
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe solvent-free mechano-chemical grinding of a bifunctional P25–graphene oxide adsorbent–photocatalyst and its configuration as porous beads†
Fatima-Ezzahra Zirarab,
Nadia Katira,
Samir Qourzalb,
Ihya Ait Ichoub and
Abdelkrim El Kadib *a
*a
aEuromed Research Center, Engineering Division, Euro-Med University of Fes (UEMF), Route de Meknes, Rond-Point de Bensouda, 30070, Fès, Morocco. E-mail: a.elkadib@ueuromed.org
bMaterials, Photocatalysis and Environment Team, Department of Chemistry, Faculty of Sciences, Ibn Zohr University, B. P. 8106, Dakhla City, Agadir, Morocco
First published on 1st August 2022
Abstract
Owing to their use in water-cleaning technology, titanium-dioxide-based nanomaterials have dominated the photocatalysis scene, with so-called Degussa (P25) being the most promising under UV light. However, this is not the case under visible light, where it is necessary to combine titanium dioxide with other photosensitising nanomaterials. Unfortunately, most of the strategies aimed in this direction are chemically non-facile, energy-intensive, economically expensive, and not suitable for large-scale production. We herein describe a straightforward solvent-free approach for accessing visible-light-activated titanium-dioxide-based photocatalysts via the mechanochemical grinding of Degussa P25 with a second solid partner. Upon comparing several solid-material benchmarks, P25–graphene oxide is the best combination. The resulting material showed efficient performance for the adsorption and photodegradation of different dye pollutants, namely methylene blue, malachite green, Congo red, and methyl orange. The recorded performance was nearly comparable to that reached using sol–gel materials, with the ultimate advantage of being more sustainable and industrially scalable. The recyclability can be improved through a porous-bead configuration using biomass waste chitosan hydrogel, an approach that can further fulfill the requirement for more sustainable photocatalyst designs.
Introduction
First discovered in 1972, titanium dioxide is the most famous photocatalyst to date,1 and it is expected to be a key player in sewage treatment and water-cleaning technology worldwide.2 While many titanium dioxide ceramics are industrially available, with so-called Degussa P25 being the most ubiquitous,3 they still have many limitations, including low surface areas, poor activities under visible light, and moderate photo-stability and electron–hole recombination properties.4 With the emergence of nanostructured hybrid materials, efforts have focused on exploring wet chemistry to closely combine titanium dioxide with other nanostructures, starting from soluble precursors.5 Substantial knowledge has been gleaned from these approaches, enabling researchers to circumvent most of the bottlenecks associated with titanium dioxide via increasing the specific surface area, improving the framework crystallinity, and expanding the working region to the most suitable visible-light region instead of the constraining UV region.6 Although there is a broad range of reported titanium-dioxide-based photocatalysts,7 most of them are prepared through non-facile and energy-intensive procedures, consequently generating little allure for large-scale production.8A popular current trend consists of the development of bifunctional adsorbent–photocatalyst nanocomposites via merging two or more components.8,9 Illustrative examples include the association of high-surface-area materials and inorganic semiconductors,10 and the creation of heterojunctions via closely combining two different photoactive materials, resulting in tuneable visible-light photoactivity.11 In this context, conductive graphitic carbon materials have shown efficiency as photosensitizing partners,12 with many precedents in the literature demonstrating the combination of titanium dioxide (including P25) with both graphene oxide (GO) and carbon nanotubes (CNTs).13
Given the prominence of the target applications, with specific consideration given to the urgent issues of sustainable energy generation and water management,14 it is highly recommended to set up straightforward, scalable, and cost-effective methods to transform already available ceramics into highly active visible-light photocatalysts.12b,15
We herein report that the minute grinding of biphasic anatase–rutile TiO2 (P25) in the presence of graphene oxide (GO) affords an active visible-light photocatalyst for dye degradation in water. Exhaustive screening reveals that TiO2@GO is the most attractive combination, outperforming a set of other attractive nanomaterials, including graphite, carbon nanotubes, active carbon, boron nitride, layered montmorillonite, and tubular halloysite. The resulting powdered material could also be configured using seafood waste chitosan, which is known to provide hydrogels. Our results bring evidence for the suitability of this configuration approach to further improve the recyclability of the as-prepared photocatalyst, which opens up more possibilities for tackling the thorny issue of the long-term use of catalysts under continuous-flow conditions.
Results and discussion
We first screened a large variety of mechanochemically ground powder samples based on the combination of titanium dioxide with carbon materials, clay, and boron nitride. In the carbon-based series, we used graphite (G), graphene oxide (GO), phosphorylated graphene (PGO), carbon nanotubes (CNTs), carbon nitride (C3N4), and activated carbon (AC) as grindable partners for Degussa P25. We also selected both montmorillonite (MMT) and halloysite (HNT) as representative clay materials, owing to their interesting lamellar and tubular topologies, respectively.16 We moreover assessed the mechanochemical mixing of titanium dioxide with lamellar boron nitride owing to the promising properties of the latter.17 For these combinations, the same ratio (45 wt% TiO2 and 55 wt% second phase) was used. The materials were denoted as TiO2@X-45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55, with X referring to the second phase, while 45 and 55 indicate the weight percentages of TiO2 and X, respectively.
:55, with X referring to the second phase, while 45 and 55 indicate the weight percentages of TiO2 and X, respectively.
After grinding and further homogenisation, the resulting solids were used for the photodegradation of methylene blue as a representative textile dye pollutant. The photocatalytic results presented in Fig. 1 show the methylene blue degradation kinetics and the overall performance, combining both adsorption and photodegradation.
