C while ketone 67 is present, to obtain the iron–Bopa-dpm catalyst 66 (Fig. 4). Two equivalents of (EtO)2MeSiH were added to the mixture and stir for 24 h to obtain the desired asymmetric hydrosilylation products 68 (Scheme 14). Excellent enantioselectivities up to 73% ee were obtained in this catalysis.
 | ||
| Fig. 4 Iron–Bopa-dpm catalyst. | ||
 | ||
| Scheme 14 Hydrosilylation of ketones using by iron–Bopa-dpm catalyst. | ||
From substrate scope studies, it is found that para-substituted phenyl ketones like dimethylamino and morpholino derivatives afforded over 80% ee values. While ketones having meta- and ortho-substituents obtained lower ee values of 50–73%. 2-Acetylnaphthalene and its substituted derivative gave 58–73% ee. They examined iron–Bopa-ip catalyst for the reaction and obtained only lesser yields with very low ee values. From the catalytic study, it is clear that in organic synthesis, hydrogen gas can be replaced by hydrosilanes.
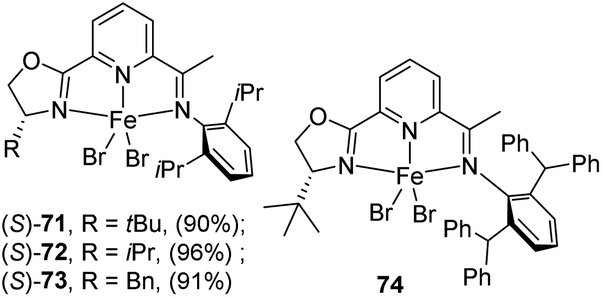
In 2015, Huang and co-workers synthesized a series of NNN type iron complexes with chiral iminopyridine-oxazoline (IPO) ligands and were tested for the asymmetric hydrosilylation reaction of aryl ketones.43 The group had developed these IPO ligands in 2014 for the first time.44 The catalysts afforded excellent product yields with higher enantioselectivity. Chiral IPO ligands when treated with anhyd. FeBr2 in THF afforded high yields of Fe(II) dibromide complexes [(S)-IPO]FeBr2 (R = tBu, (S)-71; R = iPr, (S)-72; and R = Bn, (S)-73) with various substituents at the oxazoline moieties (Scheme 15). Similarly, iron complex (S)-74 substituted with CH(Ph)2 at the 2,6-aryl positions and tBu at the oxazoline moiety was prepared in high yields.
 | ||
| Scheme 15 Synthesis of chiral iminopyridine-oxazoline iron complexes. | ||
The series of iron dibromide complexes (1 mol%) was activated using 2 equiv. of NaBEt3H to catalyse the asymmetric hydrosilylation of 4′-isobutylacetophenone 75 using Ph2SiH2 in toluene at 25 °C for 10 h (Scheme 16). The catalysts exhibited excellent activity with high enantioselectivity in the reaction.
 | ||
| Scheme 16 Asymmetric hydrosilylation of aryl ketones catalyzed by chiral iminopyridine-oxazoline iron complexes. | ||
The activity of the catalyst is further tested by reacting a range of aryl ketones 76 under the same reaction conditions using 1 mol% of (S)-74. Position of the substitution on aryl rings affects the product yields. The reaction obtained 91% ee for substrate with methoxy group at para-position. Whereas methoxy group at meta- and ortho-position resulted in 64% and 19% ee only. On contradictory to this, chloro-substitution at ortho-position afforded highest yield than para- and meta-substituted ones as 75%, 61% and 63% ee respectively. Sterically hindered 1-mesitylethanone afforded 97% yield, 93% ee. Enantioselectivity decreased with increase in steric hindrance of substitution at benzylic position. 1,1-Dialkyl and 1,1-diaryl ketones afforded products with low enantioselectivity.
They proposed a mechanism (Scheme 17) in which NaBHEt3 reduced FeBr2 is followed by coordination of ketone to the Fe centre. Silane undergo oxidative addition to this new complex 79 to form an 18 electron Fe(II) silyl hydride ketone species 80. This oxidative addition is also expected to occur prior to ketone coordinating with Fe centre. Ketone undergo migratory insertion into the Fe–H bond to form complex 81 from which ketone hydrosilylation product is the reductively eliminated.
 | ||
| Scheme 17 Plausible mechanism for chiral iminopyridine-oxazoline iron complexes catalyzed asymmetric hydrosilylation of aryl ketones. | ||
In 2017, Gade et al. synthesized PdmBOX iron(II) complex45 which acted as an excellent precatalyst along with (tmeda) Fe(CH2TMS)2 adduct for the hydrosilylation of acetophenone.46 PdmBOX (2,5-bis(2-oxazolinyldimethylmethyl)pyrrol) pincer ligands which are generated in situ were treated with FeCl2 in THF to form the precatalyst (Scheme 18).
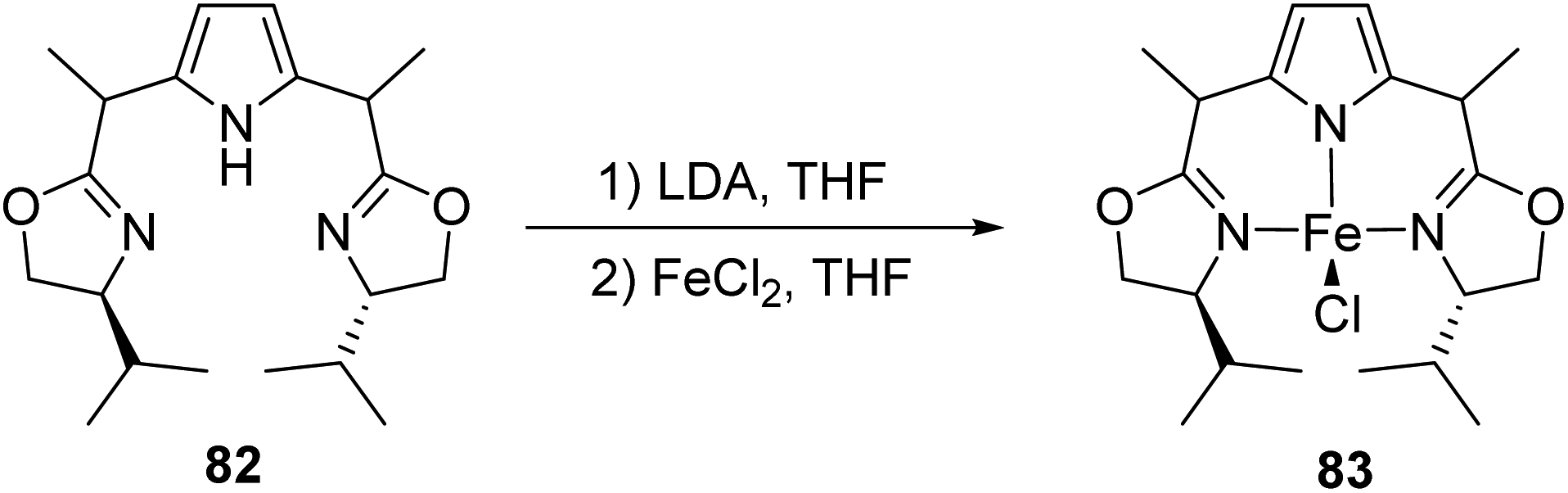 | ||
| Scheme 18 Synthesis of PdmBOX iron(II) complex. | ||
In 2009, Chirik et al. prepared a series of enantiopure pyridine bis(oxazoline) (“Pybox”) iron dialkyl compounds and bis(oxazoline) (“Box”) iron dialkyl complexes (Scheme 19).47
 | ||
| Scheme 19 PdmBOX iron(II) complex catalyzed hydrosilylation of acetophenone. | ||
(S,S)-(iPrPybox)FeCl2 86 was mixed in pentane at −35 °C for 30 min which is followed by treating with LiCH2SiMe3 at room temperature for 2 h to give Pybox iron dialkyl complex 87 with 26% yield (Scheme 20). Alternatively, the compound can be prepared by adding (py)4 FeCl2 into pentane at −35 °C for 30 min which is followed by treating with LiCH2SiMe3 at room temperature for 1–2 h and obtaining 71% yield. (S,S)-(iPrBox)FeCl2 88 is mixed in diethyl ether along with LiCH2SiMe3 at −35 °C, followed by treatment with alkyl lithium for 16 h and 89% of Box iron dialkyl complex 89.
 | ||
| Scheme 20 Synthesis of enantiopure pyridine bis(oxazoline) and bis(oxazoline) iron dialkyl complexes. | ||
The catalysts were evaluated for catalysing hydrosilylation of carbonyl compounds. At 23 °C, 0.3% of the Pybox iron dialkyl catalyst and 1 mol% of Box iron dialkyl catalyst were used along with PhSiH3 which acts as a stoichiometric reductant in toluene for asymmetric hydrosilylation of ketones. Finally, B(C6F5)3 was used to activate the catalysts and tested for the hydrosilylation reaction.
