
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico study of inhibitory capacity of sacubitril/valsartan toward neprilysin and angiotensin receptor†
Jelena Đorović Jovanović *a,
Marko Antonijevića,
Radiša Vojinovićb,
Nenad D. Filipovićc and
Zoran Markovića
*a,
Marko Antonijevića,
Radiša Vojinovićb,
Nenad D. Filipovićc and
Zoran Markovića
aDepartment of Science, Institute for Information Technologies, University of Kragujevac, Jovana Cvijića bb, 34000 Kragujevac, Republic of Serbia. E-mail: jelena.djorovic@uni.kg.ac.rs
bFaculty of Medical Sciences, University of Kragujevac, Svetozara Markovića 69, 34000 Kragujevc, Republic of Serbia
cFaculty of Engineering, University of Kragujevac, Sestre Janjić 6, 34000 Kragujevac, Republic of Serbia
First published on 17th October 2022
Abstract
Heart failure (HF) is a life-threatening condition that occurs when the heart cannot pump enough blood and oxygen to meet the body's needs. It affects mostly the elderly, commonly from the male population, especially those with obesity, diabetes, or some other chronic condition. It can be treated with different medications, and promising results were shown by a relatively new medicament called Entresto. Results obtained from molecular docking and molecular dynamics simulations to examine the inhibitory capacity of Entresto are presented in this study. Parameters obtained by the molecular docking simulations show that both parts of Entresto (sacubitril (SAC) and valsartan (VAL)) interact with targeted proteins, and inhibit their physiological function. Simulations of molecular dynamics revealed some interesting inhibitory patterns. SAC was discovered to produce structural alterations in neprilysin by binding to it, reducing neprilysin's physiological activity. In addition to blocking the active site, SAC binding causes the enzyme's structure to become less compact over time, causing changes in its biochemical characteristics and preventing the enzyme from performing its biological function. Similar to SAC, VAL also causes deviations in the structure of angiotensin receptors. The angiotensin receptor GPCR (G-protein-coupled receptors) is immersed in the lipid bilayer, and changes in the tertiary structure are only visible through RMSD and RMSF, not by examining Rg. In this regard, MD simulations validated the results of molecular docking simulations, demonstrating that both SAC and VAL had inhibitory potential towards the neprilysin and angiotensin receptors, respectively.
1. Introduction
The leading death causes in the world are cardiovascular diseases (CVDs). It is estimated that in 2019 over 18 million people died from CVDs, which accounts for 32% of all global death cases. One of the major pandemics the world is confronted in past decades is the heart failure (HF) condition.1 HF certainly has significant consequences on global health and is more common in the male population. A condition named HF is actually the continuous incapability of the heart to pump enough blood through the body to ensure a sufficient supply of oxygen.2 A more comprehensive definition is given by the American Heart Association (AHA)/American College of Cardiology,3,4 that states that HF is “a complex clinical syndrome that can result from any structural or functional cardiac disorder that impairs the ability of the ventricle to fill or eject blood.”1 When oxygen supplies are low the whole organism goes into “survival mode” by realising adrenaline and other stress hormones which additionally causes blood vessel narrowing, causing high strain on the heart muscle, and indeed inducing stress in the whole organism. To increase oxygen concentration inside tissues, the autonomic nervous system induces a rise in blood pressure, which as the consequence has increased damage to the blood vessels and straining of the heart muscle. In addition, the expression HF can lead to the conclusion that the heart is totally failing or stopped working, but indeed HF is a condition in which the heart is weaker than it should be in a healthy organism. There are various reasons for this disorder, such as old age, diabetes, high blood pressure, and being overweight. The most common symptoms of HF include shortness of breath, tiredness, swelling of the legs and ankles, chest pain, and cough.5 The usual treatment of HF includes diet and lifestyle changes, in combination with different medications. In 2015, a new class of medication that modulates neurohormonal effects, and consists of angiotensin receptor and neprilysin inhibitors (ARNIs) became available on the market. This new generation of HF pharmacotherapy involves the simultaneous inhibition of both the angiotensin and neprilysin metabolistic pathways.6–8 The latest version of this medication is entitled: “Entresto”. This medication was previously known in pre-clinical and clinical studies as LCZ696, and it represents the combination of sacubitril and valsartan (SAC and VAL).7,9 The study is known as PARADIGM-HF (Prospective Comparison of ARNI with ACEi to Determine Impact on Global Mortality and Morbidity in Heart Failure) trial10 showed that the SAC–VAL combination is more effective in the treatment of HF in comparison to enalapril.11 Also, the findings in the PARADIGM-HF trial have recognized neprilysin inhibition (in combination with angiotensin receptor blockade) as essential drug therapy for HF.The part of Entresto which is responsible for decreasing of blood pressure and inhibition of neprilysin is SAC. Neprilysin is a membrane-bound endopeptidase that alters the bioavailability of many vasoactive peptides. Initially, it was found in the kidneys. Also, neprilysin is widely distributed in the body, throughout the central nervous system, lungs, intestine, neutrophils, fibroblasts, and in endothelial cells.12 Neprilysin is the enzyme in charge of the degradation of different peptides with varying physiological roles including those with vasoactive properties (e.g., natriuretic peptides (NPs), bradykinin, substance P, adrenomedullin, glucagon, vasoactive intestinal peptide, and angiotensin II).13,14 By inhibition of the neprilysin, SAC allows higher concentrations of NPs, which are causing vasodilation lowering the “load” on the hearth muscle. The inhibition of the neprilysin alone is not sufficient, because neprilysin degrades other peptides such as angiotensin and its precursors. Because of that, it was necessary to produce medication which, besides SAC, contains at the same time an angiotensin receptor blocker. For that purpose the second part of Entresto, VAL which is an angiotensin receptor blocker, was used. With angiotensin receptors blocked, there is no synthesis of angiotensin from its precursors. In addition, the role of the VAL is to keep blood vessels from narrowing, which lowers blood pressure and improves blood flow. Originally, VAL was known under the name Diovan, and in 1996 it was approved by the US Food and Drug Administration (FDA) for the handling of hypertension.15 In the year 2002, VAL (Diovan) was approved for the treatment of heart failure.15,16 Additional studies have led to the FDA approval of VAL in the 2005 year. VAL was accepted for the treatment of reducing cardiovascular (CV) mortality in clinically stable patients after myocardial infarction.15,17 To decrease the risk of CV death and hospitalization of patients with HF the FDA approved sacubitril plus valsartan (Entresto; Novartis) on July 7, 2015.18,19
The Entresto has a dual role in the organism. It should improve the beneficial effects of the natriuretic peptide system (NPs), while concurrently lowering the detrimental effects of the renin–angiotensin–aldosterone system (RAAS).20–23 Both of these effects provide the lowering of blood pressure and spare the heart of unnecessary strain (Scheme 1).
