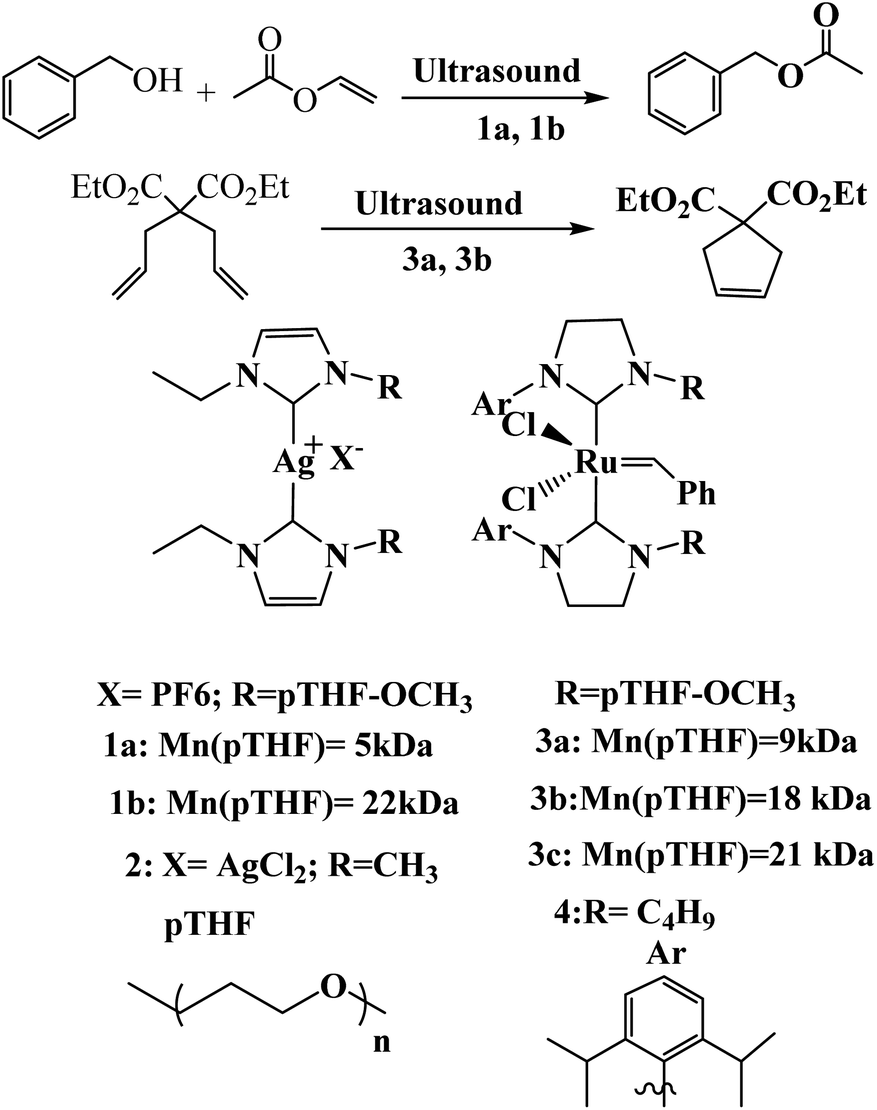
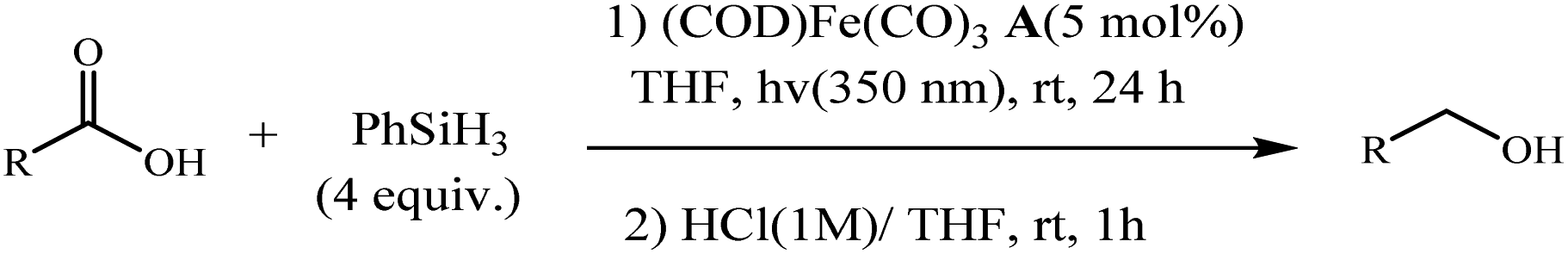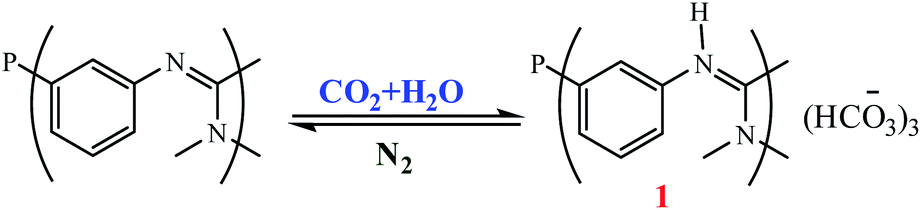DOI:
10.1039/D2RA03842K
(Review Article)
RSC Adv., 2022,
12, 23595-23617
Recent advances utilized in artificial switchable catalysis
Received
22nd June 2022
, Accepted 30th July 2022
First published on 19th August 2022
Abstract
Developing “green” catalytic systems with desirable performance such as solubility, recyclability, and switchability is a great challenge. However, inspired by nature, the studies on synthesis and activity of artificial switchable metal catalysts and organocatalysts have become an intense, fervid, and challenging field of research. The peculiarity of these catalysts is that they can be generally triggered in the “on” or “off” states by several external stimuli such as light, heat, solvents, pH change, coordination events or ion influxes, redox processes, mechanical forces, or other changes in reaction conditions. A large number of review articles are available in these areas. However, most efforts are currently focused on the invention of new types of switchable catalysts with different forms of stimuli–response units incorporated within their architectures in order to achieve control over the catalytic activity and regio-, chemo- and stereocontrol of various chemical reactions. Thus, in this review, we begin with a brief introduction to switchable catalysts, followed by discussion of types of stimuli and the influence factors on their activities in the field of biomedical engineering, and catalysis as well as related catalytic mechanisms summarized and discussed. The emphasis is on the recent advances utilized in artificial switchable catalysis.
1. Introduction
Efforts have focused on the design and improvement of a myriad of catalysts, to modulate high substrate conversions and/or high regio-, chemo-, or stereo-selectivities. The idea of modulating the activity of a catalyst merely as a response to an external stimulus, including acid–base chemistry, mechanical force, redox processes, phase switching, soluble and solid-supported and light and without the need of tedious and often expensive synthetic work constitutes an appealing concept and holds enormous potential to substantially impact the way catalysis is regarded in the future.1–12 Typically the artificial switchable catalysts are equipped with smart structural features that respond reversibly to external stimuli to induce an on/off-type regulatory function related to one or more chemical events during catalysis. With the help of this sophisticated chemistry, therefore, one can achieve a good degree of control over the outcome of catalytic reactions. Basic requirements for switches: bistability: occurrence of 2 forms of molecule that can be interconverted by an external stimulus, fast response times, thermal stability, and fatigue resistance. Significant efforts have been made in recent years to employ stimuli; including, light,13 pH,14 ion coordination,15 redox switching,16 mechanical forces,17 cooperative effects,18 steric effects,19 electronic effects,20 aggregation/dissociation,21 ligand22 and changes in reaction conditions23 in order to modulate the intrinsic activities, selectivities and minimizing energy displayed by catalysts. Although the field of ‘artificial switchable catalysis’ is young; but, flexibility of switchable materials led to researcher apply the principles of green chemistry to create switchable materials that offer potential for environmental benefit as well as innovative new materials.
Switchable materials of various kinds have the potential to address important issues in catalytic reactions such as olefin hydroformylation24 hydrosilylation,25 asymmetric olefin polymerization26 and control of enantioselectivity,27,28 separations,29 chemical CO2 recycling,30 and applications including, drug release/delivery,31,32 as mimicks of regulatory biocatalysts.33 For treatment of cancer, one of the major issues is the ineffective penetration of anticancer drugs in tumor. Existing techniques are inefficient in deep tumor drug delivery and lack on-demand drug release.
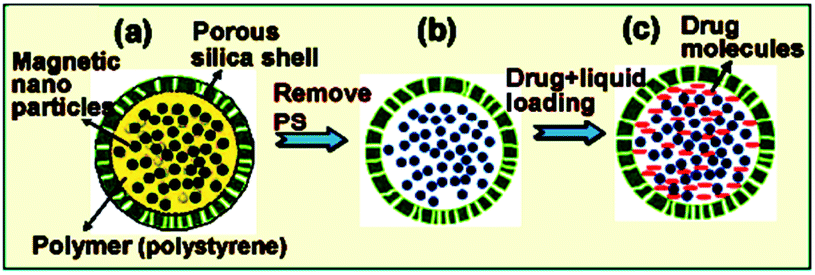
Controlled drug release by external stimuli such as temperature, electric or magnetic fields, light radiation, and changes in pH has attracted much attention that allows better drug penetration into cancer cell aggregates within tumors and subsequent switchable release is highly desirable. In this sense, Kong et al. have reported successful creation of triggerable nanocapsules which provide powerful magnetic vector for tumor penetration and on-off switchable drug release on remote RF magnetic field for on-off (Fig. 1).31
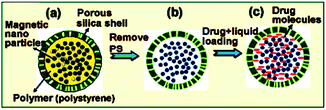 |
| | Fig. 1 Synthesis of hollow SiMNCs as a drug carrier. (a–c) Schematic illustration showing the preparation steps of drug containing hollow SiMNCs with high saturation magnetization. Reprinted by permission from ref. 31, Copyright © 2010, American Chemical Society. | |
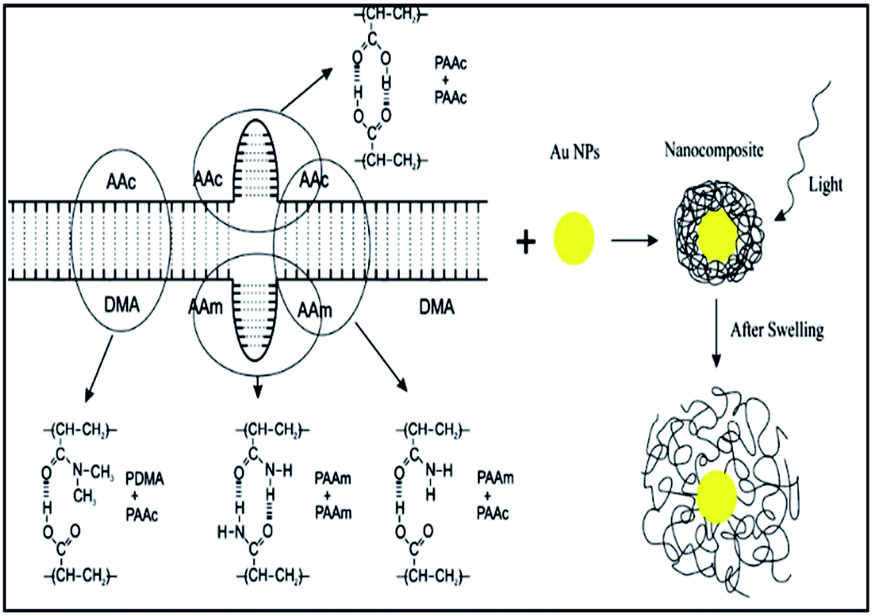
Similarly, Amoli-Diva et al. prepared a switchable dual light- and temperature-responsive drug carrier using gold nanoparticles (AuNPs)-grafted poly(dimethylacrylamide-co-acrylamide)/poly acrylic acid [P(DMA-co-AAm)/PAAc] hydrogel using free radical polymerization procedure and N,N-methylenebisacrylamide as cross-linker and ammonium persulfate as initiator. In this study, the authors investigated swelling, thermal sensitivity, thermal and optical switching properties of the prepared hydrogels in two acidic (pH = 1.2) and neutral (pH = 7.4) buffered solutions to simulate stomach and intestine body conditions. Finally, the author's evaluated loading and cumulative release (%) of ofloxacin antibiotic as model drug were considered in both thermal and optical switching conditions. The results showed that the obtained pulsatile release vehicle had the “on” state at higher temperatures and the “off” state at lower temperatures (Fig. 2).32
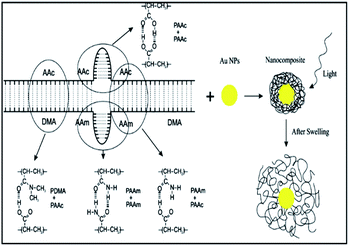 |
| | Fig. 2 Schematic of preparing procedure for the composite. Reprinted by permission from ref. 32, Copyright © 2017 Elsevier B.V. All rights reserved. | |
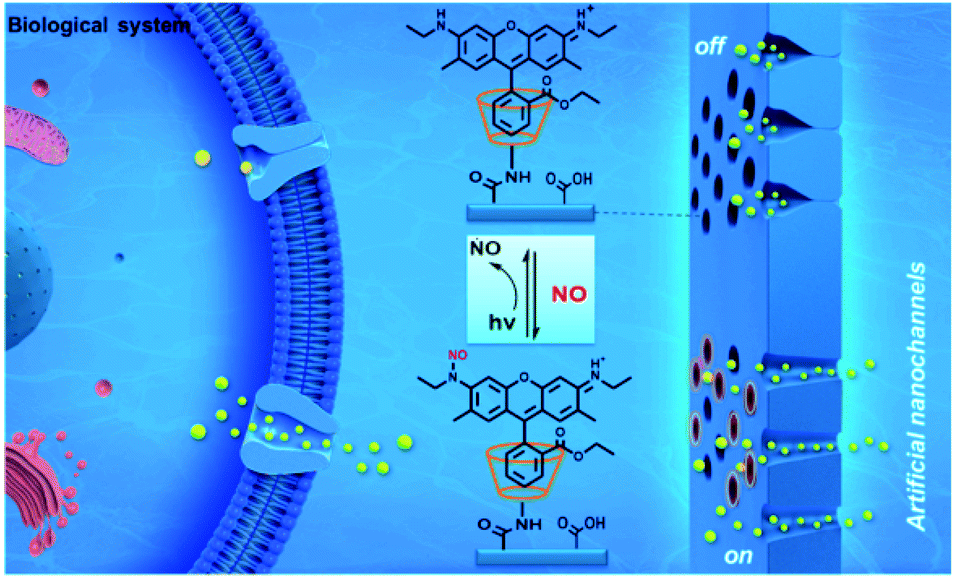
Ge and co-workers have designed a novel method for construction of switchable nanochannel-based analysis platform for the detection of bioactive gases, and would hold good promises for biomedical research, disease diagnosis and treatment.33 In this study, a switchable nitric oxide responsive nanochannel analysis platform is constructed by introducing a reversible N-nitrosation reaction of rhodamine 6G (R6G) into the artificial nanochannels. By virtue of the distinctive design, ionic current signal can handily realize reversible switching between “on” and “off” state in the presence of NO and UV light, and the system featured high stability and reproducibility (Fig. 3).
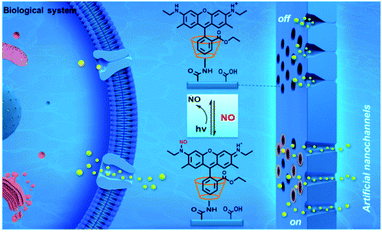 |
| | Fig. 3 Design and construction of nitric oxide activated nanochannels by a reversible N-nitrosation reaction. Reprinted by permission from ref. 33, Copyright © 2020 Elsevier B.V. All rights reserved. | |
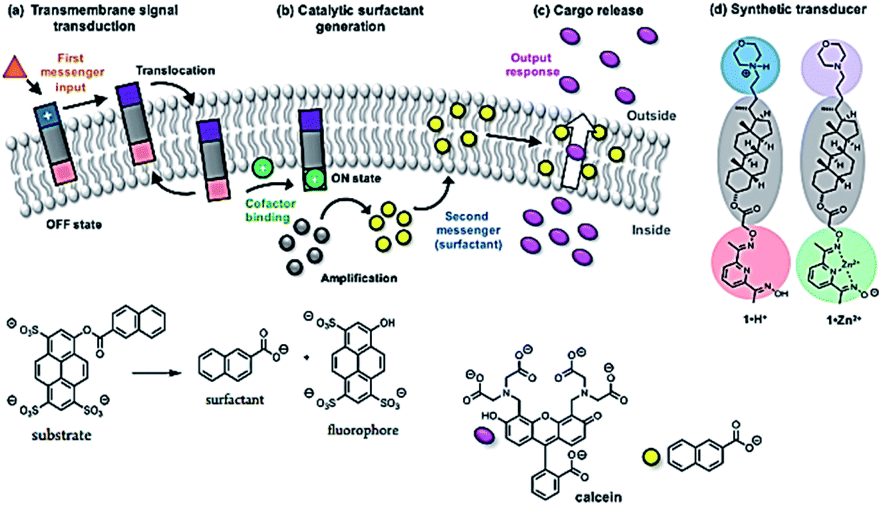
Langton and co-workers have showed that lipid bilayer vesicles (liposomes) can be triggered to release an encapsulated molecular cargo in response to an external control signal by employing an artificial transmembrane signal transduction mechanism (Fig. 4).34 An extra-vesicle input signal (pH change) employed to activate a membrane-bound artificial signal transducer, which catalyzed the intra-vesicle generation of a surfactant (2-naphthoic acid). The surfactant permeabilizes the lipid bilayer membrane to facilitate release of an encapsulated hydrophilic cargo. In the absence of the pH control signal, the catalyst is inactive, and the cargo remains encapsulated within the vesicle.
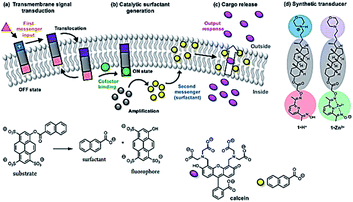 |
| | Fig. 4 Triggered cargo release from vesicles using an artificial signal transduction mechanism. (a) Transmembrane signal transduction. The input signal switches the external headgroup of a synthetic signal transducer embedded in the membrane from polar (blue) to apolar (purple), allowing it to translocate through the membrane. Binding of a charged cofactor to the inner headgroup (red) activates the catalyst (green). (b) Catalytic surfactant generation. The catalyst hydrolyzes a substrate (gray), generating a surfactant (yellow). (c) Cargo release. The surfactant enhances the permeability of the membrane to polar solutes, facilitating cargo release (pink). (d) Molecular structures of protonated signal transducer 1·H+ (OFF state) and the activated catalyst 1·Zn2+ (ON state). Reprinted by permission from ref. 34, Copyright © 2017 American Chemical Society. | |
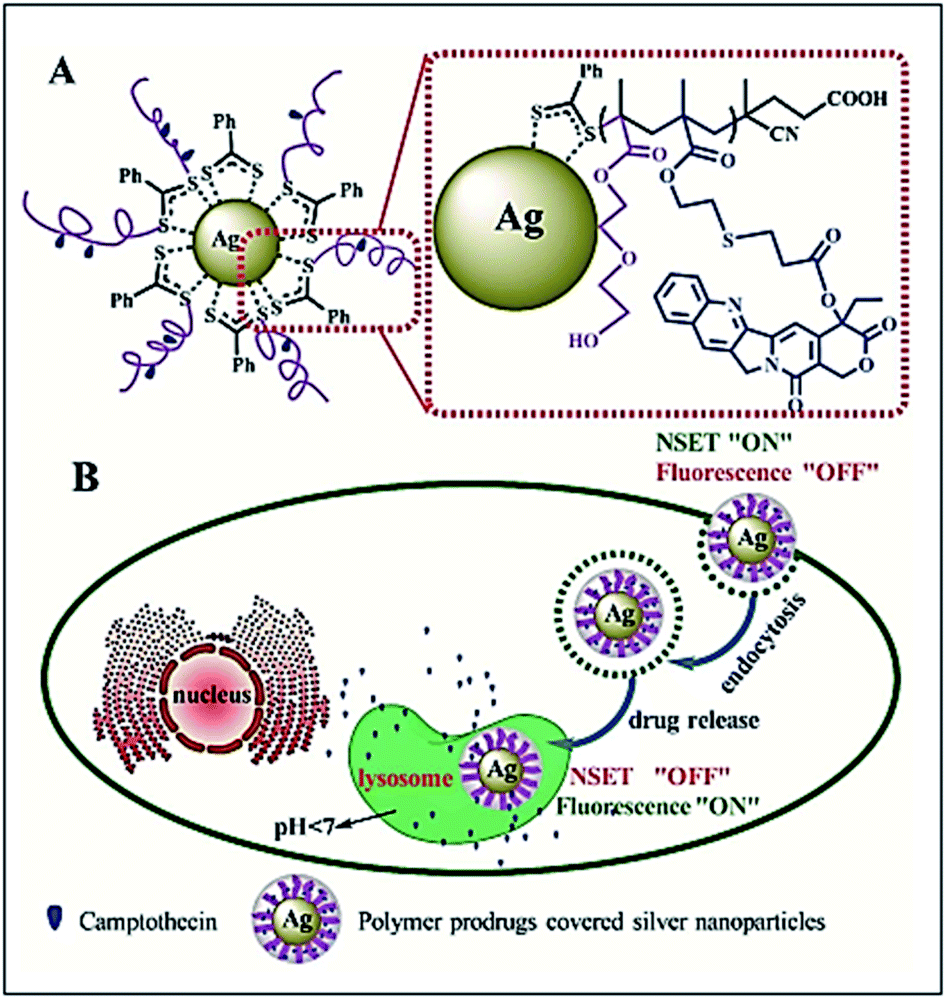
Qiu et al. have reported the preparation of the polymer prodrugs-covered Ag nanoparticle, in which silver nanoparticles (AgNPs) are covered with camptothecin (CPT)-based polymer prodrug (CPT–AgNPs) system that displays NSET effect, and this effect is strongly dependent on the CPT–AgNPs distance. By changing the distance, switching the NSET “on” or fluorescence “off” to the NSET “off” or fluorescence “on” can be achieved. This property can be used to track the CPT delivery and releasing process from the hybrid nanoparticles in a living cell. The authors noticed that fluorescence of CPT is quenched when it is linked to the polymer chain on the surface of AgNPs (NSET “ON” or Fluorescence “OFF”), while the CPT fluorescence is recovered (NSET “OFF” or fluorescence “ON”) when it is released with response to cellular acid conditions (Fig. 5).35
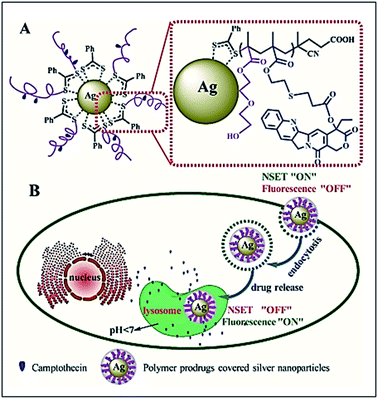 |
| | Fig. 5 Schematic illustration of the polymer prodrugs-covered Ag nanoparticle (A) and intracellular release process of CPT (B). The fluorescence of CPT is quenched when it is linked to the polymer chain on the surface of AgNPs (NSET “ON” or fluorescence “OFF”), while the CPT fluorescence is recovered (NSET “OFF” or fluorescence “ON”) when it is released with response to cellular acid conditions. Reprinted by permission from ref. 35, Copyright © 2017, American Chemical Society. | |
Homogeneous catalysts typically display often more active and selective than heterogeneous catalysts but are far more difficult to separate from the product. The development of new means to separate, recover, and recycle homogeneous catalysts is an important area of research for both industry and academia alike. One approach, biphasic solvent systems in which the catalyst is immobilized in one phase and the product distributes into the other phase. This allows for an intrinsic separation of catalyst and product. Water as the polar phase is especially helpful for separation. These systems utilize transition metals ligated by sulfonated phosphines to increase the water solubility of the catalyst, causing it to reside in the aqueous phase. After the reaction is completed, the product (organic) phase is decanted and the aqueous catalyst-bearing phase is used again.36 CO2 based-organic/aqueous tunable solvents (OATS) have also been previously published for recycling of the catalyst.37–39 In this the method catalysis can be performed in a single phase organic/switchable water co-solvent, eliminating mass transfer and partitioning issues. After catalysis, the introduction of CO2 into the system raises the ionic strength of the solution, salting out the product-containing organic phase away from the catalyst containing aqueous phase, affording a biphasic separation. Removal of CO2 from the aqueous phase regenerates the low ionic strength form of the solvent allowing for the addition of fresh solvent and reagents and recycling of the catalyst. For example, CO2-induced switchable tertiary amine-based organocatalysts were investigated for an efficient catalyst and product separation by its different partitioning between an organic and carbonated water phase. In this case study, the switching ability of tertiary amine-based catalysts between the organic and water phase by addition or removal of CO2 was investigated. The catalyst switched both nearly completely (99.9%) into the aqueous phase by addition of CO2 and effectively back into the organic phase (99.3%) by expelling CO2.40 Another method that can be used to recovery of a catalyst from a reaction, including a light-controlled phase tag to separate homogeneous catalysts that can be switched between a neutral (lipophilic) phase and a charged (lipophobic) phase through the use of a tag-centered photoreaction.41 The photoreaction results in drastic changes in the polarity and solubility of the catalyst. Thus, it is possible to optimize the solubility of the catalyst in different solvents and thereby improve extraction and separation of the catalyst from the products. In the present review, we briefly discuss the types of switchable external stimuli and, then, examine the activity of switchable catalysis.