On evaluating the substrate scope studies, it is clear that the catalyst was very active in the reaction. A TON of at least 330 h−1 was obtained when OMe and CF3 were present. Steric factor matters as in the case of 2,4,6-trimethyl acetophenone and 2,6-dimethyl-4-tBu acetophenone which had shown low turnover number. The yield of the reaction remains constant with Pybox complex according to the presence of alkyl substitution on it. Enantioselectivity of the reaction is poor with a maximum of 49 ee value.
Similar observations were received in the case of Box complex catalysis. Very low enantioselectivities were obtained. On activation with B(C6F5)3, modest improvements were obtained. For certain substrates like acetophenone and 2-hexanone, productivity was improved and for some substrates like acetophenone and α-tetralone, enantioselectivity was improved.
PNN pincer iron complexes
A dual-catalyst system which consists of a pincer-coordinated Ir and Fe catalyst was demonstrated by Huang and co-workers in 2015 for one-pot silylation of alkanes that occur via two-steps.48 The Ir complex catalyses the alkane dehydrogenation step and the Fe complex catalyses the tandem olefin isomerization–hydrosilylation reaction. The internal olefins obtained from Ir catalyzed hydrogenation are rapidly isomerized to terminal olefins and undergone anti-Markovnikov regioselective hydrosilylation by the aid of Fe catalysts 90–93 (Fig. 5) to furnish terminal silanes.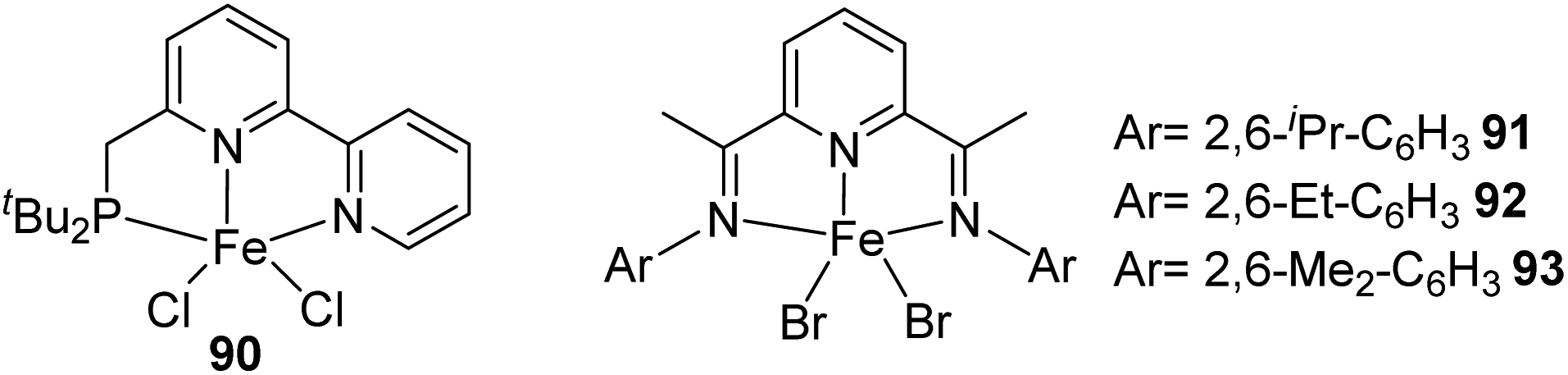 | ||
| Fig. 5 Pincer-ligated pre-catalysts of iron. | ||
Reactions of trans-3-octene and bis(trimethylsiloxy)methylsilane along with 5 mol% each of the pre-catalysts (iPrPDI)FeBr2 91–93 and (PNN)FeCl2 90 in n-hexane with 10 mol% of the activator NaHBEt3 furnished the primary alkyl silane 96 in 4% and 13% yield respectively. Substituting complex 91 by 92 or 93 which contains ethyl or methyl groups at the 2,6-positions of the N-aryl groups gave the product 96 with a much higher yield of 86% each (Scheme 21). A combination of the 5 mol% each of 92 or 93 and 0.5 mol% of 94 gave 96 in 82% yield indicating the tolerance of the Ir species (Fig. 6) by the Fe catalysts.
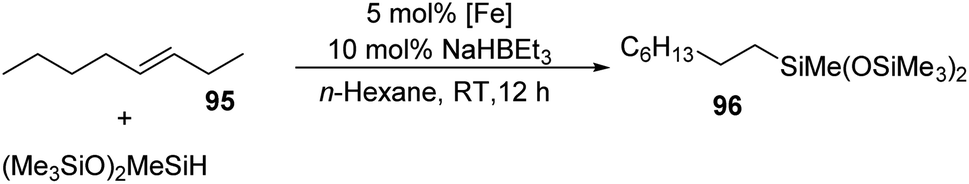 | ||
| Scheme 21 Tandem alkene isomerization–hydrosilylation catalyzed by Fe. | ||
 | ||
| Fig. 6 Ir catalytic complex. | ||
Dihalide pincer complexes of iron reacted with activators in stoichiometric amounts. NaOtBu or KOtBu results in its decomposition.49,50 Carboxylate complex or alkyl complex was formed on reaction with KOPv51 or LiCH2SiMe3 (ref. 52) respectively (Scheme 22). Yuge and co-workers in 2019 demonstrated the application of complex 98 and 99 without an activator in catalytic hydrosilylation reaction of alkenes and could obtain high TOF up to 90000 h−1 and TON up to 480000 which were the highest numbers reported in hydrosilylation reactions catalyzed by iron.53
 | ||
| Scheme 22 Reaction of quinoline-based PNN iron pincer complex with activators. | ||
Later in 2020, they developed a group of pincer iron complexes 97, 107, 108, 113, 114 and 115 (Scheme 23) of which 97 and 107 were already known and others were new.54 The application SambVca 2.1 (free web application for evaluating steric parameters of a wide variety of ligands, including pincer type ligands) was used for the correlation study of product selectivity and reaction conversion with steric parameter (% VBur) for hydrosilylation. Steric parameters were calculated separately on the site of reaction and the PNN ligand. They also analysed how steric parameters are distance-dependent.
 | ||
| Scheme 23 Synthesis of PNN iron pincer complexes. | ||
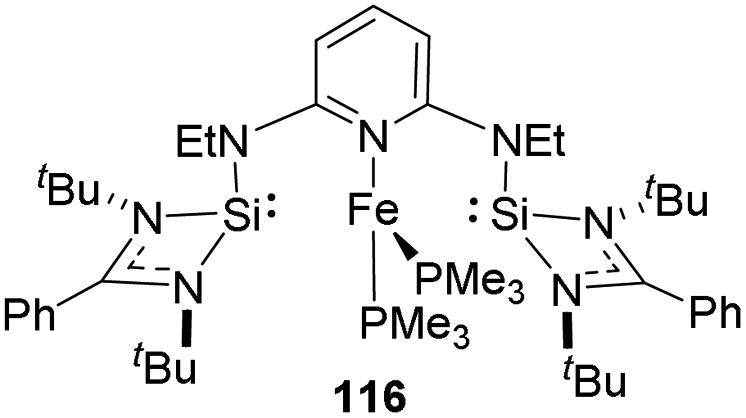
ENE pincer iron complexes
Oestreich and co-workers in 2015 established a new mechanism for hydrosilylation of carbonyls 117 catalyzed by transition metals through theoretical and experimental studies on SiNSi iron(0) pincer55,56 116 catalyzed hydrosilylation.57 A range of acetophenones when treated with 2.5 mol% of 116 at high reaction temperatures (Scheme 24). Substrates well tolerated with the method irrespective of electronic nature of substituents, but steric factor mattered negatively. | ||
| Scheme 24 SiNSi pincer complex catalysed hydrosilylation of acetophenones. | ||
They found that the reaction takes place on the periphery of the iron centre and not by outer or inner sphere mechanism and iron is not involved directly (Scheme 25). Silyl group of 119 behave as a Lewis acid. The ketone 120 forms a coordination with silyl group to form an activated intermediate 121. The hydrosilane 122 coordinated to 121 through carbonyl group and undergo hydrosilylation to release the product 124.
 | ||
| Scheme 25 Carbonyl hydrosilylation catalyzed by SiNSi iron pincer complex via peripheral mechanism. | ||
PNP and PONOP pincer iron complexes
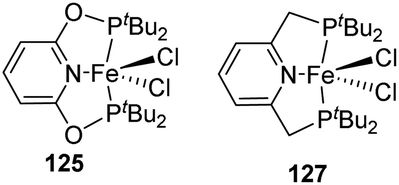
Later, the tBuPONOP (2,6-bis(di-tert-butyl-phosphinito)pyridine) iron complex (tBuPONOP)FeCl2 125 was prepared by Findlater and co-workers by reaction of anhydrous FeCl2 with tBuPONOP 124 in THF for 12 h at room temperature (Scheme 26).58 Under the same reaction conditions, (tBuPNP)FeCl2 127 was prepared using tBuPNP. | ||
| Scheme 26 Synthesis of PNP and PONOP iron complexes. | ||
The catalytic activity of 125 and 127 in hydrosilylation of carbonyls was examined (Scheme 27). It was observed that a variety of ketones with long-chain and cyclic aliphatic and aromatic groups undergo the reaction affording 40 to 80% yield. Aromatic aldehydes gave the best yields (50–96%) whereas, sterically hindered aliphatic ketones gave comparatively the least yields. Heterocyclic ketones (2-acetylthiophene), as well as aldehydes (2-thiophenecarboxaldehyde), gave moderate yields.