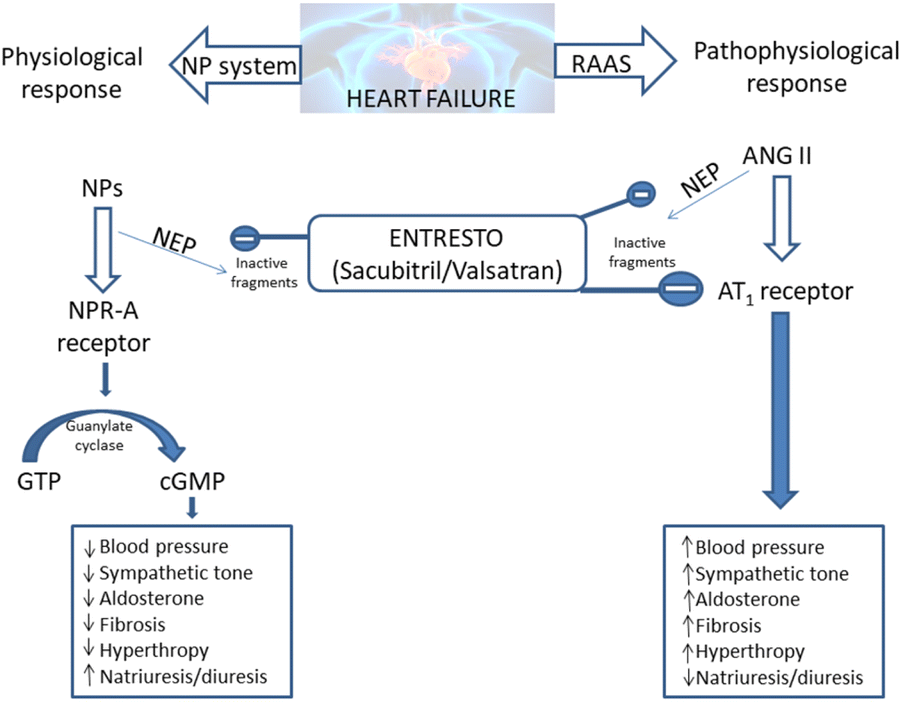 | ||
| Scheme 1 Entresto has the potential to control two counter-regulatory neurohormonal systems in HF: the renin–angiotensin–aldosterone system (RAAS) and the natriuretic peptide system (NPs).20–23 ANG: angiotensin; AT1: angiotensin type 1; cGMP: cyclic guanosine monophosphate; GTP: guanosine-50-triphosphate; NP: natriuretic peptide (e.g. atrial natriuretic peptide [ANP], B-type natriuretic peptide [BNP], etc.); NPR-A: NP receptor-A. | ||
In this study, molecular docking and molecular dynamics simulations are performed to examine and confirm inhibitory potency and binding interactions of SAC and neprilysin, as well as VAL and angiotensin receptor.