1.1 Light-driven switching
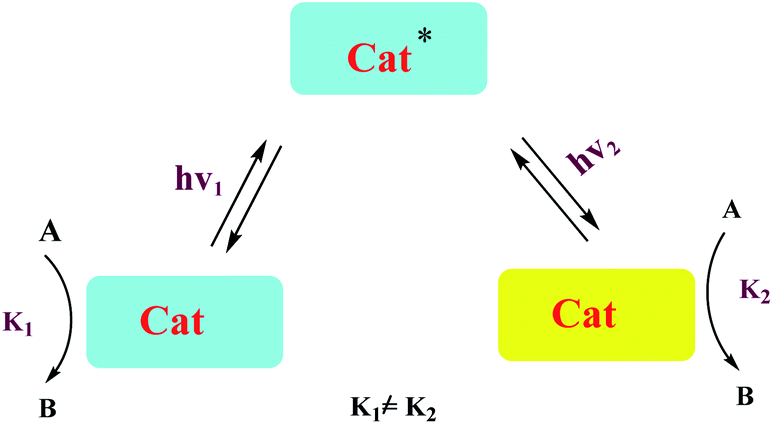
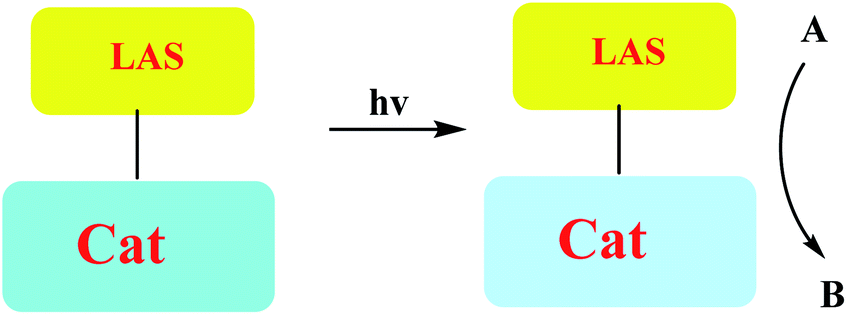
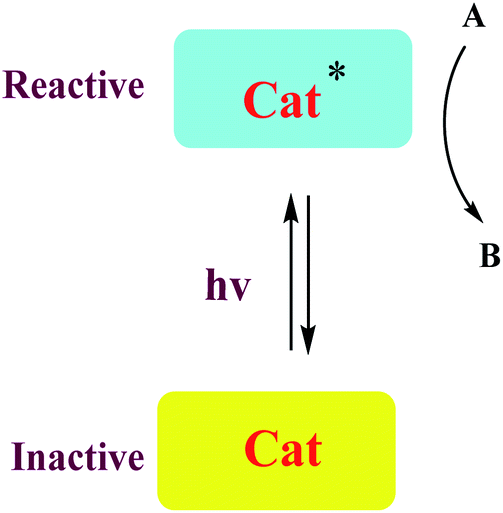
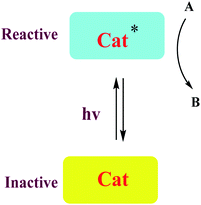
A number of conceptually different approaches have been used to realize artificial photoresponsive systems, including photocatalysis, photoactivated catalysis, and photoswitchable catalysis. (1) Photoswitchable catalysis involves a catalytically active species which undergoes a reversible photochemical transformation and, as a result, alters its intrinsic catalytic properties (Scheme 1), and (2) photocaged catalysis transforms an inactive catalyst precursor into an active species upon irradiation (Scheme 2), and (3) photocatalysis catalysis as an inactive precatalyst which is irradiated to generate a catalytically active, photoexcited state subsequently reacts with a substrate and constitutes a kind of photocontrol of catalytic activity (Scheme 3).42
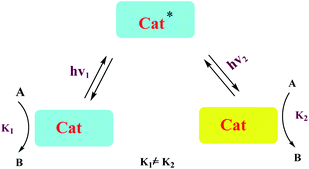 |
| | Scheme 1 Photoswitchable catalysis. | |
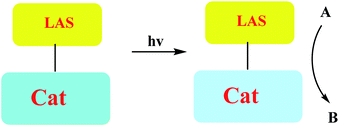 |
| | Scheme 2 Photocaged catalysis. | |
 |
| | Scheme 3 Photocatalysis. Reprinted by permission from ref. 42, Copyright © 2013, American Chemical Society. | |
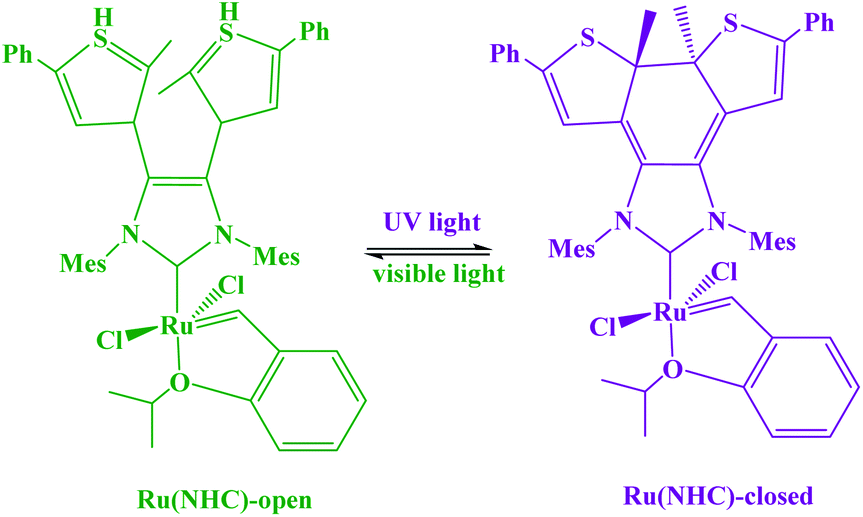
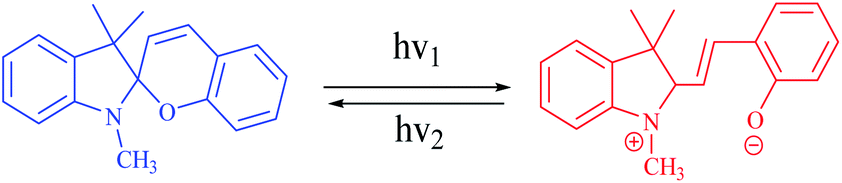
A photoswitchable catalyst involves a catalytically active species which is able to respond to light and to induce a change in the catalyst state. The use of light is an attractive stimulus due to some specific reasons, such as low-cost, ubiquitous, noninvasive, specific wavelengths, and no chemical waste in the system. Moreover, the light was used to reversibly activate/deactivate the living radical polymerization43 and to carry out the reversible ring-closing and ring-opening photoisomerization of the ligand backbone.44 A challenge for applying photoswitches is upscaling to larger volumes and higher concentrations: parameters such as the intensity of light that can cause degradation of organic compounds. In addition, the penetration depth of light cannot easily be scaled. Moreover, the lack of standardized photoreactors and light sources complicates comparison of experiments between laboratories. During recent years, a variety of molecular systems which enable its use as a photoswitch in biological systems (peptides),45 protein function46 or force generation in molecular motors.47 In addition, diarylethenes,48 stilbene49 and azobenzene,50 which undergo reversible photochemically driven E → Z isomerizations, have also been utilized to switch both of the steric and the electronic properties of catalysts. In this sense, RuII olefin metathesis, prepared by using a dithienylethene-functionalized N-heterocyclic carbine (NHC) ligand in presence light stimulus, was applied to carry out reversible ring-closing and ring-opening photoisomerization of the ligand backbone (Scheme 4).42,51 Similarly, the photoinduced changes in charge distribution that result from the ring-opening isomerization of spiropyrans may also be used to alter the catalytic activity (Scheme 5).
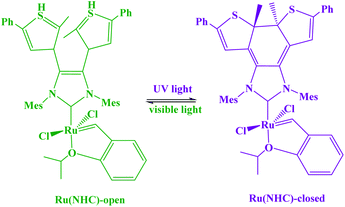 |
| | Scheme 4 Photoswitchable bifunctional olefin-metathesis catalyst. Reprinted by permission from ref. 51, Copyright © 2017, American Chemical Society. | |
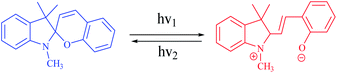 |
| | Scheme 5 Photochromic units, including spiropyrans, commonly incorporated into photoswitchable catalyst scaffolds. Reprinted by permission from ref. 42, Copyright © 2013, American Chemical Society. | |
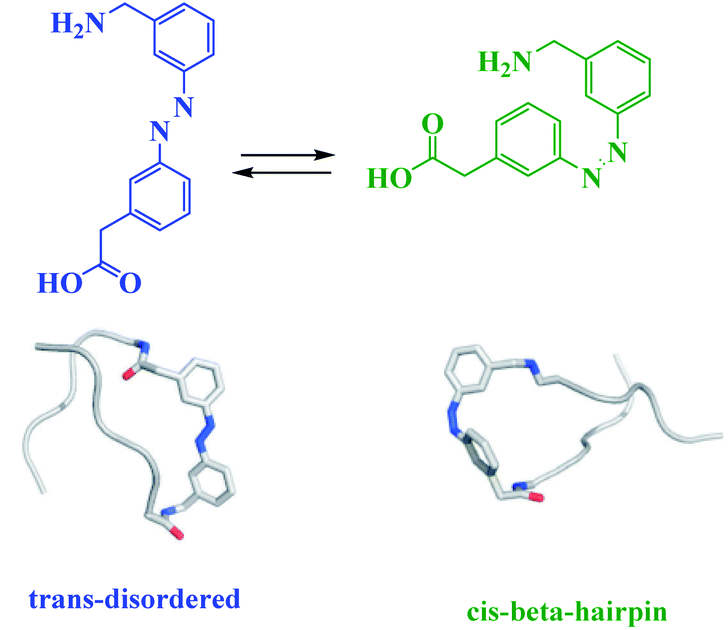
The reduction and photoisomerization of azobenzene was studied extensively.51–54 Most applications, described thus far, use azobenzene derivatives which photoisomerize from trans to cis with UV light and are affected by various factors such as substituents, solvents and temperature55,56 (Scheme 6). Moreover, azobenzene photoswitches catalyst commonly exploits cooperative, steric or electronic effects, and/or the aggregation/dissociation.
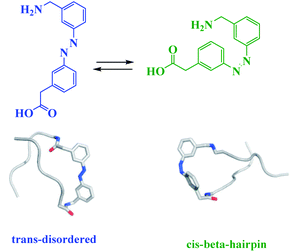 |
| | Scheme 6 A photoswitchable b-hairpin sequence using an azobenzene unit 30-(3-aminomethylphenylazo)-phenylacetic acid (AMPP). Reprinted by permission from ref. 55, Copyright © 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Reprinted by permission from ref. 56, Copyright © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
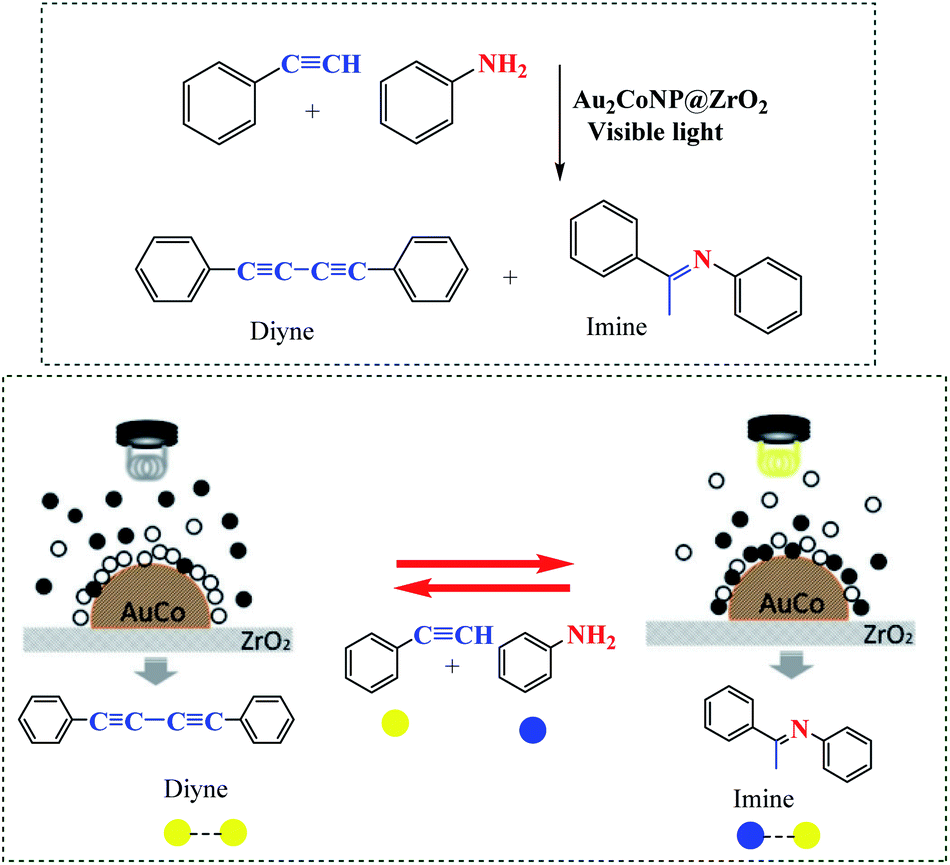
Zhu et al. demonstrated the application of Au2Co alloy nanoparticles (NPs) for selectivity of alkyne hydroamination (yielding imine (cross-coupling product of aniline and alkyne) under visible light irradiation), but 1,4-diphenylbutadiyne in the dark (Scheme 7).57 This reaction can be made switchable by a light-on/light-off.
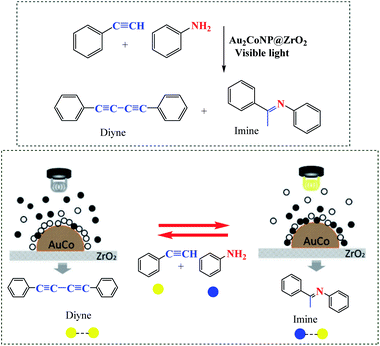 |
| | Scheme 7 Schematic illustrations of the selective trapping of aniline to alloy NPs by irradiation of the Au2Co/ZrO2 photocatalyst. Reprinted by permission from ref. 57, Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
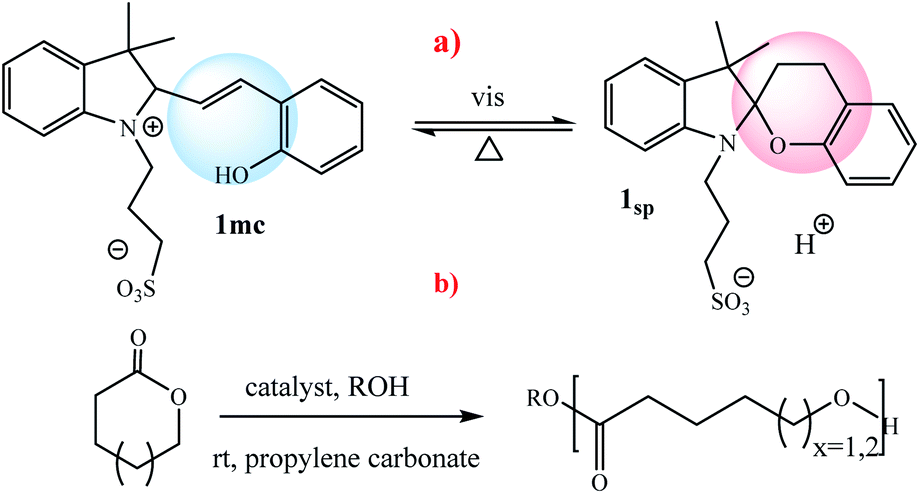
Chen et al. reported a thermally responsive gel with covalently bonded PTH photocatalyst: “Gel-PTH” that provides a transparent heterogeneous catalyst framework for photoredox CRP. This radical polymerization reaction can be made switchable by temperature “LOW”/“HIGH”, light “ON”/“OFF”, and catalyst presence “IN”/“OUT” (Scheme 8).58
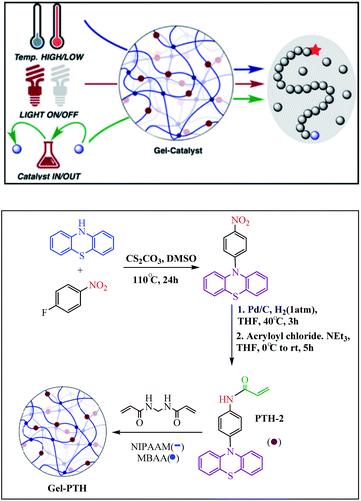 |
| | Scheme 8 Synthesis of the Gel-PTH. Reprinted by permission from ref. 58, Copyright © 2017, American Chemical Society. | |
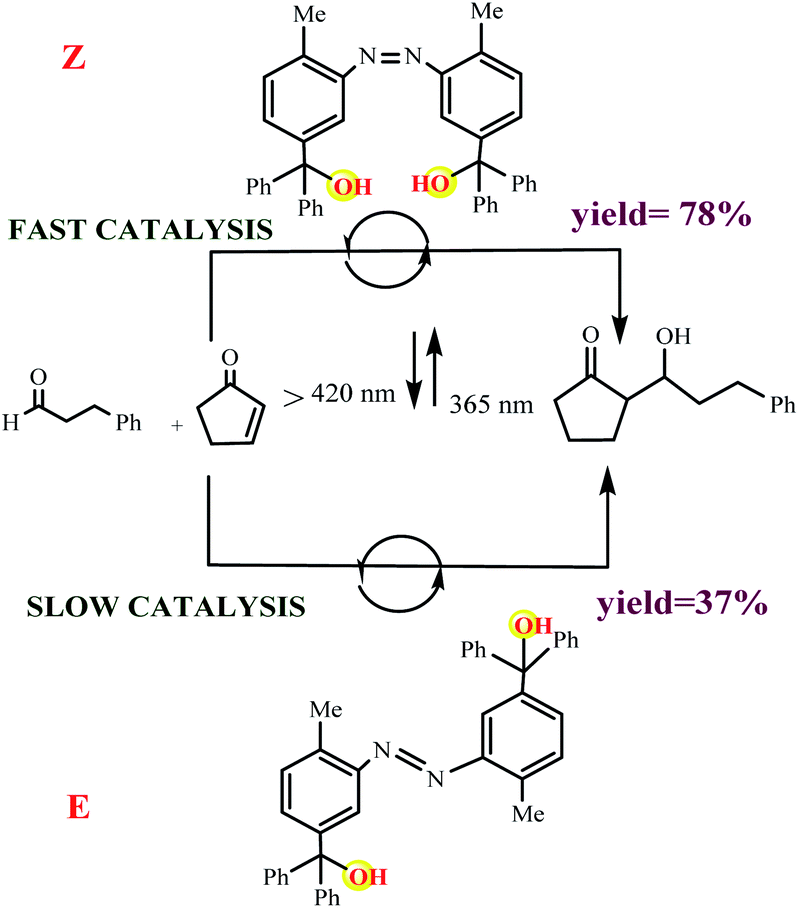
Boyer and coworkers reported an example for controlling the ROP of lactones by exploiting the spiropyran photoswitch (Scheme 9).59 In their molecular design the authors used propylene carbonate as a solvent. Irradiation of the merocyanine form 1mc with blue light (λ = 460 nm) induces the formation of the spiropyran form 1sp and releases a proton, which catalyzes the polymerization of γ-valerolactone or ε-caprolactone. It should be noted, the interconversion between the merocyanine and spiropyran forms takes place using light and thermal equilibrium. Also, the spiropyran form 1sp reverts thermally to the more stable merocyanine form 1mc, and thus recaptures the proton and pauses the polymerization.
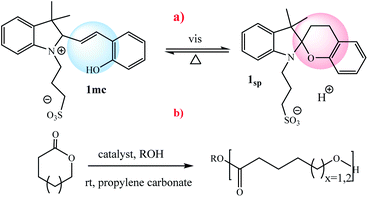 |
| | Scheme 9 (a) Photoswitching of 2 between the inactive merocyanine form 1mc and the active spiropyran 2sp. (b) ROP of γ-valerolactone or ε-caprolactone catalyzed by 1sp Reprinted by permission from ref. 59, Royal Society of Chemistry. | |
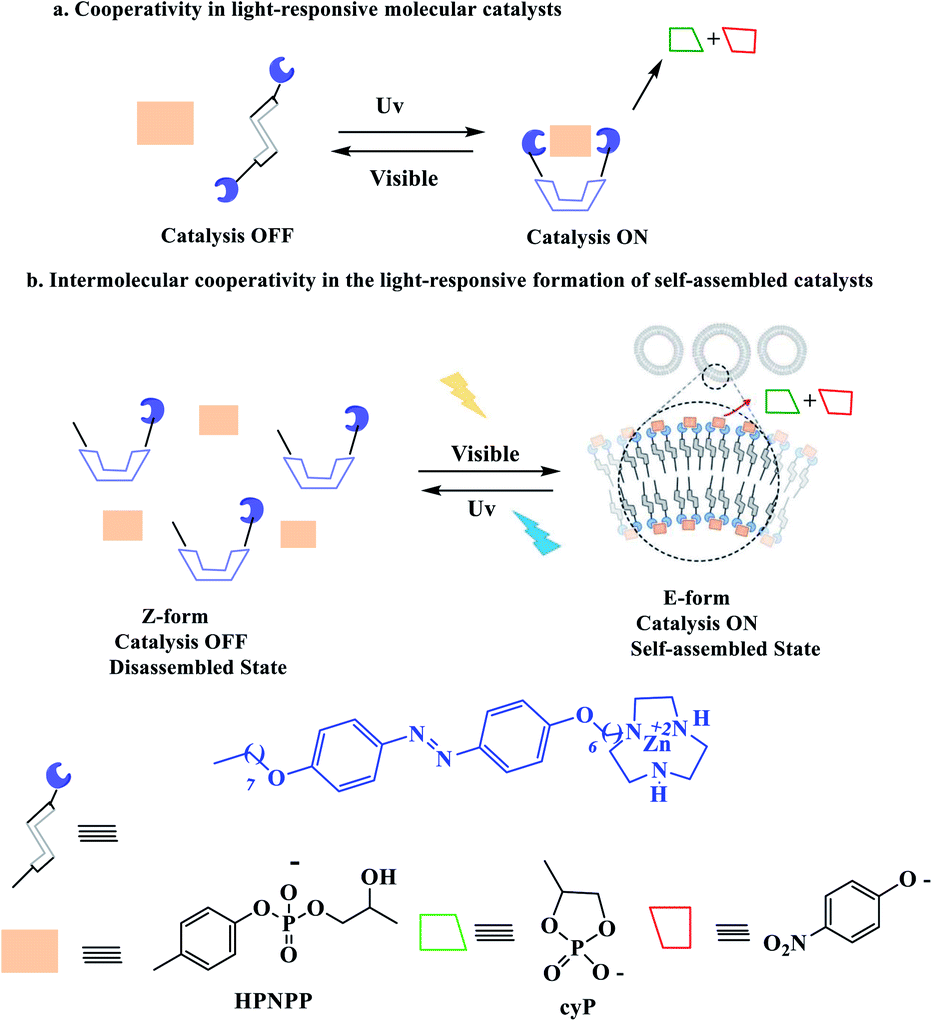
1.2 Cooperative effects
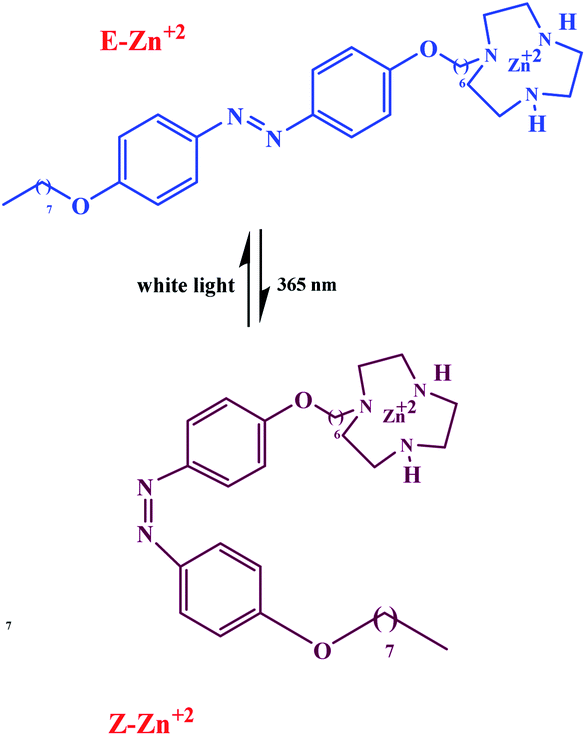
Cooperative effects in switchable catalysis involve an isomerization process which changes the distance or orientation of key sites in the catalyst. To gain control over the activity of a catalyst using this concept, a photoswitch is needed that induces a large geometrical change to shield or deshield the substrate binding site of the catalyst. In the reactions promoted by the photoactivated catalysts, the light of a different wavelength induced formation of the other state should translate into high reactivity and fast conversion. This would be reflected by a great difference in the reaction rate, selectivity, or reaction type. In order to design photoswitchable systems that are able to control a ground-state, (i.e. thermal) chemical reaction, a suitable photochromic moiety has to be incorporated into the system. The chromophore must be chosen in a manner that the structural differences between their two switching states are translated into different chemical reactivity. An example of a photoswitchable catalyst based on cooperative interactions was reported by Imahori and co-workers in 2012 (Scheme 10).60 Ligand which consists of two triaryl alcohol groups through an azobenzene moiety acts as a switchable catalyst E/Z, for the Baylis–Hillman reaction of 3-phenylpropanal and 2-cyclopenten-1-one upon exposure to light. Isomers Z and E have significantly different yields. The catalytic activity of isomers Z is significantly higher than isomers E due to bis(trityl alcohol) activated by intramolecular hydrogen bonding in isomers Z but isomers E hydroxyl groups are too far apart to engage in the intramolecular hydrogen bonding. The chromophore that are used for this processes linking two trityl alcohol moieties to an azobenzene core that lead to reactivity difference between the switched states.