 | ||
| Scheme 27 (tBuPNP)FeCl2/(tBuPONOP)FeCl2 catalyzed hydrosilylation of aldehydes and ketones. | ||
The outcome of electron-deficient and electron-rich groups on the rate of reaction of aldehydes was studied by performing competitive experiment. Reaction of equimolar mixture of the benzaldehydes 131, 133 and 135 with 2.0 equiv. of (EtO)3SiH resulted in 132, 134 and 136. Monitoring the reaction progress using GC-MS analysis proved that the siloxy products resulted in 1:2.5:4.5 ratio for aldehydes with –OMe:–H:–CF3 substitutions (Scheme 28). Thus, it was concluded that para-positioned electron-deficient substituents increases the rate of hydrosilylation of aldehydes such that the most electron-deficient aldehyde is readily reduced.
 | ||
| Scheme 28 The outcome of electron-deficient and electron-rich groups on the rate of hydrosilylation of benzaldehyde. | ||
POCOP pincer iron complexes
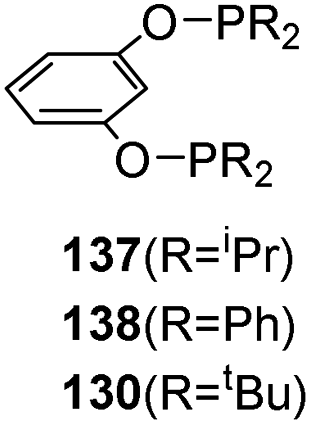
In 2011 Guan and co-workers synthesized iron hydride complexes with phophinite type ligands by cyclometalation and their application in hydrosilylation of carbonyls were explored.59 The resorcinol-derived bis(phosphinite) ligands (1,3-(R2PO)2C6H4) 137–139 were treated with Fe(PMe3)4 in equimolar amounts in THF at room temperature for 24 h to synthesize iron pincer hydride complexes (Scheme 29). The reaction obtained product with 67% and 69% yields when R = iPr and R = Ph respectively, but no product formed when 1,3-((tBu)2PO)2C6H4 was chosen. | ||
| Scheme 29 Synthesis of POCOP-pincer iron catalysts. | ||
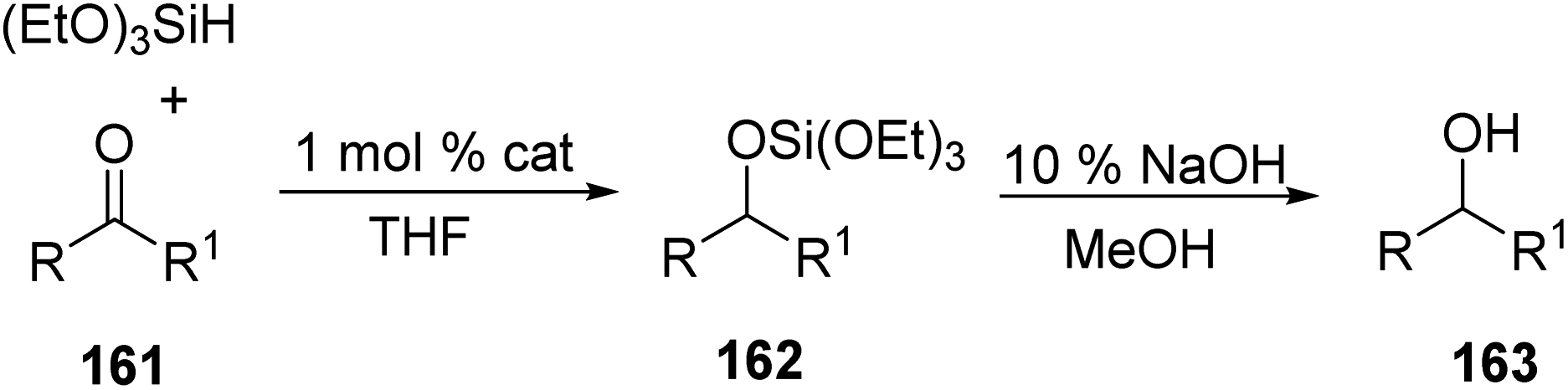
They examined the catalytic activity of 140 using PhCHO and found that with 1.1 equiv. (EtO)3SiH and 1 mol% of 140 at 50 °C in THF gave (EtO)3SiOCH2Ph in 1 h with 80–92% yield (Scheme 30). The catalytic activity was in the order 140 > 143 > 146 > 145 for complexes having same pincer ligand. Apart from (EtO)3SiH, Ph2SiH2 and PhSiH3 were also found to be appropriate silanes for hydrosilylating PhCHO, but these gave multiple hydrosilylated product due to the presence of more than one Si–H bond.
 | ||
| Scheme 30 Aldehyde hydrosilylation using 2,6-[((iPr)2PO)2C6H3]Fe(H)(PMe3)2. | ||
Studies on the substrate scope showed that fluoro-, methyl-, methoxy-, and N,N-dimethyl-benzaldehydes and aromatic aldehydes like 2-furaldehyde and 2-naphthaldehyde furnished the products in good yields but electron-rich groups make the reaction sluggish. It was also observed that only CO group is reduced and not the CC group. Moreover, ketones were found to be less reactive on comparison with aldehydes and required 80 °C for complete conversions obtaining up to 88% yield of desired products (Scheme 31). Hydrosilylation of aliphatic ketones was also successful except for bulky ketones like 2,4,6-trimethylacetophenone. Both electron-rich and electron-deficient substituents on the ketones lowered the reactivity than the ketones that are unsubstituted but methoxy-, amino- and pyridyl substituents could be tolerated.
 | ||
| Scheme 31 Ketone hydrosilylation using 2,6-[((iPr)2PO)2C6H3]Fe(H)(PMe3)2. | ||
Mechanistic studies conducted by the group show that CO insertion does not occur and the hydride remains intact throughout the cycle. Thus, they proposed a possible mechanism for the catalytic pathway that proceeds via CO or PMe3 dissociation which creates a vacant site for the coordination of silane or carbonyl to iron for activation (Scheme 32). The η2-silane σ-adduct formed by coordination of silane to the vacant site reacts with the carbonyl group. If the carbonyl occupies the vacant site giving an η1- or η2-carbonyl species, it will undergo hydrosilylation in the next step. But, the DFT calculations by Wei et al. supported carbonyl insertion to iron–H bond.60 DFT calculations of free energy changes of the most favourable proves that the addition of an Si–H bond across CO bond require a higher barrier than that for carbonyl insertion to the iron–H bond.
 | ||
| Scheme 32 Possible mechanistic pathways for carbonyl hydrosilylation catalyzed by 2,6-[((iPr)2PO)2C6H3]Fe(H)(PMe3)2. Adapted with permission from ref. 59. Copyright 2011 American Chemical Society. | ||
Wei et al. in 2014 performed the theoretical studies on POCOP-pincer iron(II) hydride to show that the hydride is participating in carbonyl hydrosilylation catalyzed by 2,6-[(R2PO)2C6H3]Fe(H)(PMe3)2.61 The mechanistic pathway of the reaction is expected to be the same as previously discussed one (Scheme 32). The carbonyl pre-coordination pathway was found to be the most favourable energetically. η1- or η2-carbonyl species formed by coordination of carbonyl to the vacant site undergoes migratory insertion to iron–hydrogen bond resulting in alkoxide intermediate. The σ-bond metathesis of Fe–O or Si–H bond of the alkoxide intermediate gave the hydrosilylated product.
Another proposed pathway was ionic hydrosilylation pathway but the theoretical studies shows that this requires ∼30.0 kcal mol−1 higher energy of activation when compared to the carbonyl pre-coordination pathway.
Again in 2014, Guan et al. published their studies on the protonation of iron hydride complexes 140 to 145 (Fig. 7) using different Brønsted acids (Scheme 33) revealing the order of basicity as 140-H > 143-H > 146-H > 145-H.62 The cationic complexes formed were found to be efficient in catalysing hydrosilylation reaction of carbonyls.
 | ||
| Fig. 7 POCOP-pincer iron hydrides. | ||
 | ||
| Scheme 33 Protonation of iron hydride complexes using different Brønsted acids. | ||
Reaction of benzaldehyde with triethoxysilane along with 1 mol% of 143+-BF4 in C6H5Cl at 50 °C was completed in 24 h. But the product could be obtained with only 15% yield when the neutral complex 143-H was used and the reaction did not proceed with 146-H. Cationic complex 145+-BF4 also had poor catalytic efficiency and gave just 10% yield of the hydrosilylation product in 48 h.
Hydrosilylation of acetophenone with triethoxysilane in C6H5Cl occurred at an elevated temperature of 80 °C and extended reaction time of 48 h with 79%, 54%, 63% and 56% conversion catalyzed by 1 mol% each of 143+-BF4, 143-H, 145+-BF4 and 145-H respectively.