2. Methodology section
The geometry optimization of the molecules of SAC and VAL is performed by applying M06-2X/6-311++G(d,p) theoretical model,24,25 implemented in the Gaussian09 software package.26 Further, carboxylate anions of SAC and VAL are optimized at the same level of theory. The frequency analysis indicated that calculations performed in Gaussian software package did not produce any imaginary frequencies. It means that the obtained geometries for all species are minima on the potential energy surface. The optimized geometries of SAC and VAL carboxylate anions have been the subject of extensive studies using molecular docking and molecular dynamics simulations.2.1. Molecular docking analysis
In this research study, molecular docking simulations are carried out, and for that purpose AutoDock 4.0 software has been used.27 The three-dimensional crystal structures of targeted proteins, neprilysin and angiotensin receptor, are provided from the Protein Data Bank (PDB 6GID and 4YAY, respectively).28,29 The preparation of protein for molecular docking simulations is carried out in the BIOVIA Discovery Studio.30 The co-crystallized ligand, water molecules, and co-factors are removed from the protein structure. The AutoDockTools (ADT) graphical user interface is used for the addition of polar hydrogen atoms and the calculation of Kollman charges. In the preparation process for molecular docking simulation, the ligand is set to be flexible, and the bonds in the ligand are set to be rotatable. The protein remains standing as a rigid structure. For protein–ligand flexible docking simulations, the Lamarckian Genetic Algorithm (LGA) is used. To predict binding sites of the known 3D structures of neprilysin and angiotensin receptor, the binding pockets are defined using AGFR (AutoGridFR) software.31 According to AGFR, the binding site with the lowest projected binding energy was utilized for molecular docking simulations between neprilysin and the carboxylate anion of SAC. The native bound ligand of the angiotensin receptor was extracted from the structure, and binding pocket investigations were performed. Furthermore, re-docking was done with carboxylate anion of VAL which is used as a ligand, and the identical docking mode was generated as it was found in the co-crystallized form of the target protein. The grid boxes center with dimensions 18.420 Å × −46.949 Å × 14.269 Å and −20.786 Å × 7.619 Å × 35.545 Å in −x, −y, and −z directions of neprilysin and angiotensin receptor were used to cover the protein binding sites and accommodate ligands to move without restraint. A grid point spacing of 0.375 Å was used for auto grid runs, and the number of GA runs are set to be 20. The molecular docking simulation is performed at a temperature of 298.15 K. Analysis of molecular docking simulations results and visualizations of binding pockets are accomplished by using the BIOVIA Discovery Studio.For the valuation of the binding affinity, AutoDock uses empirical scoring functions established on the free energy of binding (ΔGbind). It is significant to include one additional parameter for predicting a compound's inhibitory efficacy and that is the inhibitory constant (Ki). The value of Ki is calculated by AutoDock after evaluation of the free energy of binding. The inhibitory constant value is defined by the following eqn (1):
| Ki = exp(ΔGbind/RT) | (1) |
2.2. Molecular dynamics analysis
More detailed examination of protein–ligand complexes obtained in molecular docking simulations is done through molecular dynamics (MD) simulations. The behaviour of SAC and VAL with neprilysin and angiotensin receptor is investigated in the same binding pockets in the time-depended frame, using the AMBER18 software package.32,33 The topologies of the ligands and the proteins were generated applying the AMBER force field.34 Charmm-GUI server is used to generate topologies, input parameters, and coordinate files of investigated compounds.35–38 The solvation of investigated complexes is done by implementing the TIP3P solvation model.39 The VAL–angiotensin receptor complex is firstly immersed in a lipid membrane, formed from POPC (phosphatidylcholine). The upper layer of the lipid membrane is made from 200 POPC, while the lower layer is made from 198 POPC. For the solvation of the VAL–angiotensin receptor complex immersed inside the lipid membrane, 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 486 molecules of water were used. In regard to the solvation of the SAC–neprilysin complex, 17506 molecules of water have been used for the solvation of the system. The neutralization of all systems is completed by the introduction of potassium and chloride ions (0.15 M KCl) using the Monte-Carlo Ion Placing Method.40 For neutralization of VAL–angiotensin receptor complex is used 117 K+ and 126 Cl−. The neutralization of the SAC–neprilysin complex is done by adding 116 K+ and 104 Cl−. Further, the investigated protein–ligand complex structures are energetically minimized by the steepest descent algorithm and conjugate gradient algorithm during the 5000 steps. The equilibration process was carried out following NVT manner. The production steps of the MD simulation were performed in an NPT ensemble, to simulate the natural conditions with constant pressure and temperature. The MD production process is performed using the SHAKE algorithm for a 150 ns time scale, including the MC barostat (Monte-Carlo barostat) (τP = 2 ps).41 The production step of MD simulations is performed three times, to assure reproducibility. For calculations of binding energies, MM/GBSA study was employed, and average values of binding energies across the trajectory where systems are found to be in the most stable state were taken into consideration.42 In addition, for analysis system properties during and after molecular dynamics simulations, including overall stability and structural fluctuations through simulations Root Mean Square Deviation (RMSD), radius of gyration (Rg) and Root Mean Square Fluctuation (RMSF) are generated from MD output trajectories. These parameters are used to determine the stability and structural changes of the protein–ligand complex in the calculated timeframe.
486 molecules of water were used. In regard to the solvation of the SAC–neprilysin complex, 17506 molecules of water have been used for the solvation of the system. The neutralization of all systems is completed by the introduction of potassium and chloride ions (0.15 M KCl) using the Monte-Carlo Ion Placing Method.40 For neutralization of VAL–angiotensin receptor complex is used 117 K+ and 126 Cl−. The neutralization of the SAC–neprilysin complex is done by adding 116 K+ and 104 Cl−. Further, the investigated protein–ligand complex structures are energetically minimized by the steepest descent algorithm and conjugate gradient algorithm during the 5000 steps. The equilibration process was carried out following NVT manner. The production steps of the MD simulation were performed in an NPT ensemble, to simulate the natural conditions with constant pressure and temperature. The MD production process is performed using the SHAKE algorithm for a 150 ns time scale, including the MC barostat (Monte-Carlo barostat) (τP = 2 ps).41 The production step of MD simulations is performed three times, to assure reproducibility. For calculations of binding energies, MM/GBSA study was employed, and average values of binding energies across the trajectory where systems are found to be in the most stable state were taken into consideration.42 In addition, for analysis system properties during and after molecular dynamics simulations, including overall stability and structural fluctuations through simulations Root Mean Square Deviation (RMSD), radius of gyration (Rg) and Root Mean Square Fluctuation (RMSF) are generated from MD output trajectories. These parameters are used to determine the stability and structural changes of the protein–ligand complex in the calculated timeframe.