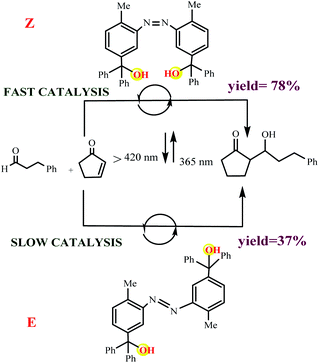 |
| | Scheme 10 Controlling the rate of the Morita–Baylis–Hillman reaction using photoswitchable catalyst E/Z. Reprinted by permission from ref. 60, Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Chen et al. have described that the catalytic system is based on amphiphilic pre-catalyst Zn2+, terminating with a Zn2+complexed 1,4,7-triazacyclononane (TACN) head group that acts as a switchable catalyst E/Z for the cleavage of the phosphodiester bond (Scheme 11).61 With HPNPP as the substrate, the transphosphorylation reaction leads to the formation of acyclic phosphate (cP) and the release of p-nitrophenolate (PNP) (Scheme 12). The resulting catalyst showed that when the E-Zn2+ isomer is used as the catalyst, the reaction is very active than that of Z-Zn2+.
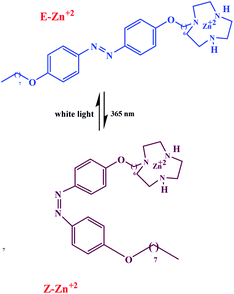 |
| | Scheme 11 Photoswitching ability of E-Zn2+ and Z-Zn2+. Reprinted by permission from ref. 61, Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
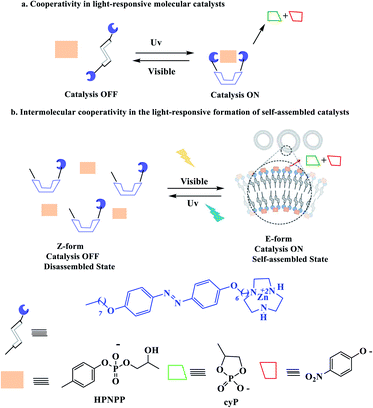 |
| | Scheme 12 Representation of (a) cooperativity in molecular photoswitchable catalysts and (b) an amphiphile exhibiting cooperative catalysis which can be switched between the “ON” and “OFF” states by irradiation with different wavelengths of light. Reprinted by permission from ref. 61, Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
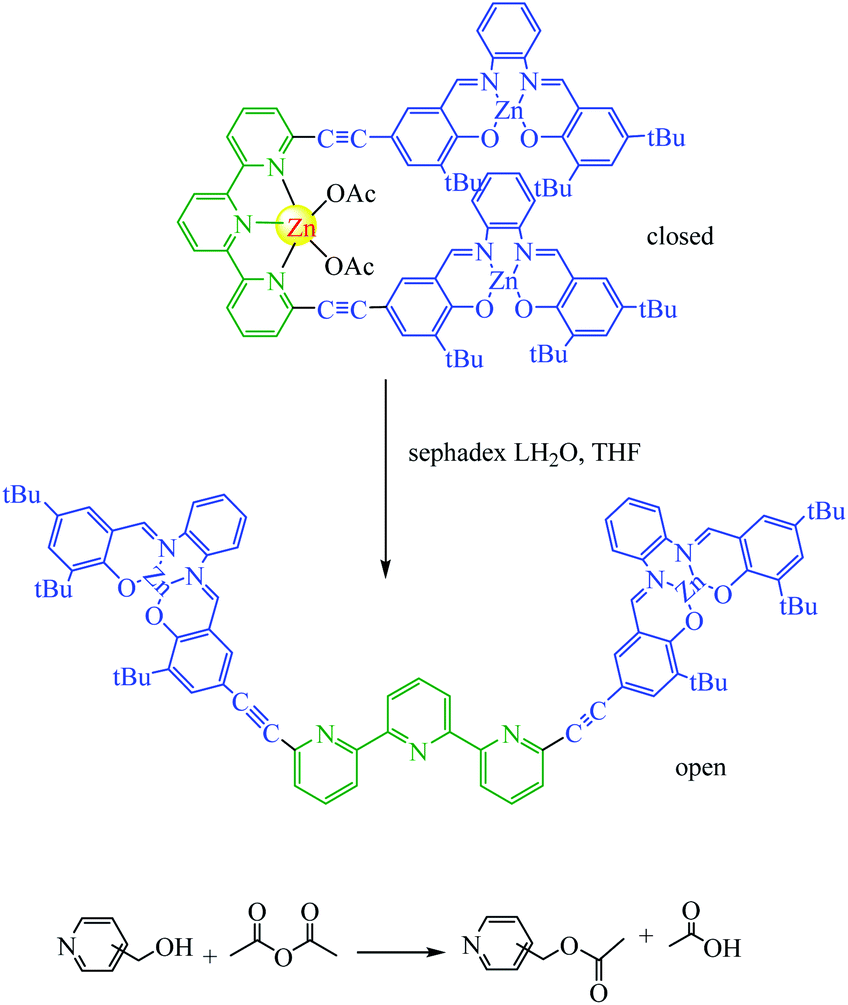
1.3 Steric effects
Almost all photoswitchable catalyst systems reported to date focus on modulating catalytic activity. For this a catalytically active molecule is functionalized with a photoswitchable entity. Ideally, the difference in catalytic activity between the two forms of the chromophore differs significantly enough, leading to an ON/OFF switching of the catalyst. An alternative strategy to regulate the activity of a catalyst through atoms that blocks access of the substrate to the active site in one state of the catalyst. A pioneering example was reported in 2019 for the acetylation of pyridinemethanol which catalysed switchable terpy (Zn-salphen)2 molecular tweezers and their metal (Scheme 13).62 The tweezers' conformation can be reversibly switched between an open and a closed form by a metal ion stimulus. Due to the fact that they both can act as a Lewis acid to activate the anhydride reagents. It should be noted that the closed tweezers show superior catalytic activity towards ortho substrates, while open tweezers present a higher rate for the acetylation of meta and para substrates. Mirkin reported the absolute rates are clearly lower than with ortho substrate (0.04 and 0.02 vs. 0.5 mM h−1 with open tweezers 1) in a similar due to ortho derivative can establish an intramolecular hydrogen bond between the OH group and the nitrogen of the pyridyl moiety that enhances its nucleophilicity compared to meta and para derivatives. The allosteric effect is surprisingly reversed for meta and para substrates compared to ortho with an increased rate.
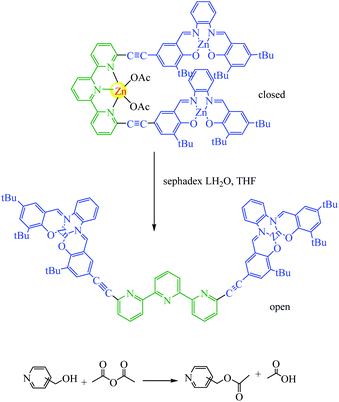 |
| | Scheme 13 Catalytic allosteric regulation by switchable molecular tweezers. A principle of the switching between open and closed conformation of the tweezers to regulate the activity of acyl-transfer reaction. Reprinted by permission from ref. 62, Copyright © 2019, The Author(s). | |
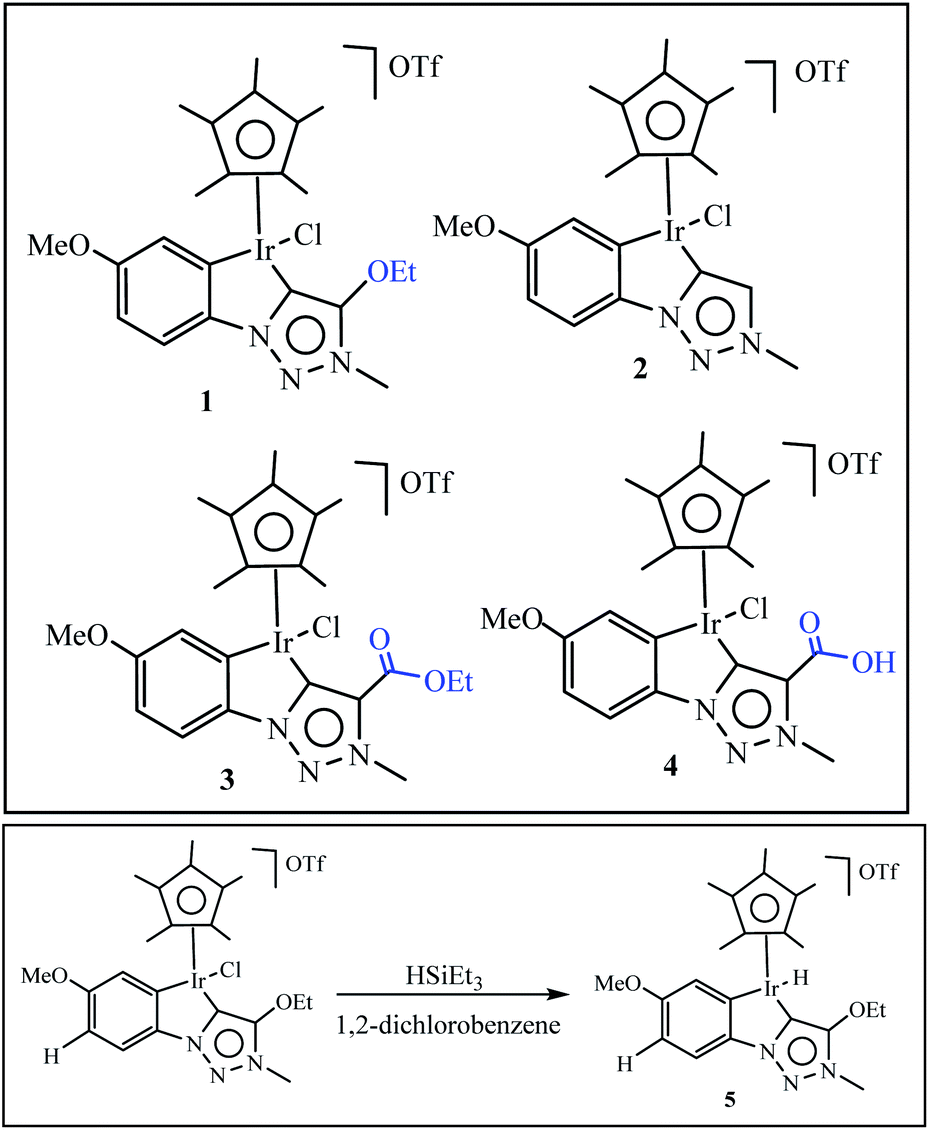
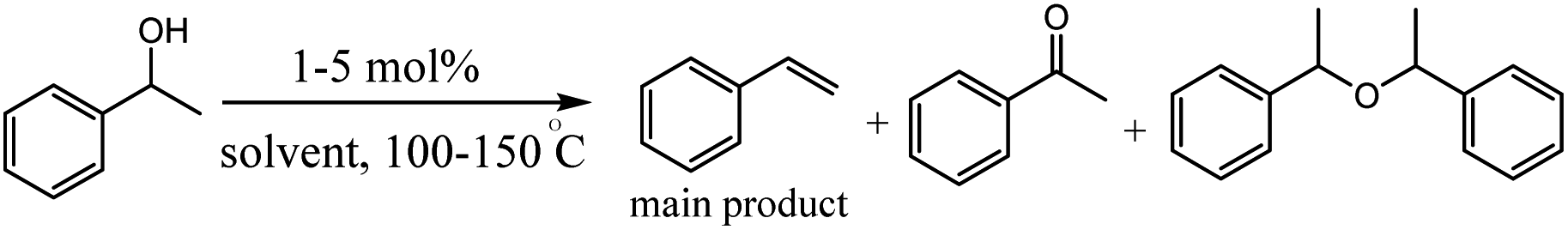
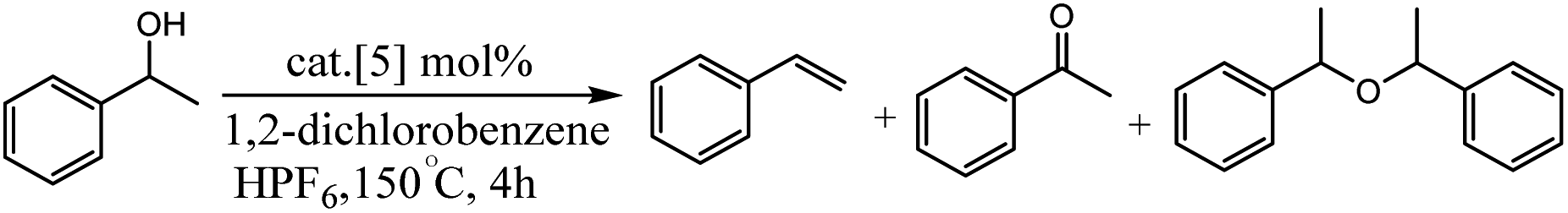
Olivares et al. have been synthesized triazolylidene iridium hydride complexes through modification of the analogous iridium chloride complexes (Scheme 14).63 After, the dehydration of alcohols was applied to examine the catalytic potential of the iridium chloride compounds and the influence of the electronic modification on the pyridyl–triazolylidene ligand scaffolds switch catalytic selectivity in the conversion of alcohols between dehydration (loss of H2O) and dehydrogenation (loss of H2), which is triggered by the presence or absence of an acid (Scheme 14). The role of the iridium complex was evaluated by comparing the catalytic activity of complex 1 to complexes 2–4 with steric and electronic modifications of the triazolylidene ligand (Table 1). Experimental studies show in presence of 1 mol% catalyst loading, complex 2 without a substituent on the triazolylidene scaffold showed slightly lower activity (48% vs. 55% with complex 1 after 30 min), while complex 3 was less active and reached only 31% conversion in the same time span (entries 6 and 7). Interestingly, complex containing a carboxylic acid group did not show any catalytic activity (entry 8), indicating that mild acids are inhibiting catalytic turnover. The increase of catalytic activity from complex 3 to 2 to 1 correlates with the substituent-induced enhanced donor properties of the triazolylidene ligand (R 1/4 COOEt, H, OEt along this series).
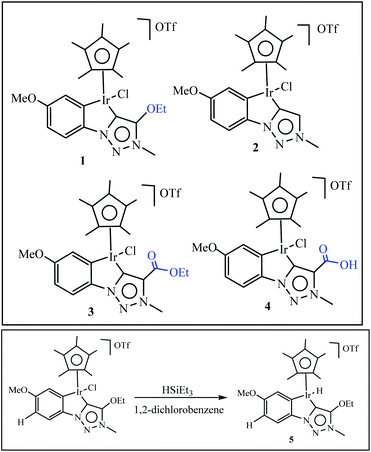 |
| | Scheme 14 Set of iridium complexes containing electronically variable substituents on the triazolylidene unit used for catalytic application. Reprinted by permission from ref. 63, Copyright © 1969, Elsevier. | |
Table 1 Catalytic activity of complexes 1–4 in dehydration catalysis of 1-phenylethanol. Reprinted by permission from ref. 63, Copyright © 1969, Elsevier
|
| Entry |
Complex |
Cat. loading |
Time (h) |
Conv. (%) |
Styrene |
Ketone |
Ether |
| 1 |
1 |
5 mol% |
1 |
91 |
77 |
10 |
5 |
| 2 |
1 |
5 mol% |
2 |
>99 |
95 |
3 |
2 |
| 3 |
1 |
5 mol% |
4 |
>99 |
9 |
— |
2 |
| 4 |
1 |
1 mol% |
0.5 |
55 |
49 |
— |
6 |
| 5 |
1 |
1 mol% |
2 |
>99 |
92 |
6 |
2 |
| 6 |
2 |
1 mol% |
0.5 |
48 |
41 |
1 |
6 |
| 7 |
3 |
1 mol% |
0.5 |
31 |
27 |
— |
4 |
| 8 |
4 |
1 mol% |
0.5 |
— |
— |
— |
— |
| 9 |
1 |
5 mol% |
4 |
65 |
30 |
20 |
15 |
In the second part of this research project, the authors examined the activity of iridium hydride complexes 5 for the dehydration as well as the dehydrogenation of alcohols. Their activity is switched by the presence/absence of HPF6 as a hydride abstractor (Table 2).
Table 2 Catalytic dehydrogenation of 1-phenylethanol. Reprinted by permission from ref. 63, Copyright © 1969, Elsevier
|
| Entry |
HPF6 |
Conv. (%) |
Styrene |
Ketone |
Ether |
Selectivity (%) |
| 1 |
— |
89 |
83 |
— |
6 |
0 |
| 2 |
3 |
>99 |
76 |
24 |
— |
24 |
| 3 |
6.2 |
93 |
37 |
53 |
3 |
57 |
| 4 |
8 |
96 |
57 |
35 |
4 |
38 |
| 5 |
10 |
96 |
59 |
34 |
3 |
35 |
| 6 |
6.2 |
96 |
52 |
40 |
4 |
42 |
| 7 |
10 |
>99 |
82 |
18 |
— |
18 |
| 8 |
6.2 |
>99 |
98 |
<2 |
— |
<2 |
| 9 |
2 + 2 |
61 |
21 |
34 |
6 |
56 |
| 10 |
6.2 |
>99 |
60 |
— |
— |
0 |
1.4 Electronic effects
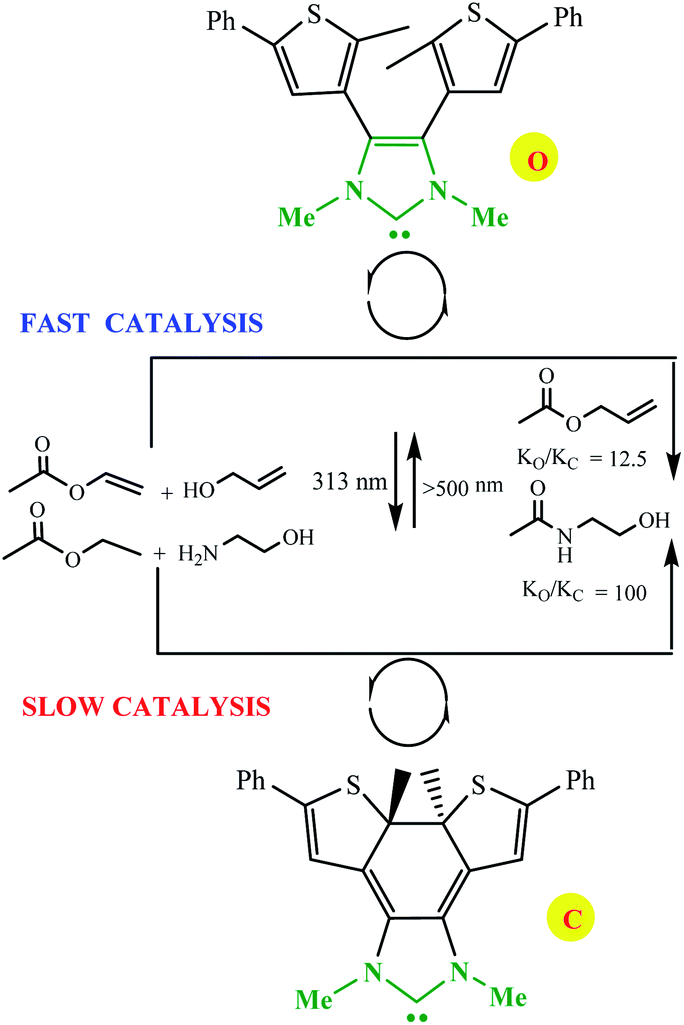
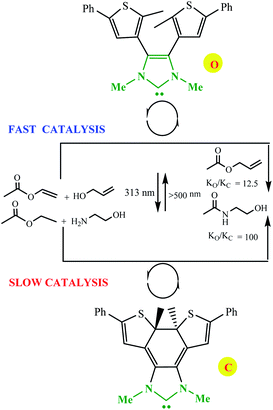
Electronic effects in switchable catalysis are often based on an inductive effect, electromeric effect, resonance effects, and hyperconjugation during an isomerization process that induces a change to alter the rate, chemo- and/or regioselectivity of catalysts. Photo-modulating catalytic activity via electronic changes was reported by Bielawski and Neilson using a switchable electronically-modulated N-heterocyclic carbene (NHC) into a dithienylethene scaffold to control transesterification and amidation reactions (Scheme 15).64 Using visible light and base the open form of the NHC catalyst, o, catalyzes the transesterification and amidation reaction. In the presence of UV irradiation to the closed form, c, the rate of both transesterification and amidation reactions is significantly decreased (ko/kc = 12.5 and 100, respectively).
 |
| | Scheme 15 Controlling the rate of transesterification and amidation reactions using switchable catalyst o/c. Reprinted by permission from ref. 65, Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
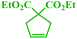
Tzur and co-workers examined the effect of the chelated heteroatom to probe electronic effects in sulfur- and nitrogen-chelated latent catalysts. The researchers synthesised novel complexes (Scheme 16).65 The authors demonstrated that the catalytic activity of nitro-substituted N-chelated catalyst 2 is significantly higher than N-chelated complex 3. In order to examine the effects of EDGs in complexes, the researchers explored the activity of the catalyst 4. Tests of the catalytic activity showed that catalyst 4 was inert to typical RCM reactions at room temperature and showed activity only at higher temperatures. In addition, the researchers tested the addition effect of a nitro group to latent S-chelated complex 5. Surprisingly, nitro-substituted complex 5 showed no activity at room temperature (Table 3).
 |
| | Scheme 16 Synthesis of N- and S-chelated complexes 2–5 by ligand exchange. Reprinted by permission from ref. 65, Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Table 3 Formation of disubstituted double bonds promoted by catalysts 2, 3 and 4. Reprinted by permission from ref. 65, Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
| Substrate |
Product |
Catalyst |
Yield (%) |
T (°C) |
T (h) |
 |
 |
2 |
18 |
55 |
2 |
| 2 |
98 |
55 |
24 |
| 3 |
37 |
24 |
2 |
| 3 |
90 |
24 |
24 |
| 4 |
8 |
55 |
2 |
| 4 |
91 |
55 |
24 |
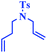 |
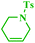 |
2 |
95 |
55 |
21 |
| 3 |
99 |
24 |
7 |
| 4 |
98 |
55 |
24 |
 |
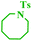 |
2 |
55 |
55 |
92 |
| 3 |
25 |
24 |
2 |
| 4 |
28 |
55 |
48 |
 |
 |
2 |
0 |
24 |
16 |
| 2 |
45 |
55 |
6 |
| 2 |
78 |
55 |
24 |
| 2 |
82 |
55 |
69 |
| 3 |
93 |
24 |
27 |
| 3 |
100 |
24 |
0.5 |
| 4 |
0 |
24 |
24 |
| 4 |
70 |
55 |
6 |
| 4 |
86 |
55 |
24 |
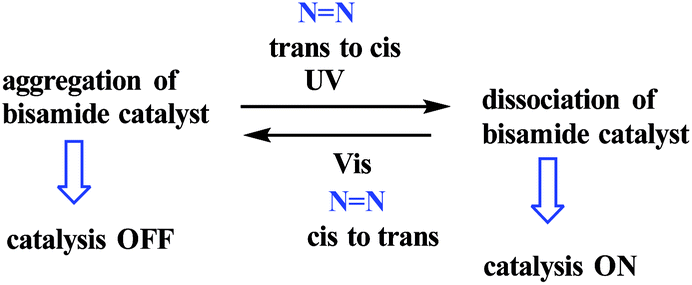
1.5 Aggregation/dissociation
The dynamic self-assembly of molecules into larger structures or ordered forms of macromolecules in biology has been exploited to induce the construction of synthetic systems with dynamic and responsive properties.