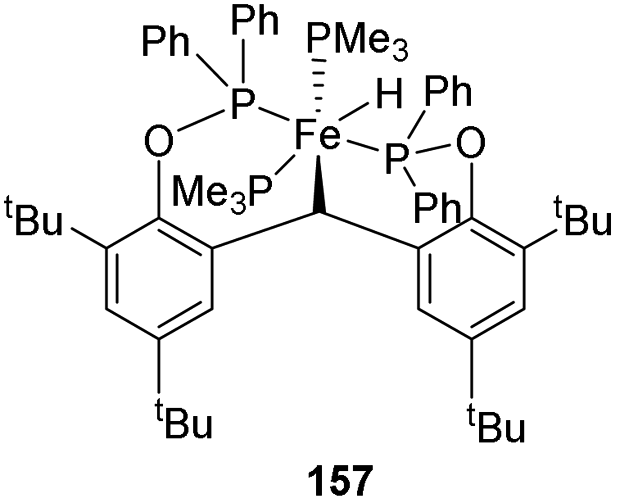
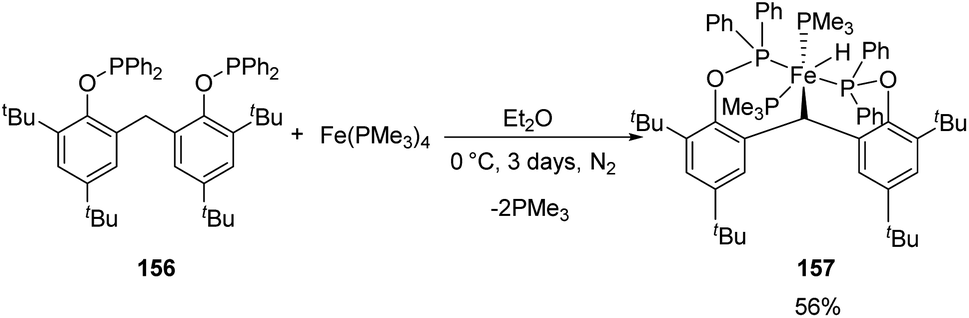
Sun and co-workers synthesized a new [POCOP]-pincer complex of iron by Csp3–H activation and discussed the application of this iron hydride in catalyzing hydrosilylation reactions.63 The pincer ligand 156 synthesized had two phenyl rings which forms a rigid backbone. The Csp3–H bond of the CH2 group upon oxidative addition to Fe(0) gave the iron hydride 157.
A solution of 156 in diethyl ether, when mixed with Fe(PMe3)4 under nitrogen atmosphere gave yellow crystals of iron hydride complex 157 with 56% yield (Scheme 34).
 | ||
| Scheme 34 Synthesis of iron hydride complex (POCHOP)Fe(H)(PMe3)2. | ||
The trimethyl phosphines are substituted by phosphorous atoms from 156 which reduces the distance between Fe(0) and the Csp3–H bonds in the CH2 group in 156. Thus, the Csp3–H bond is activated by the iron(0) centre by oxidative addition resulting from the cyclometalation. The iron(II) hydride obtained has Fe(II) centre in low-spin state and was stable for over 48 h under air exposure in room temperature.
The catalytic power of complex 157 in hydrosilylation of carbonyls were tested. Aldehydes underwent complete conversion to respective silyl ethers when treated with (EtO)3SiH at 65 °C in THF along with 1 mol% of complex 157 (Scheme 35). Base hydrolysis of products gave the corresponding alcohols. Investigation of hydrosilylation of substituted aldehydes showed that para-positioned electron-deficient substituents increased the rate of reaction, whereas ortho- groups slowed down the reaction. Also, electron-rich groups decelerated the reaction.
 | ||
| Scheme 35 Aldehyde hydrosilylation catalyzed by (POCHOP)Fe(H)(PMe3)2. | ||
Investigation of hydrosilylation of ketones (Scheme 36) showed that both ortho- and para-positioned electron-rich and electron-deficient substituents decreases the reaction rate.
 | ||
| Scheme 36 Ketone hydrosilylation catalyzed by (POCHOP)Fe(H)(PMe3)2. | ||
PPP pincer iron complexes
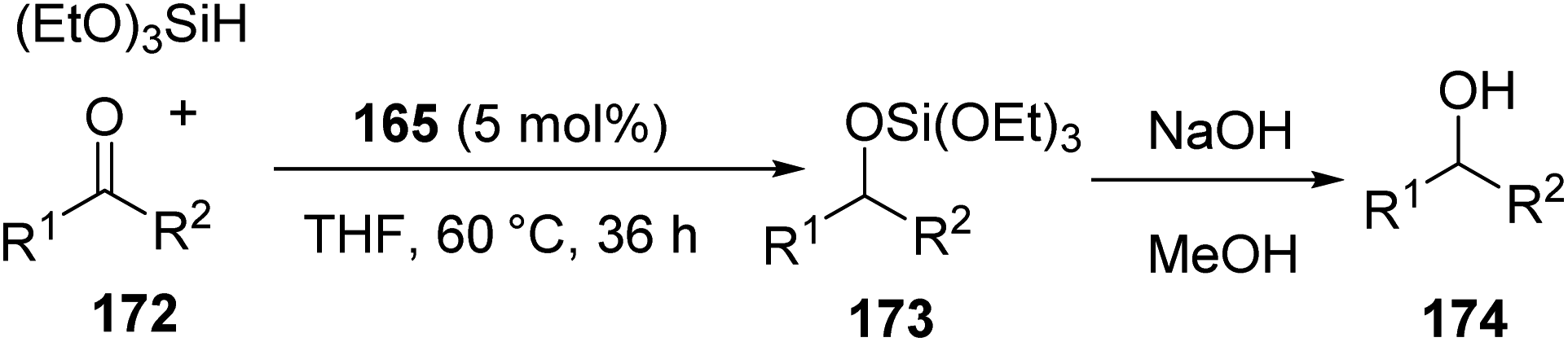
Transition metal-induced C–H activation for the synthesis of [PCP]-pincer complexes of late transition elements like Ir,64–66 Rh67,68 and Ru,69 were already known in literature. In 2018, Li et al. demonstrated a novel strategy for the synthesis of diphosphine-phosphine oxide ligand 164 (Ph2P-(C6H4))2P(O)H and resulted in [PPP]-pincer complex of iron via chelate-assisted P–H activation.70 The catalytic efficiency of the complex 165 in carbonyl hydrosilylation was examined.The complex 165 was obtained with 65% yield by reaction of solution of 164 and Fe(PMe3) in THF for 24 h at room temperature (Scheme 37).
 | ||
| Scheme 37 Synthesis of PPP-pincer iron complex [(Ph2P-(C6H4))2P(O)]Fe(H)(PMe3)2. | ||
The catalytic hydrosilylation reaction was optimized using benzaldehyde. The optimum condition was standardized to be 2 mol% of complex 165 and 1.2 equiv. triethoxysilane in THF at 60 °C, for 24 h to give good to excellent yields of corresponding alcohols (Scheme 38).
 | ||
| Scheme 38 Aldehyde hydrosilylation catalyzed by [(Ph2P-(C6H4))2P(O)]Fe(H)(PMe3)2. | ||
Ketones gave the hydrosilylated product with 5 mol% complex 165 and 1.2 equiv. triethoxysilane in THF at 60 °C for 36 h to give moderate yields of corresponding alcohols (Scheme 39).
 | ||
| Scheme 39 Ketone hydrosilylation catalyzed by [(Ph2P-(C6H4))2P(O)]Fe(H)(PMe3)2. | ||
On evaluating the substrate scope of hydrosilylation of aldehydes, at optimum conditions, electron-deficient substituents on aromatic aldehydes offered better yields than electron-rich ones, and dihalogeno aldehydes, aliphatic and aromatic substrates gave the products in moderate yields. It was also found that with an increased reaction time, this Fe catalyst exhibit high selectivity in hydrosilylation of α,β-unsaturated aldehydes.
The effect of substituents on ketones in hydrosilylation reaction showed a trend similar to that of aldehydes. The reason for the lower efficacy of 165 in hydrosilylation was accredited to the weak ability of phosphorus atom in σ-donation which decreases the electron density of Fe centre.
They also proposed a possible mechanism for the hydrosilylation catalyzed by 165 (Scheme 40), according to which insertion of the CO bond to the Fe–H bond leads to an intermediate 167 and attains equilibrium. Addition of triethoxysilane-which acts as source of hydrogen and catalytic promoter-converts the intermediate 167 to the intermediate 168. By reductive elimination 168 gives the respective siloxy compound.
