
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, crystal structure, and properties of methyl-substituted coronene amide analogue†
Kenta Rakumitsu a,
Miho Fujiia,
Sotaro Kusumotob,
Shoko Kikkawac,
Isao Azumayac and
Akihiro Yokoyama*a
a,
Miho Fujiia,
Sotaro Kusumotob,
Shoko Kikkawac,
Isao Azumayac and
Akihiro Yokoyama*a
aFaculty of Science and Technology, Seikei University, 3-3-1 Kichijoji-kitamachi, Musashino, Tokyo 180-8633, Japan. E-mail: ayokoyama@st.seikei.ac.jp
bDepartment of Material and Life Chemistry, Faculty of Engineering, Kanagawa University, 3-27-1 Rokkakubashi, Kanagawa-ku, Yokohama 221-8686, Japan
cFaculty of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan
First published on 16th September 2022
Abstract
A coronene amide analogue was synthesized in six steps using an improved method at the final biarylation step. The key to the progress of palladium-mediated biarylation involved the introduction of three methyl groups to suppress the undesired reaction and the use of tri-tert-butylphosphine as the ligand for palladium. Single-crystal X-ray analysis revealed that the core unit of the coronene analogue has a non-planar structure.
Introduction
As one of the most basic polycyclic aromatic hydrocarbons, coronene comprises one central benzene ring and six surrounding benzene rings (Fig. 1). It is characterized by D6h symmetry and a completely localized π-conjugated system.1 The size of coronene is suitable for processing techniques. Several applications have been investigated, including building blocks for self-assembling compounds2–4 and organic transistors.5–8 Doping of heteroatoms in aromatic rings is an effective approach for modulating physico-electrochemical properties, and several synthetic studies of heterocoronenes have been reported.9–20 As a coronene analogue containing amide linkages, the synthesis17 and application to organic semiconductors18 of a D2h symmetric compound 1 have been reported. Recently, our group has synthesized 2, a new C3 symmetrical coronene analogue with three peripheral amide linkages, by the cyclic trimerization of 2-bromo-4-(isobutylamino)benzoic acid, followed by palladium-mediated intramolecular biarylation.21 The novelty of our procedure is the construction of peripheral bonds of coronene analogue first, followed by the formation of central benzene unit. However, the yield of 2 was low (7%) due to biarylation between the undesired positions. That is, as shown in Scheme 1, the intramolecular biarylation of 4 between the bromo-substituted carbon atom and the neighboring aromatic carbon atom designated by red color afforded 5a, and the repetition of the same reaction afforded 2. In contrast, the intramolecular biarylation at the