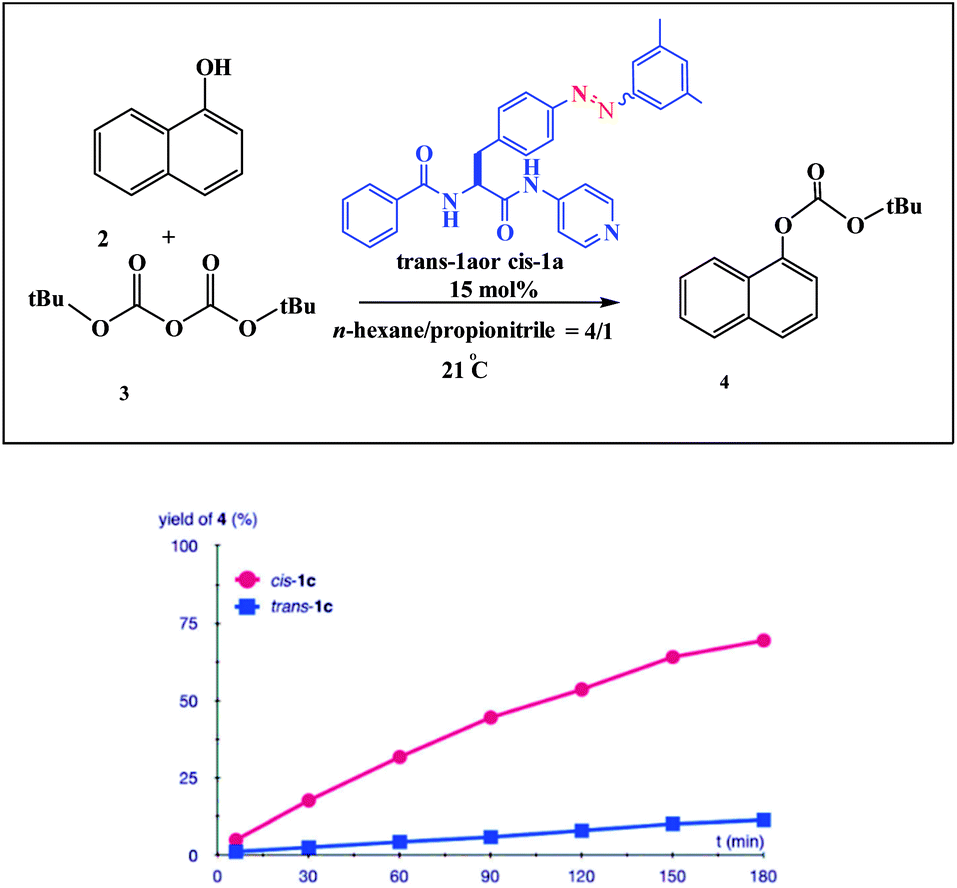
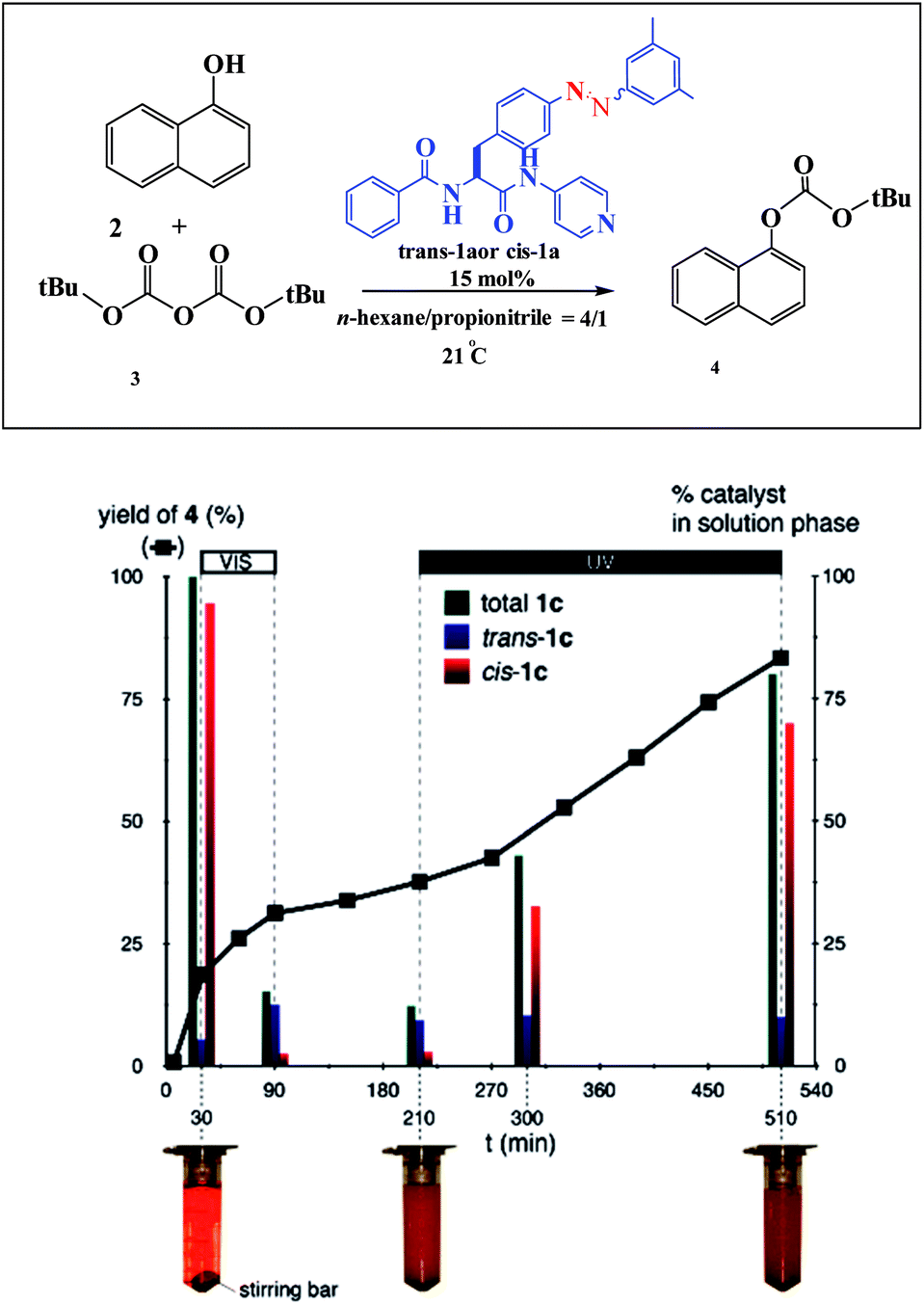
An example of a photoswitchable catalyst based on aggregation/dissociation of a diamide framework was reported by Kumagai and co-workers in 2013 (Scheme 17).66 The photoswitchable catalyst 1 was applied to interaction of 1-naphthol 2 with Boc2O (Fig. 5). The authors demonstrated that, in the presence of the soluble cis-1a, the reaction profile showed steady progress at 21 °C to give carbonate 4, and the reaction mixture remained homogeneous. When the trans-1a is used as the catalyst, the reaction barely produced 4 in the same reaction period, because of the extensive aggregation of trans-1a leading to significant deterioration of the catalytic performance. In addition, the authors demonstrated that the catalytic activity could be switched between visible light irradiation (>422 nm) and UV (365 nm) irradiation (Fig. 6). When UV (365 nm) irradiation was used to induce photoisomerization from trans-1a to cis-1a, the reaction is very slow (6/4% yield) over a 120 min period (from 90 min to 210 min) (Fig. 7). In contrast, the trans-1a, generated by light irradiation (>422 nm), afforded 4 in 33% yield after 90 min of irradiation (at 300 min). The catalytic activity of cis-1a is significantly higher than the catalytic activity of trans-1a.
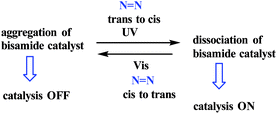 |
| | Scheme 17 Background of aggregation/dissociation property of bis(2-hydroxyphenyl)amide framework. Reprinted by permission from ref. 66, Royal Society of Chemistry. | |
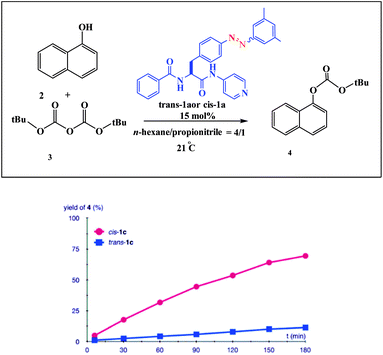 |
| | Fig. 6 Profile of the reaction of 2 with 3 promoted by trans-1a (blue square) and cis-1a (red circle). 2: 0.1 mmol, 3: 0.12 mmol, solvent 1.5 mL. Reprinted by permission from ref. 66, Royal Society of Chemistry. | |
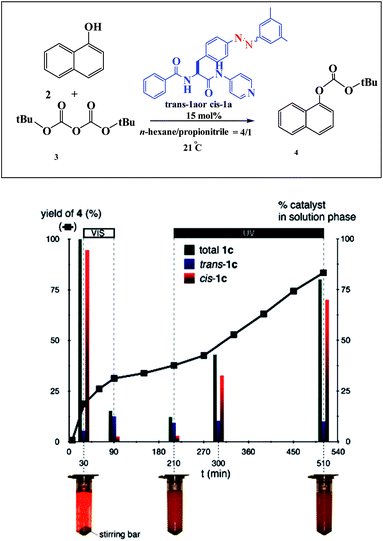 |
| | Fig. 7 Profile of the reaction of 2 with 3 promoted by 1a. Black squares represent yields of 4. 2: 0.1 mmol, 3: 0.12 mmol, solvent 1.5 mL. Green, blue, and red bars represent the amount of trans-1a + cis-1a, trans-1a, and cis-1a in the solution phase, respectively. Visible light (>422 nm) irradiation was used during the period 30–90 min. UV light (365 nm) irradiation was used during the period 210–510 min. Reprinted by permission from ref. 66, Royal Society of Chemistry. | |
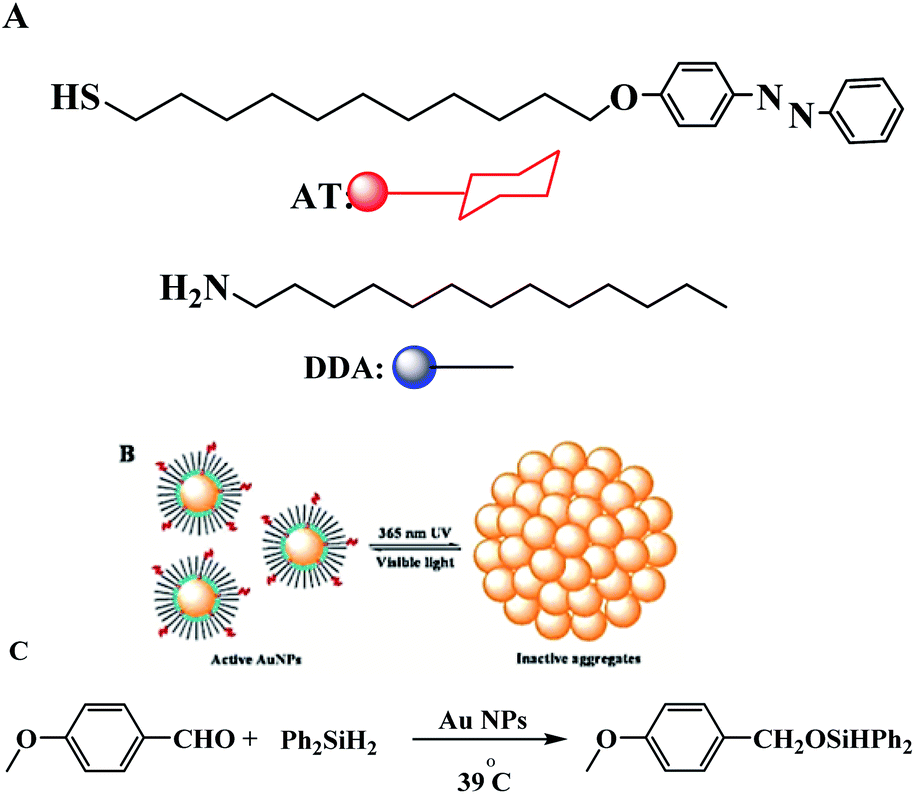
The same group described the catalytic activity of gold nanoparticles in a hydrosilylation reaction, which can turn ‘on’ or ‘off’ by irradiation with UV or visible light. On using UV light, the particles aggregated and the catalysis effectively switched “off”, while in the presence of the visible light, the particles redispersed and the catalysis proceeded (Scheme 18).67
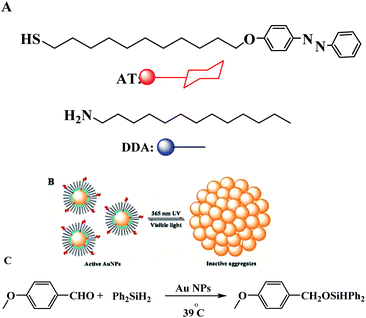 |
| | Scheme 18 (A) Molecular structures of the “background” DDA surfactant and of the photoresponsive azobenzene-thiol ligand, AT. (B) Schematic representation of a photoswitchable AuNP system. Dispersed NPs are catalytically active; aggregated NPs are catalytically inactive. (C) Hydrosilylation of 4-methoxybenzaldehyde catalyzed by AuNPs in dry toluene at 39 °C and under argon. Reprinted by permission from ref. 67, Copyright © 2010, American Chemical Society. | |
2. pH-driven switching
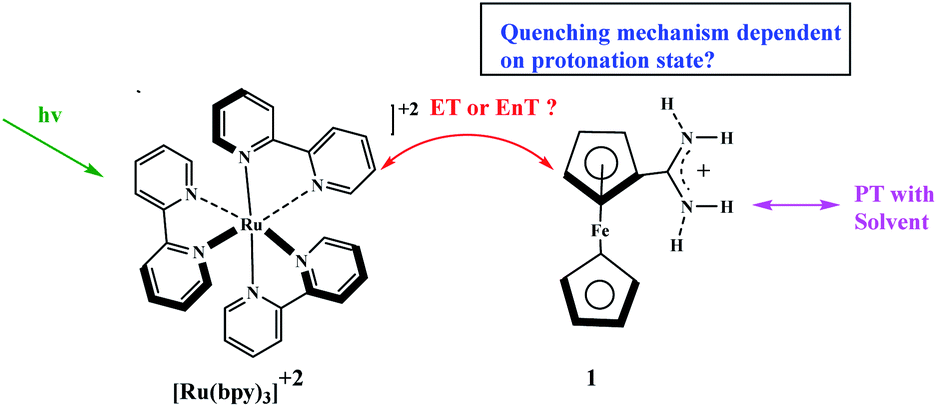
Chemically-driven processes can be switched “on” and “off” by changes in pH. Early use of a change in pH to control the catalytic activity was reported by Young and co-workers who exploited the rate constant for [Ru(bpy)3]2+ excited-state controlled under pH conditions ferrocenyl-amidinium derivative.68 By contrast, the rate constant with which the [Ru(bpy)3]2+ excited state is quenched by an analogous ferrocene derivative (ferrocenyltrimethylammonium) lacks protonic group and does not depend on pH. The principal results of this study show that the energy transfer (EnT) appears at low pH when complex 1 is protonated, and the electron transfer (ET) appears at high pH when complex 1 is deprotonated (Scheme 19). Moreover, it should be noted that complex 1 at any pH does not show evidence for charge transfer products (Scheme 19).
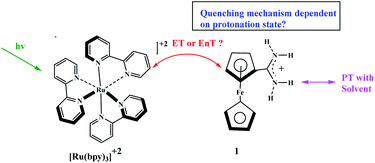 |
| | Scheme 19 Potential bimolecular, bidirectional quenching pathways between [Ru(bpy)3]2+ by either a ferrocenyl-amidinium complex (1). Reprinted by permission from ref. 68, Copyright © 2018, American Chemical Society. | |
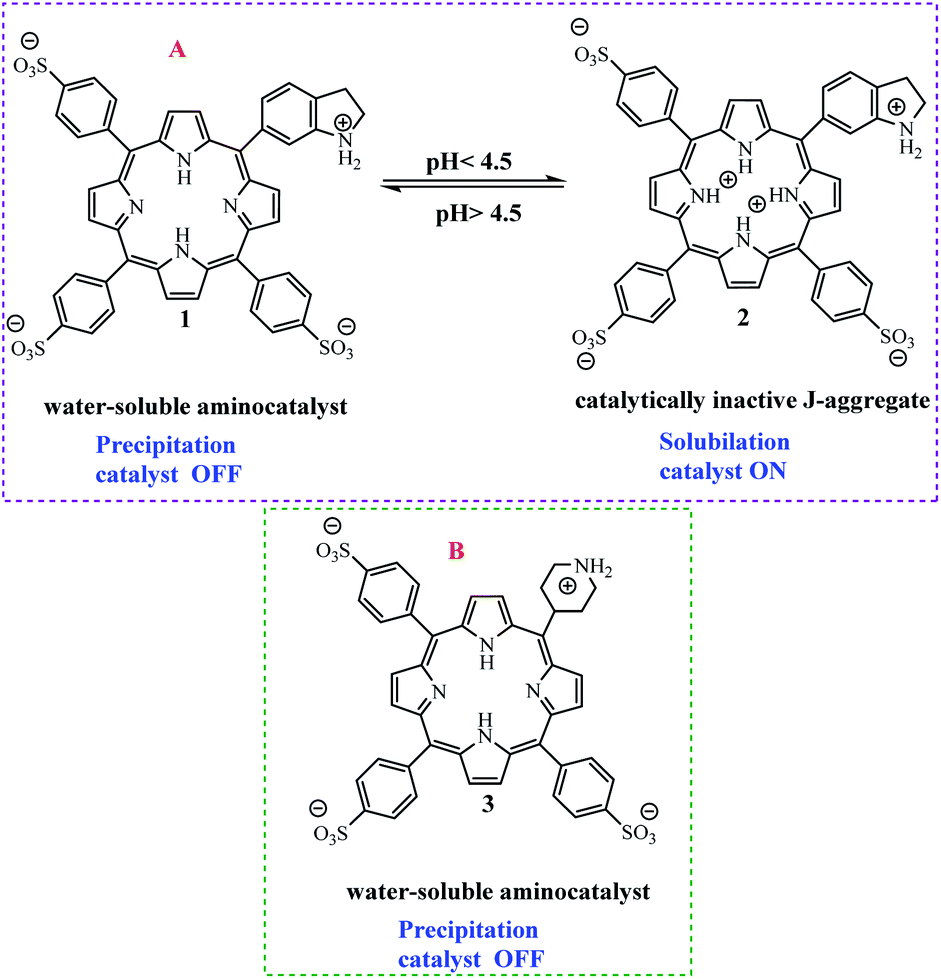
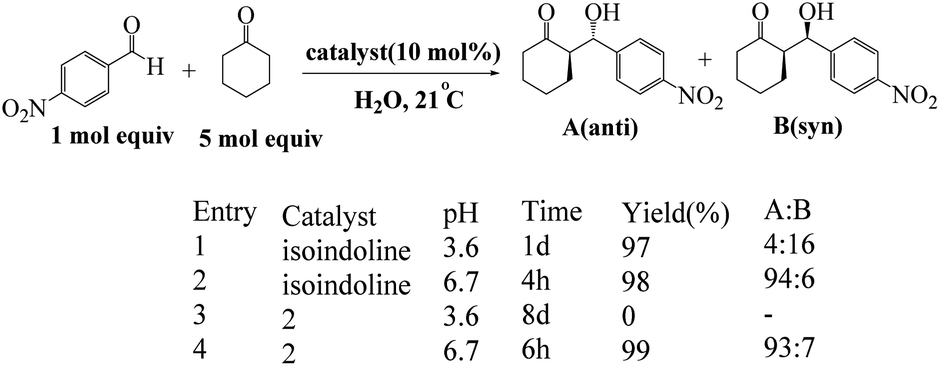
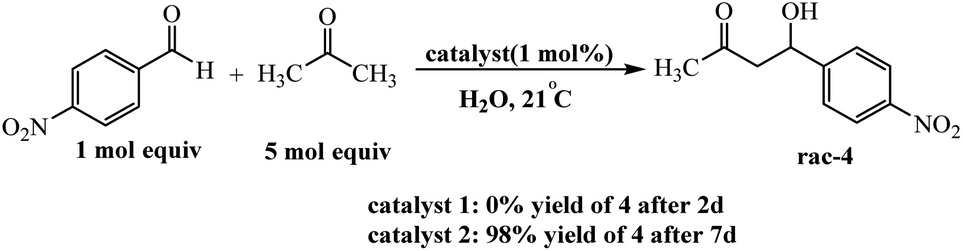
Arlegui and coworkers designed series of amphiphilic 5-(cyclic-secondary-amine)-10,15,20-tris(4-sulfonatophenyl)porphyrins as organocatalysts (Scheme 20). After, organocatalytic activity were used for Michael and aldol reactions (Schemes 21 and 22).69 Experimental studies show that the catalytic activity of organocatalysts for the aldol reaction of cyclohexanone with 4-nitrobenzaldehyde can be selectively and reversibly switched on and off by adjusting the homogeneity of its solutions through pH variations.
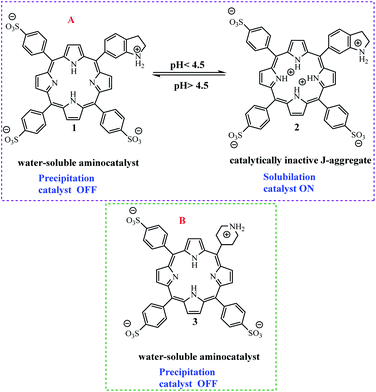 |
| | Scheme 20 (A) 5-(Piperidin-1-ium-4-yl)-10,15,20-tris(4-sulfonatophenyl)porphyrin disodium salt 2. (B) 5-(Piperidin-1-ium-4-yl)-10,15,20-tris(4-sulfonatophenyl)porphyrin disodium salt 3. Reprinted by permission from ref. 69, © 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
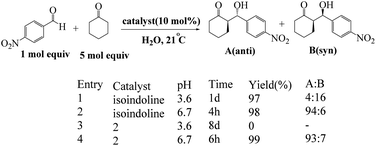 |
| | Scheme 21 Catalysis of the aqueous aldol reaction of cyclohexanone with 4-nitrobenzaldehyde by isoindoline and by the amphiphilic porphyrin. Reprinted by permission from ref. 69, © 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
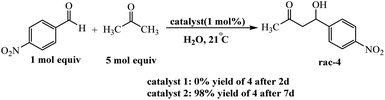 |
| | Scheme 22 Catalysis of the aqueous aldol reaction between acetone and 4-nitrobenzaldehyde by the amine–porphyrin hybrids 1 and 2. Reprinted by permission from ref. 69, © 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
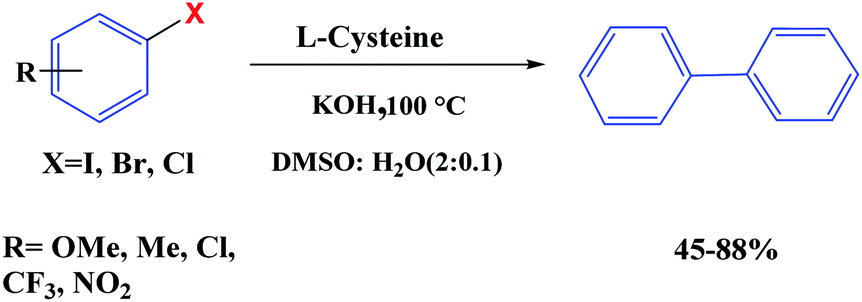
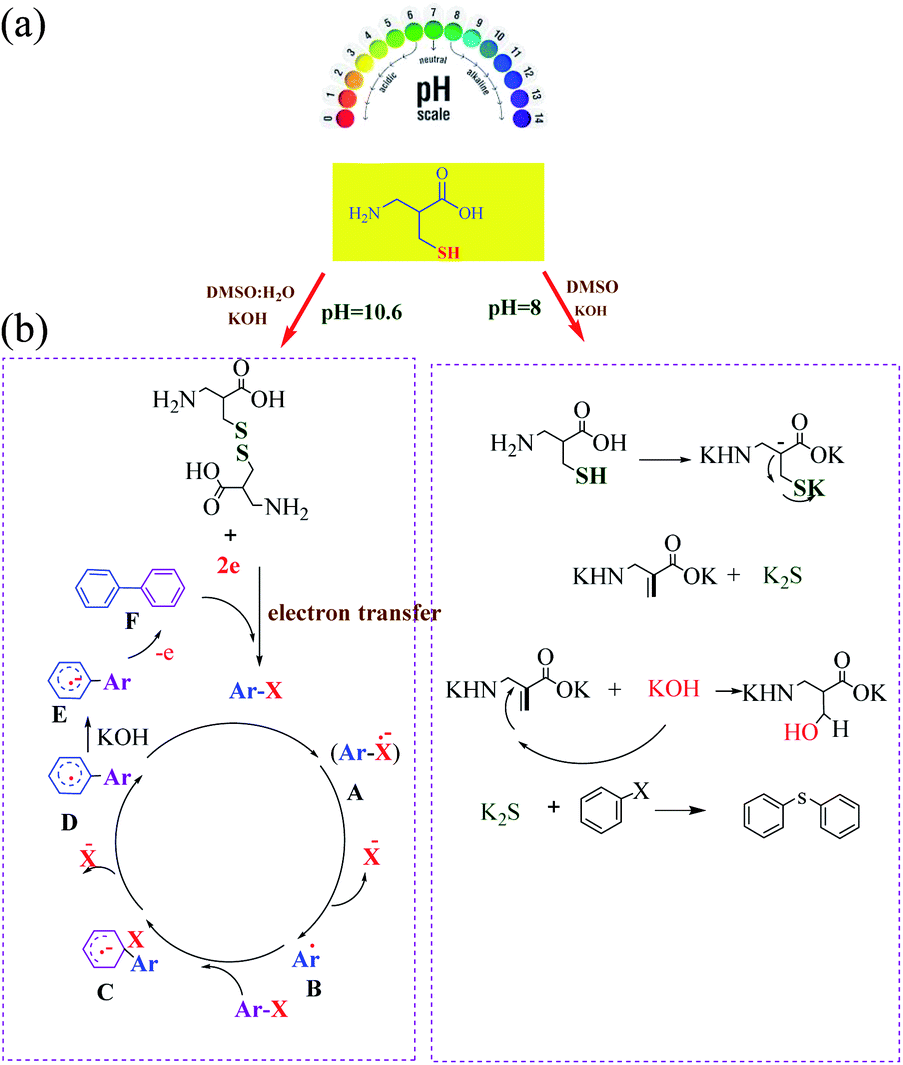
Recently, we have reported the transition metal-free homocoupling of aryl halides in the presence of L-cysteine through a homolytic aromatic substitution mechanism (defined as the replacement of a leaving group X on an aromatic ring by an attacking radical species) (Scheme 23). It is expected that elemental sulfur will be present in L-cystine as an electron-transfer route (potassium hydroxide in dimethyl sulfoxide plays a crucial role in the initiation process under transition metal free conditions).70 In this work, two reaction pathways were observed: (1) the oxidation of L-cysteine thiol groups is a dominant process and the formation is usually favored at a basic pH 8 and (2) β-elimination is usually favored at a basic pH 10.6 (Scheme 24). Therefore, the reaction of homocoupling of aryl halides in the presence of L-cysteine at pH 8 gave the desired products in excellent yields at pH. Under the optimized conditions, KOH was used as the base resulted in the homocoupling of aryl halides, and DMSO was found to be the best solvent for the reaction. While expanding the scope of the reaction, the cross-coupling of various substituted aryl halides was investigated under optimized conditions, providing the corresponding product in excellent yields.