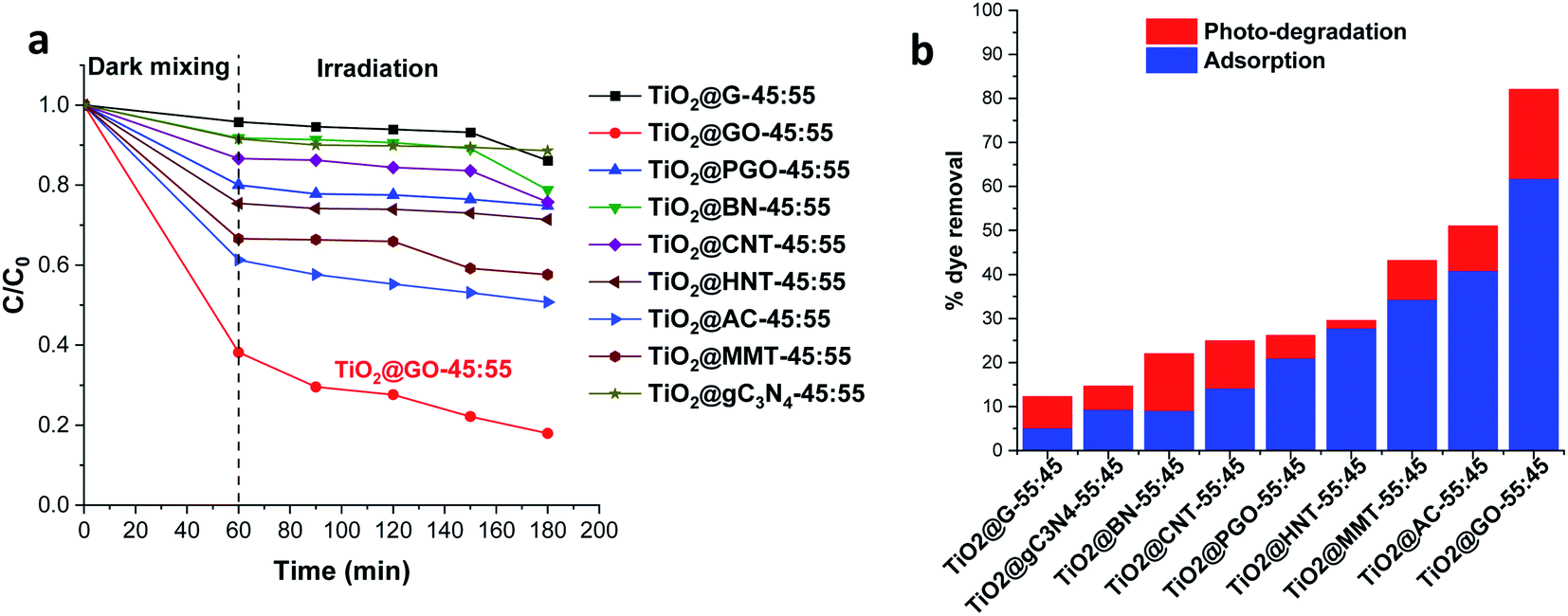 | ||
| Fig. 1 Methylene blue degradation using a set of hybrid materials built from the mechanochemical grinding of P25 Degussa with a second phase: (a) the degradation kinetics and (b) the amount of removed dye (%), combining adsorption and photocatalysis. | ||
Obviously, the most active combination is TiO2@GO-45:55, allowing 82% of the dye to be removed. The performance is very high; the second most active material, TiO2@AC-45:55, did not exceed 51%. TiO2@G-45:55 displayed poor photoactivity, enabling the degradation of only 12% of the dye. Although clay-based materials are well-known adsorbents, their performance levels (43% for TiO2@MMT-45:55 and 29% for tubular TiO2@HNT-45:55) remain far from that reached using TiO2@GO-45:55. Interestingly, TiO2@GO-45:55 works efficiently as both an adsorbent and photocatalyst, suggesting that both titanium dioxide and graphene oxide provide activity, probably in a synergistic manner. The high adsorption of methylene blue by graphene oxide has been reported in the literature, and it has been attributed to favourable π–π stacking interactions.18 Adsorption on MMT commonly occurs through cation exchange with sodium located inside of the material galleries;19 in the case of HNT, hosting occurs via the diffusion of the pollutant inside of the lumen.20 The poor performance recorded with graphite was surprising given the similarities of its molecular structure to that of graphene oxide. However, in graphite, the sheets are stacked in a layered fashion, making the diffusion of pollutants difficult. In turn, as will be discussed below, graphene oxide seems to be already exposed through sheet exfoliation induced upon mechanical grinding with titanium dioxide particles.21
Having demonstrated the superiority of TiO2@GO with respect to its competitors, we next investigated the effects of the weight ratio (TiO2@GO) on the final performance. For this purpose, we used three different materials, namely TiO2@GO-45:55, TiO2@GO-05:95, and TiO2@GO-95:05. These materials have different compositions; one is enriched with graphene oxide phase, one is enriched with titanium dioxide, and one has a good balance between the two phases.
The three materials were next characterized using X-ray diffraction (XRD), Raman spectroscopy, nitrogen sorption, and scanning electron microscopy (SEM) analyses (Fig. 2 and 3). The XRD patterns of these materials display the fingerprint of Degussa with no significant shifts or variations upon mixing with GO. Both anatase and rutile peaks are identified. In addition, a single peak assignable to the 002 plane of graphene oxide at 2θ = 10° could be observed for TiO2@GO-95:05, while it was barely visible for TiO2@GO-45:55 and invisible for TiO2@GO-95:05. The decrease and disappearance of this signal could be attributed to the exfoliation of sheets during grinding, which becomes more significant upon increasing the amount of titanium dioxide.