carbon atom designated by blue afforded 5b, which can no longer be transformed into 2. To explore the applications of coronene amide analogues, more efficient synthetic protocol should be developed. We expected that the introduction of methyl group to the 5-position of 2-bromo-4-(isobutylamino)benzoic acid unit would suppress the undesired reaction of the palladium-mediated arylation to improve the yield of the coronene amide analogue. Herein, we report the synthesis of coronene amide analogue 3 by using 6, where three methyl groups were introduced into the undesired reaction sites (Scheme 2). In addition, the crystal structure and optical properties of 3 were also reported.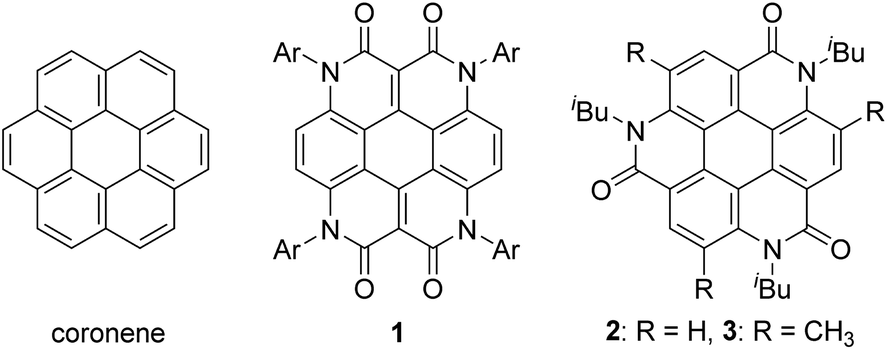 | ||
| Fig. 1 Coronene and its amide analogues 1–3. | ||
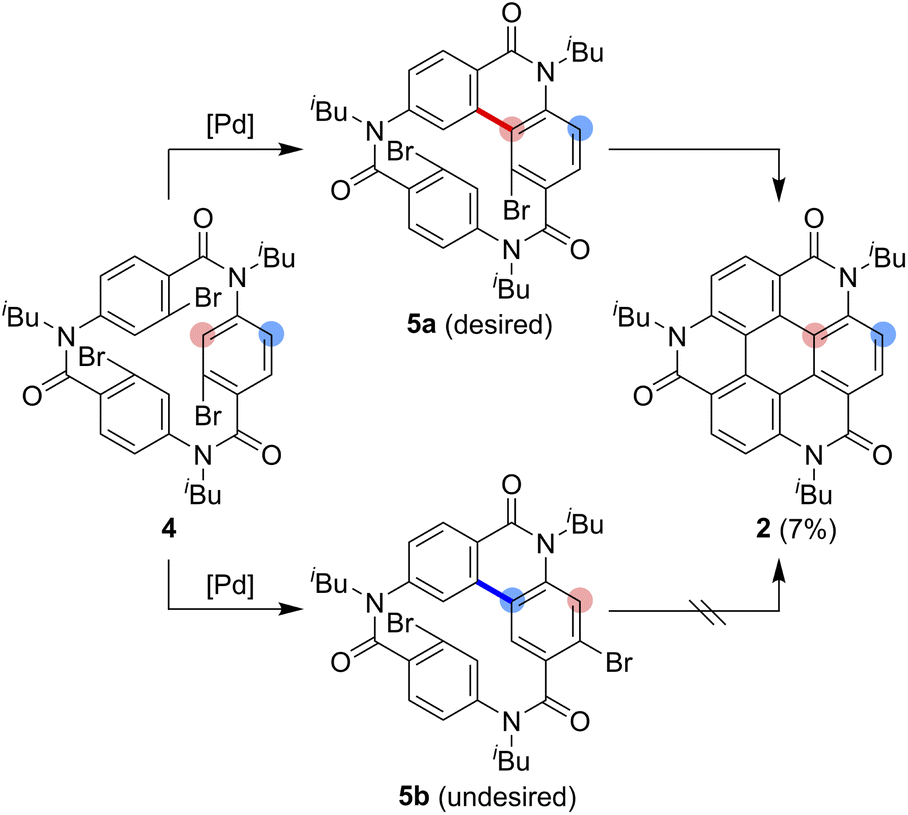 | ||
| Scheme 1 Synthesis of the coronene analogue 2. | ||
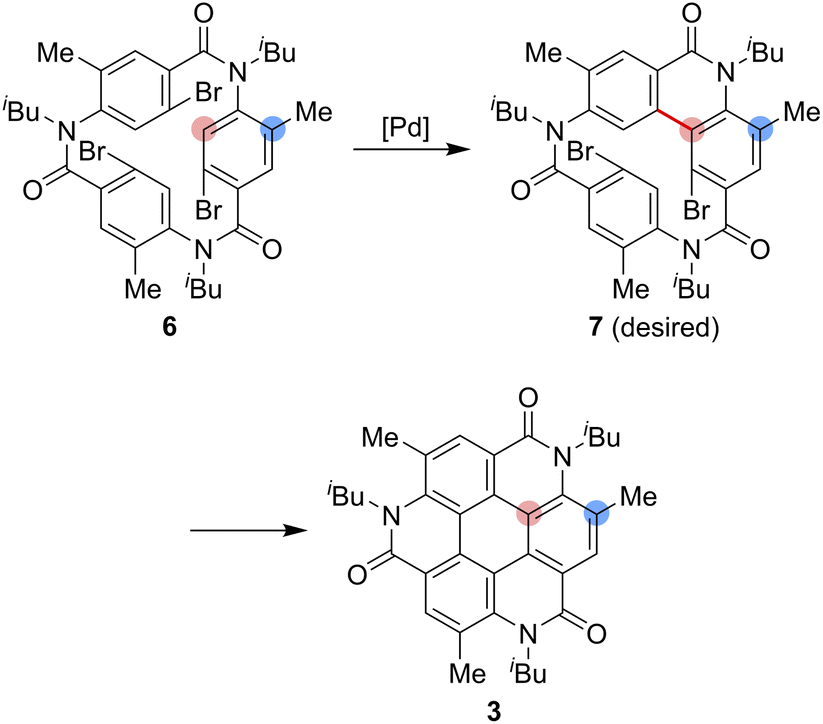 | ||
| Scheme 2 Synthesis of the coronene analogue 3. | ||
Results and discussion
Scheme 3 shows the synthesis of 6. Four-substituted benzene derivative 9 was synthesized by the regioselective bromination of commercially available methyl 3-methyl-4-nitrobenzoate (8) using palladium(II) acetate (Pd(OAc)2), N-bromosuccinimide (NBS), 1-fluoropyridinium triflate, and trifluoromethanesulfonic acid (TfOH) according to a previous study.22 Cyclic triamide 6 was expected to be synthesized from 9 by employing our previous method.21 To our delight, the introduced three methyl groups do not interfere with these reactions. That is, the reduction of the nitro group of 9 using tin chloride afforded amine 10, and the reductive amination using 10 and isobutyraldehyde afforded N-isobutyl compound 11. Carboxylic acid 12 was synthesized by the hydrolysis of 11 with potassium hydroxide. These three reactions proceeded in extremely high yields. When 12 was treated with silicon tetrachloride in pyridine at 120 °C, syn-6, in which three bromo groups are located in the same direction, and anti-6, in which one of the three bromo groups is in the opposite direction, were obtained in 51% and 21% yields, respectively.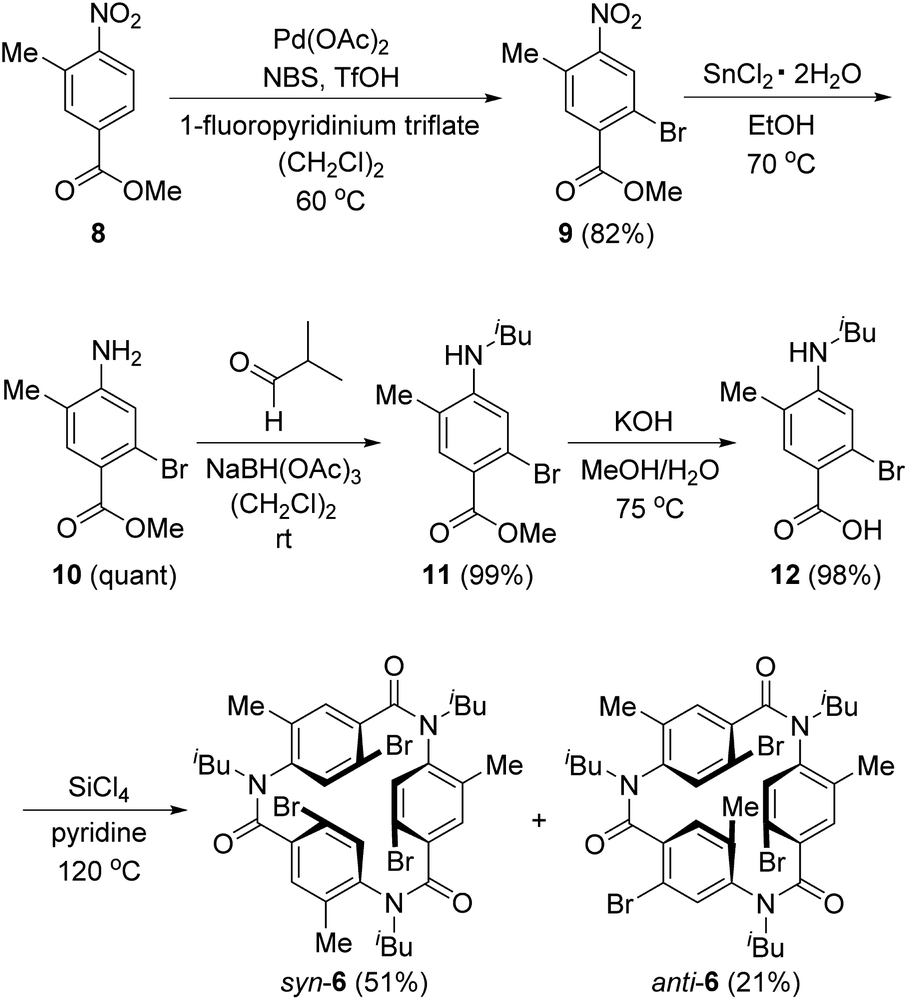 | ||
| Scheme 3 Synthesis of 6. | ||
Having completed the synthesis of biarylation precursor 6, our interest was focused on the synthesis of 3. Initially, syn-6 was used as the substrate. By employing our previously reported reaction conditions of 1 equivalent of Pd(OAc)2, 2 equivalents of triphenylphosphine (PPh3), and 20 equivalents of potassium carbonate (K2CO3) in N,N-dimethylformamide (DMF), the reaction at 100 °C for 76 h did not afford desired 3. Instead, starting material 6 was recovered in 41%, accompanied with the formation of by-product 13 in 