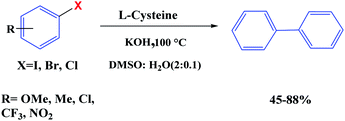 |
| | Scheme 23 Metal-free homocoupling of aryl halides. Reprinted by permission from ref. 70, Royal Society of Chemistry. | |
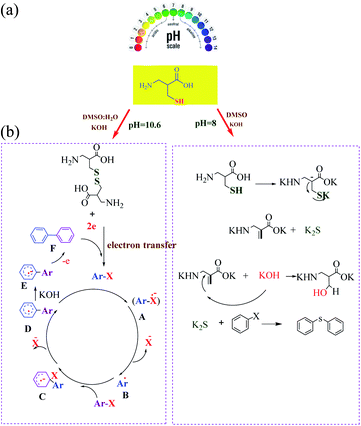 |
| | Scheme 24 (a) Switchable and proposed mechanism for the synthesis of biaryls. (b) Proposed mechanism for the synthesis of symmetrical aryl sulfide. Reprinted by permission from ref. 70, Royal Society of Chemistry. | |
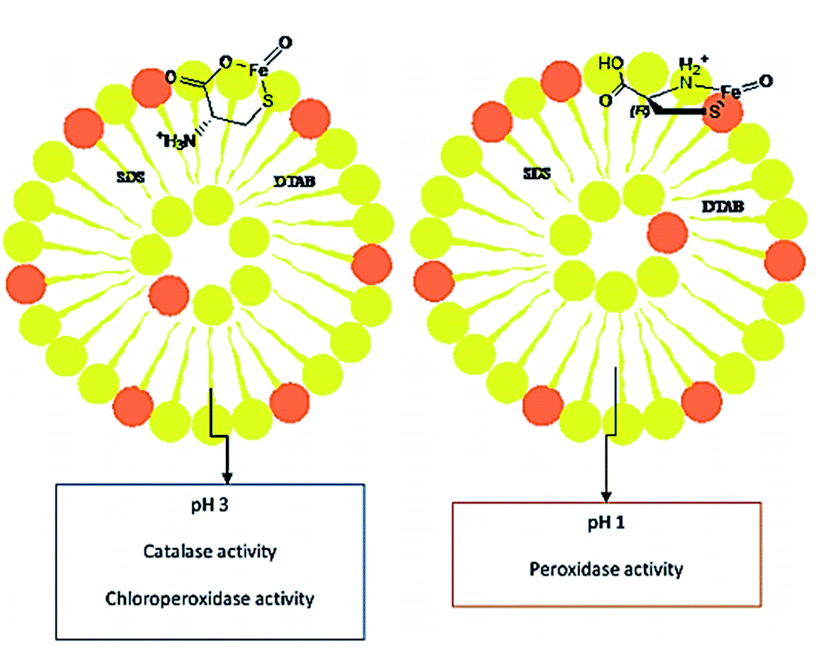
Moosavi-Movahedi et al. synthesized catalase-like biocatalyst system containing cysteine–iron complex encapsulated in a vesicular mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4; SDS/DTAB) in order to imitate a chloroperoxidase (CLP) enzyme via chlorination of thionine at pH 3. This artificial enzyme behaved both as a catalase and CLP at pH 3, and as a peroxidase at pH 1 (Fig. 8).71 These results demonstrated that the cysteine/iron(III) center acted as a multifunctional catalyst. In addition, the sulfur and ammonium moieties of L-Cys interacted in the presence of metal ion at pH 1. However, the ammonium side chain was replaced with the carboxylate group at pH 3. The shape of the metallo-vesicular catalyst at each pH value was obtained by dynamic light scattering, and was confirmed by transmission electron microscopy (TEM) (Fig. 9).
:4; SDS/DTAB) in order to imitate a chloroperoxidase (CLP) enzyme via chlorination of thionine at pH 3. This artificial enzyme behaved both as a catalase and CLP at pH 3, and as a peroxidase at pH 1 (Fig. 8).71 These results demonstrated that the cysteine/iron(III) center acted as a multifunctional catalyst. In addition, the sulfur and ammonium moieties of L-Cys interacted in the presence of metal ion at pH 1. However, the ammonium side chain was replaced with the carboxylate group at pH 3. The shape of the metallo-vesicular catalyst at each pH value was obtained by dynamic light scattering, and was confirmed by transmission electron microscopy (TEM) (Fig. 9).
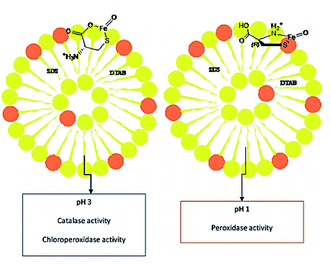 |
| | Fig. 8 Schematic interpretation of vesicular Fe–cysteine biocatalyst structures in different pH and three different enzymatic activities. Reprinted by permission from ref. 71, Copyright © 2016 Elsevier B.V. All rights reserved. | |
 |
| | Fig. 9 TEM images of the artificial enzyme at a pH level of 3 (A) and pH 1 (B). The complex was prepared from Fe3+ (10 M), cysteine (10 M), DTAB (4 mM), and SDS (1 mM). Reprinted by permission from ref. 71, Copyright © 2016 Elsevier B.V. All rights reserved. | |
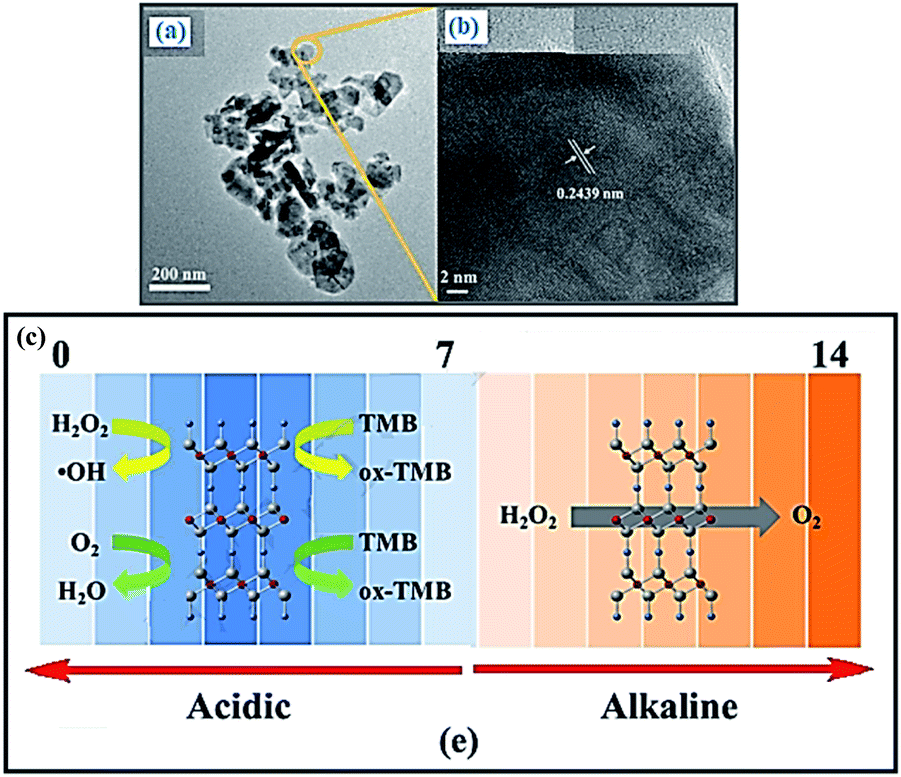
Yang et al. demonstrate that CuMnO2 nanoflakes possess pronounced intrinsic enzyme mimic properties of peroxidase, oxidase and catalase, which also can be modulated by adjusting pH value.72 Under acidic condition, CuMnO2 nanoflakes catalyze the reaction of TMB in the presence of H2O2 or O2 to produce a blue color, showing peroxidase-like and oxidase-like activity, respectively. In contrast, at alkaline pH, CuMnO2 nanoflakes catalyze the dismutation decomposition of H2O2 to produce O2, exhibiting catalase-like property (Fig. 10). Moreover, the mechanism of the catalytic reaction has also been investigated, which indicates that the peroxidase mimetic activity of CuMnO2 nanoflakes originates from their ability to generate hydroxyl radical (˙OH). Thus, based on the color reaction and the hindrance of the peroxidase-like activity of CuMnO2 nanoflakes in the presence of H2O2 and L-cysteine, a platform was constructed to detect L-cysteine concentration which also exhibits higher sensitivity compared with other bimetallic oxides.
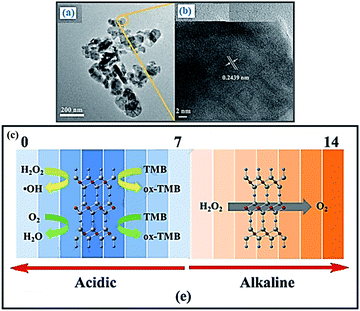 |
| | Fig. 10 TEM and HRTEM of CuMnO2 nanoflakes (a and b). (c) The enzyme mimetic activity of CuMnO2 nanoflakes is pH switchable. The catalase-like activity of CuMnO2 nanoflakes. Reprinted by permission from ref. 72, Copyright © 2018 Elsevier B.V. All rights reserved. | |
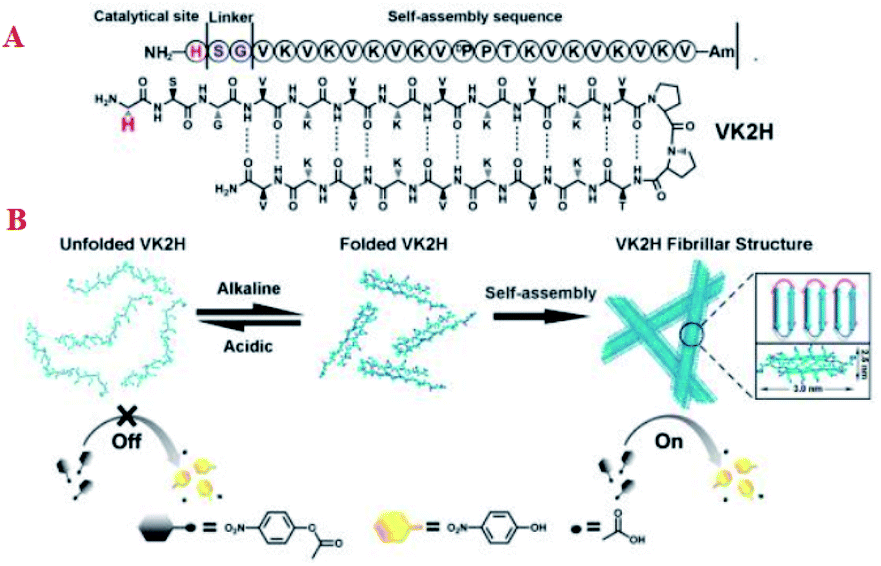
Zhang et al. developed a pH switchable artificial hydrolase based on regulating the conformation of a self-assembling β-hairpin amphiphilic oligopeptides with a hydrolytically active residue at the N-terminus of the peptide catalytic activity can be reversible switched by pH-induced assembly/disassembles of the fibrils into random coils (Fig. 11A).73 At neutral pH, the lysine groups are protonated, and this causes a repulsion which prevents the hydrogen bonding of the strands of the hairpin, so that the peptide adopts a random coil configuration. Increasing the pH to 9 reduces this repulsion as the lysine group's deprotonate, and VK2H adopts a β-sheet configuration with the two strands in antiparallel conformation (Fig. 11B).
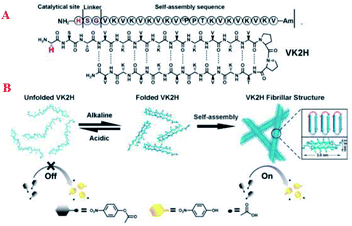 |
| | Fig. 11 Chemical structure of peptide VK2H (A); schematic representation of the pH-switched artificial hydrolase based on conformation change of VK2H and the cartoon structure of the bilayer (B). Reprinted by permission from ref. 73, Copyright © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
3. Coordination-driven switching
3.1 Ligand-based switching
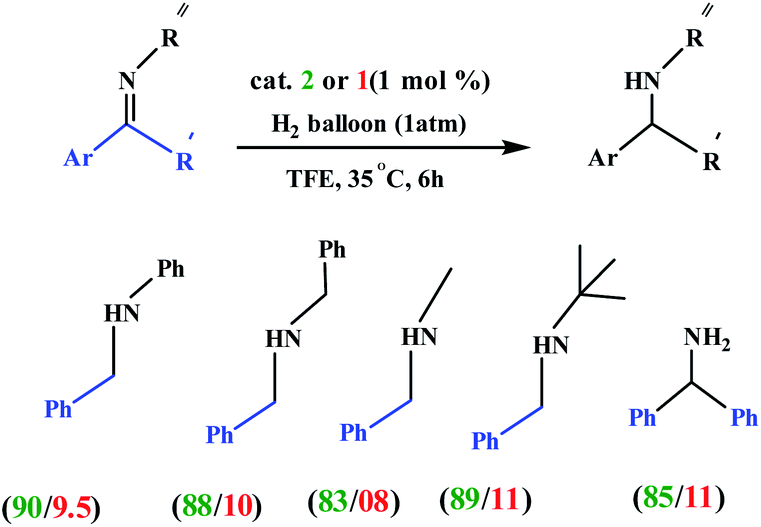
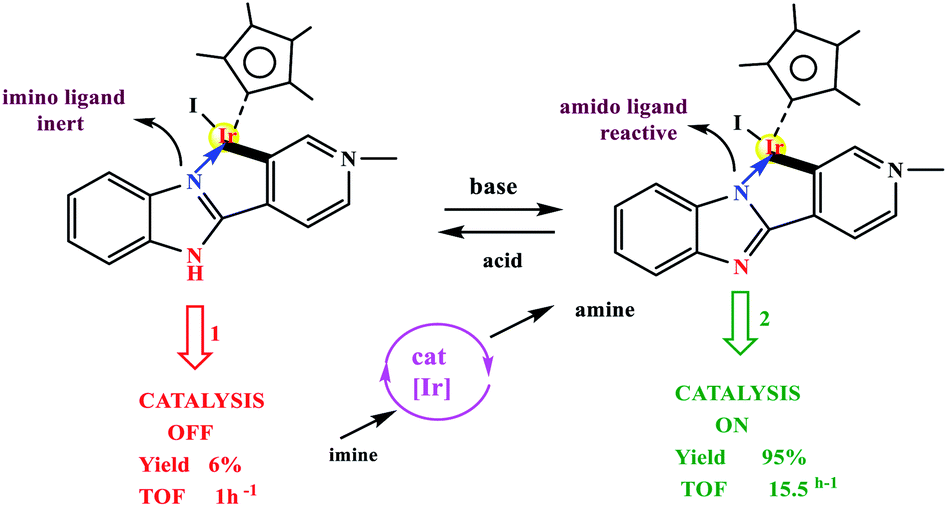
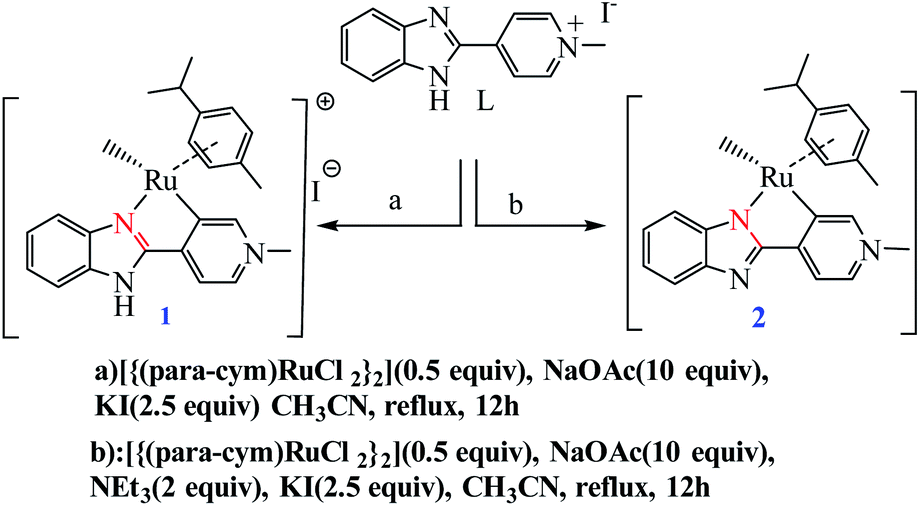
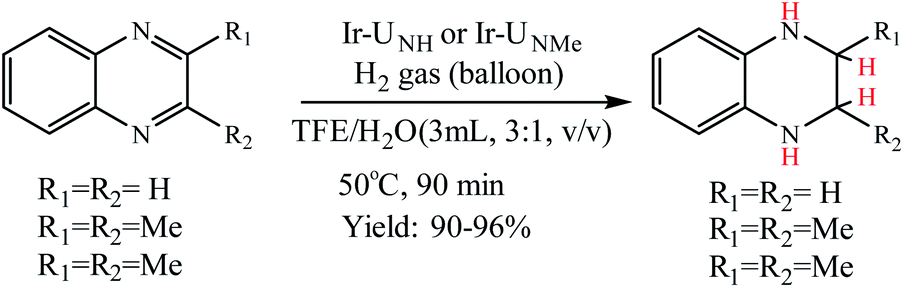
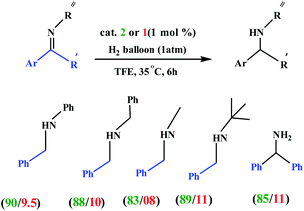
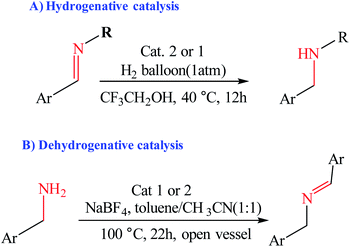
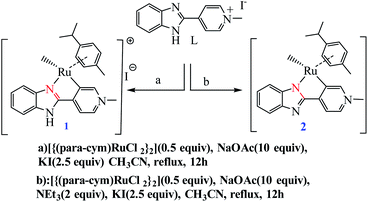
Choudhury and co-workers developed a molecular switch consisting of a hybrid pyridylidene–benzimidazole ligand bound to an IrIIICp* and used it for the catalytic hydrogenation of imines with molecular hydrogen under ambient conditions (H2 balloon, 35 °C)74 (Table 4) (Scheme 25). The catalysis could be switched OFF and ON efficiently for several cycles with the addition of acid and base, respectively (Scheme 25) (Fig. 12).
Table 4 Examples of 2 and 1-catalyzed hydrogenation of iminesa
| Conditions: 0.2 mmol of imine, H2 balloon, 1 mol% catalyst, 2 mL of TFE, 35 °C, 6 h. |
 |
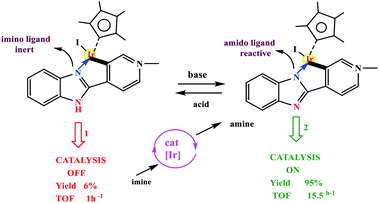 |
| | Scheme 25 (Top) Example of acid–base controlled molecular coordination-switch and (bottom) a catalysis-switch developed on a molecular coordination-switch platform. Reprinted by permission from ref. 74, Copyright © 2018, American Chemical Society. | |
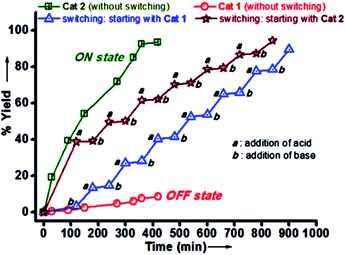 |
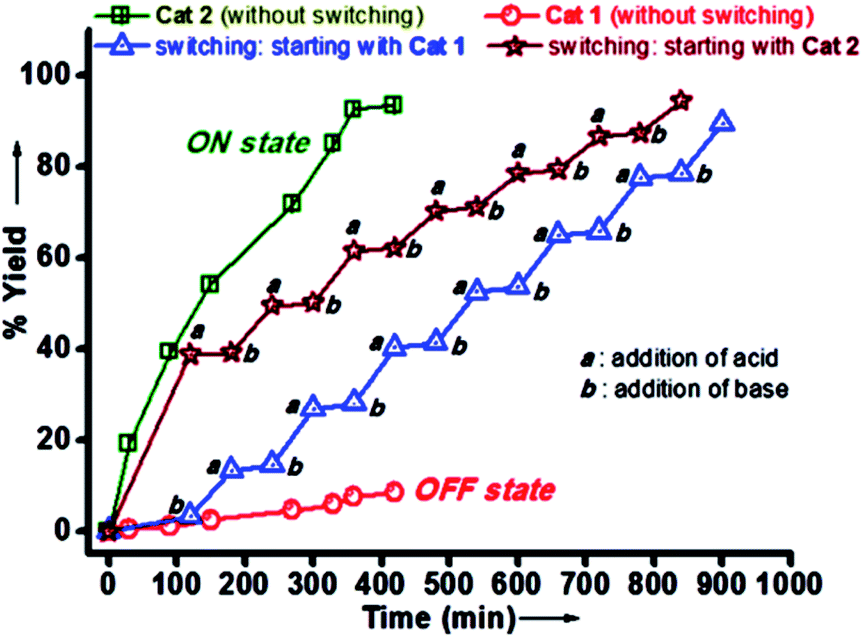
| | Fig. 12 Plot of yield (%) versus time for the hydrogenation of N-benzylideneaniline catalyzed by 1 (red curve) and 2 (green curve); blue curve represents the ON/OFF switching of the catalytic activity when started with catalyst 1 followed by consecutive addition of base (NEt3) and acid (CF3CO2H); maroon curve represents the ON/OFF switching of the catalytic activity when started with catalyst 2 followed by consecutive addition of acid (CF3CO2H) and base (NEt3). Reprinted by permission from ref. 74, Copyright © 2018, American Chemical Society. | |
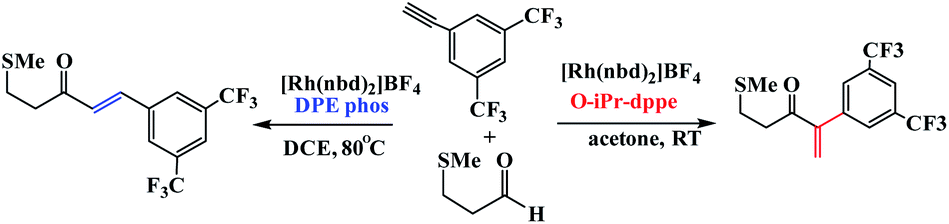
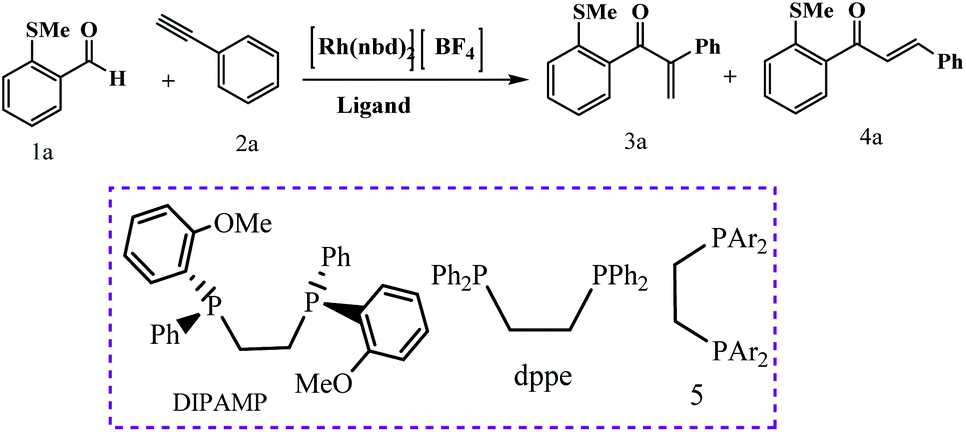
Weller et al. have described ligand control being used to switch between linear and branched products for a given substrate combination (Scheme 26).75 These ligands were used to control regioselectivity in the hydroacylation of alkynes. The principal results of this study show that when a DIPAMP-derived Rh-catalyst was employed for the combination of aldehyde 1a and phenylacetylene (2a), the ketones (3a + 4a) were obtained as a 1:4 mixture of branched and linear regioisomers (Table 5, entry 1). Moreover, in presence of bulky ortho-iPr-dppe-derived catalyst, the formation of the branched adduct was promoted.
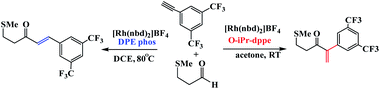 |
| | Scheme 26 Catalyst-controlled linear and branched-selective alkene and alkyne hydroacylation reactions. Reprinted by permission from ref. 75, Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Table 5 Ligand effects on the Rh-catalyzed hydroacylation reaction of aldehyde 1a and phenylacetylene.a Reprinted by permission from ref. 75, Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
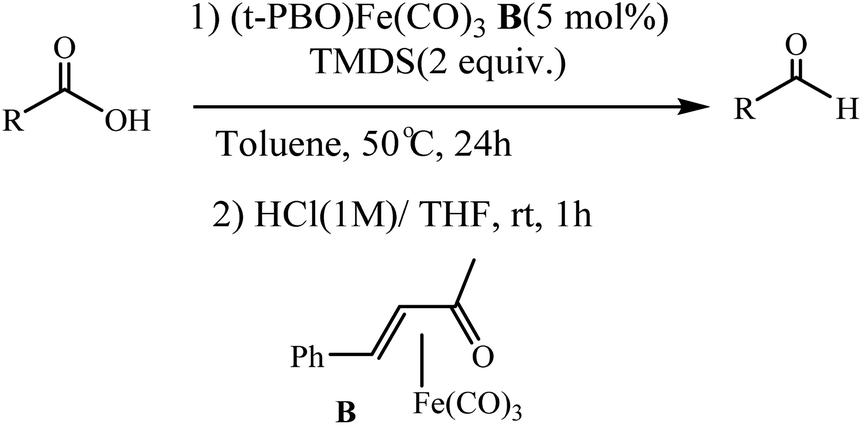
Following these strategies, in 2012, Sortais and co-workers reported that the control over the hydrosilylation of carboxylic acids using phenylsilane and (COD)Fe(CO)3 catalyst under UV-irradiation at rt, alcohols was obtained specifically in good yields, whereas aldehydes were selectively obtained using TMDS and (t-PBO)Fe(CO)3 catalyst under thermal activation (Schemes 27 and 28).76
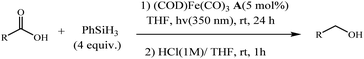 |
| | Scheme 27 Hydrosilylation of carboxylic acids into alcohols with PhSiH3 using Fe (CO)3(COD) catalyst A. Reprinted by permission from ref. 76, Royal Society of Chemistry. | |
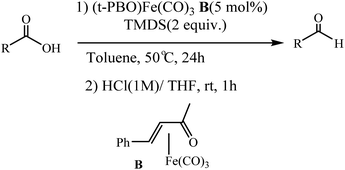 |
| | Scheme 28 Selective reduction of carboxylic acids into aldehydes with TMDS using Fe (CO)3(t-PBO) catalyst B. Reprinted by permission from ref. 76, Royal Society of Chemistry. | |
3.2 Switching promoted by the addition/removal of metal cations
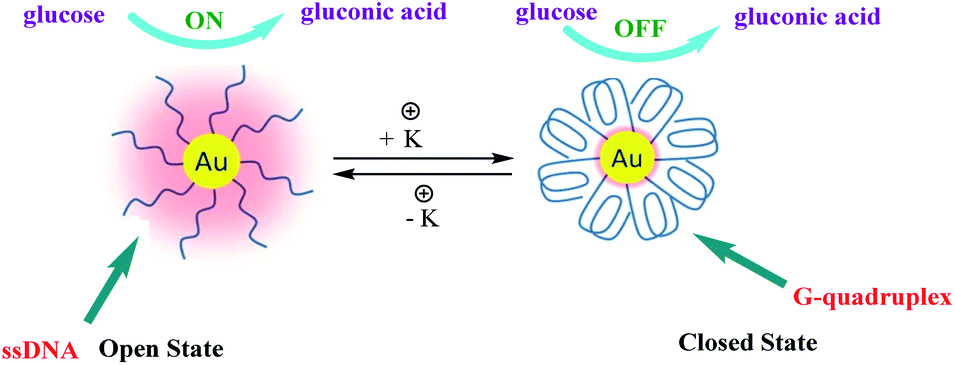
High catalytic activity of AuNPs for oxidation of glucose in the presence of O2, producing gluconic acid and H2O2, which mimic natural glucose oxidase (GOx) is well known.77–79 Moreover, the catalytic activity of AuNPs were dependent on the degree of their surface coverage with inert materials. In 2015, Zhou and co-workers developed a simple and reliable strategy for the reversible regulation of the catalytic activity of AuNPs, which was demonstrated with a glucose oxidation reaction as a proof-of-concept study80 (Scheme 29). In this study, the authors immobilized thiol-modified G-rich sequence containing four GGG stretches on AuNPs (d = 10 nm) surface through thiol–Au bonds which have glucose oxidase (GOx) like activity. A remarkable feature of this structure is its high sensitivity to alkali metal ions, especially K+. In order to the conformational change of G-rich sequences driven by K+. In the absence of K+ (open state), these G-rich strands maintain flexible single-stranded conformations (ssDNA) and if the inter-strand distance is large enough, the glucose molecules are expected to be able to interact with surface gold atoms and the reaction can go ahead. In order to alter the activity, K+ will then be introduced and under its action, each G-rich strand will fold into a G-quadruplex conformation (closed state) which has a theoretical diameter of 2.5 nm. Through alternately adding and removing K+, these DNA nanomachines will switch between “open” and “closed” states, resulting in a reversible variation of the exposed surface area of AuNPs and thus achieving a reversible regulation of their catalytic activity.
