
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAltereporenes A–E, five epoxy octa-hydronaphthalene polyketides produced by an endophytic fungus Alternaria sp. YUD20002†
Dan-Dan Xiaa,
Hao-Jie Duana,
Fei Xiea,
Tian-Peng Xiea,
Yan Zhanga,
Yue Suna,
Jian-Mei Lua,
Yu-Hong Gaoc,
Hao Zhou *a and
Zhong-Tao Ding*ab
*a and
Zhong-Tao Ding*ab
aKey Laboratory of Functional Molecules Analysis and Biotransformation of Universities in Yunnan Province, School of Chemical Science and Technology, Yunnan University, Kunming, 650091, China. E-mail: Haozhou@ynu.edu.cn; ztding@ynu.edu.cn
bCollege of Pharmacy, Dali University, Dali, 671000, China
cThe First People's Hospital of Yunnan Province, Kunming, 650034, China
First published on 10th August 2022
Abstract
Five previously undescribed epoxy octa-hydronaphthalene polyketides, altereporenes A–E (1–5) were isolated from rice culture of the endophytic fungus Alternaria sp. YUD20002 derived from the tubers of Solanum tuberosum. Their structures were determined on the basis of comprehensive spectroscopic analyses, while the absolute configurations were elucidated by the comparison of experimental and calculated specific rotations. Meanwhile, the antimicrobial, cytotoxic, anti-inflammatory and acetylcholinesterase inhibitory activities of compounds 1–5 were also investigated.
Introduction
Endophytic fungi, unique microorganisms that can asymptomatically colonize the living tissues of healthy plants,1,2 have been recognized as vital sources of novel natural products and attracted extensive attention owing to their intriguing new structures with prominent biological activities.3–8 In particular, Alternaria-related fungi have been reported to produce bioactive and structurally diverse secondary metabolites in recent years.9–11 As far as we know, previous studies on the genus Alternaria have resulted in the isolation of numerous metabolites with various novel skeletons including the hydroanthraquinone derivatives,12 benzopyrone derivatives,13 diterpenoids,14,15 α-pyrone derivatives,16 etc.In order to explore structural diversity natural products from the endophytic fungi, the chemical investigation of the secondary metabolites from Alternaria sp. YUD20002 which was derived from Solanum tuberosum (potato) was carried out. Further scale-up fermentation followed by chemical investigation of the secondary metabolites of the strain yielded five new compounds, altereporenes A–E (1–5). Their structures (Fig. 1) were characterized as complex epoxy octa-hydronaphthalene analogues coupled with different side chains by spectroscopic data analysis and quantum chemical calculated. In this work, the isolation, structural elucidation, and bioactivity of these compounds were described.
 | ||
| Fig. 1 Chemical structures of compounds 1–5. | ||
Results and discussion
Altereporene A (1) was isolated as a white amorphous powder and the molecular formula C23H34O4 was determined by its HRESIMS (m/z 397.2350 [M + Na]+, calcd for 397.2349), indicating seven degrees of unsaturation. The 13C NMR data (Table 1) indicated 23 carbon signals, and based on the DEPT spectra, these carbons were classified as four methyls, four methylenes (including an oxygenated one at δC 62.6), five olefinic methines, six sp3 methines (including three oxygenated ones at δC 60.0, 62.2, and 66.0), one oxygen-bearing quaternary carbon (δC 78.5), and three olefinic quaternary carbons. The 1H and 13C NMR