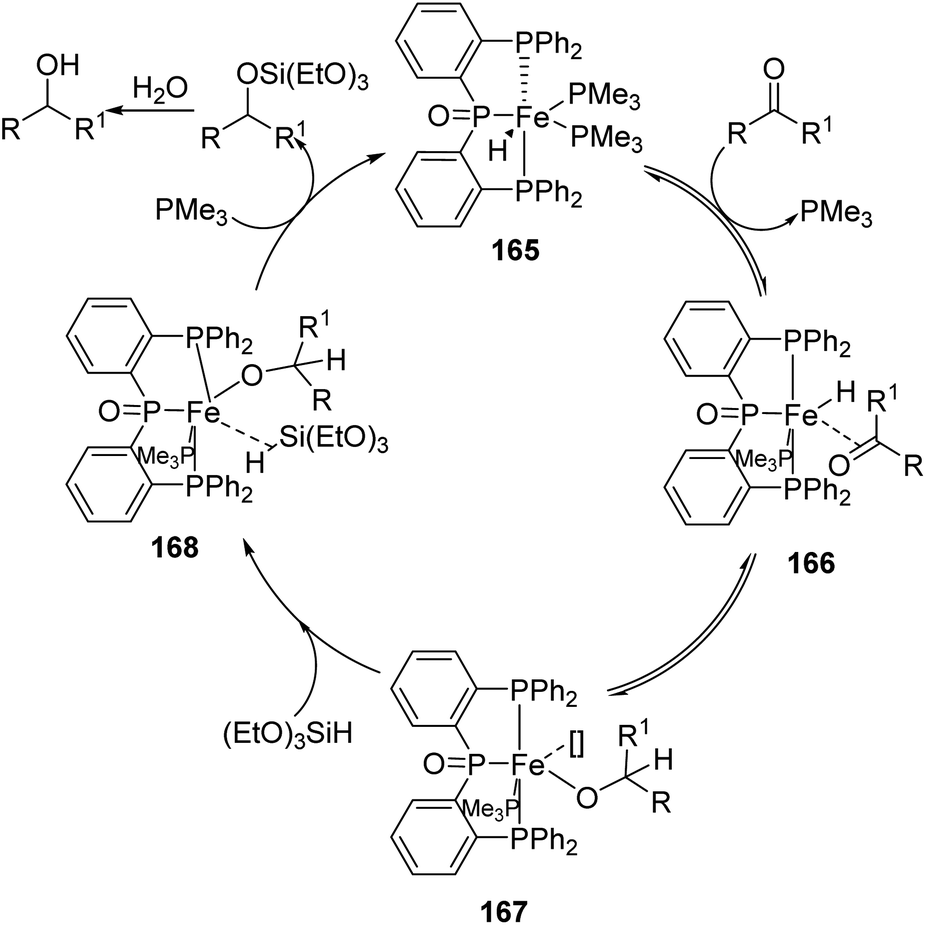 | ||
| Scheme 40 Possible mechanism for the hydrosilylation catalyzed by [(Ph2P-(C6H4))2P(O)]Fe(H)(PMe3)2. | ||
PCP pincer iron complexes
Li and co-workers synthesized new pincer complexes of iron by Csp3–H activation and their catalytic application in hydrosilylation reaction were explored.71 Treating the PCP ligand (Ph2P-(C6H4))2CH2 174 with Fe(PMe3)4 or Fe-Me2(PMe3)4 at room temperature in THF for 12 h, products [(Ph2P-C6H4)2CH]Fe(H)(PMe3)2 175 was furnished with 65% yield as a result of Csp3–H activation or [(Ph2P(C6H4))(PhP-(C6H4)2)CH]Fe(PMe3)2 176 was furnished with 45% yield as a result of Csp3–H, Csp2–H activation respectively (Scheme 41). | ||
| Scheme 41 Synthesis of new pincer complexes of iron by Csp3–H activation. | ||
The complex 175 exhibited good catalytic activity in the hydrosilylation of aldehydes and ketones with (EtO)3SiH and 0.3 to 1 mol% catalyst loading at 50 °C with a variable reaction time depending upon the substrate and obtained yields up to 90%.
Studies on substrate scope showed that electron-rich and electron-deficient substituent(s) could be tolerated in the aromatic ring by the catalyst. Reduction of substrates bearing electron-deficient groups could be achieved with 0.3 mol% of catalyst in a short time whereas that of substrates bearing electron-rich substituents was sluggish and higher loading of catalyst and extended time were required for complete conversion. Aldehydes afforded 85–90% product yields. Also, ketones showed lower reactivity when compared to aldehydes, i.e., 21–80% yield of products. From the studies, it is concluded that when polarity of carbonyl group decreases, reaction time for their hydrosilylation increases with a decrease in reaction rate.
Huang and co-workers in 2013 synthesized a group of novel pincer iron complexes 191 to 198 with good to excellent yields which contains phosphinite-iminopyridine (PNN) ligands72 that are electron-rich (Scheme 42). The novel iron complexes were found to be efficient in catalysing olefin hydrosilylation with primary, secondary, and tertiary silanes giving anti-Markovnikov selectivity.73 Moreover, the catalytic system exhibited excellent tolerance towards functional groups like ketones, amides, and esters.
 | ||
| Scheme 42 Synthesis of novel pincer iron complexes containing phosphinite-iminopyridine (PNN) ligands. | ||
With ester and amide substitutions, olefin hydrosilylation was achieved under 1 mol% catalyst loading and with ketone substitutions, alkene hydrosilylation could be attained at a catalyst loading of 2 mol%. In addition, they were successful in using 198 for synthesizing silafluofen, a new pyrethroid-type insecticide, by alkene hydrosilylation (Scheme 43).
 | ||
| Scheme 43 Synthesis of silafluofen by alkene hydrosilylation. | ||
They also observed that some internal olefins like cyclohexene and 2-hexene were non-reactive in hydrosilylation with PhSiH3 or Ph2SiH2 as silanes using (PNN)Fe-catalyst. Hydrosilylation reaction of allyl chloride and alkenes having an unprotected hydroxyl group, or primary/secondary amide substitution gave no products. Similarly, nitro, nitrile, or pyridine derivatives of styrene and allyl acetoacetate, gave no product under this catalytic system.
NCN pincer iron complexes
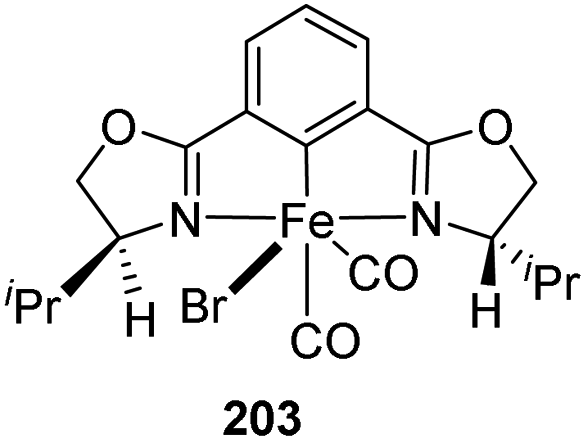
A synthetic path for novel chiral pincer complexes of iron, bearing bis(oxazolinyl)phenyl ligand was developed in 2010 by Nishiyama and co-workers.74 The [(S,S)-phebox-ip]Br 202 on reaction with Fe2(CO)9 in toluene for 3 h at 50 °C furnished phebox-Fe complex 203 with 69% yield (Scheme 44). | ||
| Scheme 44 Synthesis of [(S,S)-phebox-ip] FeBr(CO)2. | ||
The complex 203 was tested for catalyzing hydrosilylation reaction of aromatic ketones with 1.5 equiv. HSi(OEt)2Me (Scheme 45). It was found that a catalyst loading of 2 mol% with 2 mol% Na(acac) in hexane at 50 °C for 24 h could afford the hydrosilylation product with up to 99% yield and 66% ee. Na(acac) which is a strong base act as additive for the catalyst which increased the product yield within a shorter period of time. Substrates like 2-acetylnaphthalene and 2-acetylanthracene gave the products with 53% ee and 49% ee respectively. At the same time, 4-tolyl methyl ketone and 4-methoxyphenyl methyl ketone gave low enantioselectivities of 21–38% (ee).
 | ||
| Scheme 45 Hydrosilylation reaction of aromatic ketones with [(S,S)-phebox-ip] FeBr(CO)2. | ||
NCN iron pincer complexes bearing silyl, stannyl, phenyl, and methyl ligands were synthesized, characterized, and tested for catalytic activity by Nishiyama and co-workers in 2015.75 The stannyl complexes 214 to 217 and silyl complexes 208 and 209 were prepared by the oxidative addition of ligand precursors containing SnMe3−and SiMe3− (C–Sn and C–Si bonds) substitutions to Fe(CO)5. The reaction of Fe(CO)5 with (phebox-dm)SnMe3 (206 and 207) in THF in the presence of Xe lamp irradiation for 12 h under room temperature gave rise to 208 and 209 catalytic complexes with 57% and 62% yield respectively (Scheme 46). Similarly, oxidative addition of (phebox-dm)SiMe3 (210–213) to Fe(CO)5 resulted in the silyl complexes 214–217 with 60–75% yield (Scheme 47).