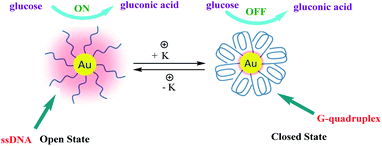 |
| | Scheme 29 Schematic illustration of the reversible regulation of the GOx-like catalytic activity of AuNPs by G-quadruplex DNA nanomachines. Reprinted by permission from ref. 80, Copyright © 2015, The Author(s). | |
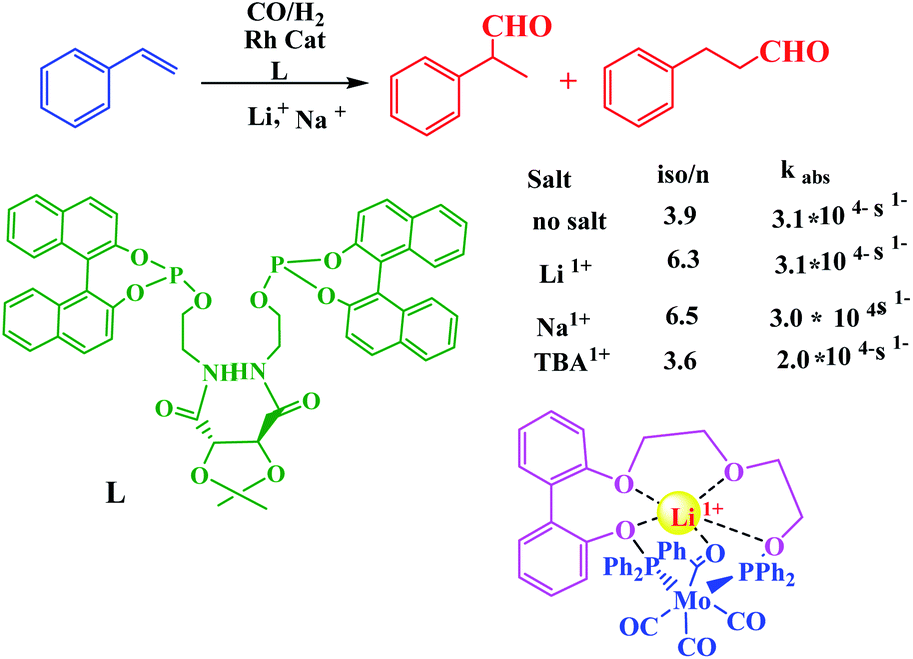
The controlled catalysis has also been achieved using Lewis acidic cation which was utilized for the activation and stabilization of substrates. In this sense, Gray group demonstrated selectivity of styrene hydroformylation by Rh enchained crown ether catalysts in the presence of Li+ or Na+ ions toward nucleophilic attack in metalla-crown ether with phosphine donors (Scheme 30).81
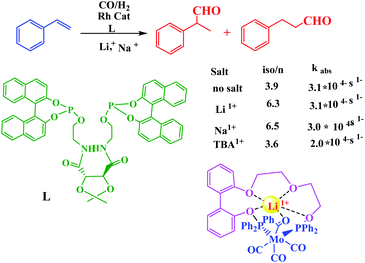 |
| | Scheme 30 Cation-modulated rhodium-catalyzed hydroformylation and a molybdenum complex that models substrate activation. Reprinted by permission from ref. 81, Copyright © 2008, American Chemical Society. | |
4. Redox switching
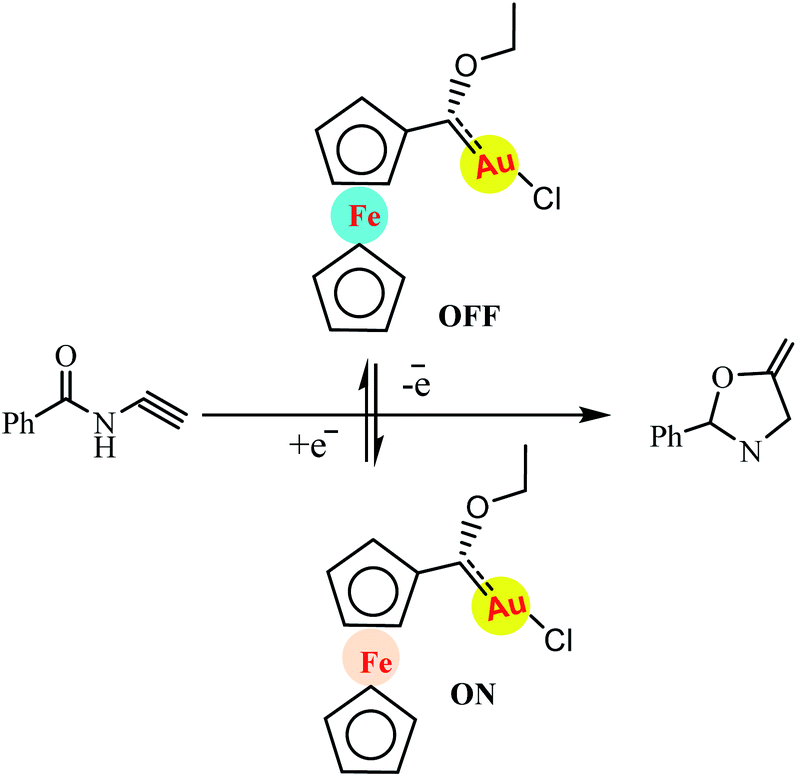
Redox switching is an attractive strategy for molecular switches that can undergo reversible structural changes upon application of external stimuli, such as light, heat, electrons, or chemical reactions. Gold(II) species catalyse the cyclisation of N(2-propyn-1-yl)benzamide to 2-phenyl-5-vinylidene-2-oxazoline without halide abstraction while the saturated gold(I) complex is inactive. Redox switching between gold(II) and gold(I) turns catalytic turnover on and of (Scheme 31).82 In a related example, Veit and co-workers reported a redox-switchable gold-catalysed cyclisation with the gold centre of the precatalyst 1 reversibly switching between highly active, presumably coordinatively unsaturated gold(II) and inactive, coordinatively saturated gold(I) centres.
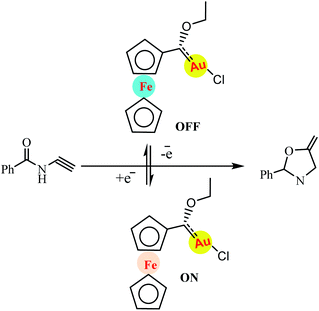 |
| | Scheme 31 Switchable gold-catalysed cyclisation of N(2-propyn-1-yl)benzamide to 2-phenyl-5-vinylidene-2-oxazoline. Reprinted by permission from ref. 82, Royal Society of Chemistry. | |
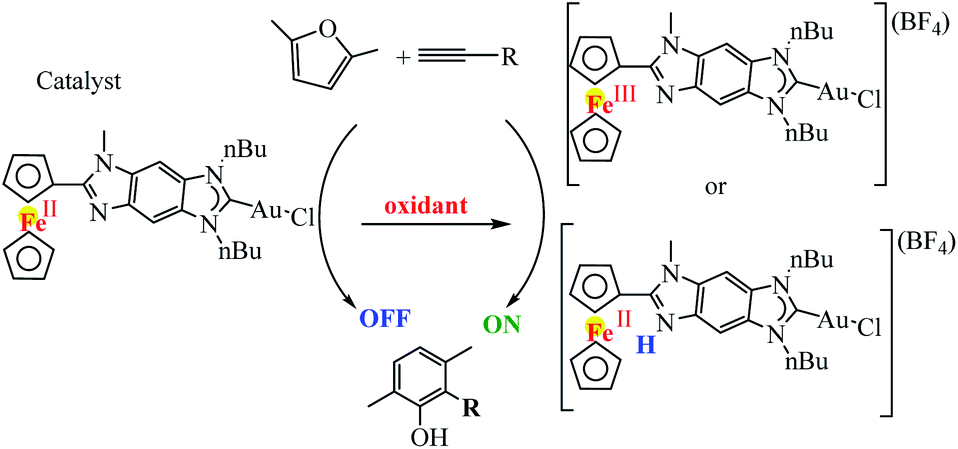
Following a similar strategy, Poyatos et al. studied the catalytic properties of the gold complex in the cyclization of 2,5-dimethylfuran with terminal alkynes (Scheme 32).83 In this study, an imidazolium salt with a fused benzoferrocenyl was synthesized and used as an N-heterocyclic carbene (NHC) precursor, and the related ferrocenyl–imidazolylidene complexes Fc–NHC–MLn (MLn = AuCl) were synthesized. The oxidation of the gold complex with acetylferrocenium tetrafluoroborate afforded the oxidized ferrocenium–NHC–AuCl (Fe(III)) species. After, the effects of the oxidation of the ligand in homogeneous catalysis were tested by using a related ferrocenyl–imidazolylidene–gold(I) complex. In the cyclization of alkynes with furans, the neutral complex was not active, while the product resulting from its oxidation produced moderate to good yields in the formation of the final products.
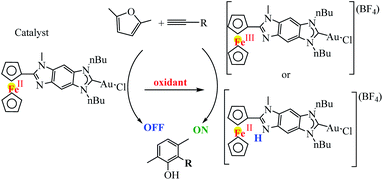 |
| | Scheme 32 Intermolecular gold(I)-catalyzed cyclization of 2,5-dimethylfuran with terminal alkynes. Reprinted by permission from ref. 83, Copyright © 2016, American Chemical Society. | |
5. Switching driven by mechanical forces
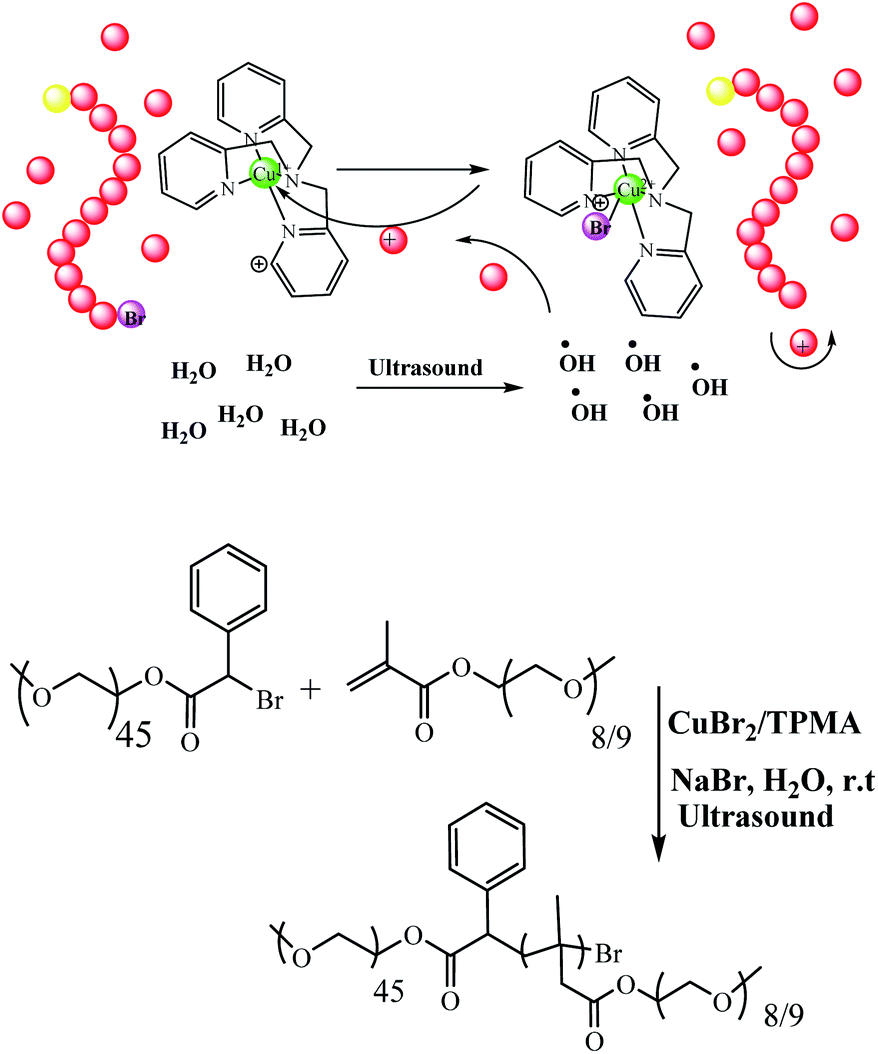
Mechanically-activated systems are triggered by mechanical forces, i.e. ultrasound, which induce hydrodynamic force fields around the collapsing cavitation bubbles. Due to the high strain rates, polymer chains uncoil and stretch under these conditions, which can cause the weakest bonds in the chain to rupture. Matyjaszewski and co-workers reported a new procedure for the polymerizations of oligo (ethylene oxide) methyl ether methacrylate (OEOMA) and 2-hydroxyethyl acrylate (HEA) in water. Moreover, it was successfully carried out in the presence of ppm amounts of CuBr2 catalyst and tris(2-pyridylmethyl)amine ligand when exposed to ultrasonication (40 kHz, 110 W) at room temperature. Temporal control over the polymer chain growth was demonstrated by switching the ultrasound on/off due to the regeneration of activators by hydroxyl radicals formed by ultrasonication (Scheme 33).84
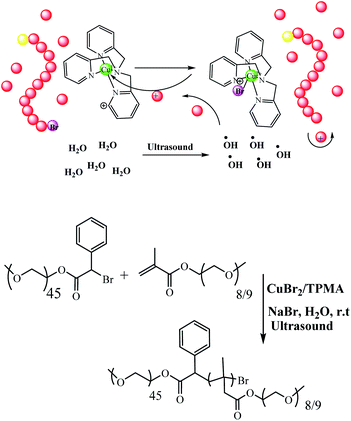 |
| | Scheme 33 Ultrasonication-induced aqueous atom transfer radical polymerization. Reprinted by permission from ref. 84, Copyright © 2018, American Chemical Society. | |
Sijbesma and co-workers reported a general method to activate silver(I) complexes of polymer-functionalized N-heterocyclic carbenes and ruthenium biscarbene complex with the mechanical force for the transesterification reaction and olefin metathesis reactions respectively (Scheme 34).85
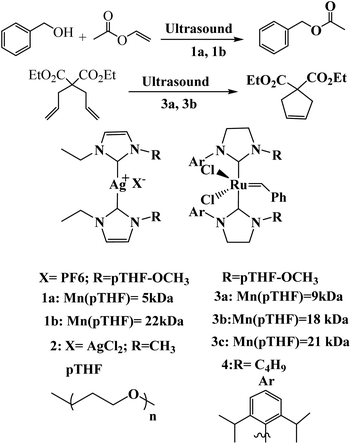 |
| | Scheme 34 Mechanochemical activation of a catalytic ring-closing metathesis (RCM) reaction. Reprinted by permission from ref. 85, Copyright © 2009, Nature Publishing Group. | |
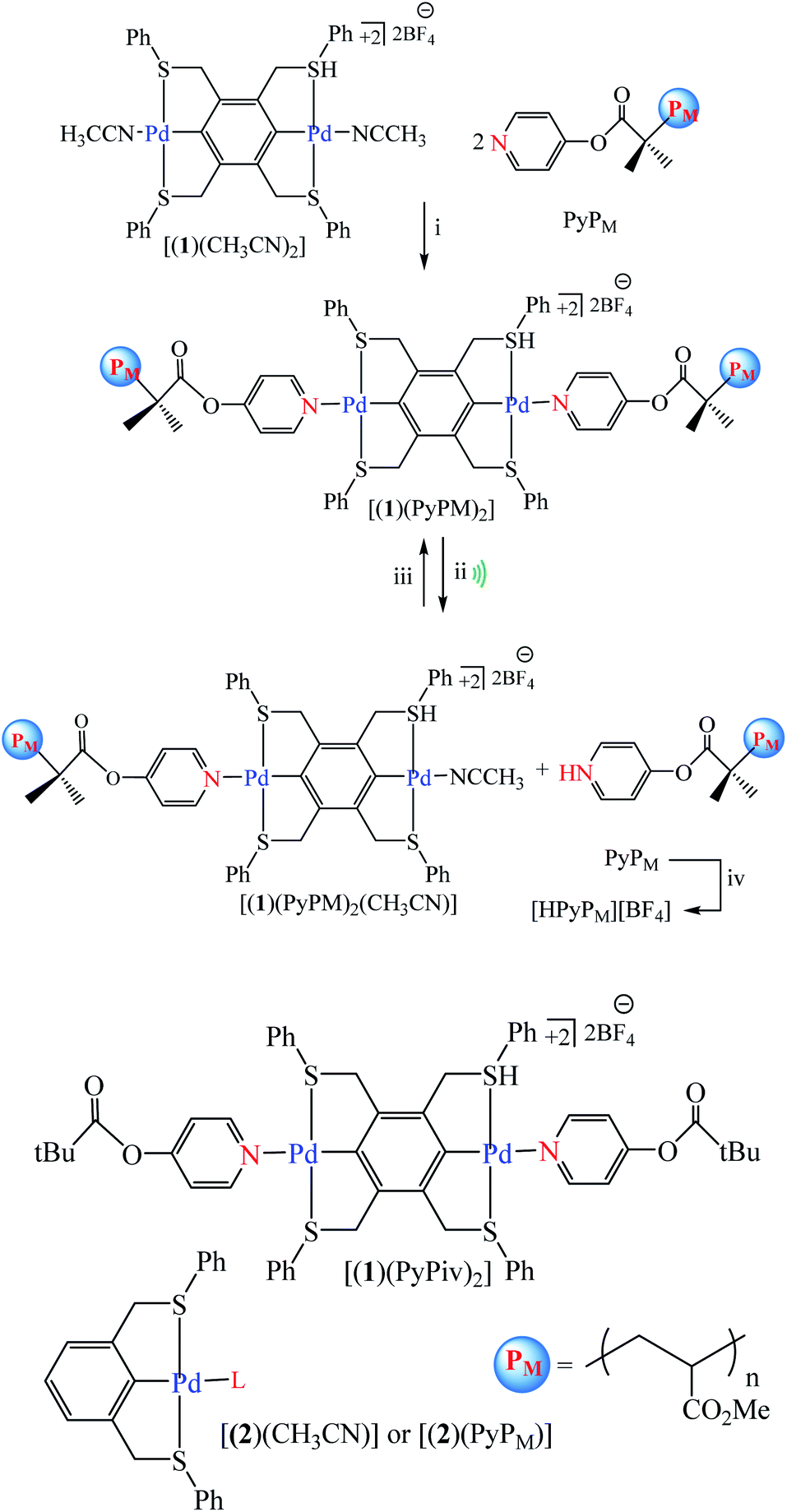
Tennyson et al. reported coupling of pyridine-capped poly(methyl acrylate), PyPM (where M corresponds to the number average molecular weight in kDa), to the SCS-cyclometalated dipalladium complex [(1)(CH3CN)2] afforded organometallic polymers [(1)(PyPM)2] with a concomitant doubling in molecular weight. Ultrasonication of solutions containing [(1)(PyPM)2] effected the mechanical scission of a palladium–pyridine bond, where the liberated PyPM was trapped with excess HBF4 as the corresponding pyridinium salt, harnessed to effect the stoichiometric deprotonation of a colorimetric indicator, or used to catalyze the anionic polymerization of R-trifluoromethyl-2,2,2-trifluoroethyl acrylate (Scheme 35).86 Experimental result show no formation of C and E was catalyzed by [(1)(PyP66)2] in the absence of sonication, consistent with negligible thermal contribution to the observed conversion (Scheme 36).86
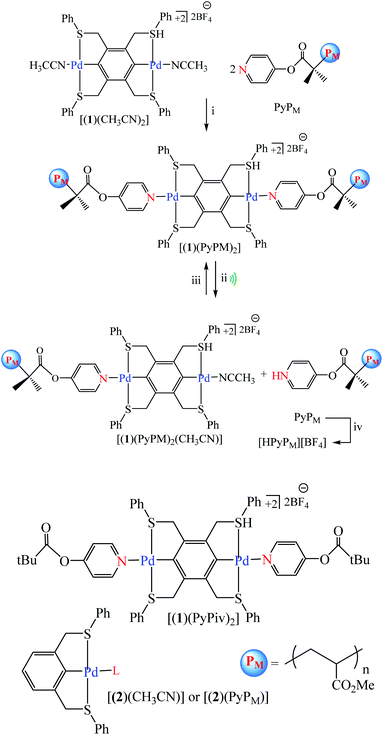 |
| | Scheme 35 Synthesis and chain scission of mechanoresponsive Pd-based polymers. Conditions: (i) DMF, room temperature, 16 h; (ii) sonication for 2 h at 4 °C of [(1)(PyPM)2] (10 mg) in CH3CN (10 mL), and (iii) recoordination of PyPM or (iv) trapping as [HPyPM][BF4] with 20 equiv. of HBF4. Reprinted by permission from ref. 86, Copyright © 2010, American Chemical Society. | |
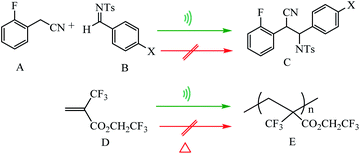 |
| | Scheme 36 Palladium-catalyzed carbon–carbon bond formation and anionic polymerization. Reprinted by permission from ref. 86, Copyright © 2010, American Chemical Society. | |
6. Switching driven by changes in size catalyst
Recently efforts have been made to employ size and morphology as a switching mechanism for catalysis. In this sense, Dominguez et al. report the synthesis of a bimetallic CoMoS composite by the hydrothermal method and the addition of cellulose fibers as a carbon source, followed by carbonization under argon atmosphere (CoMoS@C) (Fig. 13). Experimental results indicate the addition of carbon significantly enhanced the cycling stability and retained a high specific capacity of ∼715 mA h g−1 after 200 cycles at a rate of 500 mA h g−1 compared to bare-CoMoS which retained ∼102 mA h g−1 after 100 cycles.87
 |
| | Fig. 13 SEM of (a) bare-CoMoS, (b) cellulose fibers carbonized at 400 °C for 1 h, (d) CoMoS@C, and (e) CoMoS@C at higher magnification. TEM of (c) bare-CoMoS and (f) CoMoS@C. Reprinted by permission from ref. 87, Copyright © 2018, American Chemical Society. | |
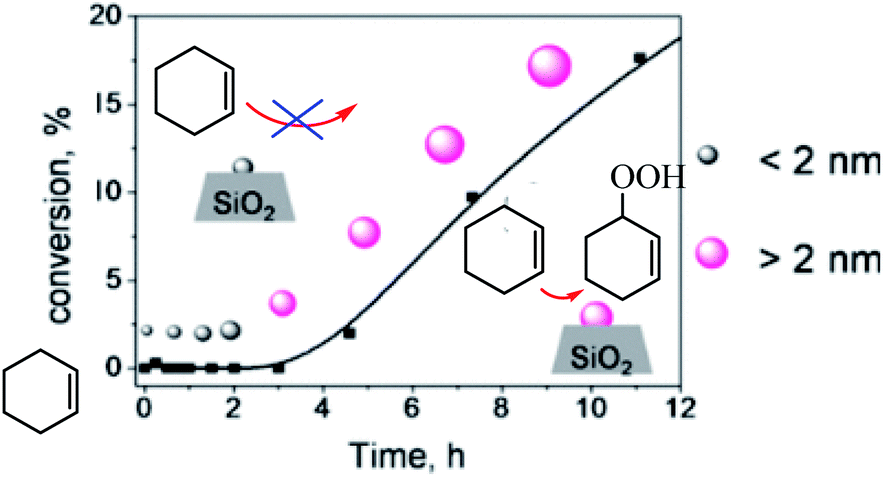
Donoeva et al. examined effect of the size of gold nanoparticles on their catalytic activity in aerobic oxidation of cyclohexene was established using supported gold nanoparticles that gradually undergo a change in size during the catalytic reaction (Scheme 37).88 The authors reported that phosphine-stabilized gold clusters and phosphine-free Au particles smaller than 2 nm are inactive in this reaction, and that catalytic activity appears only upon formation of sufficient number of metallic particles larger than 2 nm. Further increase in Au particle size results in gradual decrease in catalytic activity which correlates with the reduction of the Au surface area.