17% yield formed by two biarylation and one debromination reactions (Table 1, entry 1). Increase in the reaction temperature to the reflux temperature of DMF led to the increase in the yield of 13 (51%) and the formation of several by-products, but 3 was not obtained (entry 2). When the amounts of Pd(OAc)2, PPh3, and K2CO3 were decreased to 0.60 equivalent, 1.2 equivalents, and 12 equivalents, respectively, 6 was recovered in 38% yield, the formation of the by-products was reduced, and trace amounts of 3 were obtained (entry 3). Use of tri(o-tolyl)phosphine (P(o-tol)3) and silver carbonate (Ag2CO3) as the ligand and base,23 respectively, did not afford 3 (entry 4), but the reaction with tri-n-butylphosphine (n-Bu3P) as the ligand24 afforded 3 in 8% yield (entry 5). Next, reactions using tri-tert-butylphosphine as the ligand were attempted.25 The reaction of syn-6 with bis(tri-tert-butylphosphine)palladium(0) (Pd(t-Bu3P)2, 0.16 equivalent) and potassium acetate (KOAc, 12 equivalents) in N,N-dimethylacetamide (DMA) at 120 °C afforded 3 and 13 in ∼47% and 52% yields, respectively (entry 6). To suppress undesired debromination that afforded 13, the reaction temperature was decreased to 100 °C. However, the yields of 3 and 13 decreased (entry 7). In the above reactions, thrice the amount of Pd(t-Bu3P)2 and KOAc compared with that used in the reported procedure was used,25 as three reaction sites were present in 6. When the reaction was performed using 4 equivalents of KOAc, which is the same amount as that reported in the literature, the yield of 3 increased to 63%; hence, these reaction conditions are selected as the optimum ones (entry 8). Next, the reaction of anti-6 was attempted. Because the reactivity of anti-6 was less than that of syn-6, the reaction was performed using 0.45 equivalents of Pd(t-Bu3P)2 and 12 equivalents of KOAc. However, the yield of 3 was 19% and 13 was obtained in 28% yield. This result indicated that the rotation of the amide–Ar bond in 6 is suppressed in comparison with that of 4,26 corresponding to the substrate used in our previously reported palladium reaction without methyl groups.21 The detailed reaction mechanism for the conversion of anti-6 to 3 is unclear at this point.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | 6 | Palladium (eq.) | Ligand (eq.) | Base (eq.) | Solvent | Temperature | Time (h) | Yield (%) | ||
| 3 | 13 | 6 | ||||||||
| a Reaction of 6 (0.06 mmol) was performed using a palladium reagent, ligand, and base in a solvent under an argon atmosphere.b Estimated yield calculated by ignoring the small amount of inseparable impurities in the product. | ||||||||||
| 1 | syn-6 | Pd(OAc)2 (1.0) | PPh3 (2.0) | K2CO3 (20) | DMF | 100 °C | 76 | 0 | 17 | 41 |