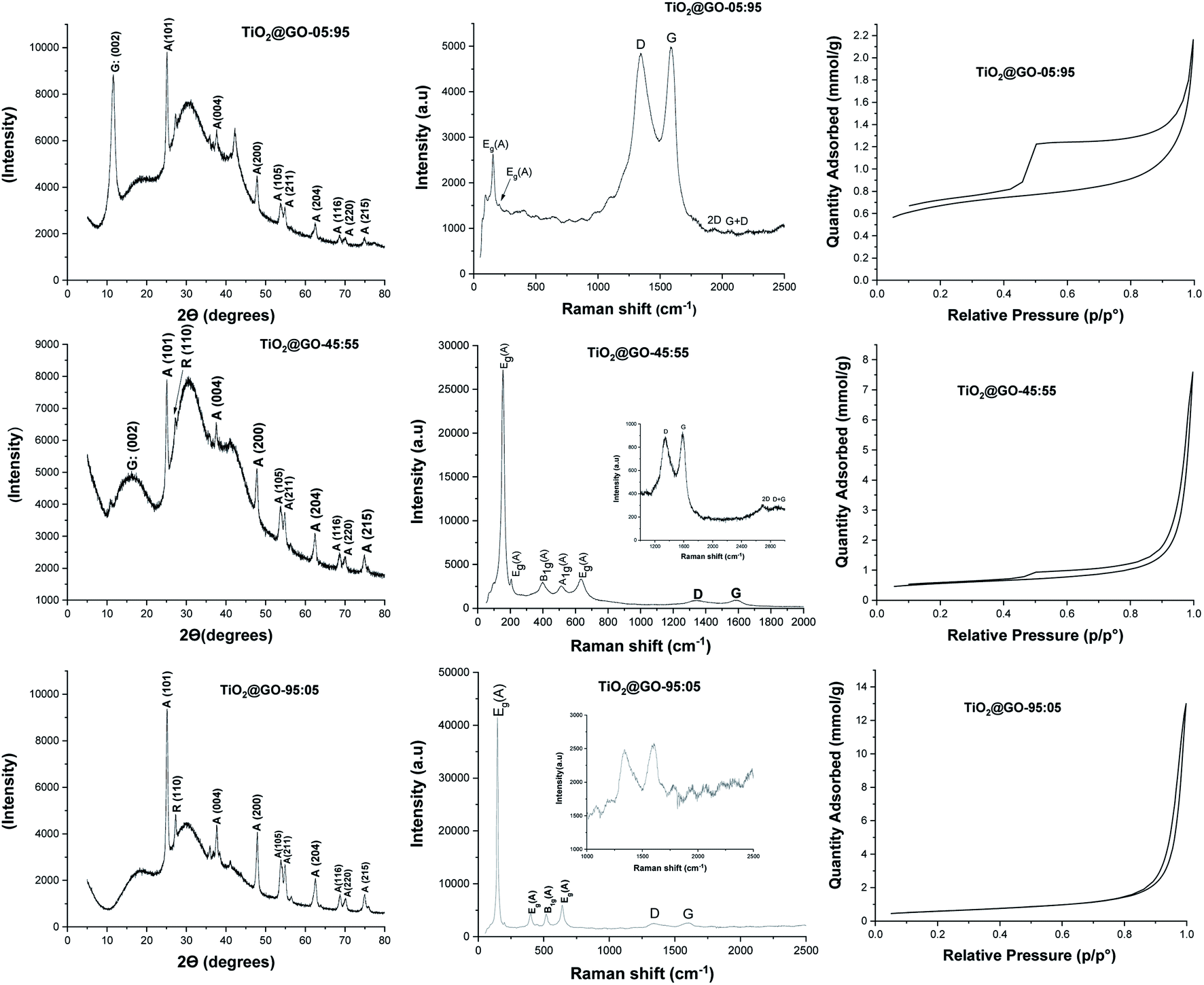 | ||
| Fig. 2 (Left column) XRD spectra, (middle column) Raman spectra, and (right column) nitrogen adsorption–desorption isotherms of the mechanically ground materials: top, TiO2@GO-05:95; middle, TiO2@GO-45:55; bottom, TiO2@GO-95:05. | ||
 | ||
| Fig. 3 Scanning electron microscopy (SEM) images of the prepared materials: from left to right, TiO2@GO-05:95; TiO2@GO-45:55; and TiO2@GO-95:05. | ||
Raman spectroscopy reveals the typical pattern of the graphitic phase, with the two characteristic D and G bands in the ranges of ∼1341 to 1349 cm−1 and ∼1588 to 1604 cm−1, respectively.22 The ID/IG ratio was found to be 0.96 for the material with the lowest amount of graphene oxide, 0.96 for the medium-level material, and 0.97 for the material enriched with graphene oxide. The intensities of the carbon-zone peaks versus the titanium-dioxide-zone peaks vary consistently with the GO:TiO2 ratio; two intense carbon signals are observed for TiO2@GO-05:95 while more crystalline peaks of titanium dioxide are observed for TiO2@GO-95:05 and TiO2@GO-45:55. Nitrogen sorption analysis shows nearly the same specific surface area for all materials (47 m2 g−1, 41 m2 g−1, and 48 m2 g−1 for TiO2@GO-95:05, TiO2@GO-45:55, and TiO2@GO-05:95, respectively). Although no significant variations in specific surface area could be observed, the isotherm profiles are quite different for the three materials, mainly in the case of TiO2@GO-05:95, which shows the pronounced development of hysteresis and an increase in total microporosity. This situation could be triggered by the dominance of the graphitic network and the presence of tactoids and small voids at the interface of carbon and the ceramic phase. In turn, the TiO2@GO-45:55 network seems to be made of large mesopores that extend to the macroporous domain as a consequence of sheet exfoliation due to titanium dioxide. Porosity seems to be brought about by titanium dioxide particles and the internal voids created by their entanglement with graphitic sheets.
SEM analyses allow for the visualisation of the networks on the microscale (Fig. 3 and S1†). Nicely, the morphology of the network depends on the predominant phase. For TiO2@GO-05:95, the typical network of graphene oxide is observed, with a few spherical P25 particles embedded. In TiO2@GO-45:55, agglomerated particles of titanium dioxide appear along with the flat layers of graphene oxide. In TiO2@GO-95:05, graphenic sheets could hardly be detected in a network that was dominated by the presence of aggregated titanium dioxide particles (Fig. 3).