spectra of 1 (Tables 1 and 2) revealed it was tricyclic and possessed four double bond to satisfy its unsaturation requirements. These data indicated that the structural features of constituent 1 arising from its 1D NMR spectra were similar to those of anthracobic acid A, a polyketide from Anthracobia sp.17 The detailed planar structure of 1 was further constructed based on the 1H–1H COSY and HMBC spectra (Fig. 2). The 1H–1H COSY spectrum indicated the presence of four independent spin systems, H2-1/H2-2/H2-3(A), H-5/H-6/H-7/H-8/H-9(H-10/H-11)/H-14/H2-13(B), H-15/H-16(C) and H-19/H3-20(D). Spin systems A and B were confirmed to be connected through the quaternary olefinic carbons (C-4) via the HMBC correlations of H3-21 to C-3/C-4/C-5 and H-3 to C-4/C-5. Meanwhile, the chemical shift of C-1 (δC 62.6) illustrated a hydroxyl group linked to C-1. The HMBC correlations, especially those from H3-22 to C-11/C-12/C-13, from Ha-13 to C-9/C-11/C-12/C-14, combined with the chemical shift of C-10 (δC 66.0) deduced the presence of a methylcyclohexene skeleton with a hydroxyl at C-10. The HMBC correlations from H-15 to C-9/C-13/C-14, from H-16 to C-8/C-15/C-17, and from H-8 to C-17 suggested the spin systems B and C were comprised of an octa-hydronaphthalene skeleton. Based on the analyses above together with the HMBC correlations from H3-23 to C-17/C-18/C-19, from H3-20 to C-18/C-19, and from H-19 to C-17, along with the chemical shift of C-17 (δC 78.5), indicating that the 2-butene moiety was connected to the oxygenated quaternary carbon (C-17). Furthermore, the presence of an epoxy group at C-15 and C-16 were assigned by the molecular formula and the chemical shifts of the 13C-NMR data of these positions. Thus, the planar structure of 1 was provided.| Pos. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a Measured at 400 MHz in methanol-d4. | |||||
| 1 | 62.6, CH2 | 177.8, C | 63.8, CH2 | 197.3, C | 179.5, C |
| 2 | 32.0, CH2 | 36.0, CH2 | 133.9, CH | 137.5, C | 130.3, C |
| 3 | 37.1, CH2 | 118.7, CH | 130.6, CH | 151.2, CH | 138.4, CH |
| 4 | 137.4, C | 139.8, C | 47.2, CH | 130.4, CH | 130.9, CH |
| 5 | 126.5, CH | 43.9, CH2 | 43.2, CH | 145.3, CH | 139.1, CH |
| 6 | 131.3, CH | 132.6, CH | 65.7, CH | 48.3, CH | 48.0, CH |
| 7 | 130.5, CH | 130.5, CH | 125.5, CH | 43.6, CH | 43.5, CH |
| 8 | 47.5, CH | 47.3, CH | 139.3, C | 66.0, CH | 65.9, CH |
| 9 | 43.8, CH | 43.3, CH | 37.0, CH | 125.5, CH | 125.6, CH |
| 10 | 66.0, CH | 66.0, CH | 30.4, CH | 139.3, C | 139.8, C |
| 11 | 125.6, CH | 125.6, CH | 59.9, CH | 36.9, CH2 | 37.0, CH2 |
| 12 | 139.2, C | 139.2, C | 62.1, CH | 30.4, CH | 30.3, CH |
| 13 | 37.1, CH2 | 37.0, CH2 | 78.3, C | 59.9, CH | 60.0, CH |
| 14 | 30.3, CH | 30.3, CH | 138.5, C | 62.2, CH | 62.2, CH |
| 15 | 60.0, CH | 60.0, CH | 123.8, CH | 78.6, C | 78.5, C |
| 16 | 62.2, CH | 62.2, CH | 13.7, CH3 | 138.1, C | 139.0, C |
| 17 | 78.5, C | 78.3, C | 23.4, CH3 | 124.4, CH | 124.0, CH |
| 18 | 138.7, C | 138.6, C | 14.8, CH3 | 13.7, CH3 | 13.7, CH3 |
| 19 | 123.6, CH | 123.7, CH | 9.4, CH3 | 13.5, CH3 | |
| 20 | 13.7, CH3 | 13.7,CH3 | 23.4, CH3 | 23.4, CH3 | |
| 21 | 16.7, CH3 | 16.8, CH3 | 14.8, CH3 | 14.8, CH3 | |
| 22 | 23.4, CH3 | 23.4, CH3 | |||
| 23 | 14.8, CH3 | 14.9, CH3 | |||
| Pos. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a Measured at 400 MHz in methanol-d4. | |||||
| 1 | 3.53, t (6.6) | 4.04, m | 9.39, s | ||
| 2 | 1.63, overlap | 2.95, d (7.4) | 5.74, dt (15.3, 6.3) | ||
| 3 | 2.10, t (7.1) | 5.39, m | 5.22, dd (15.3, 10.4) | 6.99, d (11.3) | 7.08, d (11.4) |
| 4 | 2.39, t (11.6) | 6.77, dd (14.9, 11.3) | 6.55, dd (14.8, 11.3) | ||
| 5 | 5.83, d (10.9) | 2.75, d (6.7) | 1.38, td (11.8, 3.0) | 5.91, dd (15.0, 10.6) | 5.56, overlap |
| 6 | 6.42, dd (15.0, 10.9) | 5.60, dt (15.2, 6.8) | 3.84, dd (4.2, 3.4) | 2.62, t (11.8) | 2.53, t (11.2) |
| 7 | 5.06, dd (15.0, 10.3) | 4.97, dd (15.2, 10.4) | 5.53, m | 1.51, td (11.8, 3.0) | 1.45, td (11.8,3.0) |
| 8 | 2.40, t (11.8) | 2.36, t (11.8) | 3.78, dd (6.0, 2.9) | 3.79, dd (5.0, 3.7) | |
| 9 | 1.37, td (11.8, 3.1) | 1.34, td (11.8, 3.0) | 2.20, m | 5.54, overlap | 5.53, overlap |
| 1.90, m | |||||
| 10 | 3.81, dd (5.8, 3.5) | 3.83, dd (4.4, 3.0) | 2.14, m | ||
| 11 | 5.53, d (5.6) | 5.54, m | 3.17, d (3.8) | 2.25, m | 2.23, m |
| 1.94, m | 1.93, m | ||||
| 12 | 3.08, d (3.7) | 2.20, m | 2.18, m | ||
| 13 | 2.20, m | 2.21, m | 3.21, d (3.8) | 3.19, d (3.8) | |
| 1.90, m | 1.90, m | ||||
| 14 | 2.17, m | 2.16, m | 3.11, d (3.8) | 3.10, d (3.8) | |
| 15 | 3.17, d (3.8) | 3.16, d (3.8) | 5.48, m | ||
| 16 | 3.08, d (3.8) | 3.07, d (3.8) | 1.67, overlap | ||
| 17 | 1.74, s | 5.54, overlap | 5.5, overlap | ||
| 18 | 1.68, overlap | 1.71, overlap | 1.70, overlap | ||
| 19 | 5.47, m | 5.47, m | 1.83, s | 1.93, overlap | |
| 20 | 1.68, m | 1.66, overlap | 1.75, s | 1.74, s | |
| 21 | 1.77, s | 1.68, overlap | 1.69, overlap | 1.68, overlap | |
| 22 | 1.73, s | 1.73, s | |||
| 23 | 1.67, m | 1.69, overlap | |||
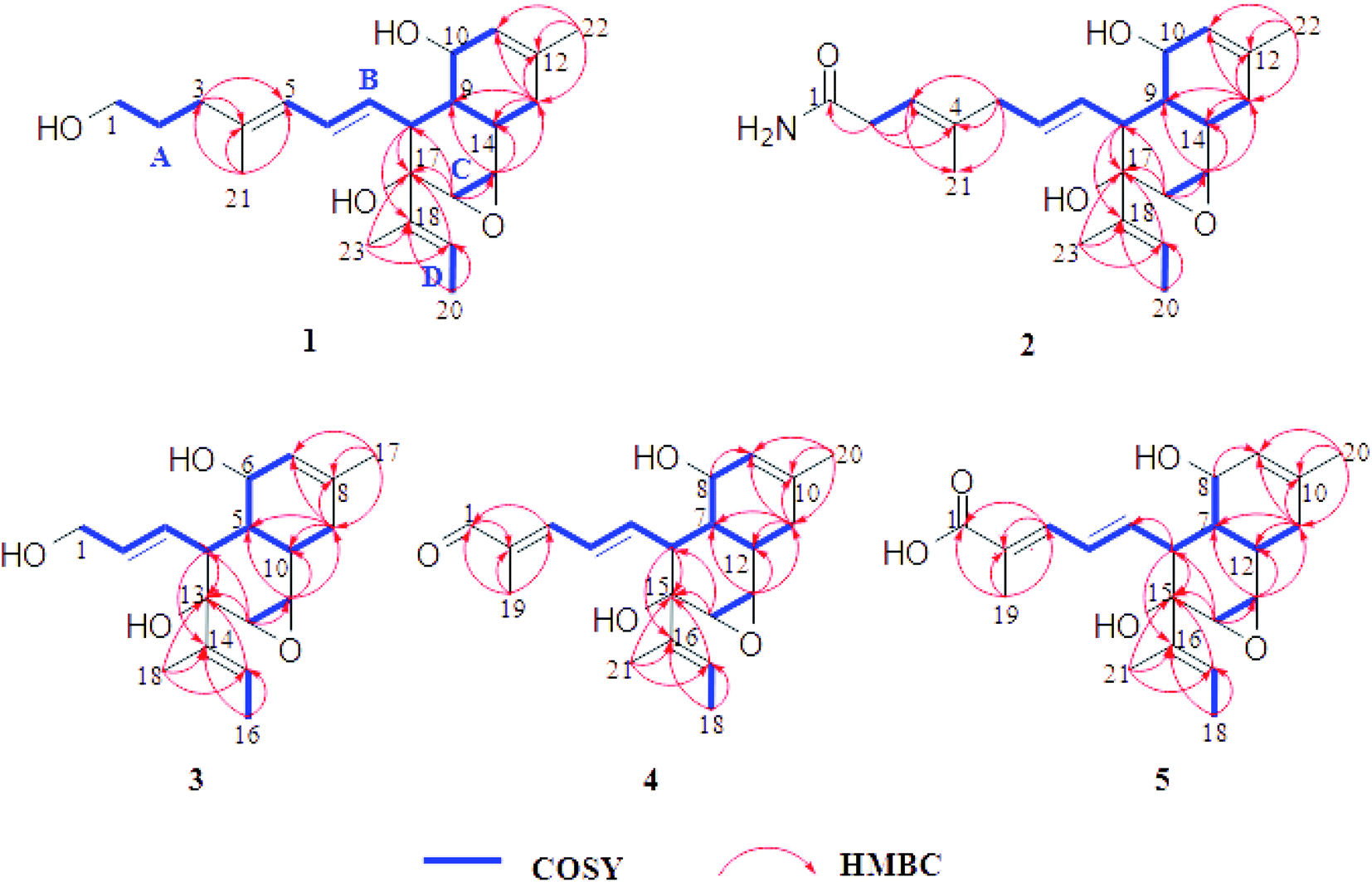 | ||
| Fig. 2 The key 1H–1H COSY and HMBC correlations of compounds 1–5. | ||
The relative configuration of 1 was established by the coupling constants and NOESY correlations (Fig. 3). The trans-orientations of C4–C5 and C6–C7 were determined by the coupling constant of JH-6/H-7 at 15.0 Hz, and NOESY correlations of H-5/H-7, H-6/H3-21. In addition, the NOESY correlation between H-19 and H-16, along with the lack of obvious correlation between H-23 and H-16, indicated that the configuration of the double bond C18–C19 should also be trans-orientation. The NOESY correlations of H-7/H-9/H-10, H-9/H-15/H-16, and H-16/H-19 suggested that the protons H-9, H-10, H-15, H-16 and H-19 were on the same side and were randomly assigned α-orientation, while the NOESY correlation between H-8 and H-14 assigned H-8 and H-14 on β-orientation of the molecule. The above analysis supported the presence of two possible isomers, (8S, 9R, 10S, 14R, 15S, 16S, 17S)-1 and (8R, 9S, 10R, 14S, 15R, 16R, 17R)-1. To determine its absolute configuration, TDDFT-ECD calculations were performed at the B3LYP/6-31g (d, p) level in methanol.18–21 However, neither of the calculated ECD curves for two possible isomers of 1 matched well with the experimental curves. Finally, the absolute configuration of 1 was elucidated by comparison of the experimental and the theoretical value of specific rotations. Each isomer was optimized at the B3LYP/6-31g (d, p) level in methanol using DFT in the Gaussian 09 program. Then, the optimized isomer was calculated using TDDFT/GIAOs at the B3LYP/6-31g* in the Gaussian 09 program to generate its specific rotation. As expected, the calculated specific rotation ([a] −223.67) was perfectly matched to the experimental one ([a] −121.0). Therefore, the absolute configuration of 1 was established as 8S, 9R, 10S, 14R, 15S, 16S, 17S, and named as altereporene A.
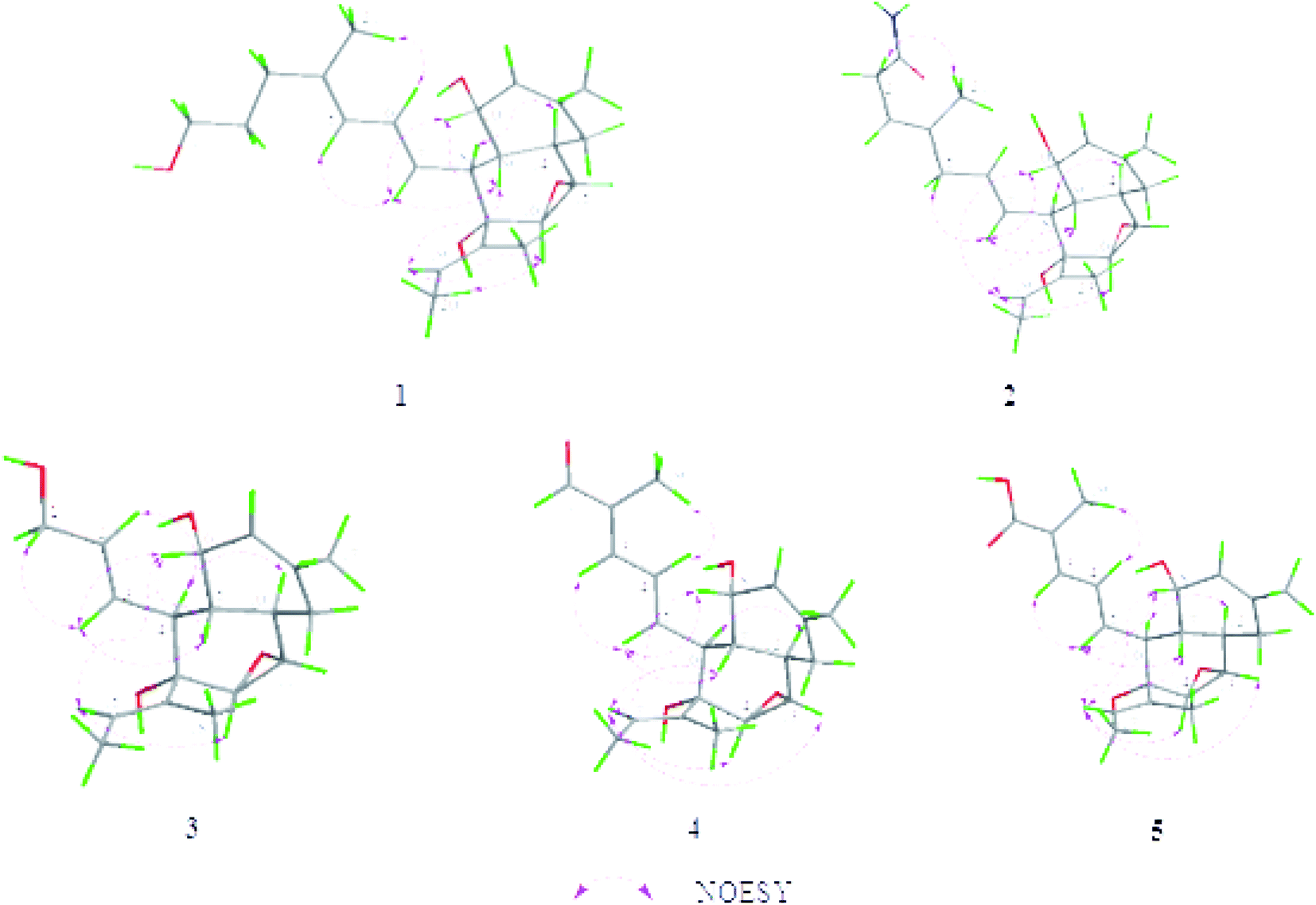 | ||
| Fig. 3 Key NOESY correlations and the lowest energy state models of compounds 1–5 at B3LYP/6-31g (d, p) in methanol. | ||
Altereporene B (2), obtained as a yellowish-green solid, possessed a molecular formula of C23H33NO4 with eight index of hydrogen deficiency based on the positive mode with an obvious HRESIMS ion peak found at m/z 410.2301 [M + Na]+ (calcd for 410.2302). The 13C NMR (Table 1) spectrum of 2 displayed resonances for 23 carbons ascribed to four methyls, three methylenes, eleven methines, and five quaternary carbons including three olefinic ones, an amide carbonyl, as well as an oxygenated carbon. A comparison of its 1D (Tables 1 and 2) and 2D NMR (Fig. 2) data with compound 1 indicated that 2 should share a very similar structural core with 1. One of the main differences between them was that the hydroxymethyl of C-1 in 1 was replaced by an amide group in 2, which could be confirmed by the key HMBC correlations (Fig. 2) from H2-2 to C-1. The HMBC correlations from H2-2 to C-3/C-4, from H-3 to C-21, and from H2-5 to C-3/C-4/C-21 suggested the double bond of C-4/C-5 in 1 migrated to C-3/C-4 in 2. Collectively, the planar structure of 2 was illustrated.
The configurations of the trans-orientations of C3–C4, C6–C7 and C18–C19 in 2 were also determined by the coupling constants of JH-6/H-7 at 15.2 Hz, the NOESY correlations of H2-2/H3-21, H-5/H-7, H-19/H-16, together with the lack of obvious NOE correlation between H-23 and H-16 (Fig. 3). In the NOESY spectrum, the key cross-peaks between H-7 and H-9/H-10, H-19 and H-9/H-10/H-16, with the addition of H-8 and H-14 indicated that H-9, H-10, H-15, H-16 and the 2-butene moiety were on the same orientation, while H-8, H-14, and the epoxy group were on the opposite orientation. The above data limited the possible enantiomers to (8S, 9R, 10S, 14R, 15S, 16S, 17S)-2 and (8R, 9S, 10R, 14S, 15R, 16R, 17R)-2. Then, the absolute configuration of 2 was also determined to be the same 8S, 9R, 10S, 14R, 15S, 16S, 17S as that of 1 by specific rotation calculation, both experimental and theoretical specific rotations of 2 were negative. Accordingly, the structure of 2 was defined as shown (Fig. 1), and named as altereporene B.
Altereporene C (3), a yellowish-green powder, was designated with the molecular formula C18H26O4, based on the HRESIMS analysis (m/z 329.1719 [M + Na]+, calcd for 329.1723) with six degrees of unsaturation. The NMR data (Tables 1 and 2) indicated the presence of a highly similar skeleton like compound 2 except for the disappearance of the substituent at C-5 in 2 and the appearance of an additional hydroxy group at C-1 in 3. This was substantiated by examination of upfield shift of C-1 (δC 63.8) in 3, and the detailed inspection of 1H–1H COSY and HMBC correlations (Fig. 2). Then, the similar coupling constants and NOESY correlations with that of 2 (Fig. 3) suggested that the double bonds were 2E and 14E in 3 and it has two possible absolute configurations of 4S, 5R, 6S, 10R, 11S, 12S, 13S-3 and 4R, 5S, 6R, 10S, 11R, 12R, 13R-3. Furthermore, with the identical specific rotations of the calculated and experimental results, the absolute configurations of 3 were assigned as 4S, 5R, 6S, 10R, 11S, 12S, 13S.
Altereporene D (4) was obtained as a white powder. The molecular formula of C21H28O4 was assigned to 4 as determined by its HRESIMS (m/z 367.1885 [M + Na]+, calcd for 367.1880), which corresponded to eight indices of hydrogen deficiency. Analysis of the 13C NMR data revealed 21 carbon signals comprising four methyls, one methylene, eleven methines, three olefinic quaternary carbons, one oxygenated quaternary carbon and one aldehyde group. The NMR spectroscopic data (Tables 1 and 2) combined with the 1H–1H COSY and HMBC correlations (Fig. 2) suggested that 4 exhibited the very similar scaffold as compound 1. The major difference was that a hydroxypropyl at C-4 (δC 137.6) in compound 1 instead by the aldehyde group at C-2 (δC 137.5) in 4, which was supported by the HMBC correlations from H-1 to C-2 and H3-19 to C-1/C-2/C-3. This assignment was further confirmed by the 1H–1H COSY and HMBC experiments (Fig. 2). E-geometry of double bonds at C2–C3, C4–C5, C16–C17 and the relative configuration of 4 were designed as the same of 1 by the analysis of coupling constants and NOE correlations (Fig. 3). The determination of its absolute configuration also depended on the specific rotation test, the calculated specific rotation of 6S, 7R, 8S, 12R, 13S, 14S, 15S-4 was in good agreement with the experimental date, allowing confirmation of its absolute configuration, as shown in Fig. 1.
Altereporene E (5) was obtained as a white powder. Its molecular formula was deduced as C21H28O5 based on its HRESIMS (m/z 383.1825 [M + Na]+, calcd for 383.1829), indicating eight degrees of hydrogen deficiency. Detailed inspection of 1D and 2D NMR data of 5 with that of 4 revealed that they were structural analogues, with the obvious difference being that the absence of the aldehyde signal in 4 and the presence of a carboxyl at C-2 in 5. It was verified by the HMBC correlations from H-3 and H-19 to C-1. A detailed 2D NMR analysis further constructed the structure of 5 (Fig. 2). Then, the absolute configuration of 5 was determined as the same of 4 to be 2E, 4E, 16E, 6S, 7R, 8S, 12R, 13S, 14S, and 15S by the analysis of NMR data and specific rotation calculation.
In the bioassays, the in vitro cytotoxic activities against five human tumor cell lines, anti-acetylcholinesterase activities and anti-inflammatory activities of compounds 1–5 were evaluated, but none showed significant inhibitory activities (details see ESI†).
Conclusions
In conclusion, a chemical investigation of endophytic fungus Alternaria sp. YUD20002 led to the isolation and identification of five new secondary metabolites altereporenes A–E (1–5). Their structures with absolute configurations were established. Further studies related to their bioactivities revealed all compounds 1–5 showed no significant inhibitory activities. However, the discovery of structurally interesting epoxy octa-hydronaphthalene derivatives extended the diversities in the family of the secondary metabolites from endophytic fungi with similar skeleton.Experimental section
General experimental procedures
Specific rotations were measured with a Jasco model 1020 digital polarimeter (Horiba, Tokyo, Japan). One-dimensional and two-dimensional nuclear magnetic resonance (NMR) spectra were obtained on Bruker DRX-400 MHz (Bruker, Karlsruhe, Germany) spectrometers with tetramethylsilane (TMS) as an internal standard. UV-vis spectra were recorded using a Shimadzu UV-2550 PC spectrometer (Shimadzu Co., Ltd, Tokyo, Japan). HRESIMS data were obtained on an Agilent G3250AA (Agilent, Santa Clara, CA, USA). Electronic circular dichroism (ECD) spectra were measured with an Applied Photophysics Chirascan spectrometer (Applied Photophysics Ltd, UK). The preparative HPLC was performed on an Agilent 1260 series equipped with a DAD detector and a Zorbax SB-C18 (250 × 9.4 mm, 5 μm) semipreparative column. Sephadex LH-20 (GE Healthcare Co, Buckinghamshire, UK) and silica gel (200–300 mesh, Qingdao Marine Chemical Group Co., Qingdao, China) were applied for column chromatography. Fractions were monitored by TLC (GF254, Qingdao Haiyang Chemical Co. Ltd), with spots were detected by spraying with 10% H2SO4 in ethanol, followed by heating.Fungal material
The fungal strain YUD20002 was isolated from the Solanum tuberosum which was collected from Kunming, Yunnan Province, China, in October 2020. The species of the strain was identified as Alternaria sp. based on the morphological method and reinforced by 18S rDNA along with internal transcribed spacer (ITS) sequences (GenBank No. ON204165). The strain YUD20002 was preserved at the School of Chemical Science and Technology, Yunnan University, China.Fermentation, extraction and isolation
The fungus was inoculated in 3 × 500 mL Erlenmeyer flasks, each containing 100 mL of potato dextrose broth. Flask cultures were incubated at 28 °C on a rotary shaker at 160 rpm for 3 days as seed culture. Large-scale fermentation was carried out in 100 Erlenmeyer flasks (1000 mL), each contained rice (65 g), which were soaked overnight before autoclaving at 121 °C for 30 min. After cooling to room temperature, those Erlenmeyer flasks were respectively inoculated 3 mL seed broth, furthermore, which were maintained for 45 days at room temperature statically for chemical investigation.The whole fermented culture was extracted with methanol, the extract was exhaustively extracted three times with EtOAc (1 day each time), after evaporating the organic solvent under vacuum, the crude extract (30 g) was obtained. The residue was subjected to silica gel column chromatography, eluted with a gradient of CH2Cl2–MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:1 v/v) to afford five fractions (Fr.1 to Fr.5).
:0 to 0:1 v/v) to afford five fractions (Fr.1 to Fr.5).
Fr.1 was subjected to Sephadex LH-20 eluted with MeOH, silica gel CC (300–400 mesh) with a gradient elution of PE-EtOAc (20:1 to 0:1 v/v) and further purified by RP-HPLC with MeOH–H2O (65:35 v/v) to obtain compound 4 (2.0 mg). Fr.3 was divided into four fractions (Fr.3-1 to Fr.3-4) by Sephadex LH-20 (in MeOH). Fr.3-3 was chromatographed on silica gel CC with a gradient elution of PE-EtOAc (10:1 to 0:1 v/v) and further isolated by RP-HPLC with MeOH–H2O (65:35–85:15 v/v) to give compound 1 (4.2 mg). Fr.3-4 was separated on silica gel CC eluted with PE–EtOAc (10:1 to 0:1 v/v) and then purified by RP-HPLC with MeOH–H2O (63:37 v/v) to provide compound 5 (3.9 mg). Compounds 2 (7.8 mg) and 3 (3.3 mg) were yield from Fr.4 through Sephadex LH-20 with MeOH elution and RP-HPLC (MeOH–H2O, 45:55–75:25 v/v).
Altereporene A (1): white powder; [a] −121.0 (c 0.14, MeOH); UV (MeOH) λmax (logε) 242 (2.44) nm; HRESIMS m/z 397.2350 [M + Na]+ (calcd for C23H34O4Na, 397.2349); 1H NMR (CD3OD, 400 MHz) and 13C NMR (CD3OD, 100 MHz) in Tables 1 and 2.
Altereporene B (2): yellowish-green solid; [a] −105.3 (c 0.14, MeOH); UV (MeOH) λmax (logε) 195 (2.47) nm; HRESIMS m/z 410.2301 [M + Na]+ (calcd for C23H34O4Na, 410.2302); 1H NMR (CD3OD, 400 MHz) and 13C NMR (CD3OD, 100 MHz) in Tables 1 and 2.
Altereporene C (3): yellowish-green powder; [a] −118.3 (c 0.12, MeOH); UV (MeOH) λmax (logε) 195 (2.38) nm; HRESIMS m/z 329.1719 [M + Na]+ (calcd for C23H34O4Na, 329.1723); 1H NMR (CD3OD, 400 MHz) and 13C NMR (CD3OD, 100 MHz) in Tables 1 and 2.
Altereporene D (4): white powder; [a] −204.9 (c 0.11, MeOH); UV (MeOH) λmax (logε) 284 (2.55) nm; HRESIMS m/z 367.1885 [M + Na]+ (calcd for C23H34O4Na, 367.1880); 1H NMR (CD3OD, 400 MHz) and 13C NMR (CD3OD, 100 MHz) in Tables 1 and 2.
Altereporene E (5): white powder; [a] −132.5 (c 0.15, MeOH); UV (MeOH) λmax (logε) 265 (2.42) nm; HRESIMS m/z 383.1825 [M + Na]+ (calcd for C23H34O4Na, 383.1829); 1H NMR (CD3OD, 400 MHz) and 13C NMR (CD3OD, 100 MHz) in Tables 1 and 2.