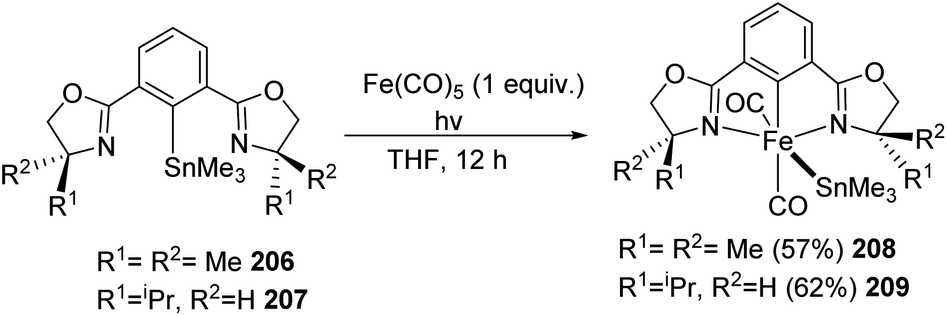 | ||
| Scheme 46 Oxidative addition of ligand precursors containing SnMe3− (C–Sn bonds) substitutions to Fe(CO)5. | ||
 | ||
| Scheme 47 Oxidative addition of ligand precursors containing SiMe3− (C–Si bonds) substitutions to Fe(CO)5. | ||
They also explained about structurally similar complexes with phenyl and alkyl substituents. The complexes 220 to 222 were prepared by transmetallation reaction of organozinc reagents with bromide complexes 218 and 219 (Scheme 48). Evaluation of catalytic activity in ketone hydrosilylation showed that complexes of silyl are the better catalyst.
 | ||
| Scheme 48 Transmetallation reaction of organozinc reagents with bromide complexes. | ||
Methyl complex 220 was obtained with 84% yield under room temperature reaction of 218 and 2 equiv. ZnMe2 for 15 min (Scheme 39). Methyl complex 221 was obtained from 219 with 55% yield by the same reaction. Treating 218 with ZnPh2 in excess resulted in phenyl complex 222 with 52% yield.
They explored the activity of the phebox-Fe complexes in asymmetric hydrosilylation reaction of aryl methyl ketone was studied. Reaction of aryl methyl ketones with 1.5 equiv. (EtO)2MeSiH, PhSiH3, Ph2SiH2, or (Me3SiO)2MeSiH along with 2 mol% catalyst in toluene for 24 h at 50 °C followed by the treatment of HCl or TBAF furnished respective (R)-alcohols. Among the three complexes, reactivity was the highest for silyl complex 215 which gave the (R)-alcohol with 98% yield and 32% ee. Stannyl complex had shown the lowest reactivity due to its highest stability itself. The phebox-Fe ligand of complex 221 undergoes decomposition when subjected to asymmetric hydrosilylation which resulted in a lower yield and poor enantioselectivity.
20 mol% of catalyst 214 gave β-silylated indole 224 with 46% yield on mixing 10 equiv. N-methylindole 223 with Ph2MeSiH at 60 °C in 3 days (Scheme 49).
 | ||
| Scheme 49 Synthesis of β-silylated indole catalyzed by NCN iron pincer complex bearing silyl group. | ||
They proposed a mechanism based on the mechanism explained by Ojima and colleagues for the Rh-catalyzed ketone hydrosilylation76 (Scheme 50).
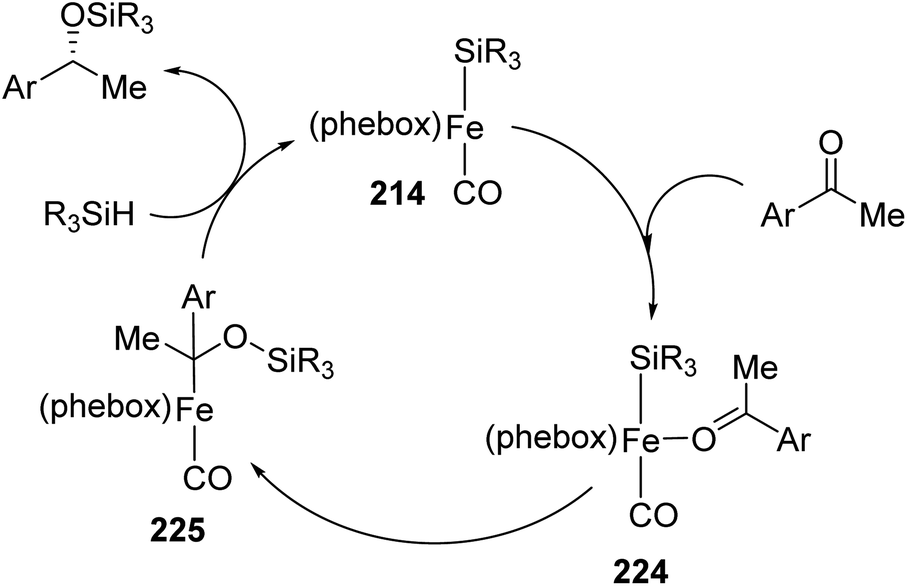 | ||
| Scheme 50 Proposed mechanism based on a mechanism from Ojima and colleagues. Adapted with permission from ref. 75. Copyright 2015 American Chemical Society. | ||
PSiP pincer iron complexes
In 2013, Sun et al. synthesized silyl iron complexes bearing [PSiP]-pincer ligand which was then utilized for hydrosilylation of aldehydes and ketones.77 κ3-(2-Ph2PC6H4)2SiMeH, [PSiP]-H 225 on treating with 1 equiv. of Fe(PMe3)4 in toluene at room temperature for 24 h formed the silyl iron complex catalyst 226 (Scheme 51). The catalyst (1 mol%) was used to explore the hydrosilylation of aldehydes and ketones using (EtO)3SiH in THF which was followed by 10% NaOH in MeOH at 60 °C to give their corresponding alcohols (Scheme 52). From the substrate scope studies, we see that benzaldehyde and furfuraldehyde afforded 100% and 92% products respectively. Methoxybenzaldehyde took a longer reaction time to give a 93% yield. Aryl and aliphatic ketones well tolerated with the reaction. | ||
| Scheme 51 Synthesis of silyl iron complex catalyst. | ||
 | ||
| Scheme 52 Hydrosilylation of aldehydes and ketones using silyl iron complex catalyst. | ||
In 2020 Li et al. employed [PSiP]-iron hydrides 230–234 (Fig. 8) for catalysing carbonyl hydrosilylation.78 Among these complexes, except 232, all other complexes were reported.79–81 Pyridine N-oxide was used in the reaction and it was unprecedentedly proved that it could bring down the temperature of reaction to 30 °C from 60 °C and promote catalytic carbonyl hydrosilylation.
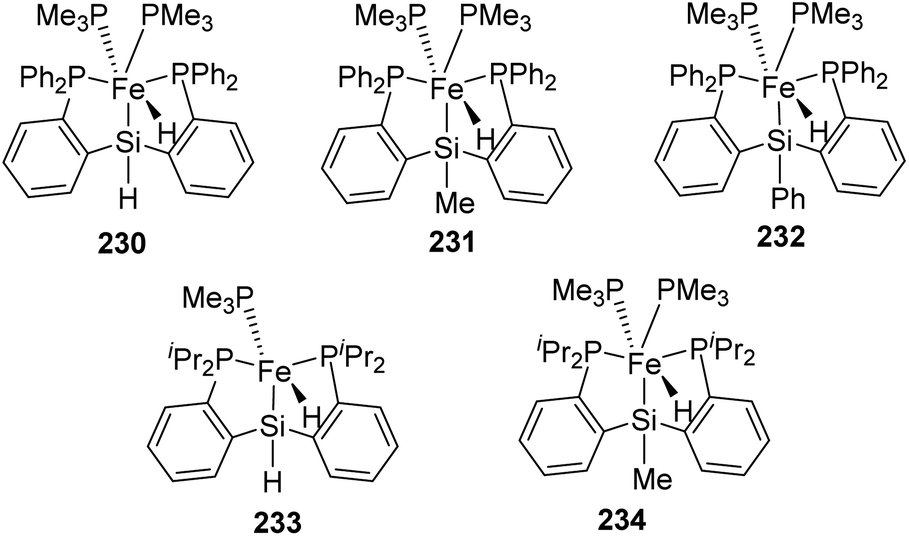 | ||
| Fig. 8 [PSiP]-iron(II) hydrides. | ||
Under the optimised reaction conditions (carbonyl substrate (1.0 mmol), (EtO)3SiH (1.2 mmol), catalyst (0.01 mmol), pyridine-N-oxide (0.05 mmol), THF (2 mL), 30 °C, 6 h), good to excellent yields of hydrosilylation products were obtained. The reaction condition for ketone substrates differ only by reaction temperature (50 °C).