It is consequently reasonable to attribute the behaviour of each material to the dominant phase. We next investigated the photocatalytic activities of the three materials toward different representative dye contaminants, including methylene blue, green malachite, Congo red, and methyl orange. The results are gathered in Fig. 4 and the kinetics are shown in the ESI (Fig. S2†). The order of performance seems to be dependent on the loading of graphene oxide. The highest photooxidation activity was observed at high graphene loading (TiO2@GO-05:95), followed by TiO2@GO-45:55 and then TiO2@GO-95:05.
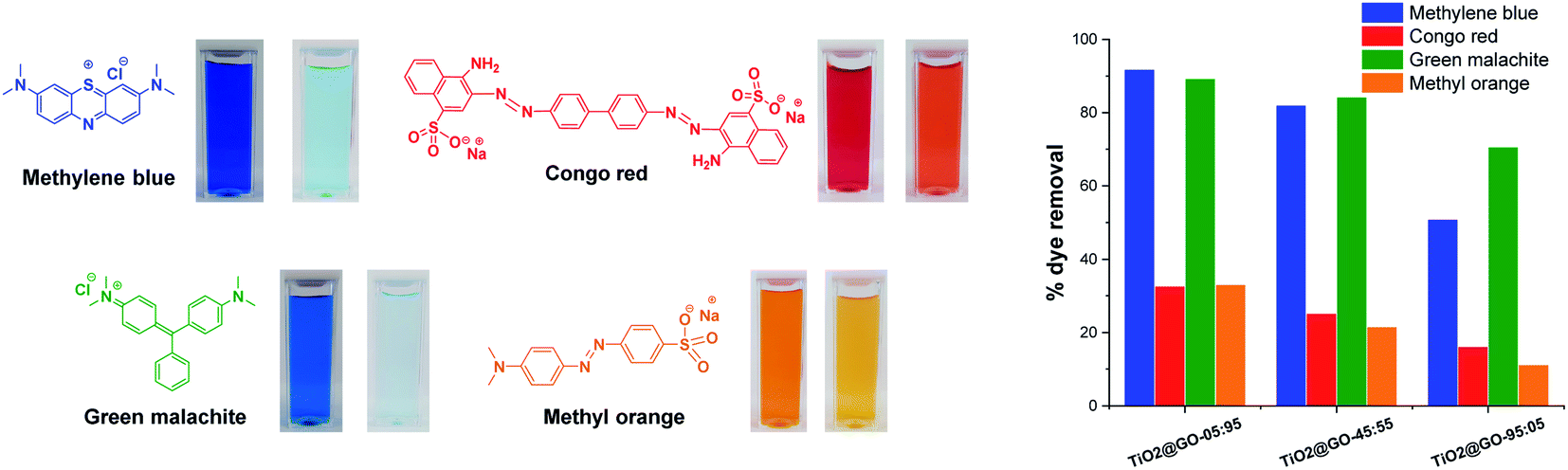 | ||
| Fig. 4 A schematic representation of the four dye contaminants investigated in this work with photos showing the colors before and after photo-treatment. The panel on the right illustrates the removal efficiencies recorded for the three materials. | ||
This trend is expected for photocatalysis under visible light, as P25 Degussa do not display any appreciable activity.23 In turn, graphene oxide brings additional adsorption sites18 and also acts as a photosensitizer for titanium dioxide.24 Within the dye series, the degradation of methylene blue and malachite green seems to be more quantitative (∼90% degradation reached with TiO2@GO-05:95). In contrast, methyl orange and Congo red are more difficult to degrade during the advanced oxidation process, as illustrated by the moderate amount of removed dye, which remains at less than 32%. Overall, the utility of mixing titanium dioxide with graphene is evident when comparing the overall performances of the hybrids with that of native Degussa P25 titanium dioxide (Fig. S3†).
Attempts to further improve the experimental protocol do not boost the catalytic performance. For instance, subjecting the mixture to sonication and wetting with ethanol (TiO2@GO_us, us refers to ultrasonication) failed to further improve the performance. Refluxing in boiling ethanol also did not improve the performance (TiO2@GO_reflux) (Fig. 5a). This indicates that close contact was already obtained during the solvent-free mechanico-chemical grinding of the two phases, with the possible exfoliation of GO sheets suspected to be the driving force behind such tight interplay.21
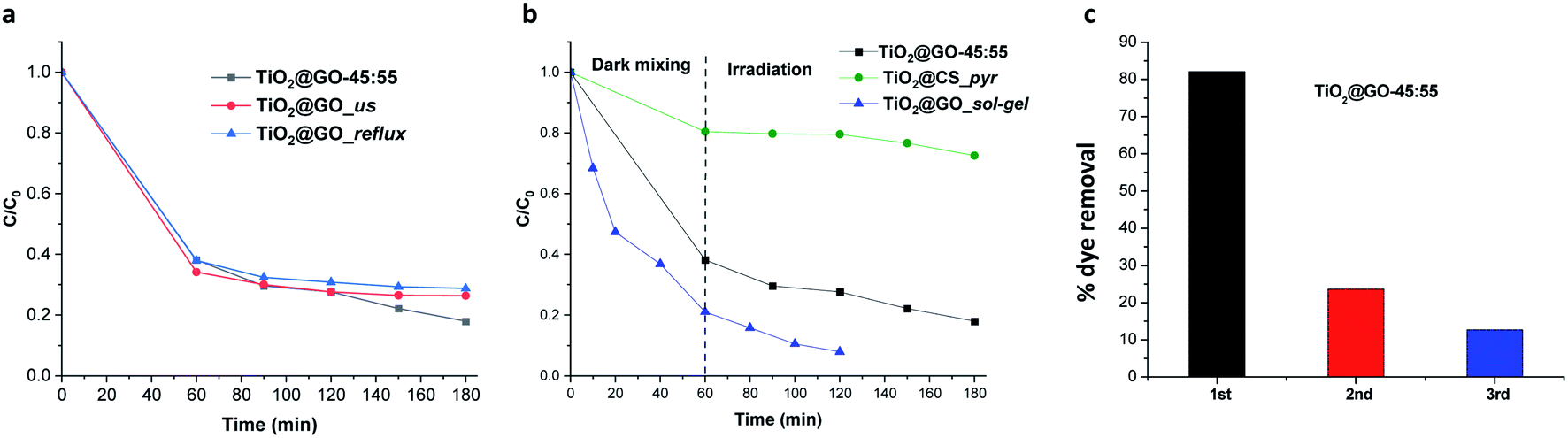 | ||
| Fig. 5 (a) The photocatalytic activity of TiO2@GO depending on the experimental procedure. (b) The photocatalytic activity of the ground material versus sol–gel and carbonized analogues. In these materials, the accessibility to titanium dioxide particles diverges substantially, making a comparison useful for probing the effects of photocatalyst exposure. (c) Recycling experiments with the ground powder, showing the fast deactivation. | ||
The accessibility of the titanium dioxide phase versus the graphene phase should also be taken into account during photooxidation. We have consequently compared the mechanically ground material to TiO2@GO_sol–gel and TiO2@CS_pyr. The first material was prepared through the post-grafting of titanium alkoxide onto graphene oxide22 followed by thermal annealing treatment to generate crystalline titanium dioxide grown on the external surface of graphene.23 The second photocatalyst was prepared via mixing chitosan and titanium alkoxide in a sol–gel process followed by carbonization under nitrogen to generate well-entangled titanium dioxide inside of the carbonaceous graphene network (see the Experimental section for details and Fig. S4† for more characterization data). In this case, chitosan serves as the carbon source25 and as a structure-directing agent for titanium alkoxide mineralization.26