Cytotoxicity assay
The in vitro cytotoxicity assay was performed according to the MTS method in 96-well microplates. The cell survival assay was carried out with the previously reported MTT method.22–24 Five human tumor cell lines, including human promyelocytic leukemia HL-60, human hepatocellular carcinoma SMMC-7721, lung cancer A-549, breast cancer MDA-MB-231, and human colon cancer SW480, were used in the cytotoxicity assays, and cisplatin was used as a positive control. All cells were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone) at 37 °C in a humidified atmosphere containing 5% CO2. The cells were seeded onto 96-well plates at 1.5 × 104 cells per well and after 24 h the compounds were added in different concentrations of 0.064, 0.32, 1.6, 8, and 40 μM for 48 h at 37 °C. Subsequently, MTS was added to the culture medium and the absorbance at 490 nm was measured with a microplate reader. The proliferation rate was calculated as the ratio of absorbance to that of the control.Acetylcholinesterase inhibitory activity
Acetylcholinesterase (AChE) inhibitory activities of the compounds 1–5 were assayed by using modified Ellman's method as described in a previous literature report.25 S-Acetylthiocholine iodide, S-butyrylthiocholine iodide, 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB, Ellman's reagent), acetylcholinesterase derived from human erythrocytes were purchased from Sigma Chemical. Compounds were dissolved in DMSO. The reaction mixture (totally 200 μL) containing phosphate buffer (pH 8.0), test compound (50 μM), and acetyl cholinesterase (0.02 U mL−1), was incubated for 20 min (37 °C). Then, the reaction was initiated by the addition of 40 μL of solution containing DTNB (0.625 mM) and acetylthiocholine iodide (0.625 mM) for AChE inhibitory activity assay, respectively. The hydrolysis of acetylthiocholine was monitored at 405 nm every 30 seconds for one hour. Tacrine was used as positive control with final concentration of 0.333 μM.Anti-inflammatory assay
The in vitro anti-inflammatory activity of compounds 1–5 were performed by evaluating the inhibition of nitric oxide (NO) level in lipopolysaccharide (LPS)-induced RAW264.7 cells.26 The cells were cultured in RPMI 1640 medium in 96-well plates at a density of 1 × 105 cells per well for 24 h, and LPS (Sigma-Aldrich, St. Louis, MO, USA) was added to the medium at a concentration of 1 μg mL−1 to induce inflammation, with L-NMMA used as the positive control. After 24 h, the cell culture supernatant was mixed with Griess reagent. The absorbance was measured at 570 nm by a microplate reader. The nitrite concentrations were calculated according to the method reported by Jin et al.27 After we removed the cell culture supernatant out for NO examination, the cell viability by comparing to living cells in untreated groups was calculated to determine the cytotoxicity of tested compounds by MTS assays.ECD calculations
The initial conformational distributions search were performed using CONFLEX software with the Merck molecular force-field (MMFF) 94 s force-field 5 kcal mol−1 above the ground state. The conformers were confirmed as energetic local minima through analytical inspection of their vibrational frequencies, the absence of imaginary frequencies confirms stationary state was found. These conformers were further optimized by the density functional theory (DFT) using Gaussian 09 at the B3LYP/6-31g (d, p) level.28,29 The solvent effects of the methanol solution were taken into account using the polarizable continuum model (PCM). The stable conformers (5 kcal mol−1 energy threshold) obtained were submitted to ECD calculation at the B3LYP/6-31g (d, p) level. The calculated ECD spectra were produced by SpecDis 1.64 software and compared to the experimental data.25Optical rotation calculations
Each isomer was optimized using DFT at the B3LYP/6-31g (d, p) level in the Gaussian 09 program. Methanol was used as a solvent with the polarizable continuum solvent model (PCM). Then, the optimized isomer was calculated using TDDFT/GIAOs at the B3LYP/6-31g* in the Gaussian 09 program to generate its specific rotation.30Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by grants from the Natural Science Foundation of China (No. 81860623), the Program for Innovative Research Team of Yunnan Province (202105AE160006), the project of Yunling Scholars of Yunnan province, a grant from the Science and Technology Project of Yunnan Provincial Department of Science and Technology (No. 202101AT070629), the Open Project of Yunnan Clinical Medical Center (2020LCZXKF-HX02) and the Science and Technology project of Yunnan Province (202101AY070001-246). Authors thank Advanced Analysis and Measurement Center of Yunnan University for the sample testing service.References
- A. H. Aly, A. Debbab and P. Proksch, Pharmazie, 2013, 68, 499–505 CAS.
- S. Kusari, C. Hertweck and M. Spiteller, Chem. Biol., 2012, 19, 792–798 CrossRef CAS PubMed.
- G. Li, S. Kusari, M. Lamshöft, A. Schüffler, H. Laatsch and M. Spiteller, J. Nat. Prod., 2014, 77, 2335–2341 CrossRef CAS PubMed.
- C. Klemke, S. Kehraus, A. D. Wright and G. M. König, J. Nat. Prod., 2004, 67, 1058–1063 CrossRef CAS PubMed.
- G. Strobel, B. Daisy, U. Castillo and J. Harper, J. Nat. Prod., 2004, 67, 257–268 CrossRef CAS PubMed.
- J. Fischer, V. Schroeckh and A. A. Brakhage, Gene Expression Systems in Fungi: Advancements and Application, Springer International Publishing, Springer, 2016, pp. 253–273. Search PubMed.
- S. Chen, H. Li, Y. Chen, S. Li, J. Xu, H. Guo, Z. Liu, S. Zhu, H. Liu and W. Zhang, Bioorg. Chem., 2019, 86, 368–374 CrossRef CAS PubMed.
