
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBarium molybdate white emitting phosphor synthesized at room temperature by co-precipitation
Soung-Soo Yia and
Jae-Yong Jung *b
*b
aDivision of Materials Science and Engineering, Silla University, Busan 46958, Republic of Korea
bResearch and Business Development Foundation, Silla University, Engineering Building, Busan 45985, Republic of Korea. E-mail: eayoung21@naver.com; Tel: +82-51-999-6441
First published on 8th August 2022
Abstract
Crystalline BaMoO4:Dy3+ and BaMoO4:Sm3+ phosphors were synthesized by co-precipitation at room temperature. The main peak (112) phase and tetragonal structure were confirmed using X-ray diffraction analysis. The lattice constant and Raman signal on d(112) were changed by the rare earth doping. A strong absorption wavelength appeared in the UV region, and BaMoO4:Dy3+ excited with UV wavelength showed a yellow spectrum. BaMoO4:Sm3+ showed a reddish orange spectrum. BaMoO4:[Sm3+]/[Dy3+] was synthesized for use as a white light phosphor, and the change in the emission characteristics of yellow, white, and reddish orange could be observed depending on the doping concentration of Sm3+ ions. The synthesized phosphor powder and PDMS polymer were mixed to form a flexible composite, and when applied on a UV-LED chip, the same color as the powder was realized, suggesting its use as an LED color filter.
1. Introduction
White light emitting phosphors doped with rare earth ions are attracting research attention for various applications such as displays, lighting, and light emitting devices.1,2 Typically, dysprosium ions are mainly used for the white light emitting phosphor. Dysprosium ions have a yellow peak (∼575 nm) and a blue peak (∼485 nm) when excited with ultraviolet light.3,4 When synthesizing Dy3+ ions into a phosphor, the intensities of the blue and yellow light emissions are adjusted to provide a perfect white color. This adjustment is necessary because the intensities of the blue light emission from the 4F9/2 → 6H15/2 transition and the yellow light emission from the 4F9/2 → 6H13/2 transition are different, because of differences in site symmetry location, depending on the host crystal when synthesized as a phosphor.There is another possible way to realize white light emission.5,6 The mainstream approach to obtain white light in existing displays and light emitting devices has been to use white light emitting diodes and tricolor display devices, appropriately mixing blue, green, red, and ultraviolet wavelengths. The representative BaMgAl10O7:Eu2+ (ref. 7) phosphor, which emits blue light, has a disadvantage in that the light emitting characteristic is deteriorated by the heat treatment process performed in oxygen atmosphere during panel manufacturing. Zn2SiO4:Mn2+,8 which is used as a green phosphor, has a problem with overlapping images because the afterimage time is too long. In the (Gd, Y)BO3:Eu3+ (ref. 9) red phosphor, the intensity of orange luminescence is stronger than the red, so there is some difficulty controlling color purity.
To compensate for these disadvantages, it would be useful to develop a phosphor doped with dysprosium ions that emits all colors rather than synthesizing and combining the phosphors of blue, green, and red respectively. To develop a phosphor doped with Dy3+ ions that has high luminous efficiency, various synthesis methods such as the solid-state reaction method,10 sol–gel method,11 hydrothermal method,12 Czochralski method,13 melt quenching method,14 and others have been used. Raju et al. synthesized a Gd3Al5O12 phosphor doped with Dy3+ ions by solvothermal synthesis and reported that the ratio of yellow and blue emission intensity was dependent on the concentration ratio of Dy3+ ions, and thus could be controlled.15 Kuang et al. prepared a SrSiO3 phosphor doped with Dy3+ ions using a solid-state reaction method. After irradiating the phosphor with a 254 nm ultraviolet lamp for 5 minutes and removing it, white light emission was observed for more than 1 hour. The white light emission was caused by a mixture of blue light emission at 480 nm and yellow light emission at 572 nm. Excited electrons in the electron trap site continuously transfer energy to the 4F9/2 energy level of Dy3+ ions, so light emission occurs for the long time reported.16
Li et al. prepared a NaLa(MoO4)2:Dy3+ phosphor with a particle size of about 1 μm with a hydrothermal method, and observed a yellow signal at 574 nm and a blue emission spectrum at 486 nm when excited at 390 nm and 456 nm. The maximum luminescence intensity was observed when the concentration of Dy3+ ions was 0.02.17
A case of co-doping with Sm3+ ions to compensate for the insufficient red color of Dy3+ ions was also reported for preparing white light phosphors. Fan et al. synthesized a warm-white light-emitting phosphor using a solid-state reaction method to co-dope Sm3+ and Dy3+ ions into a BaY2ZnO5 host matrix. It was observed that the color coordinates shifted from cold white to warm white depending on the doping amount of Sm3+ ions.18 Yu et al. co-doped Sm3+ and Dy3+ ions with crystalline Sr2SnO4 as a host matrix and the synthesized phosphor exhibited enhanced red emission compared to a sample single doped with Dy3+ upon excitation at 254 nm, after being calcined at 900 °C.19 In the present study, a barium molybdate (BaMoO4) phosphor with excellent thermal and chemical stability20 was co-doped with Dy3+ and Dy3+ and Sm3+ ions at room temperature by co-precipitation without additional heat treatment, an easy synthesis method. The structure and emission characteristics of the synthesized phosphor were subsequently investigated. By mixing the synthesized phosphor with a polymer to make a flexible composite, the phosphor could be used as an LED color filter.