Of the five catalysts, complex 233 gave the best results in catalysis with pyridine N-oxide as the promoter and complex 234 gave similar results in the absence of pyridine N-oxide. The catalytic performance of 233 and 234 is due to the isopropyl groups (electron-donating) attached to the phosphorous atoms which facilitate the PMe3 ligand dissociation and formation of the active unsaturated intermediate. From this result, it was concluded that PMe3 on dissociation give the intermediate hydride complex of iron which is a significant step of the catalytic pathway. A mechanism was proposed based on this (Scheme 53).
 | ||
| Scheme 53 Proposed catalytic pathway for [PSiP]-iron hydrides catalyzed hydrosilylation. | ||
Miscellaneous
In 2008, Gade and co-workers synthesized bpi ligands that are chiral from which iron acetato complexes 239 to 243 (Fig. 9) were prepared.82 Acetophenone (1 equiv.) on asymmetric hydrosilylation with (diethoxy)methylsilane (2 equiv.) in THF using these chiral Fe(II) complexes showed that 241 to 243 were the most stereoselective catalysts giving up to 86% enantioselectivities. From the studies on substrate scope using 243 as catalyst, they could obtain the hydrosilylated product with up to 93% enantiomeric excess for aryl(alkyl)ketones and 55–60% ee for dialkyl ketones. | ||
| Fig. 9 Iron acetato complexes containing bpi ligands that are chiral. | ||
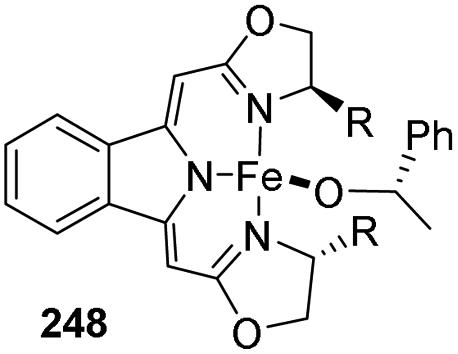
Gade and co-workers synthesized the first pre-catalyst based on iron for hydrosilylation reactions having activity along with selectivity similar to that of noble metals in 2015.83 The high reactivity and selectivity of the catalyst were due to the combined effect of alkoxide ligand and the chiral tridentate boxmi ligand. The boxmi-ligand had shown excellent activity in numerous reactions like Nozaki–Hiyama–Kishi coupling.84,85 The hydrosilylation products could be obtained with ee-values above 95% for various aryl(alkyl)ketones containing substituents offering good steric hindrance. The turnover number achieved is over 500 and at least 240 h−1 TOF was observed at −48 °C for the catalytic activity. The high activity and low temperature catalytic transformations tags it as a promising catalyst.
Reaction of boxmiH 245 and LiCH2TMS with FeCl2(py4) 244 gave the Fe(boxmi) neosyl complex 246 which on reaction with PhCH(OH)CH3 247 resulted in alkoxido complex 248 (Scheme 54).
 | ||
| Scheme 54 Synthesis of [Fe((R)-Hboxmi-Ph)((S)-OCHCH3Ph)] catalyst. | ||
They optimized the reaction conditions as catalyst loading of 5 mol% with respect to carbonyl with 2 equiv. (EtO)2MeSiH in toluene at a temperature ranging from −78 °C to RT for 6 h (Scheme 55).
 | ||
| Scheme 55 Optimized the reaction condition for [Fe((R)-Hboxmi-Ph)((S)-OCHCH3Ph)] catalyzed ketone hydrosilylation. | ||
Examination of different substrates revealed that para- substituted acetophenone does not affect the selectivity. Aryl(alkyl)ketones bearing unbranched and long alkyl chains and dodecanophenone gave the products in 95% and 99% ee respectively, but due to steric hindrance iso-butyrophenone reacted partially with a lower selectivity of 73% ee and tert-butylphenylketone gave no reaction. Also, on reduction dialkyl ketones exhibited low selectivity whereas diaryl ketones exhibited excellent selectivity.
They proposed a mechanism (Scheme 56) in which after activation of the pre-catalyst, σ-bond metathesis of alkoxido complex 252 with the silane 253 results in the silylether. The hydrido complex 254 that is left behind coordinates to the ketone rapidly followed by insertion of carbonyl to the metal-hydride bond. This step decides the stereoselectivity of hydrosilylation regenerating the alkoxido complex.
 | ||
| Scheme 56 Proposed catalytic pathway for [Fe((R)-Hboxmi-Ph)((S)-OCHCH3Ph)] catalyzed ketone hydrosilylation. | ||
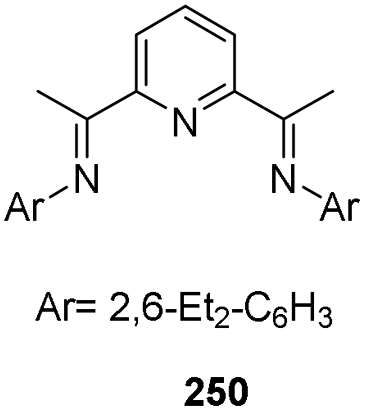
In 2014, Thomas and co-workers introduced iron-catalyzed chemo-, regio-, and stereoselective hydrosilylation of alkenes and alkynes.86 A wide range of functional groups tolerated well with the protocol. In situ generation of iron catalyst from 1 mol% each of FeCl2 and bis-(imino)pyridine ligand 250 (Fig. 10) was activated by 2 mol% of EtMgBr. The catalyst facilitated the reaction of alkenes/alkynes with phenyl silane in THF at room temperature for 1 h for the formation of regioselective hydrosilylated products with moderate to excellent yields (Scheme 57).
 | ||
| Fig. 10 Bis-(imino)pyridine ligand. | ||
 | ||
| Scheme 57 Iron-catalyzed hydrosilylation of olefins catalyzed. | ||
On evaluating the substrate scope of hydrosilylation, it is found that alkenes afforded good to excellent yields with perfect regioselectivity. Electronic nature of the functional groups of substrates have no effect on the reaction. Phenyl butane, styrene, imines and imino esters were some of them. But ketones gave chemoselective hydrosilylation at alkene without undergoing ketone reduction. Some hetero-aromatic alkenes like 2-vinylquinoline afforded quantitative yields with regioselectivity. But 4-vinylpyridine gave only 26% yield due to presence of polymerisation. Alkynes had formed vinylsilanes with excellent stereoselectivity without undergoing undesirable multiple hydrosilylation. Surprisingly internal alkyne followed the same path too. The pre-catalyst loading of 0.07 mol% corresponded to a TOF of 60000 mol h−1.
Summary and outlook
This review encapsulates applications of iron pincer complexes in hydrosilylation reactions. Iron being an earth-abundant, low-cost and environmentally benign metal can be used as a catalyst for versatile synthetic reactions. Pincer ligands stabilizes the uncontrolled reactivity of Fe centre and the iron pincer complexes formed act as an excellent catalyst for hydrosilylation reactions. The synthesis, substrate scope and proposed mechanism of various iron pincer complexes used in catalysing hydrosilylation reactions are covered in this review.The scope of employing different pincer ligands in iron complexes and the possibility of synthesizing pincer ligands of various types opens the door for iron pincer catalyzed hydrosilylation reactions. It is found that iron pincer complexes enable the catalysis of hydrosilylation reaction at ambient temperatures with high productivity with broad substrate scope when compared to the other reaction conditions. Most of the catalysts exhibited high regio-, stereo- and enantioselectivity for the reaction.
Asymmetric hydrosilylation is achieved mostly using [NNN] type catalyst complexes. Some of the iron pincer catalysts are industrially applicable as in the case of synthesizing silafluofen, an insecticide. The catalytic activity of the catalysts is expressed through TON and TOF values.
Reaction with different catalysts follows different mechanisms. Mechanistic pathways are ensured by comparing experimental data with DFT calculations. Through detailed studies, boxmi-iron(II) acetato complexes, iminobipyridine iron complexes, bis(imino)pyridine iron dinitrogen and dialkyl complexes and chiral iminopyridine-oxazoline iron complexes were found to be the best catalysts among those reviewed here for hydrosilylation reaction, which is concluded based on the productivity, substrate scope, control upon reactivity, regio-, stereo- and enantioselectivity, and easiness in catalyst synthesis and catalysis. They will outbreak the utility of iron catalysis for organic synthesis in the near future. There exist tremendous applications of employing different iron pincer complexes that can have immense applications in various reactions like hydrogenation, hydrosilylation, reductive cyclization, Csp3–H activation through double cyclometalation, under different conditions. The applications of these complexes are newly emerging for C–C bond formation, epoxidation and aziridation reactions. We presume that iron and other transition metal pincer complexes will be a solution for many unsolved catalytic problems in organic synthesis. Suitable tuning of the pincer ligands will definitely aid the unexplored arena of transition metal catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CMA thanks the Council of Scientific and Industrial Research (CSIR), New Delhi for the award of a junior research fellowship.References
- L. H. Sommer, E. W. Pietrusza and F. C. Whitmore, J. Am. Chem. Soc., 1947, 69, 188 CrossRef CAS.
- J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974–979 CrossRef CAS.
- B. D. Karstedt, US3775452A, 1973.
- L. N. Lewis, J. Stein, Y. Gao, R. E. Colborn and G. Hutchins, Platinum Met. Rev., 1997, 41, 66–75 CAS.
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS.
- S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS PubMed.
- K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694–1696 CrossRef CAS.
- I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29–31 RSC.
- I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS PubMed.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluc, in Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, Netherlands, 2009 Search PubMed.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- Á.-R. Barón, P. O. Burgos and I. Fernández, ACS Catal., 2019, 9, 5400–5417 CrossRef.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- M. Zaranek and P. Pawluc, ACS Catal., 2018, 8, 9865–9876 CrossRef CAS.
- J. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260–272 RSC.
- J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS.
- J. Guo, Z. Cheng, J. Chen, X. Chen and Z. Lu, Acc. Chem. Res., 2021, 54, 2701–2716 CrossRef CAS PubMed.