- A. H. Aly, R. Edrada-Ebel, I. D. Indriani, V. Wray, W. E. G. Müller, F. Totzke, U. Zirrgiebel, C. Schächtele, M. H. G. Kubbutat, W. H. Lin, P. Proksch and R. Ebel, J. Nat. Prod., 2008, 71, 972–980 CrossRef CAS PubMed.
- X. Lu, X.-Y. Tang, H.-X. Wang, W.-J. Huang, W.-X. Feng and B.-M. Feng, Bioorg. Chem., 2021, 116, 105309 CrossRef CAS PubMed.
- F. Li, Y. Tang, W. Sun, J. Guan, Y. Lu, S. Zhang, S. Lin, J. Wang, Z. Hu and Y. Zhang, Bioorg. Chem., 2019, 92, 103279 CrossRef CAS PubMed.
- M. M. Gamboa-Angulo, F. A. lejos-González, F. Escalante-Erosa, K. García-Sosa and L. M. Pea-Rodríguez, Novel dimeric metabolites from Alternaria tagetica, J. Nat. Prod., 2000, 63, 1117–1120 CrossRef CAS PubMed.
- C. J. Zheng, C. L. Shao, Z. Y. Guo, J. F. Chen, D. S. Deng, K. L. Yang, Y. Y. Chen, X. M. Fu, Z. G. She, Y. C. Lin and C. Y. Wang, J. Nat. Prod., 2012, 75, 189–197 CrossRef CAS PubMed.
- L. M. Abreu, R. K. Phipp and L. H. Pfernning, Tetrahedron Lett., 2010, 51, 1803–1805 CrossRef CAS.
- Z. Hu, W. Sun, F. Li, J. Guan, Y. Lu, J. Liu, Y. Tang, G. Du, Y. Xue, Z. Luo, J. Wang, H. Zhu and Y. Zhang, Org. Lett., 2018, 20, 5198–5202 CrossRef CAS PubMed.
- F. Li, W. Sun, J. Guan, Y. Lu, S. Zhang, S. Lin, J. Liu, W. Gao, J. Wang, Z. Hu and Y. Zhang, Org. Lett., 2018, 20, 7982–7986 CrossRef CAS PubMed.
- F. Li, Z. Ye, Z. Huang, X. Chen, W. Sun, W. Gao, S. Zhang, F. Cao, J. Wang, Z. Hu and Y. Zhang, Bioorg. Chem., 2021, 117, 105452 CrossRef CAS PubMed.
- Y. Shiono, Chem. Biodiversity, 2006, 3, 217–223 CrossRef CAS PubMed.
- J. Xu, F. Ji, J. Kang, H. Wang, S. Li, D. Q. Jin, Q. Zhang, H. Sun and Y. Guo, J. Agric. Food Chem., 2015, 63, 5805–5812 CrossRef CAS PubMed.
- X. Y. Wang, T. T. Xu, L. J. Sun, R. H. Cen, S. Su, X. Q. Yang, Y. B. Yang and Z. T. Ding, Bioorg. Chem., 2021, 114, 105148 CrossRef CAS PubMed.
- H. T. Li, L. H. Tang, T. Liu, R. N. Yang, Y. B. Yang, H. Zhou and Z. T. Ding, Bioorg. Chem., 2020, 95, 103503 CrossRef CAS PubMed.
- J. S. Puranen, M. J. Vainio and M. S. Johnson, J. Comput. Chem., 2010, 31, 1722–1732 CAS.
- B. Yang, W. Sun, J. Wang, S. Lin, X. N. Li, H. Zhu, Z. Luo, Y. Xue, Z. Hu and Y. Zhang, Mar. Drugs, 2018, 16, 110 CrossRef PubMed.
- A. H. Cory, T. C. Owen, J. A. Barltrop and J. G. Cory, Cancer Commun., 1991, 3, 207–212 CrossRef CAS PubMed.
- Y. Ren, J. C. Gallucci, X. Li, L. Chen, J. Yu and A. D. Kinghorn, J. Nat. Prod., 2018, 81, 554–561 CrossRef CAS PubMed.
- H. T. Li, L. Tang, T. Liu, R. Yang, Y. Yang, H. Zhou and Z. T. Ding, Org. Chem. Front., 2019, 6, 3847–3853 RSC.
- Y. Chen, Z. M. Liu, H. J. Liu, Y. H. Pan, J. Li, L. Liu and Z. G. She, Mar. Drugs, 2018, 16, 54 CrossRef PubMed.
- X. Jin, S. Q. Song, J. Wang, Q. Z. Zhang, F. Qiu and F. Zhao, Exp. Ther. Med., 2016, 12, 499–505 CrossRef CAS PubMed.
- M. E. Ochoa, P. Labra-Vázquez, N. Farfan and R. Santillan, Cryst. Growth Des., 2018, 5, 2795–2803 CrossRef.
- P. Labra-Vázquez, A. Z. Lugo-Aranda, M. Maldonado-Domínguez, R. Arcos-Ramos, M. D. P. Carreon-Castro, R. Santillan and N. Farfán, J. Mol. Struct., 2015, 1101, 116–123 CrossRef.
- H. Zhou, Y. B. Yang, R. T. Duan, X. Q. Yang, J. C. Zhang, X. G. Xie and Z. T. Ding, Chin. Chem. Lett., 2016, 27, 1044–1047 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03917f |
| This journal is © The Royal Society of Chemistry 2022 |