2. Experimental
2.1. Synthesis of BaMoO4 and BaMoO4 phosphors by co-precipitation
Starting materials: barium acetate ((CH3COO)2Ba, Sigma-Aldrich), sodium tungstate (Na2MoO4·2H2O, Sigma-Aldrich), dysprosium(III) nitrate (Dy(NO3)3·5H2O, Dy3+), and samarium(III) nitrate (Sm(NO3)3·5H2O, Sm3+).10 mmol of (CH3COO)2Ba was dissolved in beaker “A” containing 100 mL of distilled water. In beaker “B”, 10 mmol of Na2MoO4·2H2O was dissolved in 10 mL distilled water (Fig. 1). The solution that was completely dissolved in beaker “B” was slowly poured into the stirring beaker “A” and remained there for about 20 minutes. After that, powder was recovered using a centrifuge (4000 rpm, 5 min), and the powder was prepared by rinsing with distilled water 3 times to remove any remaining sodium. The white powder was dried in an oven at 80 °C for 16 hours. The phosphor was synthesized with BaMoO4 as a host, the precursor was made by simultaneously adding Dy(NO3)3·5H2O and Er(NO3)3·5H2O or Sm(NO3)3·5H2O to beaker “A”. The BaMoO4:[Sm3+]/[Dy3+] phosphor was synthesized. Rare earth [RE] ions of Dy3+ were fixed at 0.05 mmol, and the amount of added Sm3+ was changed ([Sm3+]/[Dy3+] ∼ 0.1, 0.2, 0.3, 0.4, 0.6, 0.8, 1).21
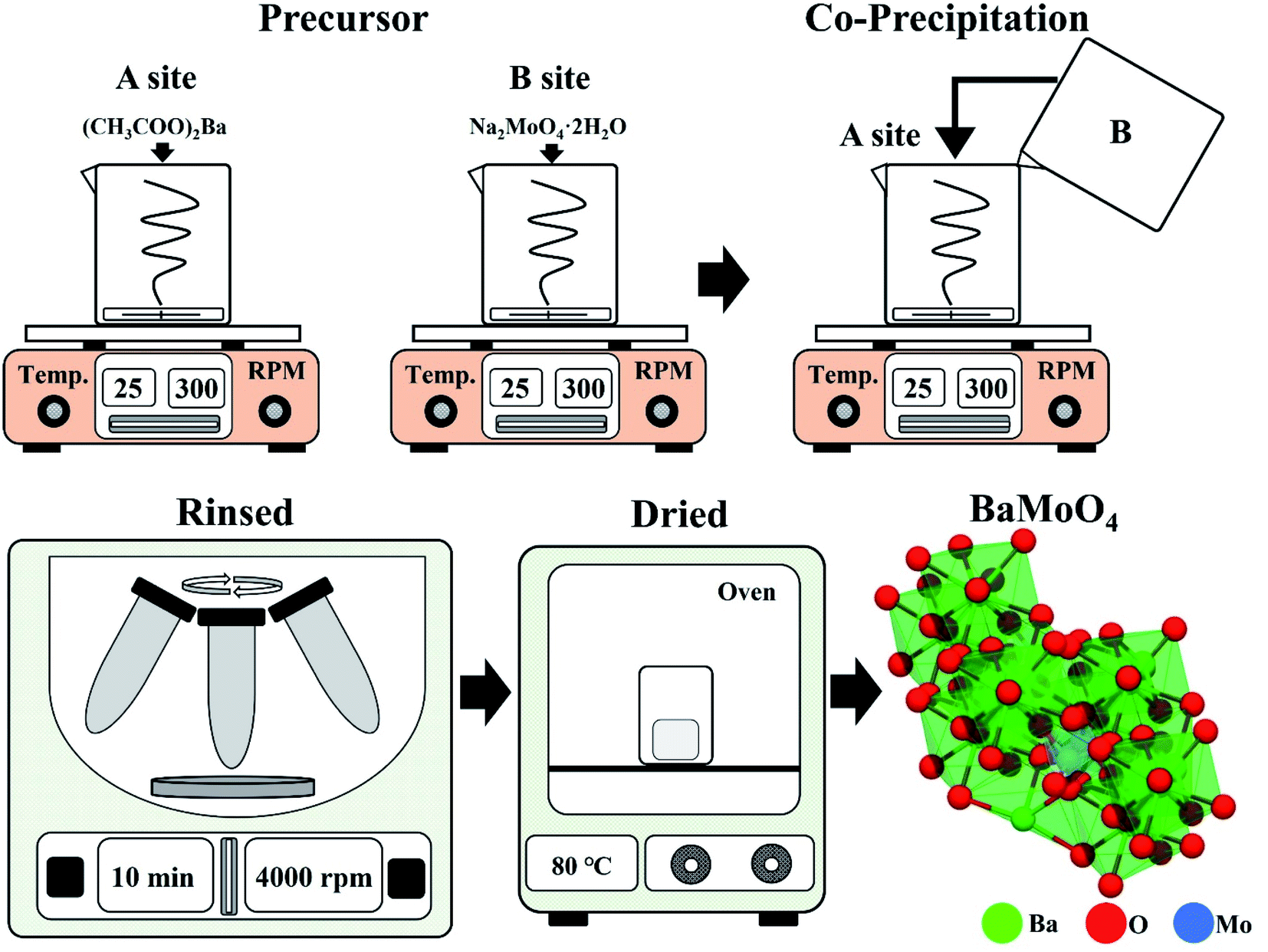 | ||
| Fig. 1 Schematic of co-precipitation experimental process. | ||
2.2. Characterization
The crystal structure of the synthesized phosphor powder was measured using an X-ray diffraction apparatus (XRD, X'Pert PRO MPD, 40 kV, 30 mA) using Cu-Kα radiation (wavelength: 1.5406 Å) at a scan rate of 4° per minute at a diffraction angle of 10° to 70°. The size and microscopic surface shape of the crystal grains were photographed with a field emission scanning electron microscope (FE-SEM, CZ, MIRA I LMH, TESCAN), and Raman spectroscope (JP/NRS-3300, 532 nm 100 mW soli-state primary laser).2.3. Fabrication of the LED color flexible composite
The synthesized BaMoO4:Dy3+, BaMoO4:Sm3+, and BaMoO4:[Sm3+]/[Dy3+] ∼ 0.1 phosphors were mixed at 0.1 g each with 1 g of polydimethylsiloxane (PDMS) and then cast in a square mold. After curing in an oven at 80 °C for 2 hours, the unique luminescent color was checked with a UV lamp, and the color reproducibility was checked by placing it on the LED chip.3. Results and discussion
3.1. Characteristics of the rare earth single doped BaMoO4
Fig. 2(a) shows the XRD diffraction patterns of a sample doped with crystalline BaMoO4 as a host material, rare earth ions single doped BaMoO4:Dy3+, and BaMoO4:Sm3+ synthesized at room temperature, respectively. Regardless of the rare earth ions doping, the samples matched BaMoO4 of ICDD no. 00-008-0455 card and had a tetragonal structure. The main peak (112) phase was strongly detected, and peaks (004), (200), (202), (114), (204), (220), (116), (312), and (224) were observed. The MXO4 (M = Ba, Sr and Ca, X = Mo and W) nanoparticles precipitated—M2+ cations as electron pair acceptors (Lewis acid) and reacted with XO42− anions as electron pair donors (Lewis base). The reaction between these two species (M2+ ← :XO42−) resulted in bonding. The lowest molecular orbital energy of the Lewis acid interacted with the highest molecular orbital energy of the Lewis base, and MXO4 nanoparticles were finally synthesized.22 To investigate the changes in crystal