3. Results and discussion
The components of Entresto, SAC and VAL are recognized as a neprilysin inhibitor and as an angiotensin receptor blocker, respectively. Sacubitril's active form (carboxylate anion of SAC), sacubitrilat (LBQ657) inhibits neprilysin, a neutral endopeptidase that would typically cleave natriuretic peptides such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and c-type natriuretic peptide (CNP). Under normal conditions, neprilysin breaks down other vasodilating peptides and also vasoconstrictors such as angiotensin I and II, endothelin-1, and peptide amyloid beta-protein. Inhibition of neprilysin results in decreased breakdown and higher concentrations of endogenous natriuretic peptides, as well as elevated amounts of vasoconstricting hormones like angiotensin II. In this manuscript, the interactions and behaviour of carboxylate anions of SAC and VAL against specific proteins are investigated. Firstly, binding pockets are predicted by applying AGFR, both for neprilysin and the angiotensin receptor (Fig. 1).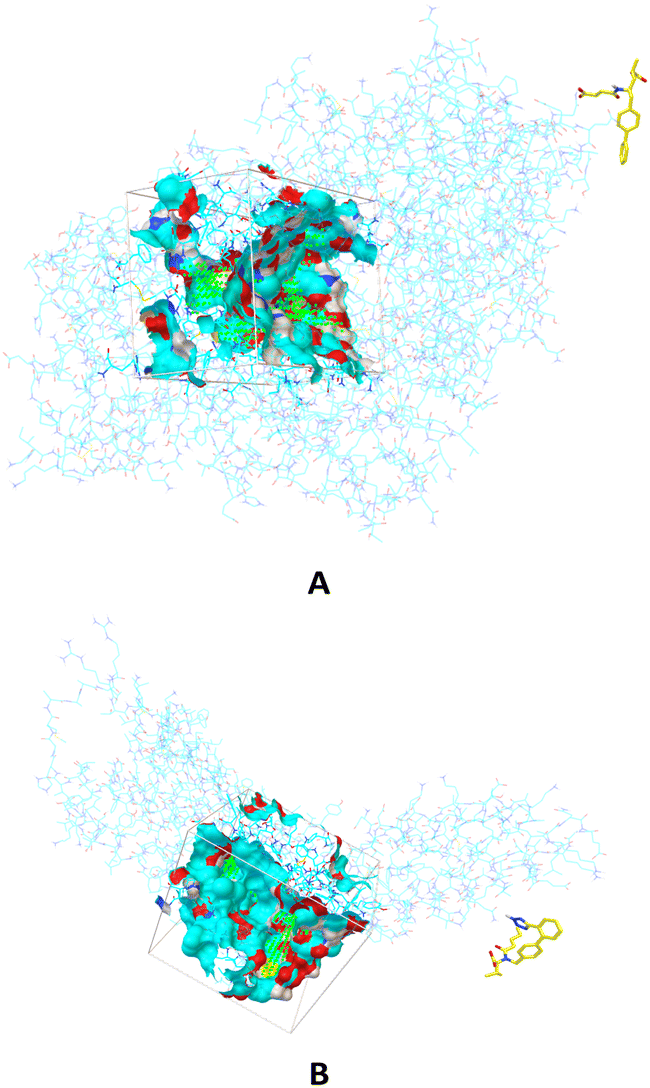 | ||
| Fig. 1 The binding sites in which are performed molecular docking simulations between carboxylate anion of SAC and neprilysin (A) and carboxylate anion of VAL and angiotensin receptor (B). | ||
The defined binding pockets are following experimental data.28,29 The molecular docking simulations are performed in predicted binding pockets in the case of both receptors. Twenty different conformations are achieved in simulations for each part of Entresto, and conformations with the highest binding affinity of both investigated compounds are further analysed. The obtained results are presented in Table 1.
| ΔGbind (kcal mol−1) | Ki (μM) | LE | BEI | |
|---|---|---|---|---|
| SAC–neprilysin | −7.45 | 3.49 | −0.27 | 13.26 |
| VAL–angiotensin receptor | −9.52 | 0.11 | −0.30 | 16.02 |
Presented outcomes show binding affinity in defined binding pockets and can be analyzed concerning values obtained for the free energy of binding (ΔGbind) and inhibition constant (Ki). The ΔGbind and Ki are thermodynamical parameters calculated in molecular docking simulations. The above-stated results indicate a link between the values of ΔGbind and Ki. Specifically, the lower value of ΔGbind is followed by the lower value of Ki, and they are indicating a higher binding affinity and inhibition potency. The results demonstrate that VAL has a greater angiotensin receptor inhibition efficacy than SAC to neprilysin. Furthermore, two additional parameters are calculated, and their values are presented in Table 1. Those are ligand efficiency (LE) and binding efficiency index (BEI). LE represents a measurement of the binding energy per atom of a ligand to its targeted protein.43 LE can be calculated mathematically by dividing the Gibbs free energy (ΔG) by the amount of non-hydrogen atoms in the ligand (eqn (2)):
| LE = (ΔG)/N | (2) |
lnKi and N is the number of non-hydrogen atoms.44 It can be converted to eqn (3):45|
LE = 1.4(−logIC50)/N
| (3) |
The other parameter which describes ligand efficiency, BEI, can be calculated following eqn (4):
| BEI = (pKi, pKd, or pIC50)/(molecular weight, kDa) | (4) |
The results shown in Table 1 reveal the correlation between the observed ΔGbind, Ki, LE, and BEI values. The interpretation of LE values shows that the lower values of LE indicate better ligand efficiency. However, when BEI values are discussed, a higher value of BEI implies better binding efficiency. Finally, the calculated values of LE and BEI for SAC and VAL confirm obtained values of ΔGbind and Ki.
Furthermore, molecular docking simulations are used to investigate the amino acids responsible for binding between targeted ligands and receptors. Amino acids from neprilysin and angiotensin receptor, that are involved in the formation of contacts with SAC and VAL, are presented in Tables S1 and S2.† Also, the distance between corresponding amino acids and investigated ligands is given in the same tables. According to the results in Table S1,† nine amino acids of neprilysin are responsible for binding with sacubitrilat. In addition, the carboxylate anion of SAC expresses its inhibitory potency toward neprilysin forming the interactions with Arg102, Arg110, Arg717, Asn542, His711, Ala543, His587, His583, and Tyr545 (Fig. 2).
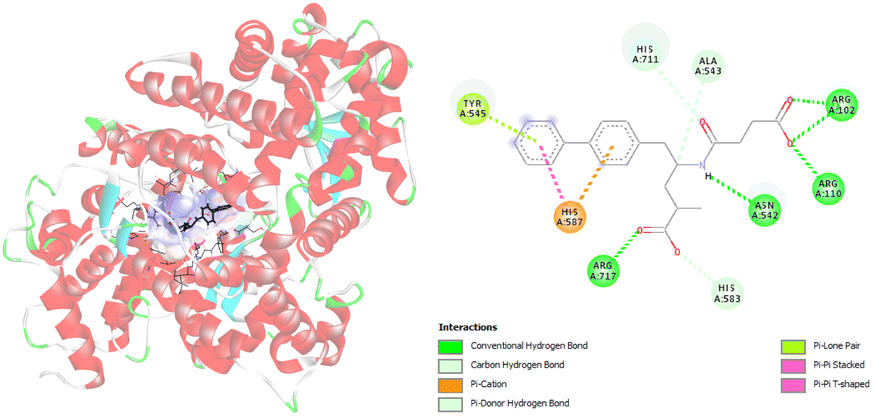 | ||
| Fig. 2 Picture showing the interaction between carboxylate anion of SAC and amino acids in neprilysin. | ||
These amino acids formed fourteen different types of interactions. Ten out of all obtained interactions are categorized as hydrogen bonds. Seven hydrogen bonds are conventional hydrogen bonds, and they are formed between SAC and Arg102, Arg110, Arg717, and Asn542. Every of mentioned Arg established two conventional hydrogen bonds with SAC. The other type of hydrogen bonds that are formed with amino acids His717 and Ala543 are carbon hydrogen bonds. The conventional hydrogen bonds established are in the range of 1.85 to 2.62 Å, while carbon hydrogen bonds are formed at distances of 2.92 and 3.50 Å (Table S1†). The amino acid His583 also established a hydrogen bond with SAC, but it forms a π–donor hydrogen bond. In addition, hydrophobic contacts are involved in the binding of SAC to neprilysin, and that is an interaction between SAC and His587. Besides this contact, His587 established with SAC π–cation electrostatic contact. The same interactions are obtained in available literature data and it is confirmed that amino acids residues such as Ala543 and Asn542 are important for neprilysin inhibition.46
The analysis of the crystal structure of substrate-free neprilysin reported by Moss et al. reveals that active site residues are Trp693, Phe106, and Arg110.28 Also, it is reported that the coordinated zinc ion and nearby residues make a small binding pocket with His583, His587, and Glu646. The results of the presented molecular docking study are consistent with the experimental data.28,46
Regarding the interactions generated between relevant angiotensin receptor amino acids and carboxylate anion of VAL during molecular docking simulations, inhibition of the angiotensin receptor is dependent on eight amino acids. Those amino acids are Arg167, Val108, Ala16, Trp84, Leu81, Tyr34, Phe77, and Tyr87 (Fig. 3, Table S2†). During molecular docking simulations different types of interactions including hydrogen bonds and hydrophobic contacts are formed. It is interesting to point out that Arg167 forms three types of interactions: hydrogen bond, hydrophobic contact, and electrostatic contact. The formed hydrogen bonds of Arg167 are conventional hydrogen bonds with atom distances close to 2 Å. The other two obtained contacts are π–cation and π–alkyl types of interactions. The most frequent interactions are hydrophobic contacts, and among themes are established alkyl, π–σ, π–π T-shaped, π–alkyl types of interactions (Table S2†). Alkyl contacts are recognized between Val108 and Leu81 and VAL. Hydrophobic contacts with π–alkyl type of interactions are formed with Arg167, Trp84, Tyr35, Phe77, and Tyr87. The amino acid residue Trp84 formed hydrophobic contact with VAL numerous times (Table S2†). Furthermore, results presented in Table S2† show that hydrophobic interactions are formed at longer distances, while hydrogen bonds are close to a distance of 2 Å.
 | ||
| Fig. 3 Picture showing the interaction between carboxylate anion VAL and amino acids in angiotensin receptor. | ||
The interactions established in molecular docking simulations between angiotensin receptor and VAL are consistent with available experimental data.29 Namely, in our research Arg167 and Trp84 amino acids with the biggest number of formed interactions, and they can be considered as responsible for binding. The same conclusion is presented in the research study of Kellici et al.47 It should be noted that one more interaction is critical for binding and that is interaction with Tyr35.29
Additionally, the detailed inspection of interactions included in binding (Tables S1 and S2†) implies that binding is related to the number and type of formed interactions. In particular, mainly hydrogen bonds are responsible for binding SAC to neprilysin. The situation is slightly different when it comes to VAL binding to the angiotensin receptor. The highest number of realized interactions are hydrophobic contacts established during molecular docking simulation. The results presented in Table 1 show that lower value of ΔGbind and Ki are obtained for binding of VAL to the angiotensin receptor. Based on the all results collected and presented in the already mentioned tables, it may be stated that a larger number of hydrophobic interactions affect and decrease the values of ΔGbind and Ki.
To investigate the interactions between SAC with neprilysin as well as VAL and the angiotensin receptor, MD simulations were performed. The structures with the lowest binding energies obtained by molecular docking simulation were chosen for the first and referent frame. To confirm the reproducibility and consistency of the obtained results, MD simulations were repeated three times in the timeframe of 150 ns.48 Binding energies were calculated using the MM/GBSA approach for the 25 ns for which RMSD values indicated the highest stabilisation of the system. In the case of both of the investigated compounds, the first 5 ns were to be disregarded, due to the initial changes in conformations and stabilisation of the system. Since all three simulations in both cases showed the most consistency in RMSD values in the timeframe from 5 to 30 ns, this was the timeframe selected for the binding energy calculations (Fig. 4 and 5).
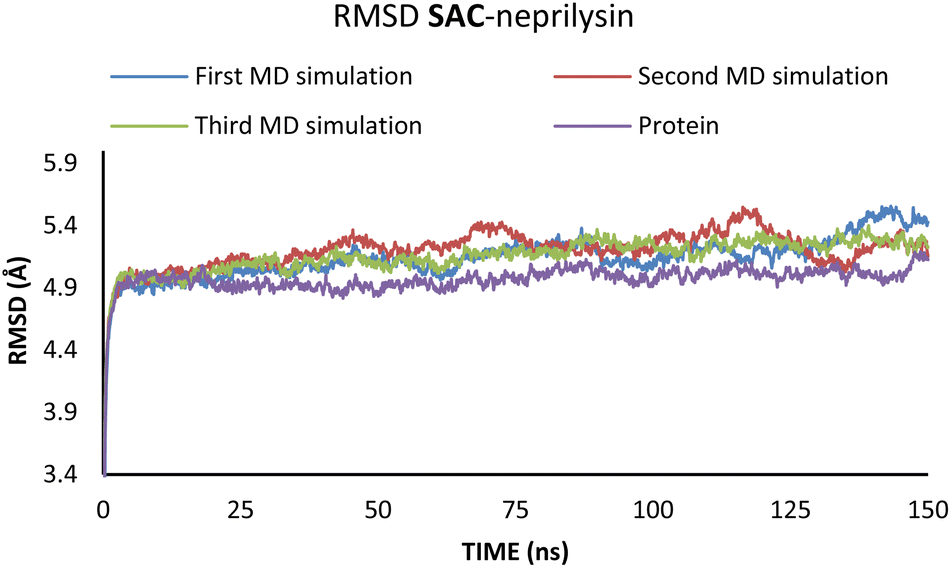 | ||
| Fig. 4 RMSD values obtained through the MD simulations of the SAC–neprilysin system. | ||
 | ||
| Fig. 5 RMSD values obtained through the MD simulations of the VAL–angiotensin receptor system. | ||
According to the results from Table 2 values for binding energies are in direct correlation with the results obtained by molecular docking simulations. The existing differences in energies are higher due to the different environments (bilayer of the lipid membrane) in which calculations were performed.49 MD simulations, on the other hand, confirm the trend shown by molecular docking. It is also worth noting that the binding site and ligand orientation inside the binding site are almost identical for the most stable position in molecular docking and molecular dynamics simulations. These two facts alone are sufficient to validate the findings of the molecular docking investigation. To better understand the inhibitory potential of the SAC and VAL towards neprilysin and angiotensin receptor it is necessary to examine the RMSD, Rg, and RMSF values. As can be seen from Fig. 4 and 5, changes in the RMSD values indicate that the binding of the ligand in the active site induces changes in the protein's primary and secondary structure. These changes are more apparent within the SAC–neprilysin system (Fig. 4).
| System | ΔGbind (kcal mol−1) |
|---|---|
| SAC–neprilysin | −20.69 |
| VAL–angiotensin receptor | −51.05 |
Additionally, changes in the tertiary structure of the protein are only noticeable for the SAC–neprilysin system, according to the Rg values presented in Fig. 6.
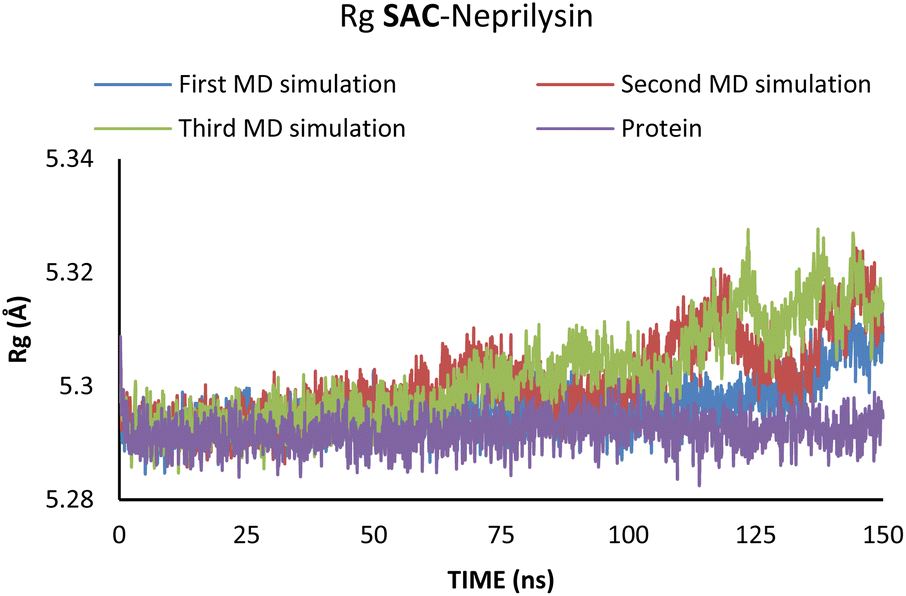 | ||
| Fig. 6 Rg values obtained through the MD simulations of the SAC–neprilysin system. | ||
According to Fig. 6, the compactness of the protein with ligand changed following the time passing, with the difference in Rg values for protein and protein–ligand complex being the highest at the end of the MD simulation. On the other hand, there is no noticeable change in the VAL-angiotensin receptor-lipid bilayer Rg values, since the compactness of the protein is determined not only by the protein structure but partly by the lipid bilayer surrounding the protein (Fig. 7). The changes in the RMSF values for both, VAL–angiotensin receptor and SAC–neprilysin systems indicate an increase in the amino acid residues fluctuations (Fig. S1 and S2†). Interestingly, based on the RMSF values, the fluctuations in the case of the SAC–neprilysin system, continuously increase with every simulation cycle. This is to be expected since we have seen from the RMSD and Rg plots that with time secondary and tertiary structure of the protein–ligand system becomes less stable. When it comes to the VAL–angiotensin receptor system, this is not the case, and RMSF values often overlap indicating different changes caused by conformational modes during the three simulation cycles. Increased RMSF values indicate that stabilising interactions between different amino-acid residues are lowering and disappearing, inducing changes not only in structure but also in the biological and physiological activity of the investigated proteins. That being said, according to the changes in the structures of the proteins indicated by the changes in values of the RMSD, RMSF, and Rg, and low binding energies it is safe to assume high inhibitory potential of SAC and VAL towards neprilysin and angiotensin receptor, respectively.