Comparing the photoactivities of the three materials shows that TiO2@GO-45:55 displays interesting photoactivity, quite comparable to TiO2@GO_sol–gel. In turn, very moderate activity was revealed for TiO2@CS_pyr, mainly during irradiation. This poor photoactivity can be correlated with the restricted exposure of titanium dioxide particles that are sequestered inside of the generated carbon network. The poor graphitisation of the network leaves abundant amorphous regions that can further worsen the photoactivity of the resulting material, as a temperature above 900 °C under argon is often claimed to be necessary to generate a uniform graphitised carbon network from chitosan polymer.27 The slight increase in the activity of TiO2@GO_sol–gel could be attributed to (i) more exposed titanium dioxide particles, (ii) favourable interfacial contact between the ceramic and the carbon phase through covalent bonding, and (iii) improved graphene quality due to the removal of oxygenated groups on the surface during thermal annealing treatment. The outstanding adsorptive capacity of TiO2@GO_sol–gel with respect to TiO2@GO-45:55 can be explained based on differences in their specific surface areas, as estimated by nitrogen sorption; the former has a value of 186 m2 g−1, while a value of only 41 m2 g−1 was recorded for the mechanically ground material. Although interesting, it is worth mentioning that TiO2@GO_sol–gel was prepared through a multistep procedure22 involving expensive precursors and calcination at high temperature,23 while a very trivial mechanical grinding procedure was applied in the case of TiO2@GO-45:55 (Fig. 5b).
Recycling experiments show the main limitations of the ground material, as fast deactivation was noticed during the second cycle, and the photoactivity almost completely vanished after further use (Fig. 5c).
We have decided to assess the photostability of these materials under similar reaction conditions. Structural analysis confirms that substantial alteration of the chemical structure occurs after irradiation. Infrared spectroscopy reveals significant differences in the patterns recorded for the fresh material compared to a sample irradiated for 60 min and 120 min (Fig. S5†). Raman spectroscopy shows that the graphene phase remains almost entirely intact while the signals from crystalline P25 completely vanished (Fig. 6). Varying the ratio of the two phases seems to trigger the same phenomena, as both TiO2@GO-05:95 and TiO2@GO-95:05 show the same alterations (Fig. 6). The total destruction of the crystalline phase could also be confirmed based on XRD, where the native peaks of Degussa totally disappeared (Fig. S6†). Indeed, in spite of the straightforwardness of the solvent-free grinding approach, the poor recyclability is the main limitation of the present approach. In addition, powdered forms are not suitable for end use and shaping should be considered.8
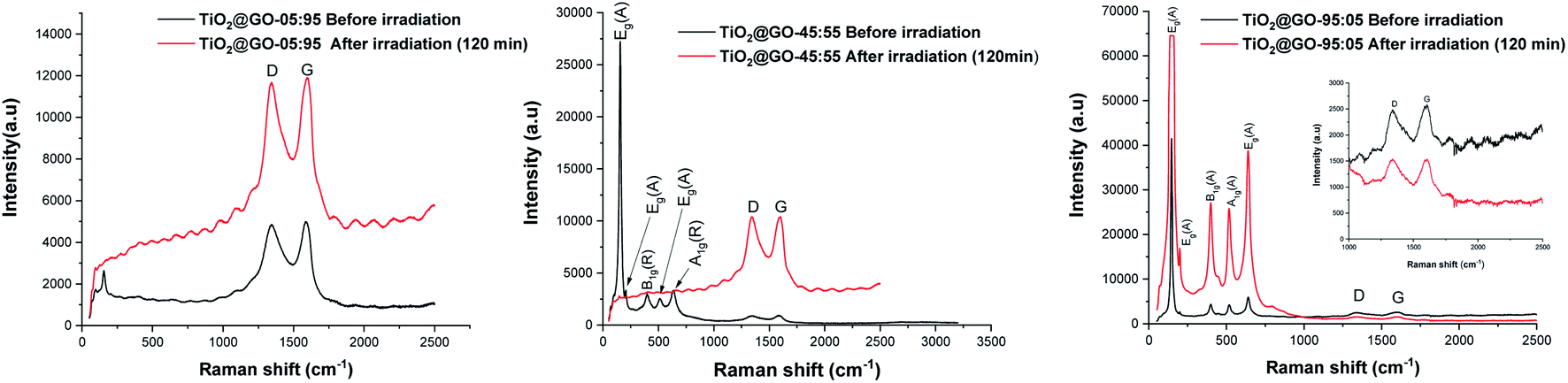 | ||
| Fig. 6 Raman spectra of the prepared materials before and after irradiation. | ||
We therefore preliminarily investigated the possible shaping of the ground formulation into porous beads using renewable biomass-derived chitosan hydrogel. We have previously used chitosan as a mould to grow and shape more sophisticated objects, including metal oxide clusters,26 metal nanoparticles,28 clay and graphene sheets,29 and metal–organic frameworks.30 We consequently attempted to shape binary TiO2@GO within singular microspheres. Interestingly, regular porous beads could be obtained using different ratios of chitosan with respect to TiO2@GO-45:55, showing that the powder does not alter the gelation memory of the polysaccharide. As illustrated in the obtained photos, both native TiO2 and native GO could also be accommodated in bead-form, and the colour of the microspheres reflects the presence of the embedded phase (Fig. 7). The advantage of these beads is that they can be recovered from the medium using a spatula, without the need for specific filters or tedious workup procedures. Once configured as porous beads, TiO2@GO_PB (PB refers to porous beads) could be recycled for an extended period of time compared to the non-shaped TiO2@GO powdered photo-material (Fig. 7). Recent studies have reported the use of hydrogels as photo-reactors,31 and our results suggest the involvement of the gelling medium to further delay the fast deactivation initially observed for the ground powder. Research in this direction is currently being undertaken to unveil the possible role of the gelling medium during the photooxidation process.