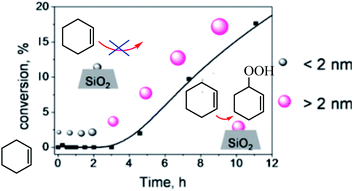 |
| | Scheme 37 Establishing Au nanoparticle size effect in the oxidation of cyclohexene. Reprinted by permission from ref. 88, Copyright © 2013, American Chemical Society. | |
7. Switching driven by changes in reaction conditions
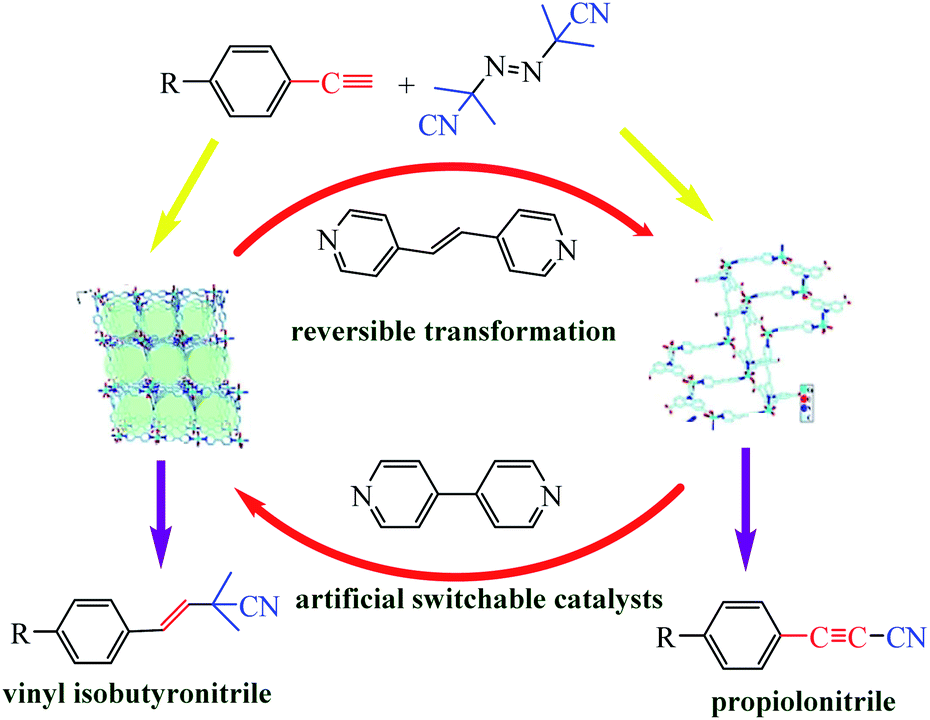
In Section of this review we examined altering reaction conditions to influence the activity of a catalyst. Huang et al. reported a dramatic transformation of an artificial switchable catalytic system upon dissolution–exchange–crystallization.89 The reversible structural transformations were distinct to the naked eye and proceeded between hierarchical anion-pillared framework {(H3O)[Cu(CPCDC)(4,4′-bpy)]}n (1; H3CPCDC = 9-(4-carboxyphenyl)-9H-carbazole-3,6-dicarboxylic acid, 4,4′-bpy = 4,4′-bipyridine), including free [H3O]+ ions as guest molecules, and a neutral 2D stair-stepping framework {[Cu(CPCDC)(4,4′-bpe)]}n (2; 4,4′-bpe = 4,4′-vinylenedipyridine) to the direct cyanation reaction of terminal alkynes and azobisisobutyronitrile (Scheme 38).
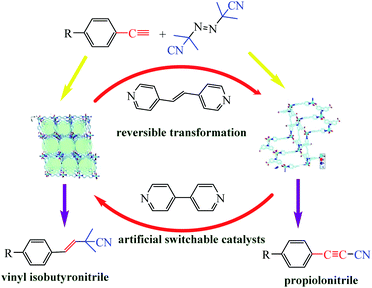 |
| | Scheme 38 Artificial switchable catalysts 1 and 2 catalyzed direct cyanation reactions to produce vinyl isobutyronitrile and propiolonitrile frameworks. Reprinted by permission from ref. 89, Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
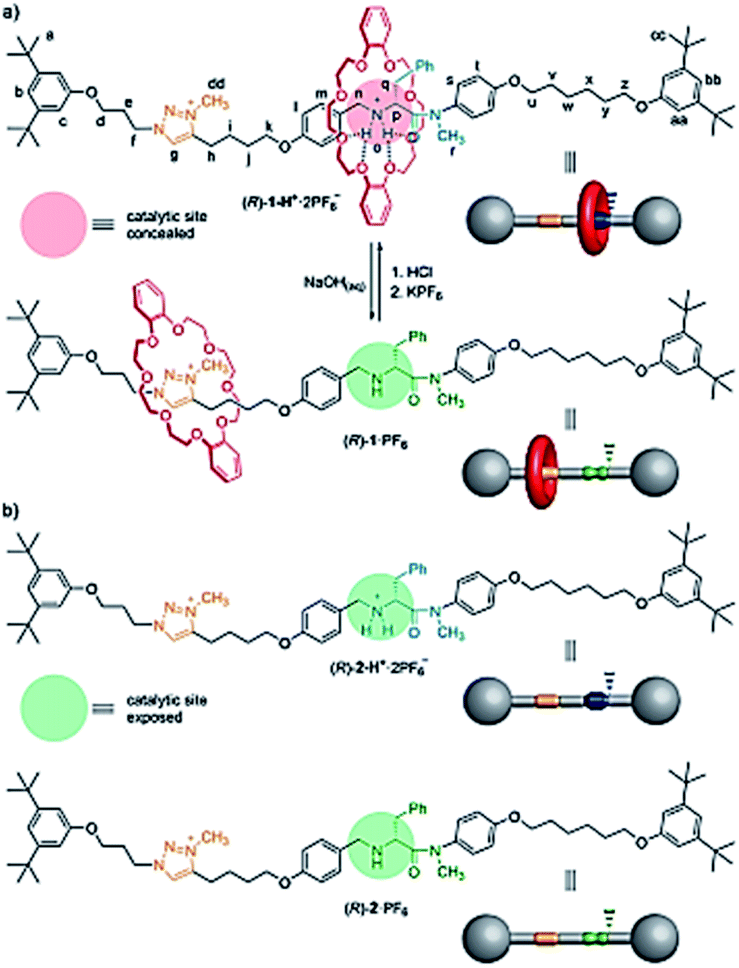
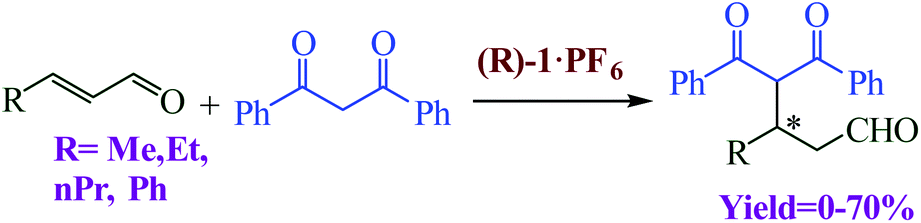
Blanco et al. designed a rotaxane-based switchable asymmetric organocatalyst (R)-1·PF6 consists of a dibenzo-24-crown-8 macrocycle and an axle bearing a triazolium ring and a chiral acyclic secondary amine derived from D-phenylalanine (Fig. 14).90 Switching of the preferred position of the macrocycle between the two binding sites is triggered by protonation/deprotonation of the amine/ammonium group. The switching mechanism of the rotaxane relies on the macrocycle preferentially encapsulating the chiral secondary ammonium group, a better binding site for the macrocycle than the triazolium ring, in the protonated form ((R)-1-H+·2PF6−; Fig. 14a). After, switchable asymmetric organocatalyst applied asymmetric michael addition (Scheme 39).
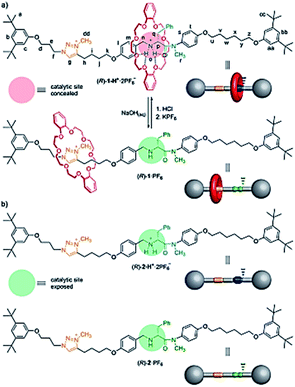 |
| | Fig. 14 (a) Acid–base switching of the position of the macrocycle in chiral rotaxane (R)-1-H+·2PF6 (catalysis ‘off’)/(R)-1·PF6 (catalysis ‘on’). (b) Structure of threads (R)-2·PF6 and (R)-2-H+·2PF6. Reprinted by permission from ref. 90, Copyright © 2014, American Chemical Society. | |
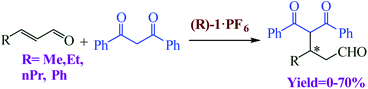 |
| | Scheme 39 Asymmetric Michael addition of 1,3-diphenyl-1,3-propanedione and α,β-unsaturated aldehydes catalyzed by (R)-1·PF6. Reprinted by permission from ref. 90, Copyright © 2014, American Chemical Society. | |
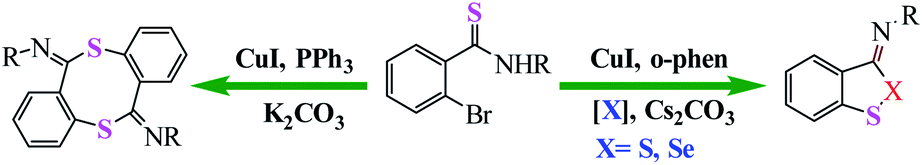
Huang et al. reported an efficient and switchable copper-catalyzed method for the synthesis of benzodithioles and benzothiaselenoles using S8 or Se as the chalcogen source is disclosed. Conducting the reaction in the absence of S8 or Se affords eight-membered dibenzodithiocine annulation products via two consecutive C(sp2)–S coupling reactions (Scheme 40).91
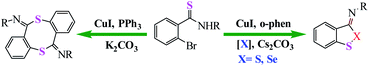 |
| | Scheme 40 Copper-catalyzed approaches to carbon–sulfur bond formation. PIP = (pyridin-2-yl)isopropyl. Reprinted by permission from ref. 91, Copyright © 2020, American Chemical Society. | |
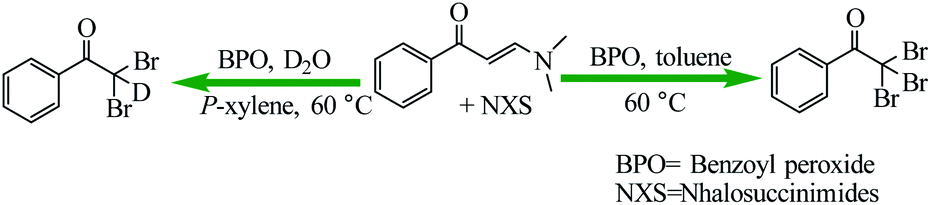
Liu et al. reported the novel free radical-based cleavage of the enaminone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond for the synthesis of α,α dihalomethyl ketones and α,α,α-trihalomethyl ketones by using N-halosuccinimides (NXS) in the presence of benzoyl peroxide (BPO) with mild heating (Scheme 41).92 Besides the tunable reaction selectivity, the metal-free operation, mild reaction conditions as well as broad scope constitute also the unique advantages of the present synthetic methods.
C double bond for the synthesis of α,α dihalomethyl ketones and α,α,α-trihalomethyl ketones by using N-halosuccinimides (NXS) in the presence of benzoyl peroxide (BPO) with mild heating (Scheme 41).92 Besides the tunable reaction selectivity, the metal-free operation, mild reaction conditions as well as broad scope constitute also the unique advantages of the present synthetic methods.
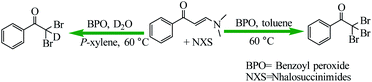 |
| | Scheme 41 Synthesis of α,α-dihaloketones and α,α,α-tribromoaryl ketones. Reprinted by permission from ref. 92, Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Semwal et al. designed a ruthenium(II)-based bi-state molecular switch, which is responsive to acid/base stimuli, was designed and exhibits highly reversible metal–ligand coordination modes. Disclosed here is a molecular switch which responds to acid–base stimuli and serves as a bi-state catalyst for two different reactions (Schemes 42 and 43).93 The two states of the switch serve as a highly active and poorly active catalyst for two catalytic reactions (namely a hydrogenation and a dehydrogenative coupling) but in a complementary manner. The “Ru-amido state” is highly active in catalytic hydrogenation of various imine substrates while the “Ru-imino state” remains poorly active in this reaction.
 |
| | Scheme 42 Hydrogenative (A) and dehydrogenative (B) catalysis performed by 1 and 2. Reprinted by permission from ref. 93, Copyright © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
 |
| | Scheme 43 Synthesis catalysts 1 and 2. Reprinted by permission from ref. 93 Copyright © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
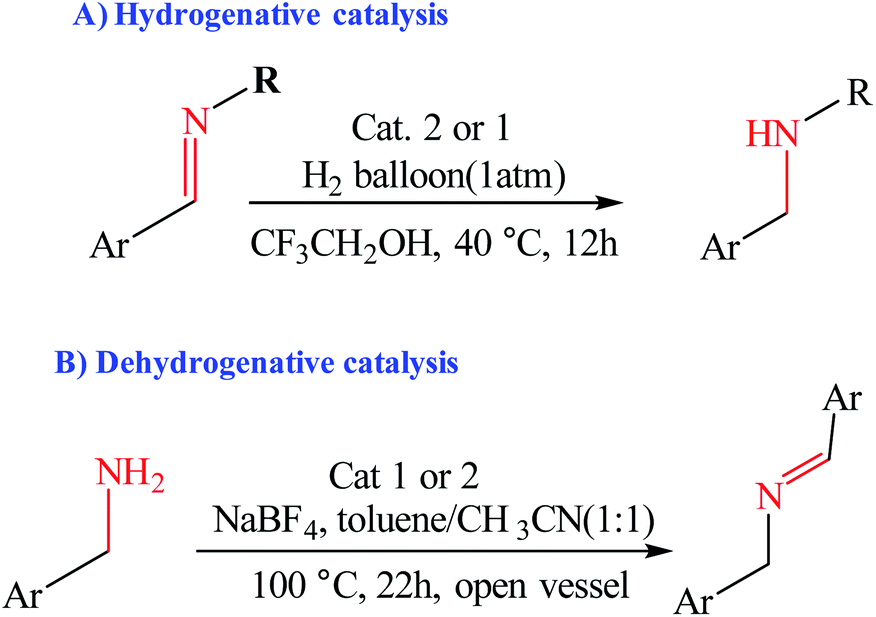
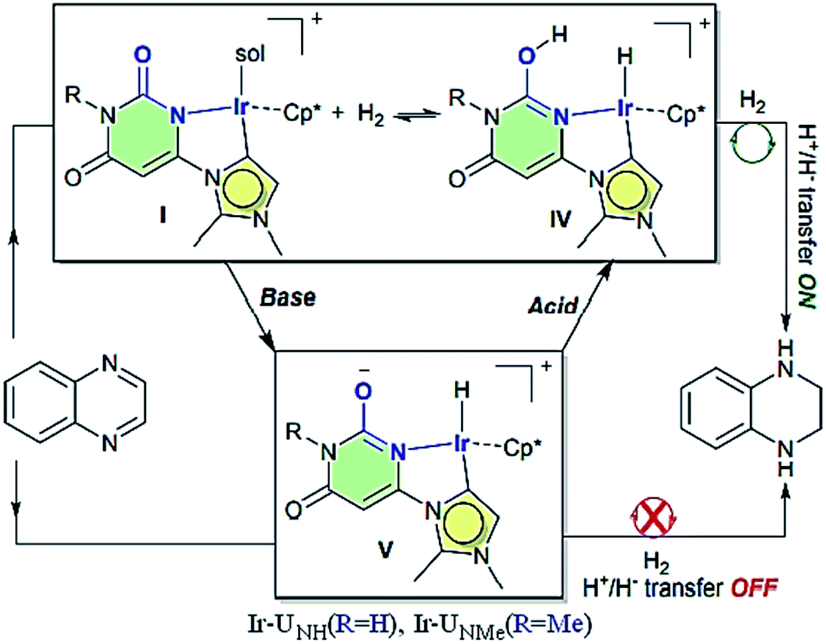
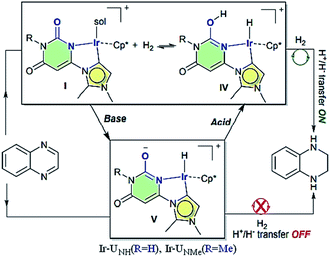
Choudhury et al. reported a switchable hydrogenation protocol with a bifunctional Ir–N-heterocyclic carbene (NHC) catalyst (Fig. 15).94 Under ambient H2 pressure, the catalyst was highly active toward hydrogenation of quinoxalines to load two molecules of H2 per molecule of the N-heteroarene (Scheme 44). Action of a base stimulus to this catalysis led to switching off the hydrogenation, while a reverse stimulus, an acid switched it on again.
 |
| | Fig. 15 The working proposal for the switchable action of the catalyst. Reprinted by permission from ref. 94, Royal Society of Chemistry. | |
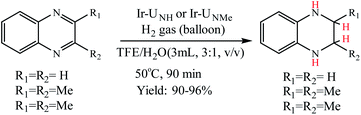 |
| | Scheme 44 General scheme for catalytic hydrogenation of quinoxalines. Reprinted by permission from ref. 94, Royal Society of Chemistry. | |
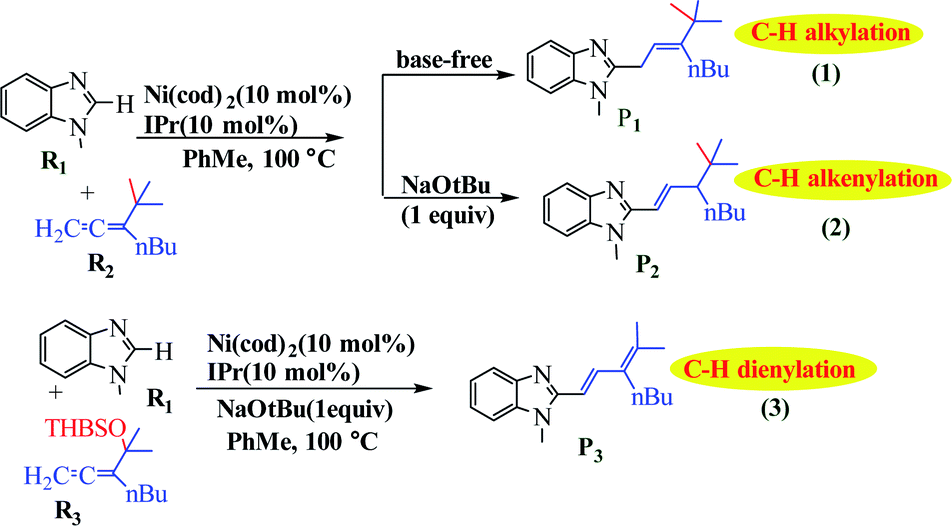
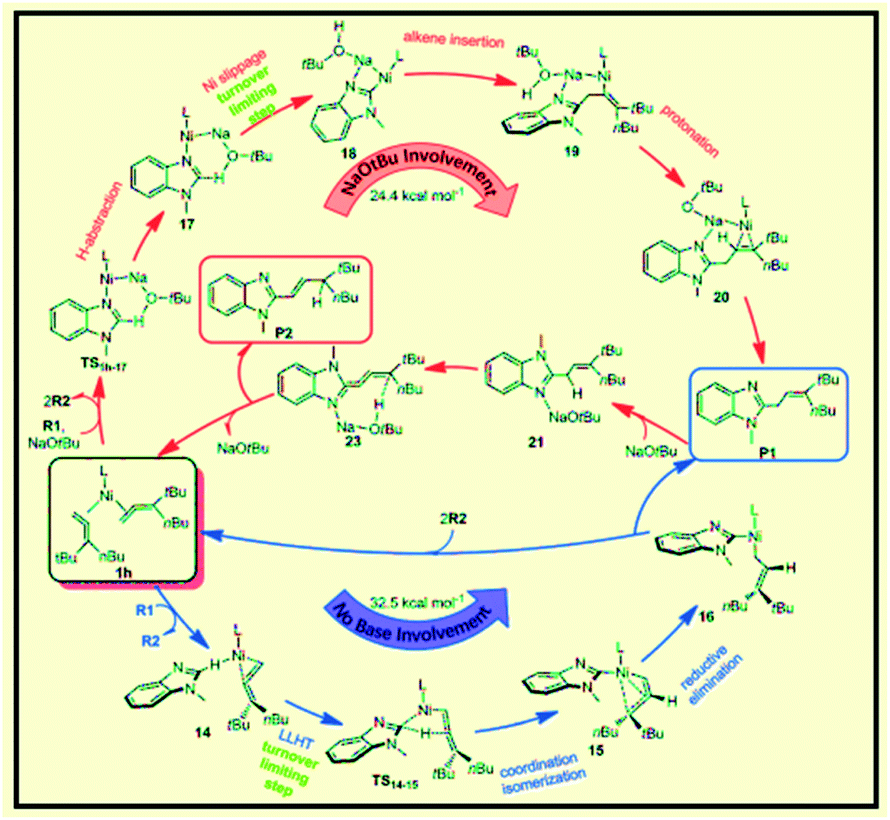
Liu et al. reported the application of switchable Ni(0) for coupling of benzimidazole with 1,1-disubstituted allenes as a new strategy for achieving controllable C–H allylations, alkenylations and dienylations (Scheme 45).95 The results using a tBu-substituted allene demonstrate that the formation of the allylated product involves a Ni-catalyzed C–H activation mechanism through ligand-to-ligand-hydrogen transfer (LLHT) under base-free conditions. In contrast, a Ni/NaOtBu co-promoted C–H activation mechanism is newly proposed in the presence of NaOtBu, which is remarkably different from the previously reported literature (Fig. 16).
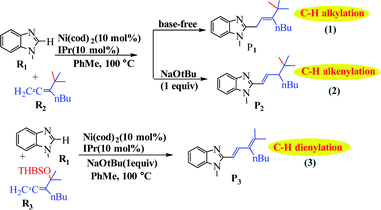 |
| | Scheme 45 Regioselectivities and chemoselectivities of Ni(0)-catalyzed couplings between benzimidazole (R1) and allenes (R2) and (R3), respectively. Reprinted by permission from ref. 95, Royal Society of Chemistry. | |
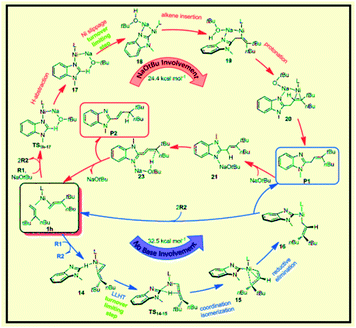 |
| | Fig. 16 Sketch of the catalytic cycles for forming P1 in the absence and presence of NaOtBu from the Ni-catalyzed couplings of benzimidazole R1 with allene R2 based on the present calculations. L = Ipr. Reprinted by permission from ref. 95, Royal Society of Chemistry. | |
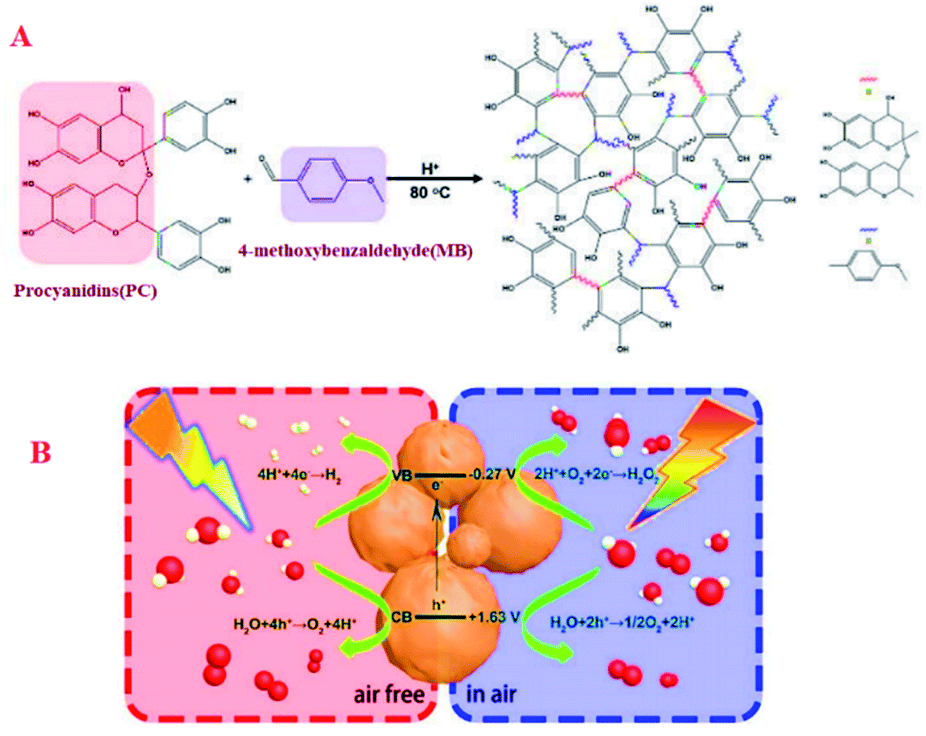
Photocatalytic water splitting to produce hydrogen and hydrogen peroxide are the core issue of photocatalysis. Thought they are both water-based reactions, it's extremely troublesome to conduct the two processes on one catalyst. For example, in the catalytic field, Wu et al. designed and synthesized the bifunctional metal-free catalyst PC-MB-3 through the polycondensation of procyanidin and 4-methoxybenzaldehyde (Fig. 14).96 The catalytic reaction pathway of the catalyst PC-MB-3 can be controlled by adjusting the reaction atmosphere, which exhibits the characteristics of switchable function and excellent catalytic activity. The catalyst developed here not only was expected to split water to produce hydrogen by four-electron reaction process in air free, but also to generate hydrogen peroxide by preferentially reducing oxygen with two-electron pathway in air (Fig. 17).
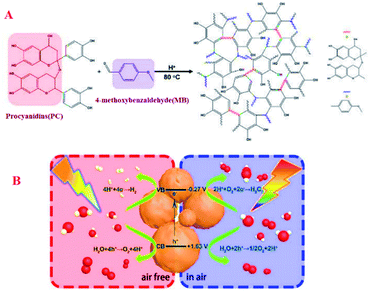 |
| | Fig. 17 Postulated schematic and the band structure of PC-MB-3 (A). The mechanism diagram of hydrogen or hydrogen peroxide produced over the PC-MB-3 through photocatalytic reaction in different (B). Reprinted by permission from ref. 96, Royal Society of Chemistry. | |
8. Switching of catalyst between soluble and solid-supported
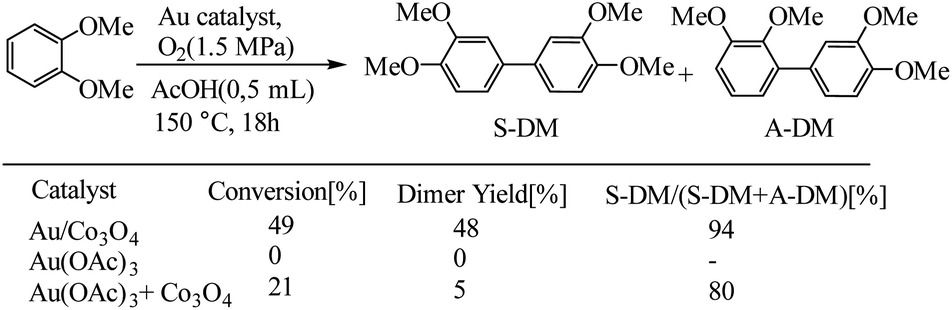
Before starting the discussion on switching of catalyst between soluble and solid-supported, it should be noted, the size single atoms, metal clusters, and metal nanoparticles due to differences in the electronic and geometric structures effects on activity and selectivity.97,98 The different catalytic behavior of mononuclear Au complex and Au nanoparticles used for oxidative coupling of dimethyl phthalate. In a related example, Ishida et al. have reported the application of Au/Co3O4 catalysts for oxidative coupling of dimethyl phthalate, as shown in Scheme 46.99 Authors reported that metallic Au nano particles were the active sites for the oxidative coupling of arenes, while mononuclear Au compounds would transform into metallic Au nanoparticles in the presence of the metal oxide support under reaction conditions.
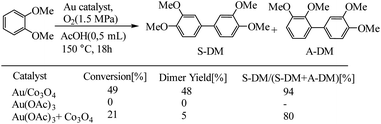 |
| | Scheme 46 Oxidative coupling of dimethyl phthalate with different Au catalysts. Reprinted by permission from ref. 99, Copyright © 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
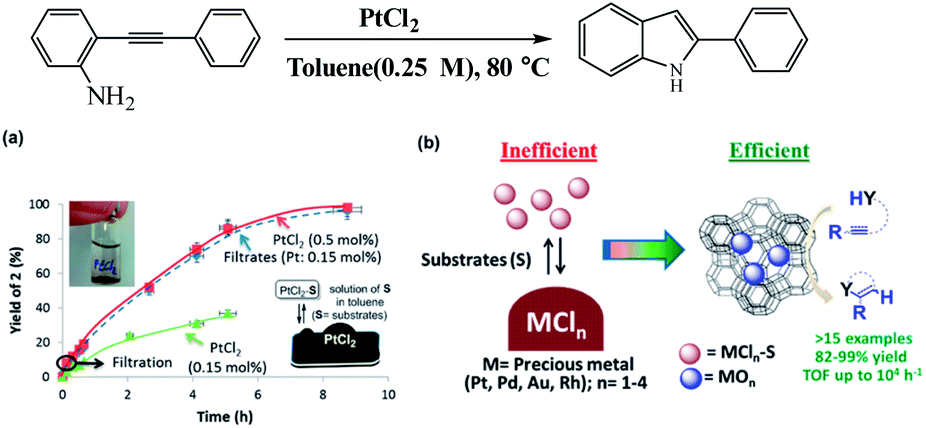
Metal species in homogeneous systems, often act as Lewis acid sites and catalyze coupling reaction. However, most of those salts are not soluble in the reaction solvents. In the work of Rubio-Marqués et al. used application of insoluble precious metal chlorides in polymeric form (i.e., PtCl2, PdCl2, AuCl, RhCl3) as catalysts for a plethora of organic reactions in solution (Fig. 18).100 The authors reported only the minor soluble fraction of these precious metal chlorides (typically 5–30%) is catalytically active for the hydroamination, hydroalkoxylation, hydrosilylation, and cycloisomerization of alkynes and alkenes, and that the resting insoluble metal is catalytically useless. To overcome such limitation of conventional metal salts, the authors generate well-dispersed Pt(II) and Pd(II) single sites on zeolite, by ion-exchange method, and they show excellent activity and recyclability for hydroamination reactions.
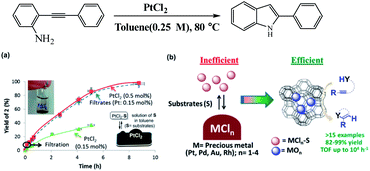 |
| | Fig. 18 (a) Yield–time profile for the hydroamination of o-(phenylacetylen)aniline catalyzed by PtCl2. The photograph on the left shows the precipitate of PtCl2 in toluene under reaction conditions, and the illustration on the right describes the equilibrium-controlled dissolution of PtCl2 with the substrates under reaction conditions. (b) Schematic illustration using metal-exchanged zeolite as substitute catalysts for hydroamination reactions with high activity. Reprinted by permission from ref. 100, Copyright © 2015, American Chemical Society. | |
9. Phase-switching
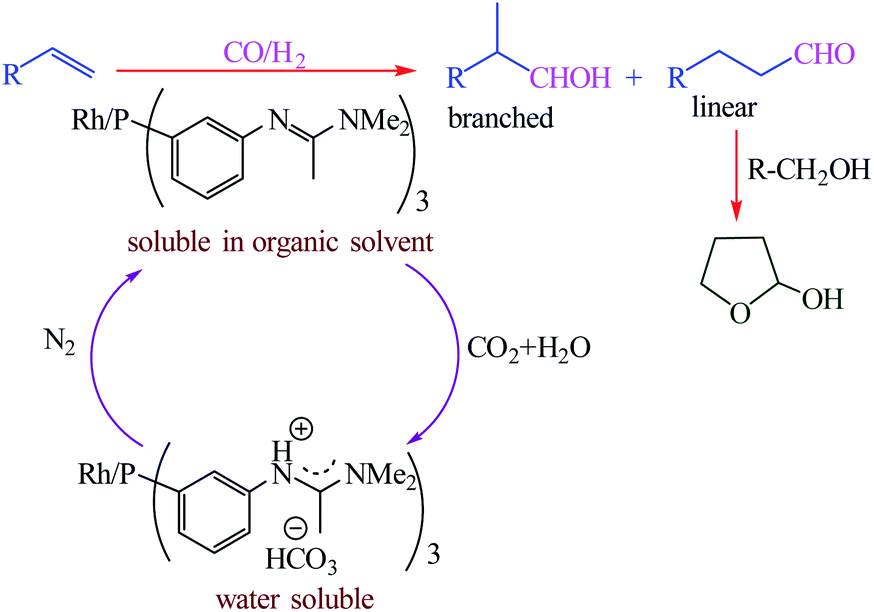
Homogeneous catalysts typically display high activity, high selectivity due to they are dissolved in the reaction medium but cannot be commercialized because of difficulties associated with separating the products from the catalyst. Desset et al. reported a new method for the separation of the products from the catalyst in homogeneous catalytic reactions of hydroformylation of alkenes (Scheme 47).101 It including using ligands which can be protonated by carbonic acid (aqueous solutions of CO2), and switching the catalyst into a separate phase by bubbling with or removal of CO2 using N2 (Fig. 19). In this method can be used effectively with low catalyst losses for substrates and products that are hydrophobic or hydrophilic.
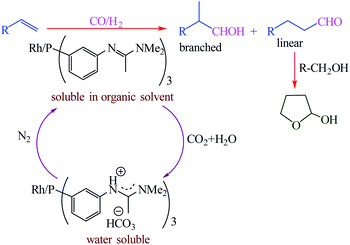 |
| | Scheme 47 Hydroformylation of alkenes (R = C6H13 (1-octene) or CH2OH (allyl alcohol)) using a catalyst that can be switched between an organic phase and water by bubbling with CO2 or N2. Reprinted by permission from ref. 101, Copyright © 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
 |
| | Fig. 19 Photographs obtained during the catalyst recycling process. (a) the crude product from cycle n (number on left of row) with added water; (b) after 10 min of stirring and CO2 bubbling; (c) after 1.5 h of stirring and CO2 bubbling, separation of the organic phase and addition of fresh toluene; (d) after 30 min of stirring at 608C and N2 bubbling. Reprinted by permission from ref. 101, Copyright © 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Mokhadinyana et al. designed ligands that are tagged with amidate groups, trisSwitchPhos 1 and an amidated xantphos ligand 2 then combined with [Rh(acac)(CO)2] for the hydroformylation of 1-octene in toluene with high selectivity and allow the catalyst to be efficiently switched from the organic phase into the aqueous phase by bubbling CO2 through the reaction mixture (Schemes 48 and 49).102 Experimental result show when ligands 1 and 2 are used for the rhodium-catalyzed hydroformylation of 1-octene, 2 controls the selectivity for the linear product, but 1 coordinates to rhodium in the resting state of the catalyst to allow successful phase switching into the aqueous phase through the formation of the bicarbonate salt with CO2. Using CO2 led to the protonation of the amidine groups through the formation of carbonic acid by reaction with water, and it also removes CO from the system, which allows 1 to coordinate to the metal atom. When 1 and 2 are both present in the coordination sphere, there are seven amidines per rhodium atom, which is sufficient to allow efficient phase switching upon protonation of the amidines (Fig. 20). The lower rate of hydroformylation of 1-octene when both ligands are present relative to when 2 is used alone, may arise because 1 can compete with the alkene for the vacant coordination site once CO is lost from [RhH(CO)2(2)] to form the active species in the catalytic cycle.
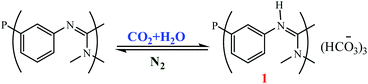 |
| | Scheme 48 Hydroformylation of 1-octene and the linear (desired) and branched products. Reprinted by permission from ref. 102, Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
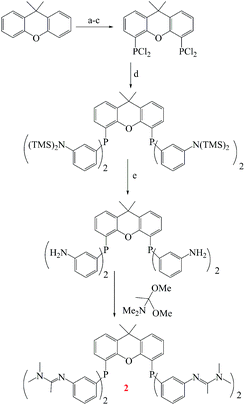 |
| | Scheme 49 Synthesis of 2: (a) THF, BuLi, −78 °C to RT, 24 h; (b) (NEt2)2PCl, −78 °C to RT, 24 h; (c) HCl(g), 08C, 15 min; (d) 3-[bis(trimethylsilyl)amino]phenylmagnesium chloride, 78 °C to RT 12 h, 55 °C, 12 h; (e) MeOH, reflux, overnight; (f) microwave, 160 °C, 1 h. TMS = trimethylsilyl. Reprinted by permission from ref. 102, Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
 |
| | Fig. 20 Phase switching after the hydroformylation of 1-octene catalyzed by rhodium complexes of (a) 2; (b) 2 and 1 combined, cycles 1a and 1b; (c) 2 and 1 combined, cycle 2. (i) Reaction solution with water added. (ii) After bubbling CO2 through the mixture for 1.5 h. (iii) After phase separation, addition of fresh toluene, and bubbling N2 through the mixture at 608C for 1.5 h. The top (organic) phase from (b iii) was used for the hydroformylation of 1-octene to give the product phase shown in (c i). Reprinted by permission from ref. 31, Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
10. Conclusions and outlook
This review described that the recent progress in the nascent field of artificial switchable catalysis is remarkable through a concise discussion on only the works developed during last couple of years. Catalytic systems based on the use of stimuli–responsive materials (such as light, pH, ion coordination, redox switching, mechanical forces and, applying acid/base-stimulated switchable metal ligand coordination modes, phase switching, and soluble and solid-supported) can be switched from an “on” active state to an “off” inactive state. Consequently, Switchable catalysis, both chemical and biological, has played a pivotal role in this ‘greening’ of the pharmaceutical industry due to offer many benefits in this respect. These features can be advantageous in the design of catalysts with unique functional properties, such as adaptability, recyclability and precise spatial and temporal control on different types of chemical reactions. The field of switchable catalysis has seen rapid development over the past three decades due to great potentials for advanced functions and solving problems which are difficult or impossible to achieve using conventional methodologies.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to thank the research facilities of Ilam University and Bu-Ali Sina University for financial support of this research project.
References
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- R. Dorel and B. L. Feringa, Chem. Commun., 2019, 55, 6477–6486 RSC.
- J. Choudhury, Tetrahedron Lett., 2018, 59, 487–495 CrossRef CAS.
- B. Qian, Z. Chang and X. H. Bu, Top. Curr. Chem., 2020, 378, 5 CrossRef CAS PubMed.
- C. Yoo, H. M. Dodge and A. Miller, Chem. Commun., 2019, 55, 5047–5059 RSC.
- J. Choudhury and S. Semwal, Synlett, 2018, 29, 141–147 CrossRef CAS.
- C. C. Chintawar, A. K. Yadav, A. Kumar, S. P. Sancheti and N. T. Patil, Chem. Rev., 2021, 121, 8478–8558 CrossRef CAS PubMed.
- A. Chirila, B. G. Das, P. F. Kuijpers, V. Sinha and B. de Bruin, Application of Stimuli-Responsive and “Non-Innocent” Ligands in Base Metal Catalysis, Wiley Press, 2018, pp. 1–31 Search PubMed.
- S. P. Ihrig, F. Eisenreich and S. Hecht, Chem. Commun., 2019, 55, 4290–4298 RSC.
- G. Romanazzi, L. Degennaro, P. Mastrorilli and R. Luisi, ACS Catal., 2017, 7, 4100–4114 CrossRef CAS.
- E. Peris, Chem. Rev., 2017, 118, 9988–10031 CrossRef PubMed.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed.
- S. L. Balof, S. J. P'Pool, N. J. Berger, E. J. Valente, A. M. Shiller and H.-J. Schanz, Dalton Trans., 2008, 42, 5791–5799 RSC.
- S. Shinkai, K. Kameoka, K. Ueda and O. Manabe, J. Am. Chem. Soc., 1987, 109, 923–924 CrossRef CAS.
- E. M. Broderick, N. Guo, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed.
- R. Groote, R. T. Jakobs and R. P. Sijbesma, Polym. Chem., 2013, 4, 4846–4859 RSC.
- F. Wuerthner and J. Rebek Jr, Angew. Chem., Int. Ed. Engl., 1995, 34, 446–448 CrossRef CAS.
- A. Ueno, K. Takahashi and T. Osa, Chem. Commun., 1981, 3, 94–96 RSC.
- D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431–5434 CrossRef CAS PubMed.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018–11020 CrossRef CAS PubMed.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- Y. H. Kim, Acc. Chem. Res., 2001, 34, 955–962 CrossRef CAS PubMed.
- A. J. Sandee, J. N. Reek, P. C. Kamer and P. W. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8468–8476 CrossRef CAS PubMed.
- T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS PubMed.
- G. Chen, J. Gui, L. Li and J. Liao, Angew. Chem., Int. Ed., 2011, 50, 7681–7685 CrossRef CAS PubMed.
- G. Romanazzi, L. Degennaro, P. Mastrorilli and R. Luisi, ACS Catal., 2017, 7, 4100–4114 CrossRef CAS.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 CrossRef CAS PubMed.
- H. Poole, J. Gauthier, J. R. Vanderveen, P. G. Jessop and R. Lee, Green Chem., 2019, 21, 6263–6267 RSC.
- E. Le Saché, L. Pastor-Perez, B. J. Haycock, J. J. Villora-Picó, A. Sepúlveda-Escribano and T. R. Reina, ACS Sustainable Chem. Eng., 2021, 8, 4614–4622 CrossRef.
- S. D. Kong, W. Zhang, J. H. Lee, K. Brammer, R. Lal, M. Karin and S. Jin, Nano Lett., 2010, 10, 5088–5092 CrossRef CAS PubMed.
- M. Amoli-Diva, R. Sadighi-Bonabi and K. Pourghazi, Mater. Sci. Eng., C, 2017, 76, 242–248 CrossRef CAS PubMed.
- L. Ge, J. Wu, C. Wang, F. Zhang and Z. Liu, Biosens. Bioelectron., 2020, 169, 112606 CrossRef CAS PubMed.
- M. J. Langton, L. M. Scriven, N. H. Williams and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 15768–15773 CrossRef CAS PubMed.
- L. Qiu, J. W. Li, C. Y. Hong and C. Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 40887–40897 CrossRef CAS PubMed.
- T. Rösler, T. A. Faßbach, M. Schrimpf, A. J. Vorholt and W. Leitner, Ind. Eng. Chem. Res., 2018, 58, 2421–2436 CrossRef.
- J. Lu, M. J. Lazzaroni, J. P. Hallett and A. S. Bommarius, Ind. Eng. Chem. Res., 2004, 43, 1586–1590 CrossRef CAS.
- J. P. Hallett, J. W. Ford, R. S. Jones, P. Pollet, C. A. Thomas, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 2008, 47, 2585–2589 CrossRef CAS.
- P. Pollet, R. J. Hart, C. A. Eckert and C. L. Liotta, Acc. Chem. Res., 2010, 43, 1237–1245 CrossRef CAS PubMed.
- J. Großeheilmann and U. Kragl, ChemSusChem, 2017, 10, 2685–2691 CrossRef PubMed.
- G. Liu and J. Wang, ACS Catal., 2013, 3, 1874–1885 CrossRef.
- B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- S. Song and N. Hu, Acc. Chem. Res., 2019, 52, 415–424 CrossRef PubMed.
- S. Kitzig, M. Thilemann, T. Cordes and K. Rück-Braun, ChemPhysChem, 2016, 17, 1252–1263 CrossRef CAS PubMed.
- Y. H. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554 CrossRef CAS PubMed.
- P. Pfaff, K. T. Samarasinghe, C. M. Crews and E. M. Carreira, ACS Cent. Sci., 2019, 5, 1682–1690 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- P. Mayorga-Burrezo, C. Sporer, J. A. de Sousa, N. Capra, K. Wurst and N. Crivillers, Chem. Commun., 2020, 56, 14211–14214 RSC.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- D. H. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- A. J. Teator, H. Shao, G. Lu, P. Liu and C. W. Bielawski, Organometallics, 2017, 36, 490–497 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- M. A. Ahsan, E. Deemer, O. Fernandez-Delgado, H. Wang, M. L. Curry, A. A. El-Gendy and J. C. Noveron, Catal. Commun., 2019, 130, 105753 CrossRef CAS.
- M. A. Ahsan, O. Fernandez-Delgado, E. Deemer, H. Wang, A. A. El-Gendy, M. L. Curry and J. C. Noveron, J. Mol. Liq., 2019, 290, 111059 CrossRef CAS.
- S. L. Dong, M. Löweneck, T. E. Schrader, W. J. Schreier, W. Zinth, L. Moroder and C. A. Renner, Chem.–Eur. J., 2006, 12, 1114–1120 CrossRef CAS PubMed.
- J. Byun, W. Huang, D. Wang, R. Li and K. A. Zhang, Angew. Chem., Int. Ed., 2018, 57, 2967–2971 CrossRef CAS PubMed.
- E. Peiris, S. Sarina, E. R. Waclawik, G. A. Ayoko, P. Han, J. Jia and H. Y. Zhu, Angew. Chem., Int. Ed., 2019, 58, 12032–12036 CrossRef CAS PubMed.
- M. Chen, S. Deng, Y. Gu, J. Lin, M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2017, 139, 2257–2266 CrossRef CAS PubMed.
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC.
- T. Imahori, R. Yamaguchi and S. Kurihara, Chem.–Eur. J., 2012, 18, 10802–10807 CrossRef CAS PubMed.
- C. Z. J. Ren, P. Solís Muñana, J. Dupont, S. S. Zhou and J. L. Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 15254–15258 CrossRef CAS PubMed.
- L. Benda, B. Doistau, C. Rossi-Gendron and L. M. Chamoreau, Chem. Commun., 2019, 2, 1–11 CrossRef.
- M. Olivares and M. Albrecht, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef PubMed.
- B. M. Neilson and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef CAS PubMed.
- E. Tzur, A. Szadkowska, A. Ben-Asuly, A. Makal, I. Goldberg, K. Woźniak and N. G. Lemcoff, Chem.–Eur. J., 2010, 16, 8726–8737 CrossRef CAS PubMed.
- A. Nojiri, N. Kumagai and M. Shibasaki, Chem. Commun., 2013, 49, 4628–4630 RSC.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018–11020 CrossRef CAS PubMed.
- C. Drolen, E. Conklin, S. J. Hetterich, A. Krishnamurthy, G. A. Andrade, J. L. Dimeglio and E. R. Young, J. Am. Chem. Soc., 2018, 140, 10169–10178 CrossRef CAS PubMed.
- A. Arlegui, P. Torres, V. Cuesta, J. Crusats and A. Moyano, Eur. J. Org. Chem., 2020, 2020, 4399–4407 CrossRef CAS.
- A. Ghorbani-Choghamarani and Z. Taherinia, New J. Chem., 2018, 42, 10989–10992 RSC.
- M. Akbarzadeh, Z. Moosavi-Movahedi, A. Shockravi, R. Jafari, K. Nazari, N. Sheibani and A. A. Moosavi-Movahedi, J. Mol. Catal. A: Chem., 2016, 424, 181–193 CrossRef CAS.
- Y. Chen, T. Chen, X. Wu and G. Yang, Sens. Actuators, B, 2019, 279, 374–384 CrossRef CAS.
- C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. G. Pappas, V. Narang and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 129, 14703–14707 CrossRef.
- S. Semwal and J. Choudhury, ACS Catal., 2018, 6, 2424–2428 CrossRef.
- C. González-Rodríguez, R. J. Pawley, A. B. Chaplin, A. L. Thompson, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 5134–5138 CrossRef PubMed.
- L. C. M. Castro, J. B. Sortais and C. Darcel, Chem. Commun., 2012, 48, 10514–10516 RSC.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- Y. Mikami, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2013, 3, 58–69 RSC.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- P. Zhou, S. Jia, D. Pan, L. Wang, J. Gao, J. Lu and H. Liu, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- S. B. Owens Jr and G. M. Gray, Organometallics, 2008, 27, 4282–4287 CrossRef.
- P. Veit, C. Volkert, C. Förster, V. Ksenofontov, S. Schlicher, M. Bauer and K. Heinze, Chem. Commun., 2019, 55, 4615–4618 RSC.
- S. Ibáñez, M. Poyatos, L. N. Dawe, D. Gusev and E. Peris, Organometallics, 2016, 35, 2747–2758 CrossRef.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS PubMed.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133 CrossRef CAS PubMed.
- A. G. Tennyson, K. M. Wiggins and C. W. Bielawski, J. Am. Chem. Soc., 2010, 132, 16631–16636 CrossRef CAS PubMed.
- N. Dominguez, B. Torres, L. A. Barrera, J. E. Rincon, Y. Lin, R. R. Chianelli and J. C. Noveron, ACS Omega, 2018, 3, 10243–10249 CrossRef CAS PubMed.
- B. G. Donoeva, D. S. Ovoshchnikov and V. B. Golovko, ACS Catal., 2013, 3, 2986–2991 CrossRef CAS.
- C. Huang, G. Li, L. Zhang, Y. Zhang, L. Mi and H. Hou, Chem.–Eur. J., 2019, 25, 10366–10374 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh, V. Marcos and J. A. Morales-Serna, J. Am. Chem. Soc., 2014, 136, 4905–4908 CrossRef CAS PubMed.
- M. Q. Huang, T. J. Li, J. Q. Liu, A. Shatskiy, M. D. Kärkäs and X. S. Wang, Org. Lett., 2020, 22, 3454–3459 CrossRef CAS PubMed.
- Y. Liu, J. Xiong, L. Wei and J. P. Wan, Adv. Synth. Catal., 2020, 362, 877–883 CrossRef CAS.
- S. Semwal and J. Choudhury, Angew. Chem., Int. Ed., 2017, 56, 5556–5560 CrossRef CAS PubMed.
- B. Maji and J. Choudhury, Chem. Commun., 2019, 55, 4574–4577 RSC.
- Y. Liu, K. Wang, B. Ling, G. Chen, Y. Li, L. Liu and S. Bi, Catal. Sci. Technol., 2020, 10, 4219–4228 RSC.
- Q. Wu, Y. Liu, J. Cao, Y. Sun, F. Liao, Y. Liu and Z. A. Kang, J. Mater. Chem. A, 2020, 8, 11773–11780 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- K. J. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 26, 3319–3329 CrossRef.
- T. Ishida, S. Aikawa, Y. Mise, R. Akebi, A. Hamasaki, T. Honma, H. Ohashi, T. Tsuji, Y. Yamamoto and M. Miyasaka, ChemSusChem, 2015, 8, 695–701 CrossRef CAS PubMed.
- P. Rubio-Marqués, M. A. Rivero-Crespo, A. Leyva-Perez and A. Corma, J. Am. Chem. Soc., 2015, 137, 11832–11837 CrossRef PubMed.
- S. L. Desset and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2009, 121, 1500–1502 CrossRef.
- M. Mokhadinyana, S. L. Desset, D. B. G. Williams and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2012, 24, 1680–1684 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Zahra Taherinia
*a and
Zahra Taherinia