- B. Cheng, W. Liu and Z. Lu, J. Am. Chem. Soc., 2018, 140, 5014–5017 CrossRef CAS PubMed.
- S. P. Thomas, Adv. Synth. Catal., 2016, 358, 2404–2409 CrossRef.
- P. J. Chirik, in Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabo, O. F. Wendt, Wiley-VCH, Weinheim, 2014, pp. 189–212 Search PubMed.
- D. Morales-Morales, Pincer Compounds. Chemistry and Applications, Elsevier, 2018 Search PubMed.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- G. Bauer and X. Hu, Inorg. Chem. Front., 2016, 3, 741–765 RSC.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166 CrossRef CAS.
- O. R. Allen, L. D. Field, M. A. Magill, K. Q. Vuong, M. M. Bhadbhade and S. J. Dalgarno, Organometallics, 2011, 30, 6433–6440 CrossRef CAS.
- A. F. Hill and C. M. A. McQueen, Organometallics, 2012, 31, 8051–8054 CrossRef CAS.
- C. Azerraf and D. Gelman, Chem.–Eur. J., 2008, 14, 10364–10368 CrossRef CAS PubMed.
- R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949–2952 CrossRef CAS.
- S. Sjövall, O. F. Wendt and C. Andersson, Dalton Trans., 2002, 1396–1400 RSC.
- S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed.
- A. M. Tondreau, E. Lobkovsky and P. J. Chirik, Org. Lett., 2008, 10, 2789–2792 CrossRef CAS PubMed.
- Y. Toya, K. Hayasaka and H. Nakazawa, Organometallics, 2017, 36, 1727–1735 CrossRef CAS.
- H.-J. Lin, S. Lutz, C. O'Kane, M. Zeller, C.-H. Chen, T. A. Assila and W.-T. Lee, Dalton Trans., 2018, 47, 3243–3247 RSC.
- K. R. D. Johnson, B. L. Kamenz and P. G. Hayes, Organometallics, 2014, 33, 3005–3011 CrossRef CAS.
- N. I. Regenauer, S. Jänner, H. Wadepohl and D.-A. Roşca, Angew. Chem., Int. Ed., 2020, 59, 19320–19328 CrossRef CAS PubMed.
- C. Chen, H. Wang, Y. Sun, J. Cui, J. Xie, Y. Shi, S. Yu, X. Hong and Z. Lu, iScience, 2020, 23, 100985–100996 CrossRef CAS PubMed.
- K. Hayasaka, K. Kamata and H. Nakazawa, Bull. Chem. Soc. Jpn., 2016, 89, 394–404 CrossRef CAS.
- J. Chen, B. Cheng, M. Cao and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 4661–4664 CrossRef CAS PubMed.
- T. Inagaki, L. T. Phong, A. Furuta, J.-i. Ito and H. Nishiyama, Chem.–Eur. J., 2010, 16, 3090–3096 CrossRef CAS PubMed.
- H. A. McManus and P. J. Guiry, J. Org. Chem., 2002, 67, 8566–8573 CrossRef CAS PubMed.
- S.-F. Lu, D.-M. Du, S.-W. Zhang and J. Xu, Tetrahedron: Asymmetry, 2004, 15, 3433–3441 CrossRef CAS.
- Z. Zuo, L. Zhang, X. Leng and Z. Huang, Chem. Commun., 2015, 51, 5073–5076 RSC.
- L. Zhang, Z. Zuo, X. Wan and Z. Huang, J. Am. Chem. Soc., 2014, 136, 15501 CrossRef CAS PubMed.
- C. Foltz, M. Enders, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2007, 13, 5994–6008 CrossRef CAS PubMed.
- J. Wenz, V. Vasilenko, A. Kochan, H. Wadepohl and L. H. Gade, Eur. J. Inorg. Chem., 2017, 5545–5556 CrossRef CAS.
- A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 3928–3940 CrossRef CAS.
- X. Jia and Z. Huang, Nat. Chem., 2016, 8, 157–161 CrossRef CAS PubMed.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS PubMed.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed.
- M. Kamitani, H. Kusaka, T. Toriyabe and H. Yuge, Bull. Chem. Soc. Jpn., 2018, 91, 1429–1435 CrossRef CAS.
- A. M. Tondreau, C. C. H. Atienza, J. M. Darmon, C. Milsmann, H. M. Hoyt, K. J. Weller, S. A. Nye, K. M. Lewis, J. Boyer, J. G. P. Delis, E. Lobkovsky and P. J. Chirik, Organometallics, 2012, 31, 4886–4893 CrossRef CAS.
- M. Kamitani, H. Kusaka and H. Yuge, Chem. Lett., 2019, 48, 1196–1198 CrossRef CAS.
- M. Kamitani, K. Yujiri and H. Yuge, Organometallics, 2020, 39, 3535–3539 CrossRef CAS.
- D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885–6897 CrossRef CAS.
- K. Kirchner, Angew. Chem., Int. Ed., 2015, 54, 4706–4707 CrossRef CAS.
- T. T. Metsanen, D. Gallego, T. Szilvasi, M. Driess and M. Oestreich, Chem. Sci., 2015, 6, 7143–7149 RSC.
- A. D. Smith, A. Saini, L. M. Singer, N. Phadke and M. Findlater, Polyhedron, 2016, 114, 286–291 CrossRef CAS.
- P. Bhattacharya, J. A. Krause and H. Guan, Organometallics, 2011, 30, 4720–4729 CrossRef CAS.
- P. Gu, W. Wang, Y. Wang and H. Wei, Organometallics, 2013, 32, 47–51 CrossRef CAS.
- W. Wang, P. Gu, Y. Wang and H. Wei, Organometallics, 2014, 33, 847–857 CrossRef CAS.
- P. Bhattacharya, J. A. Krause and H. Guan, Organometallics, 2014, 33, 6113–6121 CrossRef CAS.
- S. Huang, H. Zhao, X. Li, L. Wang and H. Sun, RSC Adv., 2015, 5, 15660–15667 RSC.
- M. C. Haibach, C. Guan, D. Y. Wang, B. Li, N. Lease, A. M. Steffens, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 15062–15070 CrossRef CAS PubMed.
- M. Wilklow-Marnell, W. W. Brennessel and W. D. Jones, Organometallics, 2017, 36, 3125–3134 CrossRef CAS.
- D. B. Lao, A. C. E. Owens, D. M. Heinekey and K. I. Goldberg, ACS Catal., 2013, 3, 2391–2396 CrossRef CAS.
- C. Crocker, R. J. Errington, W. S. Mcdonald, K. J. Odell and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1979, 101, 498–499 RSC.
- W. Weng, S. Parkin and O. V. Ozerov, Organometallics, 2006, 25, 5345–5354 CrossRef CAS.
- D. G. Gusev and A. J. Lough, Organometallics, 2002, 21, 2601–2603 CrossRef CAS.
- X. Qi, H. zhao, H. Sun, X. Li, O. Fuhr and D. Fenske, New J. Chem., 2018, 42, 16583–16590 RSC.
- H. Zhao, H. Sun and X. Li, Organometallics, 2014, 33, 3535–3539 CrossRef CAS.
- J. Zhang, M. Gandelman, D. Herrman, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Inorg. Chim. Acta, 2006, 359, 1955–1960 CrossRef CAS.
- D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154–19166 CrossRef CAS PubMed.
- S. Hosokawa, J.-i. Ito and H. Nishiyama, Organometallics, 2010, 29, 5773–5775 CrossRef CAS.
- J.-i. Ito, S. Hosokawa, H. B. Khalid and H. Nishiyama, Organometallics, 2015, 34, 1377–1383 CrossRef CAS.
- I. Ojima, T. Kogure, M. Kumagai, S. Horiuchi and T. Sato, J. Organomet. Chem., 1976, 122, 83–97 CrossRef CAS.
- S. Wu, X. Li, Z. Xiong, W. Xu, Y. Lu and H. Sun, Organometallics, 2013, 32, 3227–3237 CrossRef CAS.
- G. Chang, P. Zhang, W. Yang, S. Xie, H. Sun, X. Li, O. Fuhr and D. Fenske, Dalton Trans., 2020, 49, 9349–9354 RSC.
- S. Wu, X. Li, Z. Xiong, W. Xu, Y. Lu and H. Sun, Organometallics, 2013, 32, 3227–3237 CrossRef CAS.
- P. Zhang, X. Li, X. Qi, H. Sun, O. Fuhr and D. Fenske, RSC Adv., 2018, 8, 14092–14099 RSC.
- G. Chang, X. Li, P. Zhang, W. Yang, K. Li, Y. Wang, H. Sun, O. Fuhr and D. Fenske, Appl. Organomet. Chem., 2020, 34, e5466 CrossRef CAS.
- B. K. Langlotz, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 4670–4674 CrossRef CAS PubMed.
- T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2015, 137, 2456–2459 CrossRef CAS PubMed.
- Q.-H. Deng, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2011, 17, 14922–14928 CrossRef PubMed.
- Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 2946–2949 CrossRef CAS PubMed.
- M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584–590 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |