
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Jejuketomycins A and B, polyketide glycosides with cancer cell migration inhibitory activity from Streptomyces sp. KCB15JA151†
Jun-Pil Jang‡
a,
Gil Soo Kim‡c,
Tae Hoon Oh‡ad,
Beomcheol Parkad,
Minhee Kimad,
Gwi Ja Hwanga,
Hyeok-Won Leee,
Jin-Gyeom Leee,
Young-Soo Hongab,
Jong Seog Ahn*ab,
Sung-Kyun Ko *ab and
Jae-Hyuk Jang*ab
*ab and
Jae-Hyuk Jang*ab
aChemical Biology Research Center, Korea Research Institute of Bioscience and Biotechnology, Cheongju 28116, Korea. E-mail: jangjh@kribb.re.kr
bDepartment of Biomolecular Science, KRIBB School of Bioscience, University of Science and Technology, Daejeon 34113, Korea
cCentral Research and Development, HanpoongPharm. Co., LTD., Wanju 54843, Korea
dCollege of Pharmacy, Chungbuk National University, Cheongju, 28160, Korea
eBiotechnology Process Engineering Center, Korea Research Institute of Bioscience and Biotechnology, Cheongju, 28116, Korea
First published on 10th August 2022
Abstract
Two new polyketide glycosides jejuketomycins A (1) and B (2), were isolated from a culture of Streptomyces sp. KCB15JA151. Their chemical structures including the absolute configurations were determined by detailed analyses of the NMR and HRMS data and ECD calculations and spectral data. Compounds 1 and 2 possess an unusual 6/6/8 tricyclic ring system. Biological evaluation with the wound healing assay and time-lapse cell tracking analysis revealed that compounds 1 and 2 have significant inhibitory activities against cancer cell migration with low cytotoxicity.
Introduction
Microbial secondary metabolites provide various chemical templates for clinically useful lead compounds in the pharmaceutical industry.1,2 Actinomycetes, especially those from the genus Streptomyces, are known to be a prolific source of bioactive secondary metabolites. They produce a wide variety of products including polyketides, peptides, terpenes, and glycosides.3–6 Among them, polyketide natural products exhibit a range of functional and structural diversity and medicinally important activities, including antibiotic, anticancer, antifungal, and antiparasitic.7,8 Chemical studies and genome sequence analysis of many strains of actinomycetes have revealed the presence of biosynthetic gene clusters whose number far exceeds the number of known secondary metabolites produced by those bacteria. Usually, only a small number of the biosynthetic gene clusters are expressed under typical laboratory culture conditions to produce detectable amounts of compounds.9 This information further underscores the enormous potential of actinomycetes as a source of bioactive natural products.Cell migration is involved in a number of physiological processes including ovulation, embryonic development, tissue regeneration (wound healing), and inflammation. These migration activities of cells in vitro are thought to be related to many in vivo cellular behaviors such as tumor angiogenesis and metastasis.10 Thus, cell migration is attracting much attention as one of the alternative strategies for the development of anti-cancer chemotherapies. Several natural products such as withaferin A,11 prodigiosin,12 and migrastatin13 have been reported to inhibit cell migration. As part of our continuing efforts to find inhibitors of cell migration, Streptomyces sp. KCB15JA151, a soil-derived bacteria, caught our interest and was subjected to a systematic chemical investigation, resulting in the isolation and identification of two new polyketide glycosides, jejuketomycins A (1) and B (2) (Fig. 1). Herein, the isolation, structure elucidation, and bioactivity evaluation of these compounds are reported.
 | ||
| Fig. 1 The chemical structures of 1 and 2. | ||
Results and discussion
Streptomyces sp. KCB15JA151 were cultured in PDB media for 7 days at 28 °C, and the broth and mycelia extracts were partitioned with EtOAc. The EtOAc extracts were purified by ODS vacuum flash chromatography and reversed-phase HPLC to yield jejuketomycins A (1) and B (2). Jejuketomycin A (1) was isolated as a yellowish powder and its molecular formula was established as C28H44O9 based on the HRESIMS and NMR data. The 1H NMR spectrum (DMSO-d6) exhibited two exo-methylene protons (δH 6.15 and 5.27), six oxygen-bearing methine protons (δH 4.46, 4.07, 3.15, 3.11, 3.08, and 2.97), an anomeric proton (δH 4.17), two oxygen-bearing methylene protons (δH 3.68 and 3.44), eight methine protons (δH 3.33, 2.82, 2.57, 2.24, 2.23, 1.46, 1.41, and 1.32), six methylene protons (δH 1.91, 1.67, 1.57, 1.10, 0.80, and 0.60), and five methyl protons (δH 1.12, 1.08, 0.90, 0.87, and 0.75).The 13C NMR was combined with HSQC-DEPT data, which indicated the existence of 28 carbons, including two carbonyl carbons (δC 201.1 and 177.9), a non-protonated sp2 carbon (δC 140.5), an exo-methylene carbon (δC 122.0), an anomeric carbon (δC 101.6), six oxygen-bearing methine carbon (δC 86.2, 77.6, 77.3, 73.6, 71.0, and 70.7), an oxygen-bearing methylene carbon (δC 61.8), eight methine carbons (δC 50.1, 46.7, 45.4, 41.5, 39.3, 37.9, 35.6, and 30.1), three methylene carbons (δC 43.7, 38.1, and 34.7), and five methyl carbons (δC 24.8, 22.9, 19.5, 18.7, and 16.7) (Table 1).
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH, mult (J in Hz) | δC | δH, mult (J in Hz) | |
| a Overlapped signal. | ||||
| 1 | 177.9 | 178.0 | ||
| 2 | 50.1 | 2.82, t (8.2) | 48.6 | 2.68, t (8.2) |
| 3 | 41.5 | 1.32, ddd (13.4, 10.4, 7.7) | 44.9 | 1.48, q (10.9, 7.7) |
| 4 | 39.3 | 1.46, m | 33.5 | 1.79, m |
| 5 | 43.7 | 0.60, q (12.2), ax | 43.8 | 0.61, q (12.2), ax |
| 1.57, dq (13.4, 2.1), eq | 1.58, dq (13.2, 2.1), eq | |||
| 6 | 30.6 | 1.41, m | 26.4 | 1.81, m |
| 7 | 34.7 | 0.80, q (12.8), ax | 41.0 | 0.96, t (12.9), ax |
| 1.91, dq (13.6, 2.1), eq | 1.90, d (13.6), eq | |||
| 8 | 45.7 | 2.57, ddd (13.7, 11.3, 3.7) | 73.7 | |
| 9 | 201.1 | 198.7 | ||
| 10 | 140.5 | 140.1 | ||
| 11 | 46.7 | 3.33, m | 45.8 | 3.96, m |
| 12 | 35.6 | 2.33, m | 35.6 | 2.33, m |
| 13 | 38.1 | 1.10, ovla, ax | 37.8 | 1.08, ovla, ax |
| 1.67, ddd (14.9, 7.2, 3.4), eq | 1.66, dt (14.9, 3.4), eq | |||
| 14 | 37.9 | 2.24, m | 37.9 | 2.24, m |
| 15 | 86.2 | 4.46, dd (10.4, 1.9) | 86.1 | 4.49, dd (10.4, 1.9) |
| 16 | 71.0 | 4.07, qd (6.4, 2.0) | 71.1 | 4.07, qd (6.4, 2.0) |
| 17 | 16.7 | 1.08, d (6.4) | 16.7 | 1.08, d (6.4) |
| 18 | 19.5 | 0.75, d (6.5) | 19.2 | 0.74, d (6.5) |
| 19 | 22.9 | 0.90, d (6.5) | 22.6 | 0.88, d (6.4) |
| 20 | 122.0 | 6.15, br s | 123.5 | 6.18, br s |
| 5.27, br s | 5.36, br s | |||
| 21 | 24.8 | 1.12, d (7.6) | 25.0 | 1.12, d (7.6) |
| 22 | 18.7 | 0.87, d (6.7) | 18.7 | 0.87, d (6.4) |
| 1′ | 101.6 | 4.17, d (7.9) | 101.6 | 4.16, d (7.9) |
| 2′ | 73.6 | 2.97, t (8.2) | 73.6 | 2.98, t (8.2) |
| 3′ | 77.6 | 3.15, t (8.8) | 77.6 | 3.16, t (8.8) |
| 4′ | 70.7 | 3.08, dd (10.6, 7.7) | 70.7 | 3.09, dd (10.6, 7.7) |
| 5′ | 77.3 | 3.11, ddd (9.2, 5.8, 2.1) | 77.3 | 3.11, ddd (9.2, 5.8, 2.1) |
| 6′ | 61.8 | 3.44, dd (10.3, 5.3) | 61.8 | 3.44, dd (10.3, 5.3) |
| 3.68, d (11.2) | 3.68, d (11.2) | |||
| 8-OH | 5.47, br s | |||
The planar structure of 1 was determined by extensive 2D NMR analysis (Fig. 2). The COSY spectrum of 1 showed two spin systems. The large spin system was used in the structural elucidation of aglycone, and the small one was used in sugar unit. Ring A was established by the COSY correlation of H-3/H-4/H-5/H-6/H-7/H-8. The methyl substitutions on ring A were also determined by the COSY correlation from H-18 to H-4 and from H-19 to H-6. Ring B was determined by the COSY correlations of H-8/H-3/H-2/H-11 with the HMBC correlations from H-3 to C-9, H-8 to C-10, H-11 to C-10 and C-20, and H-20 to C-10. The COSY correlations of H-2/H-11/H-12/H-13/H-14/H-15 with the HMBC correlations of H-2 to C-1, H-11 to C-1, and H-15 to C-1 constructed ring C, which showed an eight-membered lactone. The COSY correlations of H-12/H-21, H-14/H-22 and H-15/H-16/H-17 suggested that hydroxyethyl and methyl groups were attached to ring C.
 | ||
| Fig. 2 Key 2D NMR correlations of 1 and 2. | ||
An anomeric carbon (δC 101.6) indicated the existence of a sugar unit. The sugar unit was deduced by the COSY correlation from H-1′ to H-6′ with the HMBC correlation from H-5′ to C-1′. The HMBC correlation from H-16 to C-1′ suggested that the sugar unit was bound with the aglycone via an O-glycosidic linkage. The result of the NMR analysis suggested that the planar structure of compound 1 was very similar to satosporin B14 except for the exo-methylene. Therefore, compound 1 was determined to be a new polyketide glycoside and named as jejuketomycin A.
The ROESY spectrum and coupling constants were obtained to determine the relative configuration of compound 1 (Table 1 and Fig. 3). The large coupling constant of the anomeric proton (δH 4.17, J = 7.6 Hz) suggested a β-glycosidic linkage. The other protons in the sugar unit also demonstrated a large coupling constant ranged from 8.2 to 11.2 Hz, which suggested that all of the methine protons were located on axial positions. The ROESY correlation of H-1′ to H-3′, H-1′ to H-5′, and H-2′ to H-4′ also supported the axial positions of the methine protons. Thus, the sugar unit in compound 1 was established β-glucose. The large coupling constants of H-3 (δH 1.32, J = 13.4, 7.7, 10.4 Hz), H-5ax (δH 0.60, J = 12.2 Hz), H-7ax (δH 0.80, J = 12.8 Hz), H-8 (δH 2.57, J = 13.7, 11.3, 3.7 Hz) suggested that H-3, H-4, H-5ax, H-6, H-7ax, and H-8 occupied axial positions. The ROESY correlations of H-2 to H-4, H-2 to H-8, H-4 to H-8, H-6 to H-8, H-8 to H-11, and H-11 to H-13 implied that H-2, H-4, H-6, H-8, H-11, and H-13ax were on the same side. The rigid structure by the exo-methylene and the ROESY correlation between H-8/H-11 and H-20/H3-21 with no correlation observed between H-11/H3-21 indicated H3-21 was on the opposite axial position. The ROESY correlations H-12 to H-13ax and H-13ax to H-14 in the 1, 2-axial equatorial interactions suggested H-12 and H-14 occupied equatorial positions. The large coupling constant of H-15 (δH 4.46, J = 10.4, 1.9 Hz) and the ROESY correlation between H-15 with H3-21 implied that H-15 was placed on the axial position. The coupling constant of H-16 (δH 4.07, J = 6.4, 2.0 Hz) and the ROESY correlation between H-15/H-16, H-15/H3-17, and H-16/H3-22 were very similar to those of satosporin A–C,14 which suggested an anti-configuration of the hydroxy groups between C-15 and C-16. Therefore, the relative configuration of aglycone in compound 1 was proposed to be 2S*, 3R*, 4S*, 6R*, 8R*, 11S*, 12S*, 14R*, 15R*, 16R*.
 | ||
| Fig. 3 Key ROESY correlations of 1. | ||
The time-dependent density functional theory-electronic circular dichroism (TDDFT-ECD) calculation was conducted to established the absolute configuration of compound 1.14 The aglycone structure was used to calculating the ECD spectrum to save on the computational time.15 All conformers of aglycone were geometry optimized using the B3LYP/6-311G(d) basis set.
The ECD spectra were also calculated using the B3LYP/6-311G (d) basis set from all of the optimized conformers. The experimental ECD spectra of 1 showed positive cotton effects at 220 nm and negative cotton effects at 310–340 nm. The calculated ECD curve of (2S, 3R, 4S, 6R, 8R, 11S, 12S, 14R, 15R, 16R)-1 demonstrated a similar pattern of the experimental ECD spectra (Fig. 4). Therefore, the absolute configuration of 1 was established as 3R, 4S, 6R, 8R, 11S, 12S, 14R, 15R, 16R.
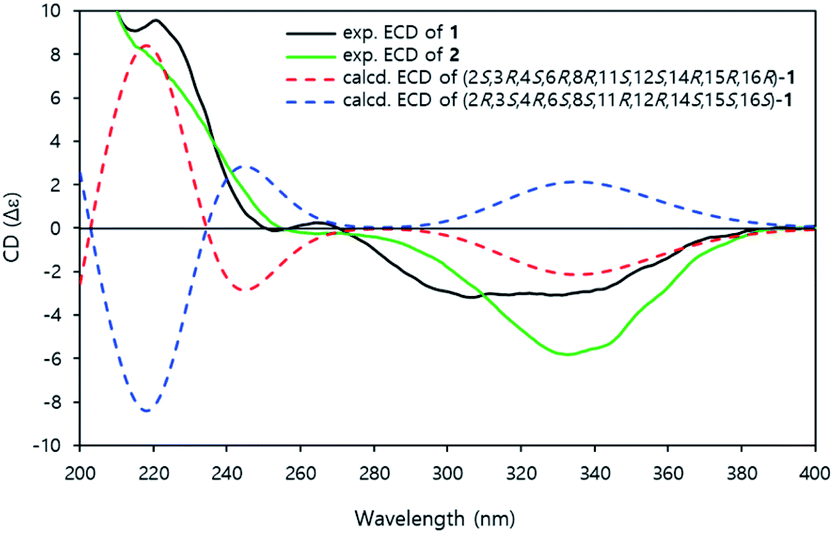 | ||
| Fig. 4 Experimental ECD and calculated ECD spectra of 1 and 2. | ||
Jejuketomycin B (2) was isolated as a yellowish powder. The molecular formula was established as C28H44O10 on the basis of the HRESIMS data. The molecular formula showed the presence of a hydroxy group, compared to 1, which indicates that 2 is an analogue of 1 with slight modifications. A comparison of the 1D NMR data (Table 1) of compounds 1 and 2 revealed that their planar structures were very similar, except for the absence of the signal of the OH proton (8-OH, δH 5.10) in 2 (Fig. 2). The identical ROESY correlations and the experimental ECD spectra, closely similar the 1H and 13C NMR data, and the presumed same biosynthetic pathway led to the conclusion that the configuration of 2 was the same as that of 1. Thus, compound 2 was named as jejuketomycin B.
Before evaluating the cancer cell migration inhibitory activity of 1 and 2, their cytotoxic effects were tested against MDA-MB-231, PC12, MEF, A549, and HeLa cells. As shown in Fig. S16,† 1 and 2 did not show any cytotoxicity up to 50 μM, whereas a slightly decreased cell number was observed in the cells treated with 100 μM. The inhibitory effect of 1 and 2 on the migration of MDA-MB-231 cells with the scratched wound healing assay was determined (Fig. 5). After treatment with the indicated concentrations of 1 and 2 for 24 h, the rate of cells migrating to the scratch wound healing area was significantly decreased 1 (100 ± 22% inhibition) and 2 (69.8 ± 19.9% inhibition). To further evaluate the effect of 1 and 2 on cell migration, time-lapse cell tracking analysis was performed by a HoloMonitor M4 time-lapse cytometer. After treating the cells with 1 and 2, they were captured every 15 min for 12 hour. The migration and motility of 1 and 2 treated cells were remarkably decreased in a dose-dependent manner (Fig. 6).