 | ||
| Fig. 7 Rg values obtained through the MD simulations of the VAL–angiotensin receptor system. | ||
4. Conclusions
Heart failure (HF) is a potentially fatal condition in which the heart is unable to pump enough blood and oxygen to meet the body's demands. It primarily affects the elderly, particularly men, who are obese, diabetic, or suffer from another chronic illness. It can be treated with a variety of drugs, including a relatively new drug called Entresto, which has shown promising results. This paper presents the results of molecular docking and molecular dynamics simulations used to investigate Entresto's inhibitory capacity. The molecular docking simulations revealed that both parts of Entresto (sacubitril (SAC) and valsartan (VAL)) bind with specific proteins (neprilysin and angiotensin receptor, respectively), inhibiting their physiological function. Molecular dynamics (MD) simulations showed very interesting modes of inhibition. It was found that SAC causes structural changes by binding to neprilysin consequently lowering neprilysin's physiological activity. Besides blocking the active site, the binding of SAC causes the structure of the enzyme to become less compact over time, which causes changes in its biochemical properties, preventing the enzyme to express its biological role. When it comes to the RMSD values, VAL shows an effect similar to the one expressed by SAC. However, the complex of the angiotensin receptor with VAL shows almost identical Rg values to the angiotensin receptor without the VAL bonded, which indicates there are no changes in the tertiary structure of the protein. This is to be expected since the whole VAL-angiotensin receptor system is immersed in the lipid bilayer of the membrane, which stabilises the protein's tertiary structure. It was also important to notice that in cases of both ligand–protein systems, fluctuations of most amino acid residues are increased by ligand binding, which indicates a difference in the protein's behaviour in the environment, prohibiting the binding of other organic molecules, and decreasing their binding affinity and consequently inhibiting the protein.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Science and Technological Development of the Republic of Serbia (Contract No. 451-03-68/2022-14/200378 and 451-03-68/2022-14/200107). This paper is supported by the project that has received funding from the European Union's Horizon 2020 research and innovation programme under gr. agreement No. 777204 (SILICOFCM).Notes and references
- V. L. Roger, Circ. Res., 2013, 113, 646–659 CrossRef CAS PubMed.
- R. P. Morrissey, L. Czer and P. K. Shah, Am. J. Cardiovasc. Drugs, 2011, 11, 153–171 CrossRef PubMed.
- S. A. Hunt, J. Am. Coll. Cardiol., 2005, 46, e1–e82 CrossRef PubMed.
- M. Jessup, W. T. Abraham, D. E. Casey, A. M. Feldman, G. S. Francis, T. G. Ganiats, M. A. Konstam, D. M. Mancini, P. S. Rahko and M. A. Silver, Circulation, 2009, 119, 1977–2016 CrossRef PubMed.
- C. D. Kemp and J. V. Conte, Cardiovasc. Pathol., 2012, 21, 365–371 CrossRef CAS PubMed.
- L. M. Ruilope, A. Dukat, M. Böhm, Y. Lacourcière, J. Gong and M. P. Lefkowitz, Lancet, 2010, 375, 1255–1266 CrossRef CAS.
- J. Gu, A. Noe, P. Chandra, S. Al-Fayoumi, M. Ligueros-Saylan, R. Sarangapani, S. Maahs, G. Ksander, D. F. Rigel, A. Y. Jeng and T. H. Lin, J. Clin. Pharmacol., 2010, 50, 401–414 CrossRef CAS PubMed.
- L. G. Hegde, C. Yu, T. Renner, H. Thibodeaux, S. R. Armstrong, T. Park, M. Cheruvu, R. Olsufka, E. R. Sandvik, C. E. Lane and J. Budman, J. Cardiovasc. Pharmacol., 2011, 57, 495–504 CrossRef CAS PubMed.
- T. H. Langenickel and W. P. Dole, Drug Discovery Today: Ther. Strategies, 2012, 9, e131–e139 CrossRef PubMed.
- J. J. McMurray, M. Packer, A. S. Desai, J. Gong, M. P. Lefkowitz, A. R. Rizkala, J. L. Rouleau, V. C. Shi, S. D. Solomon, K. Swedberg and M. R. Zile, N. Engl. J. Med., 2014, 371, 993–1004 CrossRef PubMed.
- M. Camilli, M. G. Del Buono, P. Menna and G. Minotti, Cardio-Oncology, 2020, 6, 1–4 CrossRef PubMed.
- E. G. Erdös and R. A. Skidgel, FASEB J., 1989, 3, 145–151 CrossRef.
- L. B. Daniels, J. Am. Coll. Cardiol., 2007, 50, 2357–2368 CrossRef CAS PubMed.
- N. L. Cruden, K. A. Fox, C. A. Ludlam, N. R. Johnston and D. E. Newby, Hypertension, 2004, 44, 913–918 CrossRef CAS PubMed.
- Diovan (valsartan) tablets [prescribing information], Novartis, East Hanover, NJ, Revised: August 2020, https://www.novartis.com/us-en/sites/novartis_us/files/diovan_hct_ppi.pdf Search PubMed.
- US Food and Drug Administration, FDA approves a new generic valsartan, March 12, 2019, https://www.fda.gov/news-events/press-announcements/fda-approves-new-generic-valsartan Search PubMed.
- Diovan, European Medicines Agency, May 31, 2010, https://www.ema.europa.eu/en/medicines/human/referrals/diovan-1 Search PubMed.
- FDA Approves New Heart Failure Drug, Press release, July 8, 2015, https://www.acc.org/latest-in-cardiology/articles/2015/07/08/10/00/fda-approves-new-heart-failure-drug Search PubMed.
- Entresto (sacubitril and Valsartan) tablets [prescribing information], Novartis, East Hanover, NJ, July 2015, https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/207620s018lbl.pdf Search PubMed.
- R. W. Schrier, J. G. Abdallah, H. H. Weinberger and W. T. Abraham, Kidney Int., 2000, 57, 1418–1425 CrossRef CAS PubMed.
- E. R. Levin, D. G. Gardner and W. K. Samson, N. Engl. J. Med., 1998, 339, 321–328 CrossRef CAS PubMed.
- S. Nathisuwan and R. L. Talbert, Pharmacotherapy, 2002, 22, 27–42 CrossRef CAS PubMed.
- C. J. Ferro, J. C. Spratt, W. G. Haynes and D. J. Webb, Circulation, 1998, 97, 2323–2330 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- T. Giroday, M. M. Montero-Campillo and N. Mora-Diez, Comput. Theor. Chem., 2014, 1046, 81–92 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.1, Inc., Wallingford, CT, 2013 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- S. Moss, S. Vasanta and K. R. Acharya, J. Struct. Biol., 2018, 204, 19–25 CrossRef CAS PubMed.
- H. Zhang, H. Unal, C. Gati, G. W. Han, W. Liu, N. A. Zatsepin, D. James, D. Wang, G. Nelson, U. Weierstall and M. R. Sawaya, Cell, 2015, 161, 833–844 CrossRef CAS PubMed.
- D. S. Biovia, Discovery studio modeling environment, 2017 Search PubMed.
- Y. Zhang, S. Forli, A. Omelchenko and M. F. Sanner, J. Comput. Chem., 2019, 40, 2882–2886 CrossRef CAS PubMed.
- T. S. Lee, D. S. Cerutti, D. Mermelstein, C. Lin, S. LeGrand, T. J. Giese, A. Roitberg, D. A. Case, R. C. Walker and D. M. York, J. Chem. Inf. Model., 2018, 58, 2043–2050 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke and G. Giambasu, Amber 2021, University of California, San Francisco, 2021 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- S. Jo, J. B. Lim, J. B. Klauda and W. J. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch and A. Caflisch, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi and S. Jo, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed.
- J. Lee, M. Hitzenberger, M. Rieger, N. R. Kern, M. Zacharias and W. Im, J. Chem. Phys., 2020, 153, 035103 CrossRef CAS PubMed.
- J. Zielkiewicz, J. Chem. Phys., 2005, 123, 104501 CrossRef PubMed.
- P. H. Hünenberger and J. Andrew McCammon, J. Chem. Phys., 1999, 110, 1856–1872 CrossRef.
- R. Faller and J. J. De Pablo, J. Chem. Phys., 2002, 116, 55–59 CrossRef CAS.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, Proc. Natl. Acad. Sci., 1999, 96, 9997–10002 CrossRef CAS PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Drug Discovery Today, 2004, 9, 430–431 CrossRef PubMed.
- M. D. Shultz, Bioorg. Med. Chem. Lett., 2013, 23, 5980–5991 CrossRef CAS PubMed.
- R. Sankhe, E. Rathi, S. Manandhar, A. Kumar, S. Ranganath, K. Pai, S. G. Kini and A. Kishore, J. Mol. Struct., 2021, 1224, 129073 CrossRef CAS PubMed.
- T. F. Kellici, D. Ntountaniotis, G. Liapakis, A. G. Tzakos and T. Mavromoustakos, Arabian J. Chem., 2019, 12, 5062–5078 CrossRef CAS.
- L. Wang and F. Yan, J. Chem. Inf. Model., 2018, 58, 2123–2130 CrossRef CAS PubMed.
- M. T. Matsoukas, C. Potamitis, P. Plotas, M. E. Androutsou, G. Agelis, J. Matsoukas and P. Zoumpoulakis, J. Chem. Inf. Model., 2013, 53, 2798–2811 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra04226f |
| This journal is © The Royal Society of Chemistry 2022 |