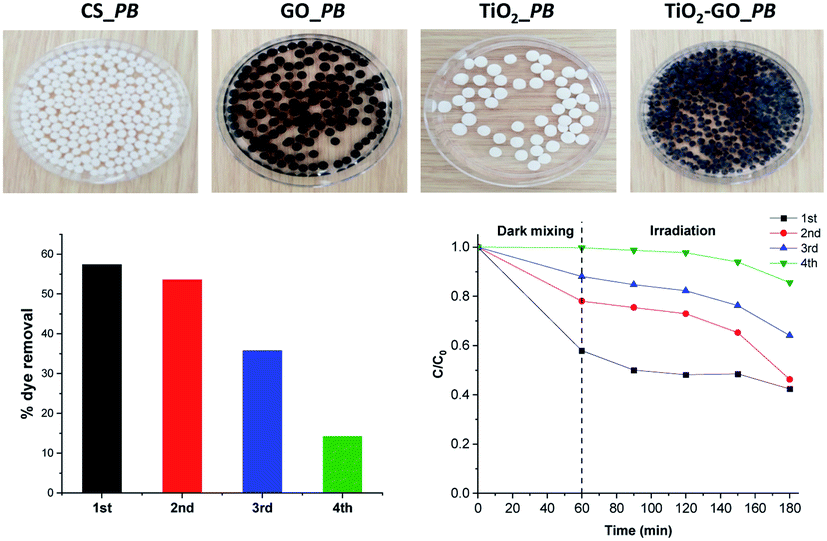 | ||
| Fig. 7 Configuring the photocatalyst into porous-bead form to improve its recyclability compared to native powder. | ||
Experimental section
General
Fourier-transform infrared (FTIR) spectra were obtained with a PerkinElmer Spectrum 100 FT-IR spectrometer using neat samples (ATR FT-IR). DR-UV spectra were recorded in the 200–800 nm range, with Spectralon as the reference, using a PerkinElmer Lambda 1050 spectrometer equipped with an integrating sphere (Labshere, North Sutton, USA). Nitrogen sorption isotherms were obtained at 77 K with Micromeritics ASAP 2010 apparatus (Micromeritics, Norcross, GA, USA). Prior to measurement, the samples were degassed for 12 h at 100 °C to remove any physisorbed species. X-ray powder diffraction (XRD) patterns were recorded using a D8 Advance Bruker AXS system (Bruker D8 Advance; Bruker Corp, Billerica, MA, USA) using CuKα radiation and a step size of 0.02° in the 2θ range from 0.45 to 87°. Scanning electron microscopy (SEM) images were acquired using a ZEISS ULTRA 55 microscope equipped with an X-ray detector (EDS). Raman spectra were recorded using a 514 nm excitation laser on a Horiba LabRAM HR Evolution spectrometer.Materials
Commercially available reagents and solvents were purchased from Acros and Sigma-Aldrich. TiO2 powder (average diameter: 30 nm; surface area: 50 m2 g−1, identical to Degussa P25) was supplied by Sigma-Aldrich. Graphene oxide (GO) was obtained via the oxidation of graphite flakes using a modified Hummers' method.32 Chitosan of medium molecular weight with a deacetylation degree of 85% was purchased from Sigma-Aldrich.Preparation of mechanochemically ground powder
The different materials were prepared via manual grinding using a mortar, for 15 min, starting from a mixture of titanium dioxide and carbon materials, clay, or boron nitride at different ratios. No solvents were used in this process.Preparation of TiO2@GO_us
A mixture of 100 mg of TiO2@GO-45:55 in 15 ml of ethanol was kept under sonication for 15 min at a frequency of 45 kHz. Then, the product was recovered after filtration and dried in an oven at 60 °C for 12 h.
Preparation of TiO2@GO_reflux
100 mg of TiO2@GO-45:55 was added to 15 ml of ethanol in a 50 ml flask. The mixture was maintained under reflux for 1 h. After filtration, the powder was dried in an oven at 60 °C for 12 h.
Preparation of TiO2@CS_pyr
2.5 ml of titanium isopropoxide was added to a solution of 1 g of chitosan in 150 ml of 1% (v/v) acetic acid solution. The reaction mixture was left under stirring for 36 h at room temperature. The mixture was then dried for 48 h at room temperature. The powder obtained was thermally annealed at 500 °C for 3 h under a N2 atmosphere at a heating ramp rate of 5 °C min−1.Preparation of porous beads
100 mg of chitosan was dissolved in 8 ml of 1% (v/v) acetic acid solution and kept under vigorous stirring for 120 min. Then, GO, TiO2, or TiO2@GO-45:55 was dispersed in 2 ml of 1% (v/v) acetic acid solution and submitted to sonication for 90 min. The suspension was gradually added to the chitosan solution and the resulting mixture was stirred for an additional 90 min. This mixture was dropped into NaOH solution (4 N) using a syringe. The beads were stored in alkaline solution for 2 h before repeated washing with distilled water until the washing water was neutral.