 | ||
| Fig. 5 Effects of scratched wound healing in MDA-MB-231 cells for (a) compound 1. (b) Compound 2. | ||
Experimental
General experimental procedures
The specific rotations were measured on a JASCO P-1020 polarimeter that uses a 100 mm glass microcell. UV spectra were obtained on a NanoDrop 2000 spectrophotometer and UltiMate DAD-3000 connected to a Thermo Scientific Dionex Ultimate 3000 Rapid Separation LC system (UPLC-PDA) using a Waters HSS T3 column (Waters, 2.1 × 150 mm, 2.5 μm). IR spectra were recorded on a Bruker VERTEX 80v FT-IR spectrometer. The NMR spectra were recorded on Bruker AVANCE HD 700 and 800 NMR spectrometers at the Korea Basic Science Institute (KBSI) in Ochang, South Korea. Chemical shifts were referenced to a residual solvent signal (DMSO-d6 δH 2.50, δC 39.51). High-resolution electrospray ionization mass spectrometry (HRESIMS) data were acquired with a Q-TOF mass spectrometer on a SYNAPT G2 (Waters) at KBSI in Ochang, South Korea. The CD spectra were obtained on a JASCO J-1500 circular dichroism spectrophotometer at KBSI in Ochang, South Korea. Liquid chromatography-mass spectrometry (LC-MS) was performed with a Thermo LTQ XL linear ion trap attached to an ESI source that was connected to a Thermo Scientific Dionex Ultimate 3000 Rapid Separation LC system (ESI-LC-MS) using a Waters HSS T3 column (Waters, 2.1 × 150 mm, 2.5 μm). Open column chromatography was performed with a silica gel (Merck, silica gel 60 (0.063–0.200 mm)). Vacuum liquid column chromatography was carried out with an ODS (Cosmosil, 75 μm). Semi preparative C18 (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) columns were used for HPLC on a YL9100 HPLC system equipped with a photodiode array detector (YL9160) that uses HPLC grade solvents (Burdick & Jackson).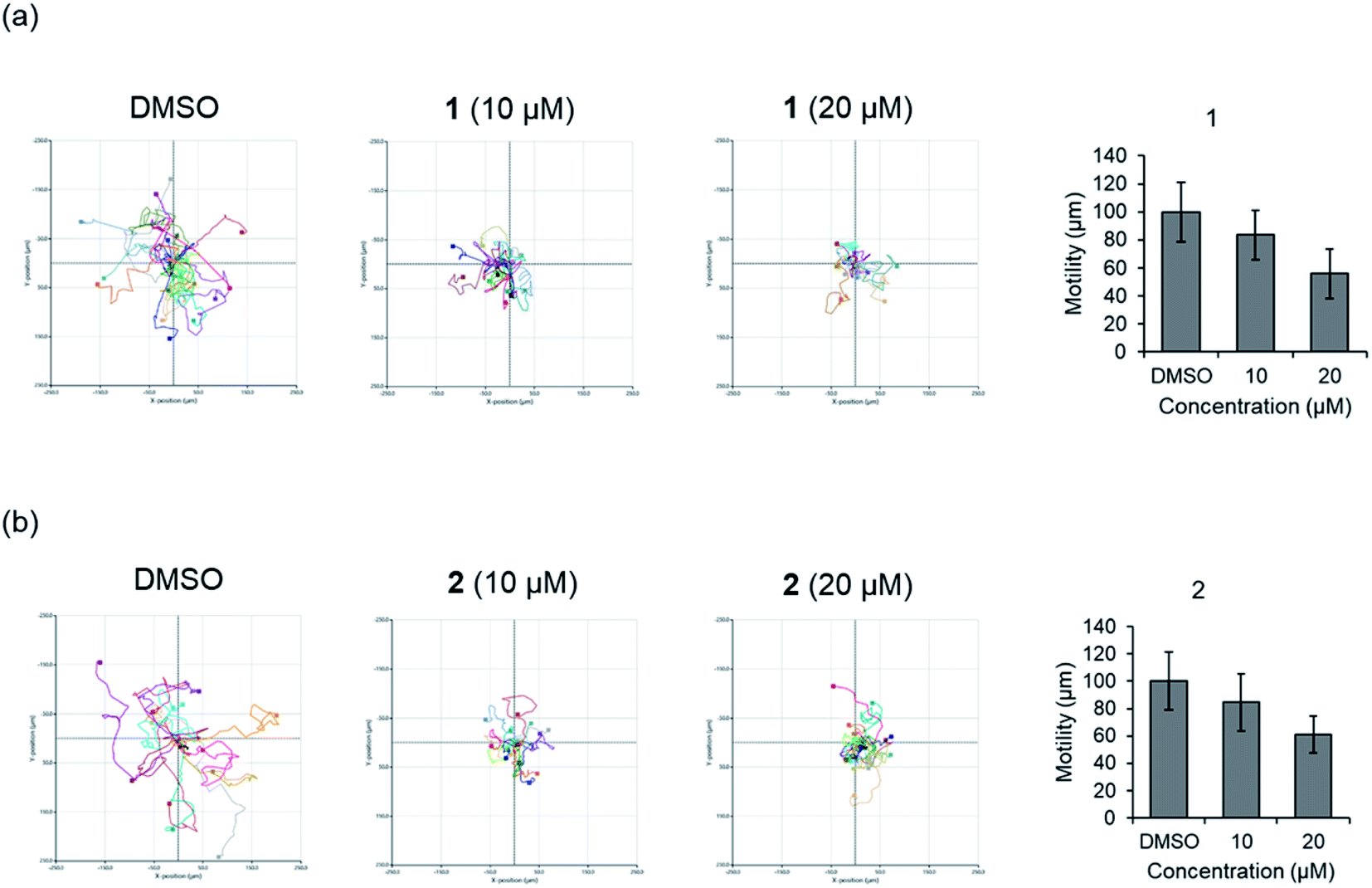 | ||
| Fig. 6 Time-lapse cell tracking analysis of cellular movement for (a) compound 1. (b) Compound 2. | ||
Microorganism collection and identification