structure due to the rare earth doping, the lattice constant was calculated using the signal position on the (112) plane, which is the main peak, and substituting it into Bragg's equation,23 as shown in Fig. 2(b). Rare earth ions with a relatively large ionic radius were doped,24,25 and a shift in the peak of the phase (112) was observed. The lattice constant of phase d(112) was increased by the rare earth ions located in the crystal lattice of BaMoO4. The full width at half maximum (FWHM) of the (112) phase was also increased by doping with rare earth ions (Fig. 2(c)). | ||
| Fig. 2 (a) XRD patterns, (b) change in d(112) spacing, (c) change in FWHM, and (d) Raman shift of synthesized BaMoO4 and BaMoO4:RE3+ powders. | ||
Fig. 2(d) shows the Raman shift spectrum obtained by excitation of the sample with a 532 nm laser, to observe the change in molecular frequency caused by the rare earth ions located in the doped BaMoO4 crystal lattice. Raman vibration modes can be divided into two groups: external and internal modes. The first one corresponds to the motion of phonons and Ba2+ cations in the rigid [MoO4]2− tetrahedron unit. The second belongs to [MoO4]2− vibration within a tetrahedral unit, considered to be the steady state of the center.26 The synthesized BaMoO4 had frequencies of 328, 362, 793, 840, and 893 cm−1. A slight change in the position of the frequencies of the BaMoO4:Dy3+ and BaMoO4:Sm3+ samples doped with rare earth ions was observed, suggesting that the vibration of molecules was affected by energy transferred from the outside by doping with rare earth ions.27 Fig. 3 shows the spectra of the absorption and emission characteristics of the BaMoO4:Dy3+ and BaMoO4:Sm3+ phosphor powders synthesized by doping with Dy3+ or Sm3+ ions, respectively. Under the 572 nm spectrum of the BaMoO4:Dy3+ sample, a broad spectrum distributed over a 230–330 nm region with a peak at 291 nm were observed, as well as several narrow absorption spectra appearing in the 330–400 nm region. Absorbance with a rather wide bandwidth is a transition signal generated by a charge transfer band (CTB) between Dy3+ cations and O2− anions, and absorbance with a narrow bandwidth is a 4f–4f transition signal generated within the 4F9 electron arrangement of Dy3+ ions.28 350 nm, which was the strongest signal among the absorption peaks with a narrow bandwidth, is a signal generated while transitioning from the 6H15/2 level, which is the ground state of the Dy3+ ions in the host lattice, to the 4P7/2 energy level, which is the excited state. In contrast, wavelengths of 323 (6H15/2 → 6P3/2), 351 (6H15/2 → 6P7/2), 365 (6H15/2 → 6P5/2), and 387 (6H15/2 → 4G11/2) nm with relatively weak absorption intensity were observed. When the BaMoO4:Dy3+ sample was excited at 291 nm, a yellow spectrum with a strong signal at 572 nm, a blue emission spectrum with a peak at 480 nm, and a red spectrum at 659 nm with a relatively weak signal were observed. These emission signals are emission spectra resulting from the 4F9/2 → 6Hj transition of Dy3+ ions.29 The yellow emission spectrum is at the 4F9/2 → 6H13/2 transition with j = 13/2, the blue emission at the 4F9/2 → 6H15/2 transition with j = 15/2, and the red emission at j = 11/2 known as the 4F9/2 → 6H11/2 transition signal.30 It is known that the size of the emission wavelength and intensity varies depending on the degree the local environment around the rare earth ion doped in the host lattice in the phosphor is deformed in the inversion symmetry. The asymmetry ratio is defined as the ratio of the light emission intensity due to the electric dipole transition and the light emission intensity due to the magnetic dipole transition. It is known that the electric dipole transition, which generates a yellow light emission signal at 572 nm when Dy3+ ions are doped, belongs to a sensitive transition, and is significantly affected by the external environment of the Dy3+ ions. Conversely, the blue emission spectrum at 480 nm is generated by magnetic dipole transition and is hardly affected by the strength of the crystal field around the Dy3+ ions. That is, when blue light emission due to magnetic dipole transition is the main peak, Dy3+ ions are located at an inversion symmetric site, and when yellow light emission due to electric dipole transition is strong, Dy3+ ions are located at non-inversion symmetry sites.31,32 In this study, since the intensity of 572 nm, which is yellow light emission, was stronger than that of 480 nm, which is the blue light emission, the Dy3+ ions located in the BaMoO4 crystal were accordingly located in non-inversion symmetry (Fig. 3(a)). Fig. 3(b) shows the absorption and emission spectra of the BaMoO4:Sm3+ phosphor single doped with Sm3+ ions. An absorption signal by CTB between Sm3+ cations and O2− anions with a wide bandwidth at 230–340 nm and a peak at 286 nm, and several absorption signals at 340–400 nm with a rather narrow bandwidth, were observed. Absorption signals with relatively narrow bandwidths had peaks at 344 (6H5/2 → 4K17/2), 361 (6H5/2 → 4H7/2), and 375 (6H5/2 → 6P7/2) nm due to f–f transitions, which were generated when they transitioned from 6H5/2 in the bottom state of Sm3+ ions, respectively.33 When the BaMoO4:Sm3+ sample was excited at 286 nm, spectra with peaks at 560, 597, 641, and 702 nm were observed. A strong red emission spectrum was exhibited, and these are the peak emission spectra due to the 4G5/2 → 6Hj (j = 5/2, 7/2, 9/2) transition of Sm3+. The yellow and orange fluorescence spectra are magnetic dipole transition signals generated by the 4G5/2 → 6H5/2 and 4G5/2 → 6H7/2 transitions, and the red fluorescence spectrum is a signal due to the 4G5/2 → 6H9/2 electric dipole transition.34,35 In the synthesized BaMoO4:Sm3+ phosphor, the ratio of the emission intensity of the spectrum emitted by the magnetic dipole transition and the electric dipole transition was relatively strong due to the reddish orange electric dipole transition. In general, it is known that the main emission wavelength intensity produced by rare earth ions is determined by competition between the magnetic dipole transition and electric dipole transition.36 As in the Dy3+-doped specimen, if the light emission intensity due to magnetic dipole transition is strong, it is located in inversion symmetry. The Sm3+ doped sample also has a stronger luminescence intensity because the red (641 nm) electric dipole transition is greater than that of the orange (597 nm) light emission, so Sm3+ ions located in BaMoO4 crystals are located in non-inversion symmetric sites.
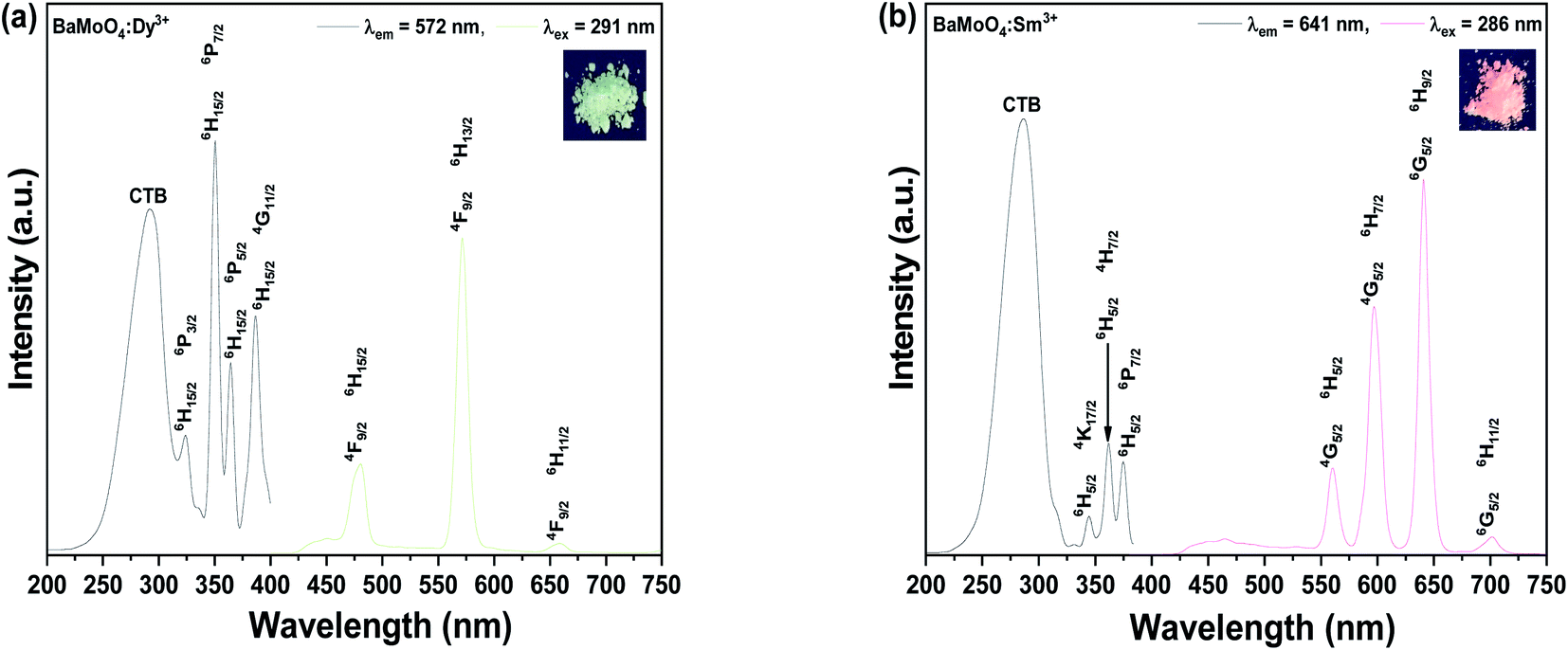 | ||
| Fig. 3 Photoluminescence properties of (a) BaMoO4:Dy3+ and (b) BaMoO4:Sm3+. | ||
3.2. Characteristics of rare earth co-doped BaMoO4
To prepare a white light-emitting phosphor, Dy3+ and Sm3+ ions were co-doped and synthesized as BaMoO4:[Sm3+]/[Dy3+]. To confirm the crystal structure of the synthesized phosphor, after the pattern according to the Sm3+ concentration change X-ray diffraction analysis is shown in Fig. 4(a). The synthesized phosphor was consistent with the ICDD no. 00-008-0455 card, and the tetragonal structure of BaMoO4 was confirmed. A signal on the main peak (112) was strongly detected in the synthesized BaMoO4:[Sm3+]/[Dy3+] phosphor, and (004), (200), (112), (213), (204), (220), (116), (312), (224) phases were observed. The amount of added Dy3+ ions was fixed, and as the doping concentration of Sm3+ ions increased, a small shoulder was observed on the left side of the main peak (112). From the BaMoO4:[Sm3+]/[Dy3+] ∼ 6 sample, a shoulder was observed at the position indicated by diamonds. This was confirmed to be the Sm2O3 phase, which appeared due to the excessive doping of Sm3+ ions and oxidized rare earth ions. To investigate the particle size and formation of the BaMoO4:[Sm3+]/[Dy3+] phosphor synthesized at room temperature by co-precipitation, FE-SEM imaging was performed, and the image is shown in Fig. 4(b). The synthesized particles of the phosphor powder had a long octahedral shape in the longitudinal direction. The size was about 1.08 μm in the horizontal direction and about 2.63 μm in the longitudinal direction. Muhammad et al. synthesized crystalline Co3O4 nanoparticles at a temperature of 60 °C in a process like the co-precipitation method used in this study and observed plate-shaped particles of several to several tens of microns using FE-SEM.37 F. Sedighi et al. synthesized silver doped crystalline SrWO4 at 70 °C by co-precipitation to observe the shape of a flower and particles such as a star, and reported a change in the shape of the particles depending on the amount of added Na(B(C6H5)).38 Fig. 5(a) illustrates the PL spectrum of the co-doped BaMoO4:[Sm3+]/[Dy3+] phosphor after fixing the concentration of Dy3+ ions and adjusting the amount of added Sm3+ ions. When the samples were excited at 291 nm, a signal at 572 nm, the main peak of Dy3+, was strongly detected, but the intensity of the signal decreased as the doping concentration of Sm3+ ions increased. From the BaMoO4:[Sm3+]/[Dy3+] ∼ 0.3 sample, blue light emission at 480 nm (4F9/2 → 6H15/2) and yellow light emission at 572 nm (4F9/2 → 6H13/2) of Dy3+ ions, orange light emission at 597 nm (4G5/2 → 6H7/2) and red light emission at 641 nm (4G5/2 → 6H9/2) of Sm3+ ions were observed together. As shown in Fig. 5(b), the yellow signal at 572 nm, the main peak of Dy3+, decreased as the concentration of Sm3+ ions increased, but the red signal at 641 nm increased. This phenomenon means that the luminescence energy was converted from Dy3+ ions located in the BaMoO4, the host crystal, to Sm3+ ions. The energy transfer from Dy3+ ions to Sm3+ ions can be expressed by eqn (1).39| η = 1 − I/I0 | (1) |
 | ||
| Fig. 4 (a) XRD patterns of BaMoO4:[Sm3+]/[Dy3+] according to Sm3+ ion concentration and (b) FE-SEM image of BaMoO4:[Sm3+]/[Dy3+]. | ||
 | ||
| Fig. 5 BaMoO4:[Sm3+]/[Dy3+] of (a) PL spectra under 291 nm, (b) change of 572 nm (Dy3+) and 641 nm (Sm3+), (c) energy transfer efficiency, and (d) CIE coordinate of the BaMoO4 phosphors. | ||
The critical distance RC was calculated using the following equations:40
| RC ≈ 2[3V/4πxcN]1/3 | (2) |
 | ||
| Fig. 6 Schematic energy level structure of the Dy3+ and Sm3+ ions. | ||
3.3. Flexible composite for LED color filter
When the synthesized BaMoO4:Dy3+, BaMoO4:Sm3+, BaMoO4:[Sm3+]/[Dy3+] ∼ 0.1 phosphor powder was placed under a UV lamp, yellow, white, and reddish orange emission colors could be observed as shown in Fig. 7. The BaMoO4:Dy3+ phosphor showed a yellow emission wavelength, but green emission was seen in photography. It looked yellowish close to white with the naked eye, but it was photographed as green due to a camera filter. Composites were made by mixing individual powders with PDMS, a silicon-based polymer, which were easily bent and exhibited a unique color emission like that of the powder. To determine whether the manufactured composite could be used as an LED filter, a voltage of about 10 V was applied to the LED chip by placing the composite on a UV-LED chip. The manufactured composite exhibited colors similar to those in UV lamps, and this suggests that the BaMoO4 phosphor synthesized by doping with rare earth ions could be used as an LED color filter. | ||
| Fig. 7 Photographs of synthesized phosphors powder and flexible composite under UV lamp, and composite applied on the UV-LED chip. | ||
4. Conclusion
Crystalline BaMoO4 was synthesized at room temperature by co-precipitation. Using the BaMoO4 as a phosphor host, Dy3+ and Sm3+, which are rare earth ions, were doped to synthesize the phosphor. Diffraction analysis confirmed a tetragonal structure, and a strong diffraction peak on the main peak (112). After doping with rare earth ions with relatively large ionic radii, the lattice constant of the (112) phase and the position of the Raman signal were changed. BaMoO4:Dy3+ and BaMoO4:Sm3+ phosphors excited with UV wavelengths with strong absorbance in the UV region showed yellow and reddish orange spectra. A sample was prepared, BaMoO4:[Sm3+]/[Dy3+], by co-doping with Dy3+ and Sm3+ ions for white light fluorescence, and the main peak (112) phase was also confirmed by X-ray diffraction analysis. However, as the concentration of Sm3+ ions was increased, a secondary phase Sm2O3 peak was observed. When the sample was excited at 291 nm, the intensity of the reddish orange emission spectrum in the yellow emission spectrum increased, as the concentration of Sm3+ ions increased. The synthesized phosphor, BaMoO4:[Sm3+]/[Dy3+] ∼ 0.1 showed white light emission, and a composite was prepared by mixing it with a PDMS polymer for use as an LED color filter. The prepared composite was easily bent, and when applied on a UV-LED chip, the light emission of yellow, white, and reddish orange, which are the intrinsic colors of the synthesized phosphor, were confirmed.Data availability statement
The data presented in this study are available on request from the corresponding author.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1F1A1072676).References
- G. B. Nair, H. C. Swart and S. J. Dhoble, Prog. Mater. Sci., 2020, 109, 100622, DOI:10.1016/j.pmatsci.2019.100622.
- G. Li, N. Yang, J. Zhang, J. Si, Z. Wang, G. Cai and X. Wang, The Non-Concentration-Quenching Phosphor Ca3Eu2B4O12 for WLED Application, American Chemical Society (ACS), 2020 Search PubMed.
- H. Kuo, C. Hung, H. Chen, K. Chen, C. Wang, C. Sher, C. Yeh, C. Lin, C. Chen and Y. Cheng, Opt. Express, 2011, 19(4), A930–A936, DOI:10.1364/OE.19.00A930.
- N. T. Tran and F. G. Shi, J. Lightwave Technol., 2008, 26, 3556–3559, DOI:10.1109/JLT.2008.917087.
- Y. Lin, M. Karlsson and M. Bettinelli, Top. Curr. Chem., 2016, 374, 21, DOI:10.1007/s41061-016-0023-5.
- L. Yang, Z. Mu, S. Zhang, Q. Wang, D. Zhu, Y. Zhao, D. Luo, Q. Zhang and F. Wu, J. Mater. Sci.: Mater. Electron., 2018, 29, 6548–6555, DOI:10.1007/s10854-018-8638-7.
- S. Zhang, H. Liu, D. Pan, J. Tian, Y. Liu and A. A. Volinsky, RSC Adv., 2014, 5, 1113–1119, 10.1039/c4ra12879f.
- R. P. Sreekanth Chakradhar, B. M. Nagabhushana, G. T. Chandrappa, K. P. Ramesh and J. L. Rao, J. Chem. Phys., 2004, 121, 10250–10259, DOI:10.1063/1.1808420.
- C. Xiangzhong, Z. Weidong, Z. Xiying, X. Tian, L. Zhen, Y. Zhijian, Z. Chunlei and H. Xiaowei, Red Emitting Phosphor (Y, Gd)BO3:Eu3+ for PDP Prepared by Complex Method, Elsevier BV, 2006 Search PubMed.
- I. P. Sahu, D. P. Bisen and N. Brahme, Luminescence, 2015, 30, 526–532, DOI:10.1002/bio.2771.
- L. Jiang, C. Chang, D. Mao and B. Zhang, Mater. Lett., 2004, 58, 1825–1829, DOI:10.1016/j.matlet.2003.11.014.
- L. Han, G. Liu, X. Dong, J. Wang, X. Wang and Y. Yang, J. Mater. Sci.: Mater. Electron., 2017, 28, 16519–16526, DOI:10.1007/s10854-017-7564-4.
- G. Dominiak-Dzik, W. Ryba-Romanowski, R. Lisiecki, P. Solarz, B. Macalik, M. Berkowski, M. Głowacki and V. Domukhovski, Cryst. Growth Des., 2010, 10, 3522–3530, DOI:10.1021/cg100429b.
- L. Zur, J. Mol. Struct., 2013, 1041, 50, DOI:10.1016/j.molstruc.2013.02.036.
- G. Raju, J. Y. Park, H. C. Jung, H. K. Yang, B. K. Moon, J. H. Jeong and J. H. Kim, Opt. Mater., 2009, 31, 1210–1214, DOI:10.1016/j.optmat.2008.12.015.
- J. Kuang, Y. Liu and J. Zhang, J. Solid State Chem., 2006, 179, 266–269, DOI:10.1016/j.jssc.2005.10.025.
- L. Li, W. Zi, G. Li, S. Lan, G. Ji, S. Gan, H. Zou and X. Xu, J. Solid State Chem., 2012, 191, 175–180, DOI:10.1016/j.jssc.2012.03.003.
- B. Fan, J. Liu, W. Zhao and L. Han, J. Lumin., 2020, 219, 116887, DOI:10.1016/j.jlumin.2019.116887.
- X. Yu, X. Xu and J. Qiu, Mater. Res. Bull., 2011, 46, 627–629, DOI:10.1016/j.materresbull.2010.12.028.
- V. Nassif, R. E. Carbonio and J. A. Alonso, J. Solid State Chem., 1999, 146, 266–270, DOI:10.1006/jssc.1999.8352.
- J. Jung, J. Kim, Y. Shim, D. Hwang and C. S. Son, Materials, 2020, 13, 4165, DOI:10.3390/ma13184165.
- T. Thongtem, S. Kungwankunakorn, B. Kuntalue, A. Phuruangrat and S. Thongtem, J. Alloys Compd., 2010, 506, 475–481, DOI:10.1016/j.jallcom.2010.07.033.
- C. G. Pope, J. Chem. Educ., 1997, 74, 129, DOI:10.1021/ed074p129.
- F. Boulc'h and E. Djurado, Solid State Ionics, 2003, 157, 335–340, DOI:10.1016/S0167-2738(02)00230-8.
- F. F. Sene, J. R. Martinelli and L. Gomes, J. Non-Cryst. Solids, 2004, 348, 63–71, DOI:10.1016/j.jnoncrysol.2004.08.127.
- V. Panchal, N. Garg and S. M. Sharma, J. Phys.: Condens. Matter, 2006, 18, 3917–3929, DOI:10.1088/0953-8984/18/16/002.
- M. Guo, J. Lu, Y. Wu, Y. Wang and M. Luo, Langmuir, 2011, 27, 3872–3877, DOI:10.1021/la200292f.
- B. Vengala Rao, K. Jang, Ho S. Lee, S. Yi and J. Jeong, J. Alloys Compd., 2010, 496, 251–255, DOI:10.1016/j.jallcom.2009.12.175.
- F. Zhang, Y. Wang, Y. Wen, D. Wang and Y. Tao, Opt. Mater., 2011, 33, 475–479, DOI:10.1016/j.optmat.2010.10.035.
- K. Mishra, S. K. Singh, A. K. Singh and S. B. Rai, Mater. Res. Bull., 2012, 47, 1339–1344, DOI:10.1016/j.materresbull.2012.03.017.
- S. Liu, S. Liu, J. Wang, P. Sun, Y. Zhong, J. H. Jeong, B. Deng and R. Yu, Mater. Res. Bull., 2018, 108, 275–280, DOI:10.1016/j.materresbull.2018.08.026.
- Y. Tian, B. Chen, B. Tian, R. Hua, J. Sun, L. Cheng, H. Zhong, X. Li, J. Zhang, Y. Zheng, T. Yu, L. Huang and Q. Meng, J. Alloys Compd., 2011, 509, 6096–6101, DOI:10.1016/j.jallcom.2011.03.034.
- M. Kumar and A. S. Rao, Opt. Mater., 2020, 109, 110356, DOI:10.1016/j.optmat.2020.110356.
- M. P. Demesh, O. P. Dernovich, N. V. Gusakova, A. S. Yasukevich, A. A. Kornienko, E. B. Dunina, L. A. Fomicheva, A. A. Pavlyuk and N. V. Kuleshov, Opt. Mater., 2018, 75, 821–826, DOI:10.1016/j.optmat.2017.12.001.
- L. Liu, J. Zhang, F. Xu, F. Du, F. Yang, J. Peng and X. Ye, J. Rare Earths, 2021, 39, 140–145, DOI:10.1016/j.jre.2020.03.004.
- G. Phaomei, W. Rameshwor Singh and R. S. Ningthoujam, J. Lumin., 2011, 131, 1164–1171, DOI:10.1016/j.jlumin.2011.02.023.
- J. Muhammad Ramzan Saeed Ashraf, Open Chem., 2019, 17, 865–873, DOI:10.1515/chem-2019-0100.
- F. Sedighi, A. Sobhani-Nasab, M. Esmaeili-Zare and M. Behpour, J. Nanostruct., 2019, 9, 331–339, DOI:10.22052/JNS.2019.02.015.
- N. Liu, L. Mei, L. Liao, J. Fu and D. Yang, Sci. Rep., 2019, 9, 1–9, DOI:10.1038/s41598-019-51915-1.
- V. R. Kharabe, A. H. Oza and S. J. Dhoble, Luminescence, 2015, 30, 432–438, DOI:10.1002/bio.2756.
- B. Bondzior, D. Stefańska, A. Kubiak and P. J. Dereń, J. Lumin., 2017, 190, 123–127, DOI:10.1016/j.jlumin.2017.05.017.
- C. Chen, C. Yu, F. Xu, Q. Li and Y. Zhang, Ceram. Int., 2021, 47, 1–9, DOI:10.1016/j.ceramint.2020.08.190.
| This journal is © The Royal Society of Chemistry 2022 |