Photodegradation studies
The photodegradation experiments were carried out via combining 80 ml of methylene blue, green malachite, Congo red, or methyl orange solution (C0 = 10−3 mmol l−1) and 50 mg of photocatalyst. The mixture was kept under constant magnetic stirring in the dark during an hour so that the dye adsorption equilibrium was established on the catalyst surface. Then, we exposed the reaction mixture to radiation. Experiments were performed at room temperature. The samples were filtered with a Millipore filter with a pore size of 0.45 μm. The remaining concentration of dye in solution (C) was measured using a UV-vis spectrophotometer that had been previously calibrated. The wavelength of maximum absorption (λmax) values are 664 nm (methylene blue), 617 nm (green malachite), 500 nm (Congo red), and 464 nm (methyl orange). Between each catalytic run, the powder was centrifuged, washed with pure water, and dried at 60 °C for 12 h before the next run.Conclusions
The simple and fast grinding of P25 Degussa with graphene oxide under solvent-free conditions affords an efficient visible-light-activated photocatalyst, showing a performance contrast with native Degussa, which is devoid of noticeable photoactivity. Broad screening reveals the suitability of graphene oxide for such a purpose, as it outperforms analogues such as P25–graphite, P25–CNT, and P25–AC. This photoactivity enhancement may be rooted in the presence of plentiful oxygenated groups in GO, allowing for more interplay with titanium dioxide Degussa and providing the driving force for sheet exfoliation. Grinding P25 with graphite does not result in spectacular performance, probably because of the difficulty of achieving graphite-sheet exfoliation, making the stacked interlayer space inaccessible to pollutants. Grinding P25 with clay-based absorbents like layered montmorillonite and nanotubular halloysite failed to achieve the same performance, which further evidences the role of GO as a photosensitizer for titanium dioxide particles. This allows the active region of the heterojunction to be expanded to visible light, where native Degussa is traditionally not active. Our preliminary recycling experiments reveal interesting behavior during the entrapment of the photocatalyst inside of chitosan hydrogels, and this suggests that the performance can be further improved through additional optimization. The timesaving nature and simplicity of the protocol provide a driving force to peruse further efforts to implement this technology on a large scale.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- C. R. Holkar, A. J. Jadhav, D. V. Pinjari, N. M. Mahamuni and A. B. Pandit, J. Environ. Manage., 2016, 182, 351–366 CrossRef CAS PubMed.
- (a) A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS; (b) X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed; (c) J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- (a) V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS; (b) H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- (a) V. Subramanian, E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 11439–11446 CrossRef CAS; (b) P. D. Cozzoli, E. Fanizza, R. Comparelli, M. L. Curri, A. Agostiano and D. Laub, Appl. Catal., B, 2004, 108, 9623–9630 CAS; (c) M. Besançon, L. Michelin, L. Josien, L. Vidal, K. Assaker, M. Bonne, B. Lebeau and J.-L. Blin, New J. Chem., 2016, 40, 4386–4397 RSC; (d) B. J. Aronson, C. F. Blanford and A. Stein, Chem. Mater., 1997, 9, 2842–2851 CrossRef CAS; (e) H. Safardoust-Hojaghan and M. Salavati-Niasari, J. Cleaner Prod., 2017, 148, 31–36 CrossRef CAS; (f) Y. Deng, M. Chen, G. Chen, W. Zou, Y. Zhao, H. Zhang and Q. Zhao, ACS Omega, 2021, 6, 4247–4254 CrossRef CAS PubMed; (g) Y. Brahmi, N. Katir, A. Hameau, A. Essoumhi, E. M. Essassi, A.-M. Caminade, M. Bousmina, J.-P. Majoral and A. El Kadib, Chem. Commun., 2011, 47, 8626–8628 RSC; (h) Y. Brahmi, N. Katir, M. Ianchuk, V. Collière, E. M. Essassi, A. Ouali, A.-M. Caminade, M. Bousmina, J. P. Majoral and A. El Kadib, Nanoscale, 2013, 5, 2850–2856 RSC; (i) Y. Brahmi, N. Katir, J. A. M. Agullo, A. Primo, M. Bousmina, J. Pierre Majoral, H. Garcia and A. El Kadib, Dalton Trans., 2015, 44, 15544–15556 RSC; (j) N. Katir, Y. Brahmi, J. P. Majoral, M. Bousmina and A. El Kadib, Chem. Commun., 2015, 51, 17716–17719 RSC; (k) N. Katir, N. Marcotte, S. Michlewska, M. Ionov, N. El Brahmi, M. Bousmina, J. P. Majoral, M. Bryszewska and A. El Kadib, ACS Appl. Nano Mater., 2019, 2, 2979–2990 CrossRef CAS.
- M. Dahl, Y. Liu and Y. Yin, Chem. Rev., 2014, 114, 9853–9889 CrossRef CAS PubMed.
- (a) D. Fattakhova-Rohlfing, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487–9558 CrossRef CAS PubMed; (b) L. Liu and X. Chen, Chem. Rev., 2014, 114, 9890–9918 CrossRef CAS PubMed; (c) H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- F. Fresno, R. Portela, S. Suárez and J. M. Coronado, J. Mater. Chem. A, 2014, 2, 2863–2884 RSC.
- F. Matter and M. Niederberger, Adv. Sci., 2022, 9, 2105363 CrossRef CAS PubMed.