The strain KCB15JA151 was isolated from a soil sample from an agricultural field of garlic on Jeju Island, Korea. A soil sample was spread onto selective medium (24.0 g of Potato Dextrose Broth and 17 g of agar per 1 L of distilled water, pH 5.6) supplemented with Chloramphenicol (50 ppm) and Kanamycin (100 ppm). Strain KCB15JA151 was isolated from the selective media and transferred onto potato dextrose agar (PDA). Strain KCB15JA151 (GenBank accession no. MW282889) was identified as Streptomyces sp. by 16S rRNA gene sequencing, which showed a 99.7% similarity to Streptomyces roietensis WES2.Fermentation, extraction, and isolation
The Streptomyces sp. KCB15JA151 was grown in potato dextrose broth (PDB) for 7 days at 28 °C on a rotary shaker operating at 165 rpm in a baffled Erlenmeyer flask. The cultured broth (45 L) was divided into the filtered broth and mycelia by Büchner funnel. The filtered broth was extracted with an equal volume of ethyl acetate three times. The mycelia were extracted with an equal volume of 80% acetone in water followed by evaporation under reduced pressure to remove the organic solvent. The aqueous solution of the acetone extract was extracted three times with an equal volume of ethyl acetate. The combined extracts (14.49 g) from the supernatant and mycelia were fractionated by silica gel column chromatography (70 i.d. × 320 mm) and eluted with a stepwise gradient of CHCl3–MeOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 30:1, 20:1, 10:1, 7:1, 5:1, 3:1, 2:1, 1:1, and 0:100 (v/v)). The bioactive fraction 4 (CHCl3–MeOH, 10:1, 1.25 g) was separated by reversed-phase C18 vacuum liquid column chromatography (50 i.d. × 250 mm) eluted with a stepwise gradient of MeOH in water (20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, and 100%). The active fractions 4–5 (60% MeOH, 95.2 mg) were purified by reversed phase HPLC (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) that used a linear gradient condition (35–70% MeCN–H2O containing 0.05% formic acid, flow rate 3 mL min−1) to yield compounds 1 (8.7 mg) and 2 (2.7 mg).