| 2 | syn-6 | Pd(OAc)2 (1.0) | PPh3 (2.0) | K2CO3 (20) | DMF | reflux | 74 | 0 | 51 | 0 |
| 3 | syn-6 | Pd(OAc)2 (0.60) | PPh3 (1.2) | K2CO3 (12) | DMF | reflux | 68 | trace | 30 | 38 |
| 4 | syn-6 | Pd(OAc)2 (0.58) | P(o-tol)3 (2.0) | Ag2CO3 (5.8) | DMF | reflux | 24 | 0 | 43 | 0 |
| 5 | syn-6 | Pd(OAc)2 (0.61) | n-Bu3P (1.9) | K2CO3 (5.9) | DMF | reflux | 25 | 8 | <25b | 34 |
| 6 | syn-6 | Pd(t-Bu3P)2 (0.16) | — | KOAc (12) | DMA | 120 °C | 26 | <47b | <52b | 0 |
| 7 | syn-6 | Pd(t-Bu3P)2 (0.16) | — | KOAc (12) | DMA | 100 °C | 25 | <39b | <34b | 29 |
| 8 | syn-6 | Pd(t-Bu3P)2 (0.16) | — | KOAc (4) | DMA | 120 °C | 24 | 63 | 37 | 0 |
| 9 | anti-6 | Pd(t-Bu3P)2 (0.45) | — | KOAc (12) | DMA | 120 °C | 24 | 19 | 28 | 0 |
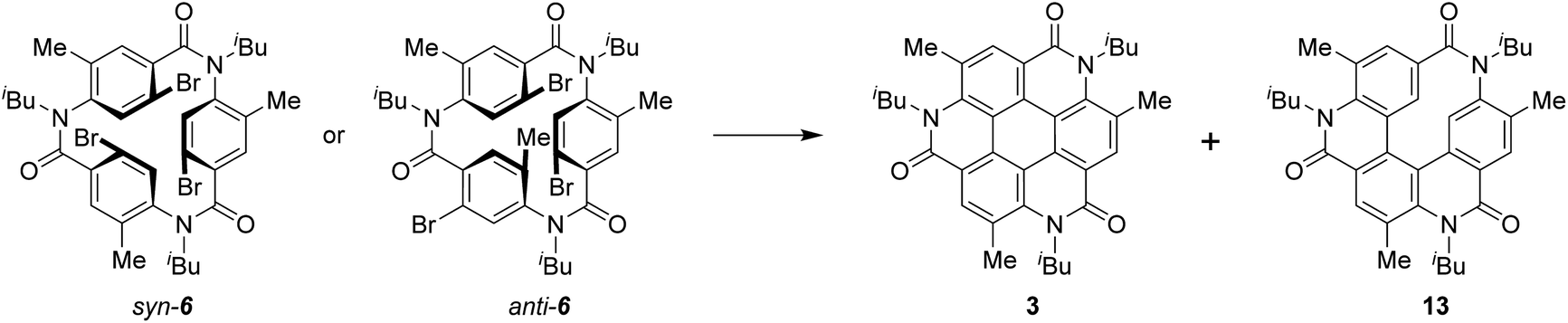
Pale brown plate-like crystals of 3 suitable for X-ray diffraction measurements were obtained by the slow evaporation of a solution of 3 in CH2Cl2/hexane at room temperature. Fig. 2a shows the crystal structure of 3. The unit-cell dimensions of 3 were a = 11.26590(10), b = 25.57580(10), c = 11.44520(10) Å, and β = 116.5650(10)° (Table S1, ESI†).27 Four molecules were present in the unit cell (Z = 4), and solvent molecules were absent in the crystal lattice. Compound 3 crystallized as a kryptoracemate in the Sohncke space group P21 with two enantiomeric molecules in the asymmetric unit (Z′= 2).28,29 Compared to the core of 3 with those of other coronene analogues,14–17,19,20 it exhibited low planarity with a maximum distance of 0.304 Å between the peripheral atom of the core and 2D plane based on the central ring (Fig. 2b). The non-planar structure might be related to the steric repulsion between the N-isobutyl group of the amide bond and methyl group of the adjacent benzene ring. In its structure, just two molecules were stacked separately by π–π stacking (d = 3.518–3.525 Å, Fig. 2c) and multiple CH⋯O bonds (dC–O = 3.177–3.553 Å) (Fig. S1, ESI†). Each of the three isobutyl groups was directed in the opposite direction, and only two molecules in 3 exhibited π–π stacking, just not as some other coronene derivatives with continuous π–π interaction in a one-dimensional columnar structure (Fig. 2e and S2, ESI†).14–16,20 Given the structure of 3, alternations of the C–C bond lengths in the central hexagon (1.411–1.441 Å) represented lower aromaticity than that of the benzene ring (Fig. 2d).
 | ||
| Fig. 2 The crystal structure of 3; (a) top view and the numbering of peripheral atoms (nitrogen and oxygen atoms are indicated in blue and red, respectively), (b) the distance between the selected atom and 2D plane based on the central ring, (c) the dimerized molecules with the perpendicular distance (Å) from the center of the central benzene ring in one molecule to the plane of the central benzene ring in the other molecule, and (d) the bond lengths. (e) Packing structure of molecules of 3 viewed down the c-axis. | ||
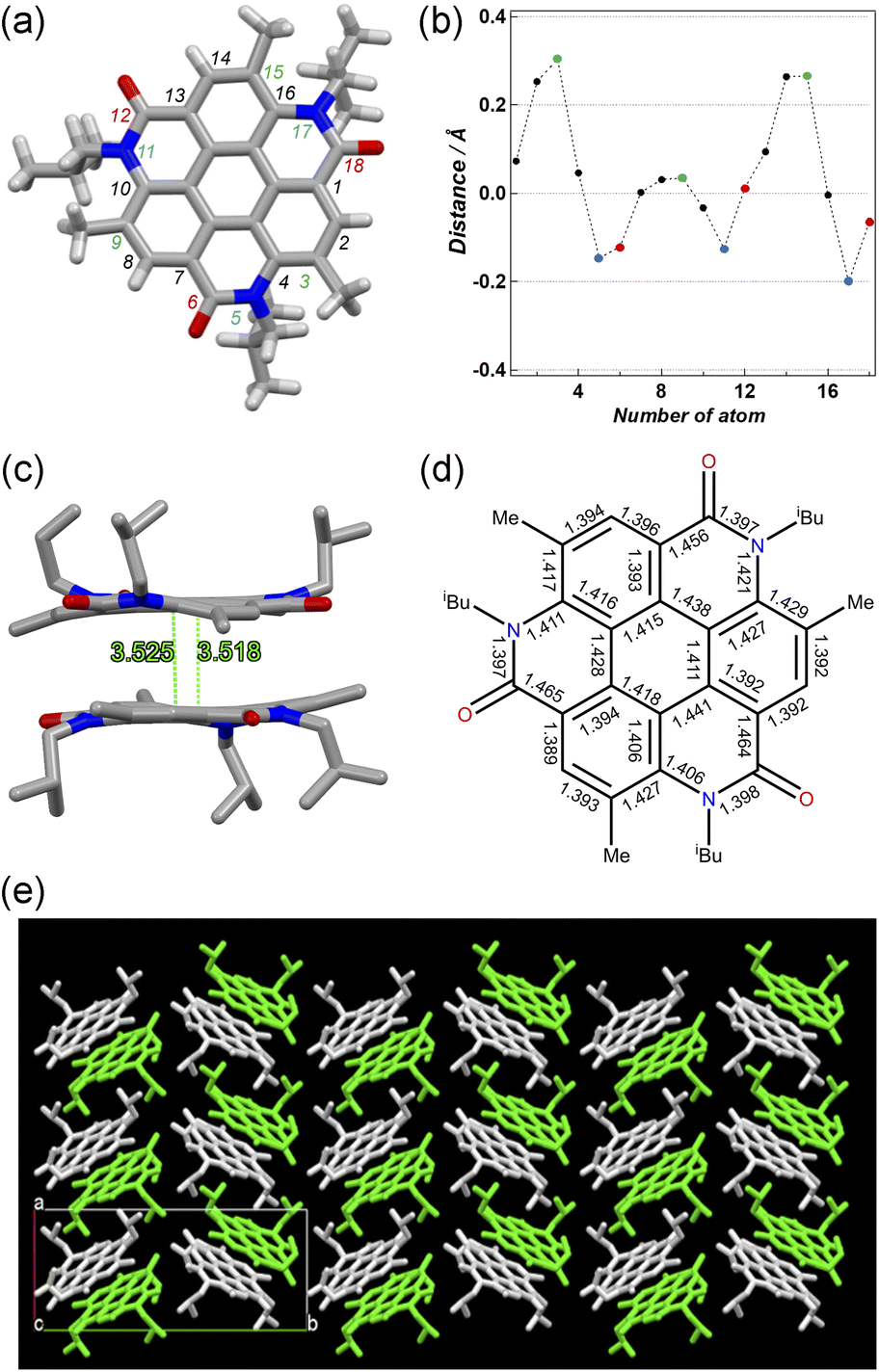
Hirshfeld surfaces and energy framework analyses were applied to visually and quantitatively evaluate the intermolecular interactions of 3 in the solid state. CrystalExplorer 17.5 was applied to generate Hirshfeld surfaces and energy framework using the cif file obtained from single crystal X-ray diffraction data of 3.30,31 The analysis was visualized by the normalized contact distance (dnorm), which was obtained using a high surface resolution with a static color scale of −0.222 Å (red) to 1.823 Å (blue). The dnorm is a symmetric function of distances to the surface between nuclei inside and outside the Hirshfeld surface (di and de, respectively),30,31 relative to their respective van der Waals (vdW) radii. The red areas show the intermolecular contacts less than their vdW radius of the atom, while the blue areas show intermolecular contacts longer than their vdW radii. White areas are the same of their vdW radii. As shown in Fig. 3a, the red areas on the Hirshfeld surface of 3 represent a close intermolecular CH⋯O distance between two coronene molecules. Because the white color represents the distance identical to vdW radii, the white area at the core unit of 3 indicates that the π–π contacts appear over the entire plane of the molecule. The molecular surface corresponding to the isobutyl groups, on the other hand, is represented by blue, indicative of a relatively far distance between them (Fig. 3b). Energy framework analysis supporting these results provides quantitative visualization of intermolecular interactions (Fig. S3, ESI),† which includes the mapping of an interaction between two molecules in the crystal based on the strength of interaction energies (IE). Energy frameworks were constructed from pairwise intermolecular interaction energy calculations using the HF/3-21G in CrystalExplorer 17.5.30,31 The calculated total interaction energy includes electrostatic, polarization, dispersion, and exchange-repulsion terms. This is represented as a cylindrical tube, the thickness of which revealed the intensity of the interactions between two molecules (Fig. S4, ESI†). It is most energetically stabilized as IE = −149.3 kJ mol−1 when the two molecules interacted closely based on the π–π and CH⋯O interactions (Fig. S3, ESI).† In particular, the dispersion energy provides a strong contribution to its stabilization (IE = −200.4 kJ mol−1). The total energy with the other molecules, on the other hand, is considerably weaker and greater than −66.5 kJ mol−1.
 | ||
| Fig. 3 Hirshfeld surface of 3 mapped with dnorm viewed from (a) the core side and (b) isobutyl group side (mapped over a fixed color scale of −0.222 (red) to 1.823 (blue) a.u.). | ||
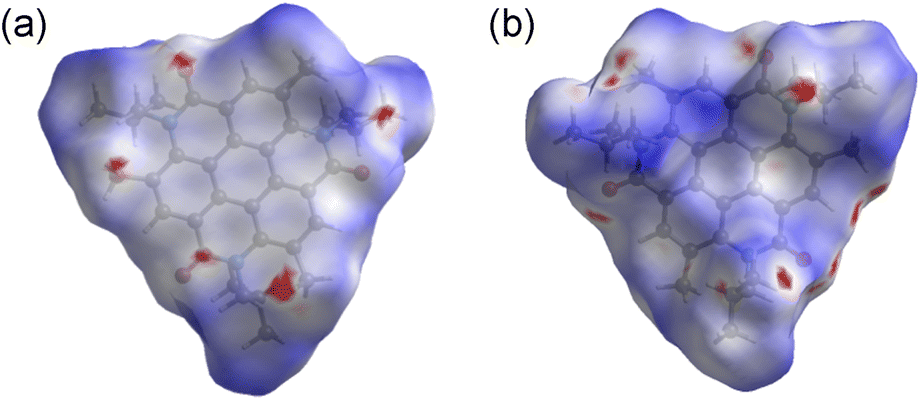
Fig. 4 shows the UV-vis absorption spectra of 2 and 3 and the emission spectrum of 3 in CHCl3 solutions. Compound 3 exhibited an absorption band at 324 nm, and a low-intensity shoulder peak at ∼390 nm in the lowest-energy region. Luminescence with emission maxima at 429, 455, and 484 nm was observed after excitation at 323 nm, corresponding to a Stokes shift of ca. 100 nm. The absorption spectrum of 2 without methyl substituents exhibited maxima at 319 and 305 nm and weak maxima at 350 and 379 nm.21 The absorption spectrum of 3 was similar to that of 2, indicating the similar electronic structure of these compounds. The UV-vis absorption spectra of 3 in tetrahydrofuran and ethyl acetate were similar to that in chloroform, and a notable solvent effect was not observed (Fig. 5). In the cyclic voltammetry (CV) shown in Fig. S5 (ESI†), 3 did not exhibit any oxidation or reduction peak in the range of −2.1 V to +1.0 V vs. Fe/Fe+, indicating that 3 has a higher energy level LUMO than the reported amide bond-containing coronene 1.17 This was probably due to the reduction of the π-conjugation induced by the low planarity of 3 or by the lack of four benzene units linked to amide nitrogen in 3 compared to 1.
 | ||
| Fig. 4 UV-vis absorption (blue line) and fluorescence spectra (red line, excitation at 323 nm) of 3 in CHCl3 (5.0 × 10−4 M), and UV-vis absorption spectrum of 2 (black line) in CHCl3 (1.3 × 10−5 M). Inset: photograph of purple luminescence of the solution of 3 in CHCl3 (5.0 × 10−4 M) under a 365 nm UV lamp. | ||
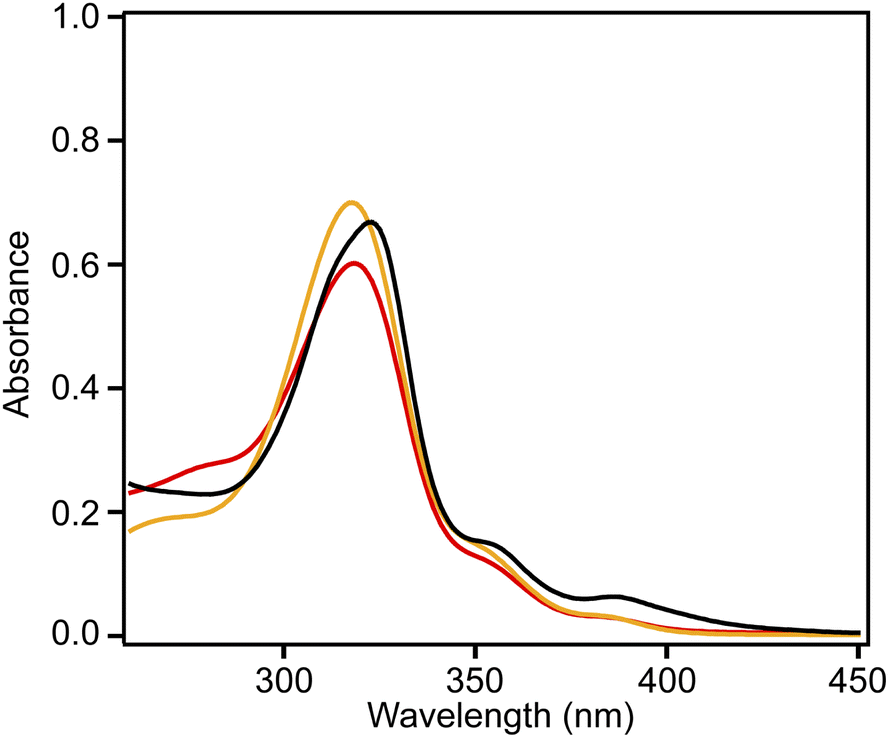 | ||
| Fig. 5 Absorption spectra of 3 in chloroform (black), tetrahydrofuran (red), and ethyl acetate (orange) (5.0 × 10−4 M). | ||
Conclusions
The coronene amide analogue 3 was efficiently synthesized and its properties were discussed. Tetrasubstituted aromatic compound 9 was prepared by the regioselective bromination of 8. Subsequent four-step transformations according to our previous study afforded syn-6 and anti-6. The palladium-mediated intramolecular biarylation of 6 proceeded with Pd(t-Bu3P)2 and KOAc in DMA to afford coronene amide analogue 3 in good yield. Single-crystal X-ray analysis revealed that the core structure of 3 is not planar, possibly derived from steric repulsions between isobutyl and methyl groups. The UV-vis absorption spectra indicated that 2 and 3 had the similar electronic structure. The sequential cyclic amide formation/intramolecular biarylation procedure could be applied to the synthesis of other amide bond-containing polycyclic aromatic compounds. Currently, the synthesis of various polycyclic aromatic compounds using this method and studies on their structure and properties are underway.Experimental section
General experimental methods
Column chromatography was performed on silica gel (neutral silica gel 60N, 63–210 μm, Kanto Chemical) with a specified solvent. 1H and 13C NMR spectra were obtained on a JEOL ECA-500 instrument. The internal standards of the 1H and 13C NMR spectra were tetramethylsilane (0.00 ppm) and the midpoint of CDCl3 (77.0 ppm) in CDCl3, respectively. IR spectra were recorded on a JASCO FT/IR-470 plus. Electrospray ionization (ESI) mass spectra were recorded on a Thermo Fisher Scientific Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer. UV-vis spectra were recorded on a JASCO V-630 spectrophotometer. Luminescence spectra were recorded on a SHIMADZU RF-5300PC spectrofluorophotometer. Cyclic voltammetry was performed on an ALS Model 600C electrochemical analyzer. X-ray data were collected on a Rigaku XtaLAB P200 diffractometer with multi-layer mirror monochromated Cu Kα radiation and a hybrid photon counting detector (PILATUS 200K) at −180 °C. The data were corrected for Lorentz and polarization effects. An empirical absorption correction based on the multiple measurement of equivalent reflections was applied using CrystalClear program. The structures were solved by direct method (SHELXT32) and refined by full-matrix least squares fitting on F2 (SHELXL Version 2018/3 (ref. 33,34)) using all data. All non-hydrogen atoms were refined anisotropically. The aromatic protons of 3 were located from difference syntheses and ride on the parent atoms. The other hydrogen atoms were located on the calculated positions. All calculations were performed using CrystalStructure crystallographic software package except for refinement, which was performed using SHELXL. The results are shown in Table S1 (ESI).† The absolute configuration could not be decided from the Flack parameters.35,36 Crystallographic data have been deposited with the Cambridge Crystallographic Data Center as supplementary publication number CCDC 2130884. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via http://www.ccdc.cam.ac.uk/structures/.syn-6: 1H NMR (500 MHz, CDCl3) δ 7.64 (s, 3H), 6.85 (s, 3H), 4.18 (dd, J = 9.7 and 13.2 Hz, 3H), 2.77 (dd, J = 4.9 and 13.5 Hz, 3H), 2.12 (s, 9H), 1.87–1.79 (m, 3H), 1.14 (d, J = 6.6 Hz, 9H), 0.93 (d, J = 6.6 Hz, 9H); 13C NMR (126 MHz, CDCl3) δ 167.9, 141.1, 138.8, 134.3, 132.9, 127.8, 115.9, 54.7, 26.9, 20.6, 20.4, 17.1; HRMS (ESI+) calculated for [C36H4279Br81Br2N3O3Na+] 826.0649 [M + Na]+, found 826.0652; IR (KBr) 3854, 3735, 3469, 2958, 1657, 1599, 1558, 1487, 1402, 1336, 1297, 1162, 1126, 892 cm−1; mp 313.0–314.8 °C.
anti-6: 1H NMR (500 MHz, CDCl3) δ 7.67 (s, 1H), 7.19 (s, 1H), 7.16 (s, 1H), 6.98 (s, 1H), 6.89 (s, 1H), 6.68 (s, 1H), 4.39 (dd, J = 9.5 and 13.2 Hz, 1H), 4.05 (dd, J = 8.7 and 13.3 Hz, 1H), 3.68 (dd, J = 7.9 and 13.3 Hz, 1H), 3.45 (dd, J = 6.9 and 13.5 Hz, 1H), 3.08 (dd, J = 6.0 and 13.2 Hz, 1H), 2.79 (dd, J = 5.3 and 13.3 Hz, 1H), 2.36 (s, 3H), 2.26 (s, 3H), 1.98 (s, 3H), 1.95–1.79 (m, 3H), 1.07–1.04 (m, 6H), 1.02 (d, J = 6.6 Hz, 3H), 1.00–0.98 (m, 6H), 0.93 (d, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 168.2, 168.0, 167.8, 141.4, 140.9, 140.5, 138.3, 137.0, 136.13, 136.11, 135.7, 135.6, 135.2, 134.3, 133.5, 131.9, 131.6, 131.0, 115.7, 114.7, 114.1, 56.3, 55.4, 54.3, 27.3, 27.1, 26.9, 20.62, 20.57, 20.5, 20.4, 20.3, 20.2, 18.6, 17.9, 17.6; HRMS (ESI+) calculated for [C36H4279Br81Br2N3O3Na+] 826.0649 [M + Na]+, found 826.0657; IR (KBr) 3735, 2959, 1655, 1597, 1558, 1487, 1398, 1332, 1295, 1164, 1125, 1035, 893, 736, 643 cm−1; mp 191.6–193.9 °C.
Compound 3: 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 3H), 4.77 (br s, 6H), 2.99 (s, 9H), 1.87 (m, 3H), 0.69 (m, 18H); 13C NMR (126 MHz, CDCl3) δ 163.6, 139.7, 133.4, 125.5, 124.0, 117.2, 112.6, 51.9, 27.7, 24.9, 19.4; HRMS (ESI+) calculated for [C36H39N3O3Na+] 584.2884 [M + Na]+, found 584.2887; IR (KBr) 3854, 3839, 3735, 3676, 3649, 3567, 1716, 1698, 1647, 1617, 1558, 1541, 1508, 1457, 419 cm−1; mp 358.2–360.4 °C.
Compound 13: 1H NMR (500 MHz, CDCl3) δ 8.44 (d, J = 0.6 Hz, 1H), 7.79 (d, J = 0.6 Hz, 1H), 6.17 (d, J = 8.9 Hz, 1H), 5.99 (dd, J = 1.4 and 8.8 Hz, 1H), 5.71 (t, J = 1.4 Hz, 1H), 4.32–4.28 (m, 1H), 3.98–3.87 (m, 3H), 3.75 (dd, J = 9.6 and 13.9 Hz, 1H), 2.92 (dd, J = 6.2 and 13.9 Hz, 1H), 2.69 (s, 3H), 2.53 (s, 3H), 2.30–2.23 (m, 1H), 2.14–2.04 (m, 1H), 1.84–1.79 (m, 1H), 1.48 (d, J = 1.4 Hz, 3H), 1.07–1.00 (m, 12H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 178.6, 168.6, 163.9, 146.1, 144.9, 144.3, 136.3, 134.8, 132.6, 130.2, 129.1, 127.2, 126.7, 126.6, 124.9, 123.4, 121.2, 120.9, 117.7, 68.4, 57.5, 52.0, 49.2, 48.6, 29.4, 28.4, 26.6, 22.9, 20.8, 20.3, 19.71, 19.69, 19.62, 15.4; HRMS (ESI+) calculated for [C36H41N3O3Na+] 586.3041 [M + Na]+, found 586.3047; IR (KBr) 3735, 3446, 2959, 1702, 1653, 1547, 1457, 1383, 1156, 1047, 913, 806, 748, 680, 575, 417 cm−1; mp 124.2–126.5 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant number JP18K05220 and by a grant from Seikei University.Notes and references
- J. M. Robertson and J. G. White, J. Chem. Soc., 1945, 607–617 RSC.
- I. Destoop, E. Ghijsens, K. Katayama, K. Tahara, K. S. Mali, Y. Tobe and S. D. Feyter, J. Am. Chem. Soc., 2012, 134, 19568–19571 CrossRef CAS PubMed.
- C. Kulkarni, P. A. Korevaar, K. K. Bejagam, A. R. A. Palmans, E. W. Meijer and S. J. George, J. Am. Chem. Soc., 2017, 139, 13867–13875 CrossRef CAS PubMed.
- Y. Xiao, J. Tao, X. Peng, Y. Song, P. Lei, H. Xu, X. Xiao, B. Tu and Q. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 17129–17138 CrossRef CAS PubMed.
- I. Diez-Perez, Z. Li, J. Hihath, J. Li, C. Zhang, X. Yang, L. Zang, Y. Dai, X. Feng, K. Müellen and N. Tao, Nat. Commun., 2010, 1, 31–35 CrossRef PubMed.
- X. Wan, K. Chen, D. Liu, J. Chen, Q. Miao and J. Xu, Chem. Mat., 2012, 24, 3906–3915 CrossRef CAS.
- H.-D. Wu, F.-X. Wang, M. Zhang and G.-B. Pan, Nanoscale, 2015, 7, 12839–12842 RSC.
- J. Zhang, G. Liu, Y. Zhou, G. Long, P. Gu and Q. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 1183–1188 CrossRef CAS PubMed.
- S. Tokita, K. Hiruta, K. Kitahara and H. Nishi, Bull. Chem. Soc. Jpn., 1982, 55, 3933–3934 CrossRef CAS.
- S. Tokita, K. Hiruta, K. Kitahara and H. Nishi, Synthesis, 1982, 229–231 CrossRef CAS.
- J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209–8213 CrossRef CAS.
- L. Hao, W. Jiang and Z. Wang, Tetrahedron, 2012, 68, 9234–9239 CrossRef CAS.
- Y. Zhang, Z. Zhao, X. Huang, Y. Xie, C. Liu, J. Li, X. Guan, K. Zhang, C. Cheng and Y. Xiao, RSC Adv., 2012, 2, 12644–12647 RSC.
- G. Li, W.-W. Xiong, P.-Y. Gu, J. Cao, J. Zhu, R. Ganguly, Y. Li, A. C. Grimsdale and Q. Zhang, Org. Lett., 2015, 17, 560–563 CrossRef CAS PubMed.
- X.-Y. Wang, F.-D. Zhuang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, Chem. Commun., 2015, 51, 4368–4371 RSC.
- Q. Tan, H. Chen, H. Xia, B. Liu and B. Xu, Chem. Commun., 2016, 52, 537–540 RSC.
- R. Duan, D. Schollmeyer, K. Müllen and C. Li, J. Mater. Chem. C, 2018, 6, 1334–1337 RSC.
- J. De, I. Bala, S. P. Gupta, U. K. Pandey and S. K. Pal, J. Am. Chem. Soc., 2019, 141, 18799–18805 CrossRef CAS PubMed.
- Y.-X. Sun, X.-G. Wang, G.-D. Shen, T. Yang, Y.-H. Yang, J. Li, M.-Y. Yang, H.-M. Sun and J.-F. Wei, Adv. Synth. Catal., 2020, 362, 1651–1656 CrossRef CAS.
- Y. Sun, T. Yang, C. Chen, B. Yang, Y. Yang, J. Li, H. Sun and J. Wei, Org. Lett., 2021, 23, 5616–5620 CrossRef CAS.
- A. Yokoyama, A. Ishii, T. Ohishi, S. Kikkawa and I. Azumaya, Tetrahedron Lett., 2021, 62, 152704 CrossRef CAS.
- X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440–4444 CrossRef CAS PubMed.
- T. Harayama, T. Akiyama, H. Akamatsu, K. Kawano, H. Abe and Y. Takeuchi, Synthesis, 2001, 444–450 CrossRef CAS.
- T. Harayama, T. Sato, Y. Nakano, H. Abe and Y. Takeuchi, Heterocycles, 2003, 59, 293–301 CrossRef CAS.
- Q. Hu, T. Gao, Y. Shi, Q. Lei and L. Yu, RSC Adv., 2018, 8, 13879–13890 RSC.
- R. Yamakado, S. Matsuoka, M. Suzuki, D. Takeuchi, H. Masu, I. Azumaya and K. Takagi, Chem. Commun., 2015, 51, 5710–5713 RSC.
- The numbers in parentheses represent standard deviations..
- T. Rekis, Acta Crystallogr., 2020, B76, 307–315 Search PubMed.
- Y.-Y. Zhang, J.-T. Yu, B. Li, D.-J. Li, Z.-G. Gu, X.-F. Sun, H.-L. Cai, G. E. Kostakis and G. Peng, CrystEngComm, 2018, 20, 4582–4589 RSC.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2014, A70, C1437 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef.
- H. D. Flack and G. Bernardinelli, Acta Crystallogr., 1999, A55, 908–915 CrossRef CAS.
- H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2130884. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04035b |
| This journal is © The Royal Society of Chemistry 2022 |