DOI:
10.1039/D2RA03859E
(Paper)
RSC Adv., 2022,
12, 24156-24162
Structural stability, electronic, optical, and thermoelectric properties of layered perovskite Bi2LaO4I
Received
23rd June 2022
, Accepted 22nd July 2022
First published on 25th August 2022
Abstract
Layered perovskites are an interesting class of materials due to their possible applications in microelectronics and optoelectronics. Here, by means of density functional theory calculations, we investigated the structural, elastic, electronic, optical, and thermoelectric properties of the layered perovskite Bi2LaO4I within the parametrization of the standard generalized gradient approximation (GGA). The transport coefficients were evaluated by adopting Boltzmann semi-classical theory and a collision time approach. The calculated elastic constants were found to satisfy the Born criteria, indicating that Bi2LaO4I is mechanically stable. Taking into account spin–orbit coupling (SOC), the material was found to be a non-magnetic insulator, with an energy bandgap of 0.82 eV (within GGA+SOC), and 1.85 eV (within GGA+mBJ+SOC). The optical-property calculations showed this material to be optically active in the visible and ultraviolet regions, and that it may be a candidate for use in optoelectronic devices. Furthermore, this material is predicted to be a potential candidate for use in thermoelectric devices due to its large value of power factor, ranging from 2811 to 7326 μW m−1 K−2, corresponding to a temperature range of 300 K to 800 K.
Introduction
Perovskites are multifunctional materials with various fascinating properties, such as dielectric,1 pyroelectric, piezoelectric, ferroelectric,2 ferromagnetic, half-metallic,3 superconducting,4 thermoelectric,5 optoelectronic,6 photovoltaic behavior,7 etc. Perovskites are a broad family of materials and have a crystal structure similar to that of CaTiO3, the first known perovskite.8 The typical stoichiometric structure for the perovskite family is of the form ABX3, where A and B are two different types of cations bound together by an anion X.9 Generally, cation A has a larger radius which is more electropositive, having a coordination number 12, while cation B with a smaller radius are more electronegative and has a coordination number 6. B are bound to ABX3 in the form of octahedra.10,11
Layered perovskites are materials formed by the association of several perovskite-like structures. In these structures, perovskite-like slabs are separated by anionic layers.12 The layered perovskite Bi2LaO4I is a bismuth-based oxyhalide perovskite that consists of triple-fluorite blocks formed by the combination of LaO2 slabs and fluorite-type Bi2O2 layers.13 Experimentally, materials like Bi2MO4X (M = Y, La–Lu; X = Cl, Br, I) are prepared by allowing various solids to react with each other under some external conditions.14 In such materials, the valence band has a major contribution from O and possesses a unique band structure, different to that of oxyhalides of the form BiOX (X = Cl, Br, I).15 The special features of layered perovskites include an optimal and tunable bandgap,16 excellent carrier mobility,17 and their potential as candidates for microelectronics, optoelectronics,18 and photovoltaic applications.19 A recent study showed that these materials (for example, Bi2YO4Cl and Bi2LaO4Cl) have potential for visible-light photocatalysis,20,21 ferroelectricity,22 excellent photoconductivity,23 and photocatalytic activity for water splitting.24,25
The elastic properties explain the mechanical behavior of a material, which is calculated by using the second derivative of polynomial fits of total energy versus applied strain.26 The optical properties of a compound describe its interaction with electromagnetic energy, given by the complex dielectric function ε(ω), which is the sum of real ε1 and imaginary parts ε2, as given in eqn (1). The dielectric function plays a significant role in determining physical properties, and describes the collective excitation of the Fermi sea and the overall band behavior of the solid.27,28
| | |
ε(ω) = ε1(ω) + iε2(ω)
| (1) |
where
ε1 gives information about the ability of the material to store photon energy, whereas
ε2 provides information on the electron loss function.
Thermoelectric (TE) materials are very useful for converting waste energy into a useable form by converting heat into electricity, and electricity into cooling, based on the Seebeck effect and the Peltier effect.29 These materials are used as power generators,30 coolers,31 temperature sensors,32,33 optical detectors,34,35 temperature stabilizers,36 etc. The performance of layered perovskites in TE applications is directly related to the transport parameters:29 the Seebeck coefficient (S), electrical conductivity (σ), and thermal conductivity (κ) at some absolute temperature (T) and expressed by the TE figure of merit as,
| |
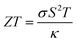 | (2) |
where
κ includes both electron (
κe) and phonon (
κl) contributions.
29,37–39 With the above-mentioned points, we were motivated to perform density functional calculations for the determination of the structural, elastic, electronic, optical, and TE properties of Bi
2LaO
4I.
Computational details
The various properties of Bi2LaO4I have been calculated using the density functional theory (DFT) approach, the quantum mechanical treatment of Kohn–Sham equations of state for many-body systems.40,41 For the computational calculations, we used the WIEN2k code, an augmented plane-wave plus local-orbital program.42,43 The standard generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof has been used for parametrization on account of the electronic exchange-correlation functional.44 In addition to this, the modified Becke–Johnson (mBJ) potential has been used for the bandgap estimation.45 An 11 × 11 × 4 k-mesh, generated by a set of 600 k-points, was used in the whole Brillouin Zone (BZ), which corresponds to 63 k-points in the irreducible BZ. The self-consistent calculations were achieved with an energy convergence criterion of 10−5 Ry and a charge convergence of 10−4e. The elastic properties were calculated using the IRelast package for tetragonal symmetry.26 The transport coefficients were computed using BoltzTraP,46 which works under the Boltzmann semi-classical equation under the constant relaxation time approximation (CRTA) and rigid band approximation (RBA).47,48 For this calculation, a dense k-mesh of 39 × 39 × 16 was sampled, generated by a set of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 k-points.