:0, 30:1, 20:1, 10:1, 7:1, 5:1, 3:1, 2:1, 1:1, and 0:100 (v/v)). The bioactive fraction 4 (CHCl3–MeOH, 10:1, 1.25 g) was separated by reversed-phase C18 vacuum liquid column chromatography (50 i.d. × 250 mm) eluted with a stepwise gradient of MeOH in water (20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, and 100%). The active fractions 4–5 (60% MeOH, 95.2 mg) were purified by reversed phase HPLC (Cosmosil 5C18-MS-II, 5 μm, 10 × 250 mm) that used a linear gradient condition (35–70% MeCN–H2O containing 0.05% formic acid, flow rate 3 mL min−1) to yield compounds 1 (8.7 mg) and 2 (2.7 mg).
Characterization of compounds 1 and 2
ε) 220 (4.30) nm; CD (MeOH) Δε 220 (9.56), 264 (0.25) 306 (−3.18); IR (ATR) νmax 3376, 2917, 1729, 1377, 1070, 1023, 596 cm−1; 1H and 13C NMR spectroscopic data, see Table 1; HRESIMS m/z 547.2882 [M + Na]+ (calcd for C28H44O9Na, 547.2883).ε) 220 (4.23) nm; CD (MeOH) Δε 220 (7.95), 330 (−5.98); IR (ATR) νmax 3439, 2916, 1729, 1606, 1375,1062, 531 cm−1; 1H and 13C NMR spectroscopic data, see Table 1; HRESIMS m/z 539.2851 [M − H]− (calcd for C28H43O10, 539.2861).ECD calculation
The conformational search was conducted using Avogadro 1.2.0 with the MMFF94 force field.16 The geometry optimization of all conformers were calculated using Gaussian 09 with the B3LYP/6-311g(d) basis set. TD-DFT method was performed at the B3LYP/6-311g(d) level of theory using Gaussian 09 to obtain the calculated ECD spectrum of each optimized conformers. The Boltzmann-weighted ECD spectrum was generated from each calculated spectrum using SpecDis 1.71.Cell culture
PC12 (pheochromocytoma of rat adrenal medulla), HeLa (Human cervix cancer), A549 (Human lung cancer), MEF (Mouse Embryonic Fibroblasts), and MDA-MB-231 (Human breast cancer) cells were purchased from the American Type Culture Collection (ATCC). The cells were routinely cultured in DMEM (Dulbecco's modified Eagle's medium; Welgene, Korea, LM 001-05) containing 10% FBS (fetal bovine serum; Welgene, Korea, S001-07), 100 units penicillin, and 100 μg mL−1 streptomycin (Gibco, USA, 15140-122) at 37 °C in humidified atmosphere containing 5% CO2.Cell viability assay
Cell viability was determined using the EZ-Cytox colorimetric assay (Daeil Lab service, 0793). For the cytotoxicity, PC12, HeLa A549, MEF, and MDA-MB-231 cells were cultured in 96-well plates (1.0 × 104 cells per well) for 12 h. The cells were treated with test compounds at the indicated concentrations for 24 h. After the treatment for 24 h, media were replaced with EZ-Cytox solution and incubated with the cells for 1 h. Live cells were read in a microplate reader (Molecular Devices, Spectra Max 190) at 450 nm. Cell viability was normalized to the control group.Scratch wound healing assay
MDA-MB-231 cells were cultured in 24-well plates (9.0 × 104 cells per well) for 12 h. Each well was artificially wounded by creating a scratch in the center with a 200 μL tip and cultivated in DMEM with 10 μg mL−1 mitomycin C for 3 h. The cells were treated with the test compounds at the indicated concentrations for 24 h. Cells were fixed in 4% paraformaldehyde for 12 min, stained with 0.2% crystal violet, captured by a microscope and analyzed with the ZEN software (ZEISS, Germany). The wound closure areas were measured with ImageJ (Software 1.48q, Rayne Rasband, National Institutes of Health, USA).Time-lapse cell tracking analysis
MDA-MB-231 cells (9 × 103 cells/100 μL) were seeded in μ-Slide I(ibidi, Germany, 80106). After 3 h, 900 μL of DMEM were added and incubated overnight. The test compounds with the indicated concentrations were treated and directly transferred to the HoloMonitor M4 time-lapse cytometer (Phase Holographic Imaging) kept in a 37 °C incubator. The live cells were captured every 15 min for 12 h, and the migration was analyzed with the HoloStudio M4 software.Statistics