- (a) W. K. Jo, S. H. Shin and E. S. Hwang, J. Hazard. Mater., 2011, 191, 234–239 CrossRef CAS PubMed; (b) P. Bhavani, M. Hussain and Y.-K. Park, J. Cleaner Prod., 2022, 330, 129899 CrossRef CAS; (c) F. Wang, M. Hao, W. Liu, P. Yan, B. Fang, S. Li, J. Liang, M. Zhu and L. Cui, Nano Mater. Sci., 2021, 3, 205–212 CrossRef CAS; (d) L. Cui, M. Hao, F. Wang, B. Fang, J. Liang, M. Zhu and X. Xie, Int. J. Photoenergy, 2020, 2020, 8868782 CrossRef.
- (a) Y. Hu, C. Zhou, H. Wang, M. Chen, G. Zeng, Z. Liu, Y. Liu, W. Wang, T. Wu, B. Shao and Q. Liang, Chem. Eng. J., 2021, 414, 128795 CrossRef CAS; (b) P. Shandilya, A. Guleria and B. Fang, J. Environ. Chem. Eng., 2021, 9, 106461 CrossRef CAS; (c) G. Liao, C. Li, X. Li and B. Fang, Cell Rep. Phys. Sci., 2021, 2, 100355 CrossRef CAS; (d) F. Yi, J. Ma, C. Lin, L. Wang, H. Zhang, Y. Qian and K. Zhang, J. Alloys Compd., 2020, 821, 153557 CrossRef CAS.
- (a) Y. Yao, G. Li, S. Ciston, R. M. Lueptow and K. A. Gray, Environ. Sci. Technol., 2008, 42, 4952–4957 CrossRef CAS PubMed; (b) N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- (a) Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS PubMed; (b) H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed; (c) Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS PubMed.
- R. Das, C. D. Vecitis, A. Schulze, B. Cao, A. F. Ismail, X. Lu, J. Chen and S. Ramakrishna, Chem. Soc. Rev., 2017, 46, 6946–7020 RSC.
- (a) Y. Liu, D. Zhang, Y. Shang, W. Zang and M. Li, RSC Adv., 2015, 5, 104785–104791 RSC; (b) Z. Qianqian, B. Tang and H. Guoxin, J. Hazard. Mater., 2011, 198, 78–86 CrossRef PubMed.
- (a) S. Frindy, A. Primo, A. e. k. Qaiss, R. Bouhfid, M. Lahcini, H. Garcia, M. Bousmina and A. El Kadib, Carbohydr. Polym., 2016, 146, 353–361 CrossRef CAS PubMed; (b) B. Boumhidi, N. Katir, J. El Haskouri, K. Draoui and A. El Kadib, New J. Chem., 2020, 44, 14136–14144 RSC.
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. Tang and C. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed.
- (a) P. Montes-Navajas, N. G. Asenjo, R. Santamaría, R. Menéndez, A. Corma and H. García, Langmuir, 2013, 29, 13443–13448 CrossRef CAS PubMed; (b) L. Chen, C. Batchelor-McAuley, B. Rasche, C. Johnston, N. Hindle and R. G. Compton, Appl. Mater. Today, 2020, 18, 100506 CrossRef.
- H. Ennajih, R. Bouhfid, E. M. Essassi, M. Bousmina and A. El Kadib, Microporous Mesoporous Mater., 2012, 152, 208–213 CrossRef CAS.
- Y. Li, X. Yuan, L. Jiang, H. Dai, Y. Zhao, X. Guan, J. Bai and H. Wang, Environ. Sci.: Nano, 2022, 9, 841–866 RSC.
- (a) H. Xue, Y. Wu, Y. Zou, Y. Shen, G. Liu, Q. Li, D. Yin, L. Wang and J. Ming, Adv. Funct. Mater., 2020, 30, 1910657 CrossRef CAS; (b) A. Puthirath Balan, S. Radhakrishnan, C. F. Woellner, S. K. Sinha, L. Deng, C. d. l. Reyes, B. M. Rao, M. Paulose, R. Neupane, A. Apte, V. Kochat, R. Vajtai, A. R. Harutyunyan, C.-W. Chu, G. Costin, D. S. Galvao, A. A. Martí, P. A. van Aken, O. K. Varghese, C. S. Tiwary, A. Malie Madom Ramaswamy Iyer and P. M. Ajayan, Nat. Nanotechnol., 2018, 13, 602–609 CrossRef CAS PubMed.
- A. Anouar, N. Katir, A.-S. Mamede, A. Aboulaich, K. Draoui, S. Royer and A. El Kadib, Mater. Chem. Front., 2019, 3, 242–250 RSC.
- F.-E. Zirar, A. Anouar, N. Katir, I. A. Ichou and A. El Kadib, RSC Adv., 2021, 11, 28116–28125 RSC.
- M. Tamtaji, A. Tyagi, C. Y. You, P. R. Galligan, H. Liu, Z. Liu, R. Karimi, Y. Cai, A. P. Roxas, H. Wong and Z. Luo, ACS Appl. Nano Mater., 2021, 4, 7563–7586 CrossRef CAS.
- N. Hammi, S. Chen, F. Dumeignil, S. Royer and A. El Kadib, Mater. Today Sustain., 2020, 10, 100053 CrossRef.
- A. El Kadib and M. Bousmina, Chem.–Eur. J., 2012, 18, 8264–8277 CrossRef PubMed.
- S. Frindy, A. El Kadib, M. Lahcini, A. Primo and H. García, ACS Catal., 2016, 6, 3863–3869 CrossRef CAS.
- (a) A. El Kadib, M. Bousmina and D. Brunel, J. Nanosci. Nanotechnol., 2014, 14, 308–331 CrossRef CAS PubMed; (b) A. Anouar, N. Katir, A. El Kadib, A. Primo and H. García, Molecules, 2019, 24, 3290 CrossRef CAS PubMed.
- A. El Kadib, Chem. Rec., 2020, 20, 753–772 CrossRef CAS PubMed.
- N. Hammi, S. Chen, A. Primo, S. Royer, H. Garcia and A. El Kadib, Green Chem., 2022, 24, 4533–4543 RSC.
- R. R. Mansurov, V. S. Zverev and A. P. Safronov, J. Catal., 2022, 406, 9–18 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04017d |
| This journal is © The Royal Society of Chemistry 2022 |