000 k-points.
Results and discussion
Structural properties
Layered perovskite Bi2LaO4I has a tetragonal crystal structure with optimized lattice parameters a = 4.046 Å, and c = 9.865 Å, belonging to space group P4/mmm. The structure consists of cationic slabs of [Bi2LaO4] separated by anionic layers of iodine (I), resulting in a layered sequence of the type –I–[Bi2LaO4]–I–; the interlayer distance is 2.32 Å. The cation ordering in this structure looks like –Bi–O2–La–O2–Bi–, where Bi is co-ordinated to both oxide and iodide layers, while La is only co-ordinated to oxide layers. The bond lengths between various atoms, such as Bi–O, La–O and Bi–I, are found to be 2.28 Å, 2.52 Å, and 3.68 Å, respectively. Fig. 1 illustrates the crystal structure of Bi2LaO4I. The calculated lattice parameters for Bi2LaO4I, Bi2LaO4Br and Bi2LaO4Cl are as listed in Table 1.
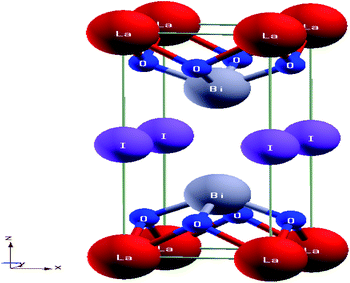 |
| | Fig. 1 The tetragonal crystal structure of layered perovskite Bi2LaO4I. | |
Table 1 The optimized lattice parameters for Bi2LaO4I, Bi2LaO4Br and Bi2LaO4Cl
| Compounds |
a (Å) |
c (Å) |
Calc. (comp.) |
| Bi2LaO4I |
4.046 |
9.865 |
Calc. |
| Bi2LaO4Br |
4.015 |
9.510 |
Calc. |
| Bi2LaO4Cl |
3.996 |
9.271 |
Calc. |
| 3.954 |
9.127 |
(Ref. 14) |
Elastic properties
The elastic properties of the material can be determined by the elastic tensor or the elastic matrix, which are different for different crystal structures. For the tetragonal symmetry (P4/mmm, type I) of layered perovskite Bi2LaO4I, only six components (C11, C12, C13, C33, C44 and C66) of the symmetric elastic tensor (C) are unique out of thirty-six components. In terms of the second-order variation of energy, the components of matrix C are given by
Here, Ve is the volume in the absence of strain (S), i.e., the equilibrium volume, E is the energy of the crystal, and Sp and Sq represent the components of strain. Particularly, for the homogeneous deformation of the crystal structure by the application of strain, the quadratic variation of energy is given by| |
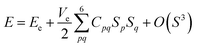 | (3) |
where Ee is the energy corresponding to volume Ve.49,50 The calculated values of the elastic constants and elastic parameters of Bi2LaO4I corresponding to volume Ve = 161.49 Å3 are listed in Tables 2 and 3, respectively.
Table 2 Unique components of the elastic tensor for Bi2LaO4I with units of GPa
| C11 |
C12 |
C13 |
C33 |
C44 |
C66 |
| 180.36 |
68.61 |
61.72 |
132.37 |
44.54 |
60.55 |
Table 3 Various calculated elastic coefficients: bulk modulus (B), shear modulus (G), Young’s modulus (Y), Pugh’s ratio (B/G), Poisson’s ratio (ν), transverse velocity (vt), longitudinal velocity (vl), average velocity (vm), Debye temperature (θD), and melting temperature (θm) of Bi2LaO4I
| B (GPa) |
G (GPa) |
Y (GPa) |
B/G |
ν |
| 96.21 |
49.41 |
126.57 |
1.94 |
0.28 |
| vt (m s−1) |
vl (m s−1) |
vm (m s−1) |
θD (K) |
θm (K) |
| 2520.11 |
4564.37 |
2808.14 |
308.26 |
1093.65 ± 300.00 |
The mechanical or elastic stability of the crystal structure is determined by the Born stability criteria. For a tetragonal crystal structure of type I, the Born stability criteria are defined as:
| C11 − |C12| > 0, C44 > 0, C66 > 0 |
| C33 > 0, 2C132 < C33(C11 + C12) |
The calculated elastic constants of Bi2LaO4I are found to satisfy the above stability criteria. Hence, Bi2LaO4I is predicted to be a stable structure. These elastic constants are used to derive expressions for the bulk and shear modulus of the solid by considering various theories. The Voigt theory expresses the bulk modulus (B) and shear modulus (G) in terms of the components of the elastic matrix, whereas the Reuss theory expresses them in terms of the components of the inverse matrix of the elastic matrix.51,52 For any material, the value of the Voigt modulus surpasses the value of the Reuss modulus. However, the actual value lies between them. To account for the true value, according to Hill theory, the actual values of the bulk and shear modulus are the average of their corresponding values derived using the Voigt and Reuss theories. In this consideration, the expressions for the Young’s modulus (Y) and Poisson’s ratio (ν) are given by
| |
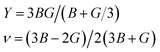 | (4) |
The magnitude of the bulk modulus reflects the reluctance of the material to change shape under the effect of applied pressure. The greater the magnitude of the bulk modulus for a material, the lower its compressibility, therefore, that material may have the ability to resist high pressure. The shear modulus characterizes the hardness of the material, i.e., its ability to withstand an applied shear force. Similarly, the Young’s modulus describes the stretchiness or compressibility of the material, indicating the stiffness of the material under the effect of uniaxial tension.52,53 Pugh’s ratio (B/G) is one of the important parameters describing the elastic nature of the material.54 The critical value of this ratio is approximately 1.75; a ratio less than 1.75 indicates a brittle nature, whereas a value above this indicates a ductile nature. The calculated value of Pugh’s ratio (1.94) suggests that the compound is ductile. Poisson’s ratio estimates the compressibility of the compound. If the ν value is close to 0.5, it represents an incompressible system.55 In this work, the determined value of Poisson’s ratio is 0.28, therefore, the material has low compressibility and is stable under deformation. Furthermore, the magnitude of Poisson’s ratio describes the nature of bonding in the compound. The typical values of Poisson’s ratio that describe covalent, ionic, and metallic materials are in the order of 0.1, 0.25, and 0.5, respectively.56 On this basis, ν for Bi2LaO4I is close to that of an ionic material. The ionic nature, ductile behavior, and Poisson’s ratio are found to be consistent with those for Bi2LaO4Cl.21 The Debye temperature (θD) is directly related to many properties, including thermal conductivity (κ). The larger the value of θD, the larger κ will be. This can be calculated from the elastic constants in terms of the average wave velocity (vm).57 The average velocity is obtained from the longitudinal 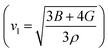 and transverse
and transverse 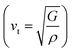 components of the velocity. The calculated values of θD and the velocities are given in Table 3. The θD value is found to be large. We also calculated the melting temperature as 1093.65 ± 300.00 K.
components of the velocity. The calculated values of θD and the velocities are given in Table 3. The θD value is found to be large. We also calculated the melting temperature as 1093.65 ± 300.00 K.
Electronic properties
To determine the ground state of the compound, we considered two types of configurations: non-magnetic and magnetic (La↑, La↓). The total energy of each state was found to be the same up to the convergence limit for the calculation of total energy, with zero magnetic moments in the case of spin-polarized calculations. This suggests that the ground state is non-magnetic. Furthermore, the symmetrical nature of the density of states (DOS) in the spin-up and spin-down channels favors the non-magnetic ground state. The total DOS calculated using the GGA+mBJ functional and considering the effect of spin–orbit coupling (SOC) are shown in Fig. 2. The main contributions in the valence region near the Fermi level (Ef) come from the 5p orbital of iodine and the 2p orbital of oxygen, whereas the contribution in the conduction region comes from the 6p orbital of bismuth, as shown in Fig. 3 (left). The peaks in the conduction region at around 5 eV are due to the 5d orbital of lanthanum. In both the total DOS and partial DOS, the effect of SOC is significant and shifts the electronic states near the Fermi level. From the band structure calculations along the Γ–X–M–Γ–Z–R–A–Z high-symmetry points within GGA+mBJ and GGA+mBJ+SOC (see Fig. 3 (right)), the insulating state is obtained with energy bandgaps of 2.17 eV and 1.85 eV, respectively. The bandgaps obtained with various functionals are reported in Table 4. Due to SOC, the bands in the conduction region are pushed down towards the Fermi level (Ef), thereby reducing the bandgap noticeably. In both cases, the top of the valence band lies at X and R, and the bottom of the conduction band lies at Γ, therefore, the material is predicted to be an indirect bandgap insulator.
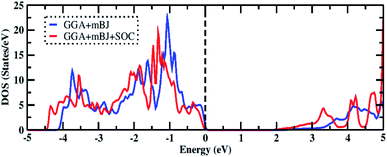 |
| | Fig. 2 The total DOS of Bi2LaO4I using the GGA+mBJ and GGA+mBJ+SOC methods. The vertical dotted line represents the Fermi level. | |
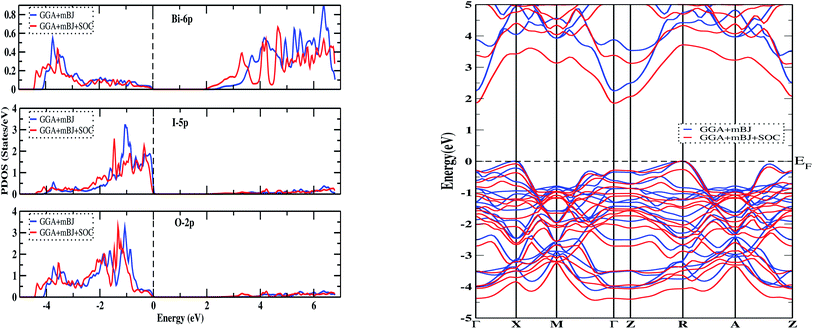 |
| | Fig. 3 The partial DOS (left) and band structure (right) of Bi2LaO4I using the GGA+mBJ and GGA+mBJ+SOC methods. The vertical and horizontal dotted lines represent the Fermi level. | |
Table 4 Bandgap (in eV) of Bi2LaO4I for various functionals
| Functional |
GGA |
GGA+SOC |
GGA+mBJ |
GGA+mBJ+SOC |
| Bandgap |
1.2 |
0.82 |
2.17 |
1.85 |
Optical properties
The two components of the dielectric function and other optical constants were calculated using the Kramers–Kronig relations.58 The optical parameters: refractive index n(ω), absorption coefficient α(ω), electron energy loss function L(ω), reflectivity R(ω), and optical conductivity σ(ω) were calculated using components (ε1 and ε2) of the dielectric function.28,59 Due to the tetragonal symmetry of Bi2LaO4I, there are only two independent components (x and z) of the optical elements. Fig. 4 (left) demonstrates the fluctuation of the computed components of the dielectric function in response to incident electromagnetic energy. The slow rise of ε1(ω) when increasing the energy from 0 to 2 eV is an indication of interactions between electrons in the material and photons. The peak represents an interband transition of electrons from the valence region to the conduction region. Following this, ε1(ω) drops as the photon energy increases. It also has zero values and negative values, which may be due to the plasmonic type oscillation. The static values of ε1(ω) in the x and z-directions are ∼7 and ∼5.8, respectively. The material is anisotropic in the entire photon energy range. The complex part of the dielectric function describes optical absorption and plasmon excitation in the system. There is a rapid increase in ε2(ω) in the energy range of 3 to 5.20 eV after the threshold energy. In that energy range, there is one sharp peak at 5.2 eV. Upon further increasing the photon energy, ε2(ω) decreases with some fluctuations. The peak values of both parts of the dielectric function are found to lie in the visible and ultraviolet regions, and the optical response occurs in the range of 0.50 to 9 eV.
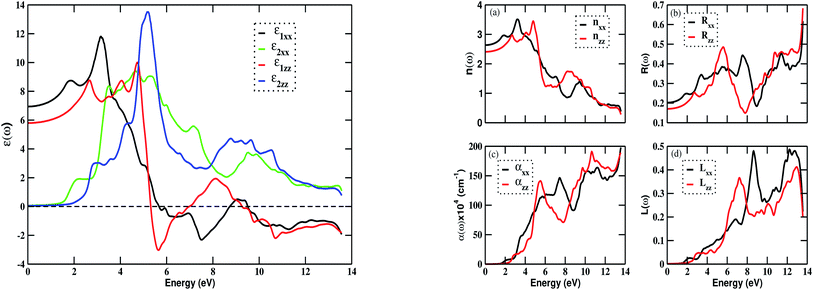 |
| | Fig. 4 The variation of dielectric function (left) and optical parameters (right): (a) refractive index n(ω), (b) reflectivity R(ω), (c) absorption coefficient α(ω), and (d) electron energy loss function L(ω) of Bi2LaO4I as a function of incident photon energy. | |
The plots of the frequency-dependent optical parameters: n(ω), R(ω), α(ω), and L(ω) are shown in Fig. 4 (right). The refractive index function is also a complex function whose real part gives a refractive index, whereas the imaginary part provides an extinction coefficient. In the visible and ultraviolet regions of the frequency, the refractive index increases with increasing photon energy, peaking at about 3.20 eV in the x-direction and 4.8 eV in the z-direction, as shown in Fig. 4(a) (right). Here, we noticed that the refractive index follows the trend of ε1(ω). The zero-frequency limits of the refractive index are nxx(0) = 2.65 and nzz(0) = 2.45, following the relation n2(0) ≈ ε1(0). The maximum values of the refractive index and extinction coefficient are found to be 3.50 and 2.54, respectively. The reflectivity spectra are given in Fig. 4(b) (right), showing a rapid variation of reflectivity with photon energy, which may be suitable for various optical applications. The calculated values of reflectivity in the zero-frequency limit are Rxx = 0.20 and Rzz = 0.17. The maximum value of reflectivity corresponds to a negative value of ε1(ω).
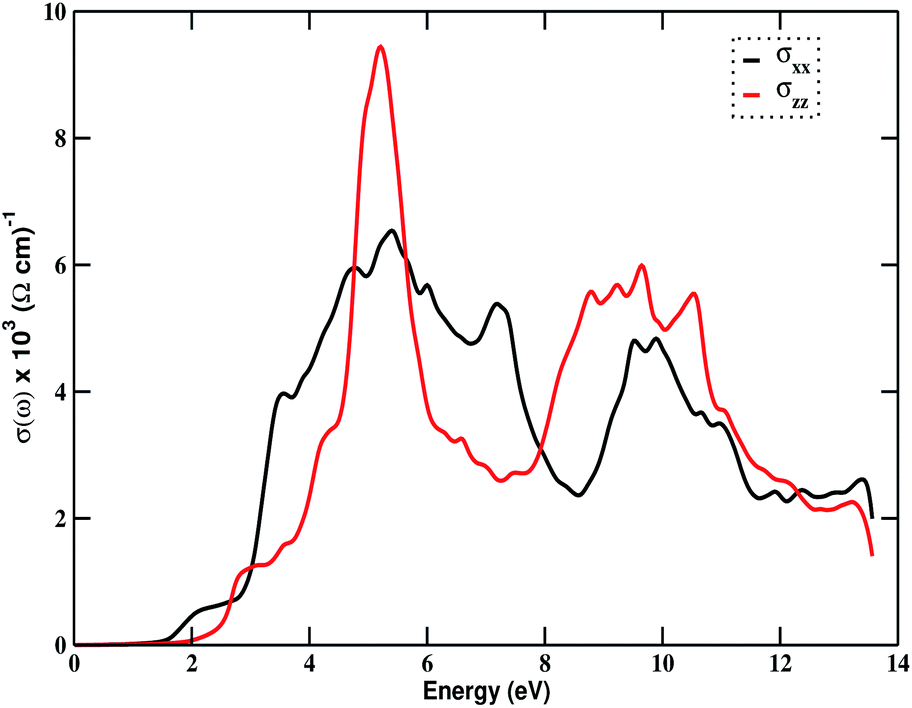
Fig. 4(c) (right) shows the variation of the absorption coefficient with incident photon energy. The absorption edge starts from around 1.50 eV of energy. Absorption peaks appear around 5.50 eV and 10.50 eV for the z-component, and 7.50 eV and 11.20 eV for the x-component. This shows that the compound Bi2LaO4I is optically active and is a potential candidate for optoelectronic applications in the ultraviolet region. The maximum value of α(ω) in the entire photon energy range is 190 × 104 cm−1. Fig. 4(d) (right) displays energy loss due to the scattering of electrons from the material. The loss is greater for photons with energy greater than 6 eV in both directions, with peak values at around 7 eV and 13 eV in the x-direction, and at around 9 eV and 12.20 eV in the z-direction. These peaks may have arisen due to the mixing of single-electron excitation and collective excitation. The optical behavior of the material is reflected by the plot of optical conductivity, shown in Fig. 5. It begins responding to the incident photons with an energy of 1.80 eV, but the response is good in the range 3 to 12 eV, indicating that the material is optically active in the visible and ultraviolet region, with a maximum optical conductivity of 9.44 × 103 (Ω cm)−1 at an energy value of 5.20 eV.
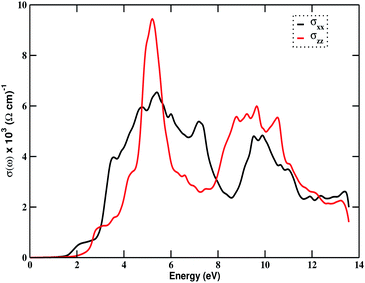 |
| | Fig. 5 The variation of optical conductivity in the photon energy range of 0–14 eV for Bi2LaO4I. | |
Thermoelectric properties
Many types of research show that large-bandgap insulating materials are promising candidates for TE applications. To determine the TE properties of the material, we calculated the Seebeck coefficient (S), electrical conductivity (σ), power factor (S2σ), and electronic thermal conductivity (κe) based on Boltzmann theory. Due to the limitations of BoltzTraP, the lattice contribution is neglected. In CRTA, σ, κe and S2σ are expressed in terms of relaxation time (τ), whereas S is independent of τ. The application of CRTA and RBA in the Boltzmann transport equation gives the electrical conductivity, thermal conductivity, and Seebeck coefficient as a function of the Fermi function.27,49
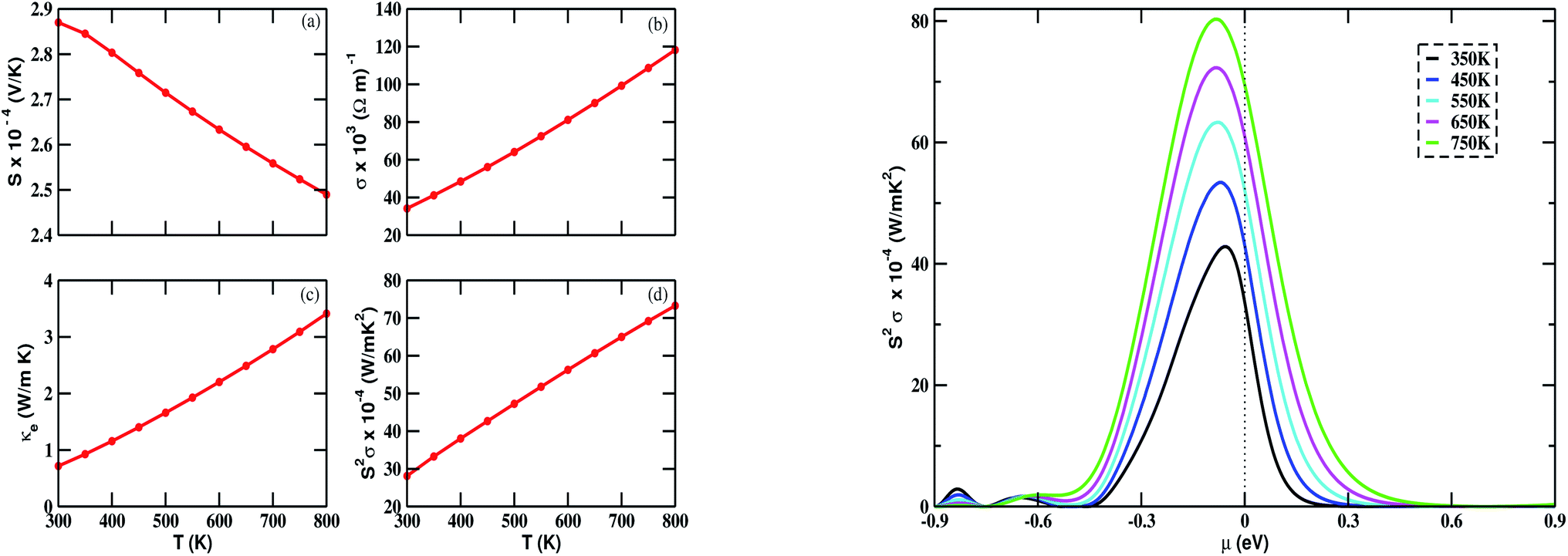
The calculated value of S at 300 K is 287 μV K−1. We observed from Fig. 6(a) (left) that the Seebeck coefficient steadily decreases as the temperature is increased from 300 to 800 K. At 800 K, it has the lowest value of 248 μV K−1. The positive value of S in the whole temperature range suggests that the transport phenomena are due to p-type charge carriers. The electrical conductivity is found to increase as the temperature is increased, as in Fig. 6(b) (left). The calculated value of σ lies in the range of 34.13 × 103 to 118.21 × 103 (Ω m)−1. κe is found to increase as well. From Fig. 6(c) (left), we noticed that the relationship between κe and temperature is nearly linear. The calculated power factor, which is the product of S2 and σ, is shown in Fig. 6(d) (left). Though S is seen to decrease with increasing temperature, the power factor increases with temperature. This is due to the contribution from σ. The maximum calculated value of the power factor is 73.26 × 10−4 W m−1 K−2 at 800 K.
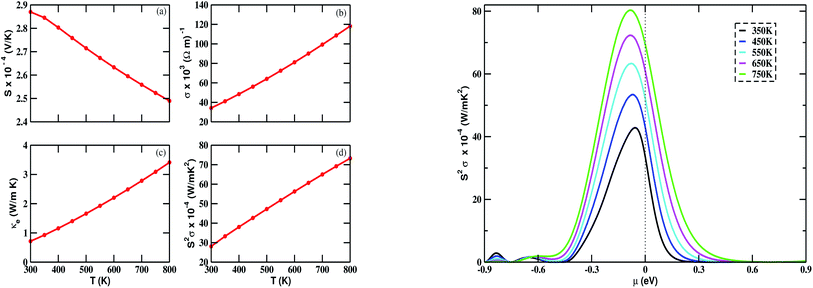 |
| | Fig. 6 Variation of (a) Seebeck coefficient (S), (b) electrical conductivity (σ), (c) electronic thermal conductivity (κe), and (d) power factor S2σ with temperature (T) ranging from 300 K to 800 K (left); and power factor (S2σ) vs. chemical potential (μ) (right) for Bi2LaO4I with τ = 10−14 s. | |
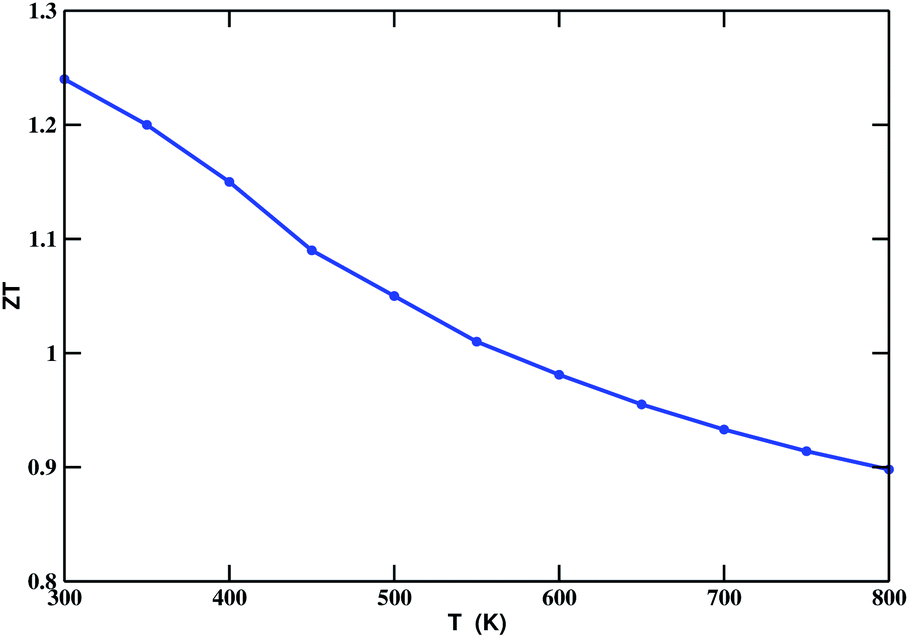
The chemical potential (μ) dependence of the power factor at various temperatures is shown in Fig. 6 (right). The dotted vertical line corresponding to zero chemical potential is an indication of the middle of the energy gap. The negative values of μ correspond to p-type doping, whereas the positive values correspond to n-type doping. In all cases, the peak of the curve lies in the range of negative values of μ. Thus, in this case, we can enhance the power factor and, hence, the TE performance, by doping with an appropriate amount of p-type charge carriers. By considering κe, we calculated the TE figure of merit, shown in Fig. 7. The calculated values lie between 1.24 and 0.89. The decreasing value of ZT with increasing temperature may be due to an increase in κe.
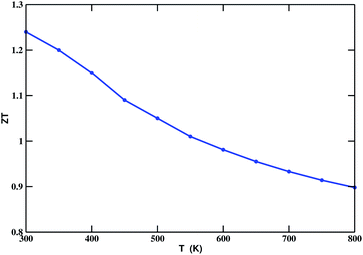 |
| | Fig. 7 ZT as a function of temperature for Bi2LaO4I (considering the κe component of κ). | |
Conclusions
The structural, elastic, electronic, optical, and TE properties of the layered perovskite Bi2LaO4I were studied by means of density functional theory. It was found that the material is a non-magnetic insulator with an indirect bandgap of 1.2 eV. Calculation of the elastic constants showed that the material is stable. We also calculated the optical elements, i.e., dielectric function, refractive index, reflectivity, absorption coefficient, loss function, and optical conductivity, which suggest that the material is optically active. Boltzmann transport theory was used to calculate the TE properties. By considering the electronic thermal conductivity, calculated power factor, and figure of merit, the results indicate that Bi2LaO4I is a promising TE material, whose potential may be further enhanced by appropriate hole doping.
Author contributions
R. K. J. and S. R. B. performed the theoretical calculations, while M. P. G. assisted and supervised the project. All authors contributed in writing the manuscript.
Conflicts of interest
There is no conflict of interest.
Acknowledgements
We are grateful to Prof. R. Nagarajan of University of Delhi for fruitful discussions and careful reading of the manuscript. R. K. J. acknowledges the Nepal Academy of Science and Technology, Lalitpur, Nepal for the MSc fellowship. M. P. G. acknowledges the Alexander von Humboldt Foundation, Germany for the equipment grants and IFW-Dresden, Germany for providing the large-scale computer nodes for scientific computations.
Notes and references
- C. Kim, G. Pilania and R. Ramprasadt, J. Phys. Chem. C, 2016, 120, 14575 CrossRef CAS.
- K. Uchino, Ferroelectric devices, 2nd edn, 2009, ISBN:978-1-4398-0375-2 Search PubMed.
- B. Bouadjemi, S. Bentata, A. Abbad, W. Benstaali and B. Bouhaf, Solid State Commun., 2013, 168, 6 CrossRef CAS.
- R. M. Hazen, Sci. Am., 1988, 258, 74 CrossRef CAS.
- T. Wu and P. Gao, Materials, 2018, 11, 999 CrossRef PubMed.
- J. Liang and Z. Jin, Sol. RRL, 2017, 1, 1700086 CrossRef.
- H. J. Snaith, Nat. Mater., 2018, 17, 372 CrossRef CAS PubMed.
- P. Gao, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2014, 7, 2448 RSC.
- A. Chilvery, S. Das, P. Guggilla, C. Brantley and A. Sunda-Meya, Sci. Technol. Adv. Mater., 2016, 17, 650 CrossRef CAS PubMed.
- H. Tanaka and M. Misono, Curr. Opin. Solid State Mater. Sci., 2001, 5, 381 CrossRef CAS.
- R. Mohamed, X. Cheng, E. Fabbri, P. Levecque, R. Kötz, O. Conrad and T. Schmidt, J. Electrochem. Soc., 2015, 162, F579 CrossRef CAS.
- G. Caruntu, L. Viciu, L. Spinu, W. Zhou and J. Wiley, MRS Online Proc. Libr., 2002, 718, 34 CrossRef.
- M. Schmidt, H. Oppermann, R. W. Henn, E. Gmelin, N. Soger and M. Bin-Newies, Z. Anorg. Allg. Chem., 2000, 626, 125 CrossRef CAS.
- C. J. Milne, P. Lightfoot, J. D. Jorgensen and S. Short, J. Mater. Chem., 1995, 5, 1419 RSC.
- D. Kato, K. Hongo, R. Maezono, M. Higashi, H. Kunioku, M. Yabuuchi, H. Suzuki, H. Okajima, C. Zhong and K. Nakano, et al., J. Am. Chem. Soc., 2017, 139, 18725 CrossRef CAS PubMed.
- G. Eperon, T. Leijtens, K. Bush, R. Prasanna, T. Green, J. Wang, D. Mcmeekin, G. Volonakis, R. Milot and R. May, et al., Science, 2016, 354, 861 CrossRef CAS PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger and K. Katsiev, et al., Science, 2015, 347, 519 CrossRef CAS PubMed.
- D. Sirbu, F. Balogun, R. Milot and P. Docampo, Adv. Energy Mater., 2021, 11, 2003877 CrossRef CAS.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682 CrossRef CAS.
- J. Di, J. Xia, H. Li, S. Guo and S. Dai, Nano Energy, 2017, 41, 172 CrossRef CAS.
- A. Nakada, M. Higashi, T. Kimura, H. Suzuki, D. Kato, H. Okajima, T. Ya-Mamoto, A. Saeki, H. Kageyama and R. Abe, Chem. Mater., 2019, 31, 3419 CrossRef CAS.
- A. Moure, Appl. Sci., 2018, 8, 62 CrossRef.
- A. Nakada, D. Kato, R. Nelson, H. Takahira, M. Yabuuchi, M. Higashi, H. Suzuki, M. Kirsanova, N. Kakudou and C. Tassel, et al., J. Am. Chem. Soc., 2021, 143, 2491 CrossRef CAS PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201 CrossRef CAS PubMed.
- K. Chatterjee, R. Dos Reis, J. K. Harada, J. K. Mathiesen, S. L. A. Bueno, K. M. Jensen, J. M. Rondinelli, V. Dravid and S. E. Skrabalak, Chem. Mater., 2021, 33, 347 CrossRef CAS.
- M. Jamal, M. Bilal, I. Ahmad and S. Jalali-Asadabadi, J. Alloys Compd., 2018, 735, 569 CrossRef CAS.
- D. P. Rai, Sandeep, A. Shankar, A. P. Sakhya, T. P. Sinha, P. Grima-Gallardo, H. Cabrera, R. Khenata, M. P. Ghimire and R. K. Thapa, J. Alloys Compd., 2017, 699, 1003 CrossRef CAS.
- B. Amin, I. Ahmad, M. Maqbool and R. Ahmad, J. Appl. Phys., 2011, 109, 023109 CrossRef.
- Y. Du, J. Xu, B. Paul and P. Eklund, Appl. Mater. Today, 2018, 12, 366 CrossRef.
- J. Yang and T. Caillat, MRS Bull., 2006, 31, 224 CrossRef CAS.
- D. Zhao and G. Tan, Appl. Therm. Eng., 2014, 66, 15 CrossRef CAS.
- M. Massetti, F. Jiao, A. J. Ferguson, D. Zhao, K. Wijeratne, A. Würger, J. L. Blackburn, X. Crispin and S. Fabiano, Chem. Rev., 2021, 121, 12465 CrossRef CAS PubMed.
- Y. Xie, T.-M. Chou, W. Yang, M. He, Y. Zhao, N. Li and Z.-H. Lin, Semicond. Sci. Technol., 2017, 32, 044003 CrossRef.
- P. X. Zhang, G. Y. Zhang, C. T. Lin and H. Habermeie, Egypt. J. Sol., 2004, 27, 1 CrossRef.
- D. Xu, Y. Wang, B. Xiong and T. Li, Front. Mech. Eng., 2017, 12, 557 CrossRef.
- M. Jaegle, Multiphysics simulation of thermoelectric systems-modeling of peltier cooling and thermoelectric generation, in COMSOL conference 2008 Hannover, 2008 Search PubMed.
- D. P. Rai, A. Shankar, M. P. Ghimire, R. Khenata and R. K. Thapa, RSC Adv., 2015, 5, 95353 RSC.
- A. LaLonde, Y. Pei, H. Wang and G. Snyder, Mater. Today, 2011, 14, 526 CrossRef CAS.
- Y. Pei, X. Shi, A. LaLonde, H. Wang, L. Chen and G. Snyder, Nature, 2011, 473, 66 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka, and J. Luitz, WIEN2K: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties, Technische Universität Wien, Vienna, Austria, 2001, ISBN 3-9501031-1-2 Search PubMed.
- K. Schwarz, P. Blaha and G. K. H. Madsen, Comput. Phys. Commun., 2002, 147, 71 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- F. Tran and P. Blaha, Phys. Rev. Lett., 2009, 102, 226401 CrossRef PubMed.
- G. K. H. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67 CrossRef CAS.
- J. M. Ziman, The Theory of Transport Phenomena in Solids, 1960, ISBN-13: 9780198507796 Search PubMed.
- J.-H. Han and K.-S. Kim, Phys. Rev. B, 2018, 97, 214206 CrossRef CAS.
- M. Born, Math. Proc. Cambridge Philos. Soc., 1940, 36, 160 CrossRef CAS.
- G. Grimvall, B. Magyari-Kope, V. Ozolis and K. A. Perssonf, Rev. Mod. Phys., 2012, 84, 945 CrossRef CAS.
- H. Dong, C. Chen, S. Wang, W. Duan and J. Li, Appl. Phys. Lett., 2013, 102, 182905 CrossRef.
- R. Yu, X. Chong, Y. Jiang, R. Zhou, W. Yuan and J. Feng, RSC Adv., 2015, 5, 1620 RSC.
- D. Chattaraj, C. Majumder and S. Dash, J. Alloys Compd., 2014, 615, 234 CrossRef CAS.
- S. F. Pugh, London, Edinburgh Dublin Philos. Mag. J. Sci., 1954, 45, 823 CrossRef CAS.
- P. H. Mott, J. R. Dorgan and C. M. Roland, J. Sound Vib., 2008, 312, 572 CrossRef.
- J. Haines, J. Leger and G. Bocquillon, Annu. Rev. Mater. Res., 2001, 31, 1 CrossRef CAS.
- O. L. Anderson, J. Phys. Chem. Solids, 1963, 24, 909 CrossRef CAS.
- B. Velický, Czech. J. Phys. B, 1961, 11, 787 CrossRef.
- C. M. I. Chukwuemeka, J. Phys.: Condens. Matter, 2003, 15, 5945 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
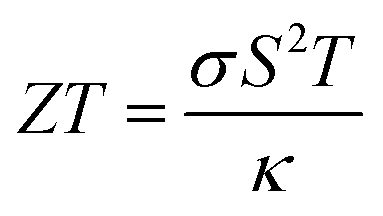


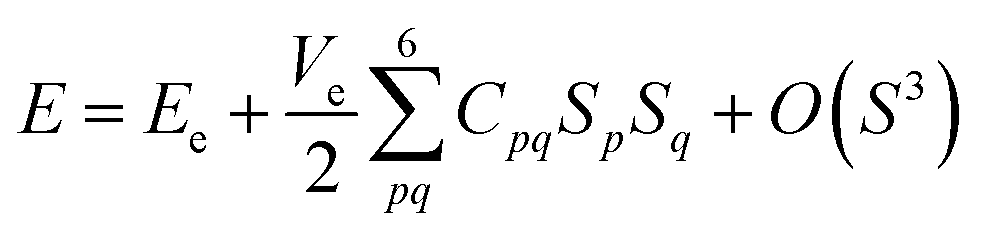

 and transverse
and transverse  components of the velocity. The calculated values of θD and the velocities are given in Table 3. The θD value is found to be large. We also calculated the melting temperature as 1093.65 ± 300.00 K.
components of the velocity. The calculated values of θD and the velocities are given in Table 3. The θD value is found to be large. We also calculated the melting temperature as 1093.65 ± 300.00 K.