Data are presented as the mean ± standard error of the mean (SEM) of at least three independent experiments (n = 3). Statistical analyses were performed using GraphPad Prism (GraphPad Software, 9.4.0, San Diego, CA, USA). The differences were considered statistically significant at *p < 0.05 and, **p < 0.01, when compared to the DMSO control group.Conclusions
Jejuketomycins A (1) and B (2) are structurally unique glycosylated polyketides with an unusual 6/6/8 tricyclic ring system. Satosporin produced by Kitasatospora griseola is the most similar compound with a 6/6/8 tricyclic ring. However, the jejuketomycins exhibit a different exo-methylene group at the C-10 position. In previous reports, it was estimated that satosporins are likely biosynthesized by a type I polyketide synthase using D-lactate as an unusual starting unit.14 However, we were unable to identify candidate biosynthetic genes matching with the proposed satosporine polyketide assembly in the draft genome sequencing data of Streptomyces sp. KCB15JA151. Therefore, these new satosporin derivatives with the exo-methylene residue are expected to be synthesized by a rare biosynthetic machinery.17 In particular, the mechanism of forming tricyclic rings with the exo-methylene residues will be an interesting topic. The discovery of the jejuketomycins expands the diversity of the structural and biological aspects of polyketide natural products and also demonstrates that actinobacteria may have significant potential in the biosynthesis of bioactive organic molecules with unique skeletal frameworks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (2021M3H9A103743912), the KRIBB Research Initiative Program (KGM5292221 and KGM1222211) funded by the Ministry of Science ICT (MSIT) and the Basic Science Research Program (2021R1I1A204970412) of the Ministry of Education of the Republic of Korea. We thank the Korea Basic Science Institute, Ochang, Korea, for providing the NMR (700 and 800MHz), HRESIMS, and CD data.
References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- J. V. Pham, M. A. Yilma, A. Feliz, M. T. Majid, N. Maffetone, J. R. Walker, E. Kim, H. J. Cho, J. M. Reynolds, M. C. Song, S. R. Park and Y. J. Yoon, Front. Microbiol., 2019, 10, 1404 CrossRef.
- J. J. Hug, C. D. Bader, M. Remskar, K. Cirnski and R. Muller, Antibiotics, 2018, 7, 44 CrossRef.
- S. Fotso, T. Mahmud, T. M. Zabriskie, D. A. Santosa, Sulastri and P. J. Proteau, J. Nat. Prod., 2008, 71, 61–65 CrossRef CAS.
- Y. Sheng, P. W. Lam, S. Shahab, D. A. Santosa, P. J. Proteau, T. M. Zabriskie and T. Mahmud, J. Nat. Prod., 2015, 78, 2768–2775 CrossRef CAS PubMed.
- T. Mahmud, Curr. Opin. Chem. Biol., 2009, 13, 161–170 CrossRef CAS.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS.
- D. G. Kingston, J. Nat. Prod., 2011, 74, 496–511 CrossRef CAS.
- D. A. Hopwood, Heredity, 2019, 123, 23–32 CrossRef PubMed.
- P. Carmeliet, Nat. Med., 2003, 9, 653–660 CrossRef CAS.
- J. Lee, E. R. Hahm and S. V. Singh, Carcinogenesis, 2010, 31, 1991–1998 CrossRef CAS.
- J. Zhang, Y. Shen, J. Liu and D. Wei, Biochem. Pharmacol., 2005, 69, 407–414 CrossRef CAS.
- K. Nakae, Y. Yoshimoto, T. Sawa, Y. Homma, M. Hamada, T. Takeuchi and M. Imoto, J. Antibiot., 2000, 53, 1130–1136 CrossRef CAS.
- J. C. Arens, F. Berrue, J. K. Pearson and R. G. Kerr, Org. Lett., 2013, 15, 3864–3867 CrossRef CAS PubMed.
- A. Mandi and T. Kurtan, Nat. Prod. Rep., 2019, 36, 889–918 RSC.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- F. Zhang, H. Y. He, M. C. Tang, Y. M. Tang, Q. Zhou and G. L. Tang, J. Am. Chem. Soc., 2011, 133, 2452–2462 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic data of compounds 1 and 2. See https://doi.org/10.1039/d2ra04039e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |