DOI:
10.1039/D2RA03763G
(Paper)
RSC Adv., 2022,
12, 26556-26564
Indirect aqueous carbonation of CaSO4·2H2O with aspartic acid as a recyclable additive
Received
18th June 2022
, Accepted 7th September 2022
First published on 20th September 2022
Abstract
Calcium leaching using additives is the most critical step in the indirect aqueous carbonation process of CaSO4·2H2O. However, recovery of the soluble additives from the sulfate-rich carbonation filtrate limits the large-scale industrial implementation of current carbonation technologies. To address this issue, we employed aspartic acid (Asp) as a leaching additive. The dissolution capability of CaSO4·2H2O in aqueous ammonia was found to improve significantly owing to the complexation effect between Asp and the Ca2+ ions. The maximum amount of dissolved CaSO4·2H2O was determined according to the competitive relationship between the complexing effect and the inhibitory effect of free ammonia molecules on the dissociation of CaSO4·2H2O, and the solution pH influences such competition. The precipitation of CaCO3 was examined by monitoring the variations in the pH and conductivity of the carbonation reaction system. As a result, the shift in the Asp dissociation equilibrium extended the induction period, and the growth period was divided into three stages according to the relative difference between the consumption and formation rates of CO32−. Moreover, it was determined that the carbonation products consisted of stable spherical vaterite particles. The recovery of Asp was also demonstrated at its isoelectric point, with a recovery efficiency of >80% being achieved, and recycling experiments confirmed the stability of the recycled Asp. Finally, the amount of dissolved CaSO4·2H2O and the total carbonation efficiency during cycling were determined as 16.3 ± 0.4 g L−1 and 46.5 ± 1.9%, respectively.
1. Introduction
Aqueous carbonation is a promising approach for CO2 capture, utilisation, and storage, and this process can be carried out via either a direct or an indirect route.1 Remarkably, the indirect pathway has received increasing attention in the last two decades because of its mild reaction conditions, high conversion efficiency, and capability to obtain high-purity carbonate products.2 The large-scale dumping of the industrial byproduct gypsum (BG) in open air, such as phosphogypsum (PG) and flue gas desulfurization gypsum (FGDG), has caused severe environmental pollution and resource wastage.3 According to chemical analysis, the main component of BG is CaSO4·2H2O with a theoretical CaO content of 32.5 wt%, which can store about 0.26 kg ton−1 of CO2.4 Furthermore, the annual output of BG is more than 430 million tons, and thus almost 120 million tons of CO2 would be converted into CaCO3 by carbonation reaction every year.4–6 Therefore, the exploration of efficient and economically feasible indirect aqueous carbonation methods based on the use of CaSO4·2H2O would be expected to promote the industrial development of the flue gas CO2 carbonation process using BG. Furthermore, such an approach could synergistically reduce CO2 emissions and utilise the waste BG.7
Conventionally, the indirect aqueous carbonation route using CaSO4·2H2O mainly involves two successive steps: (a) calcium leaching from CaSO4·2H2O using additives to obtain a calcium-rich leachate solution, and (b) the precipitation of CaCO3 via the gas–liquid carbonation reaction of CO2 (g) with Ca2+ (aq.) after adjusting the above leachate solution to an alkaline pH by the addition of a basic medium.8 It is well known that the solubility of CaSO4·2H2O in pure water is extremely low (i.e., 0.26 g per 100 g H2O at 25 °C). This low solubility limits the yield of CaCO3 during the subsequent gas–liquid carbonation reaction because of the inadequate calcium content in the liquid phase. Therefore, the use of additives in the leaching step is critical for improving the solubility of CaSO4·2H2O in aqueous media.
Based on the salt and complex effects, researchers have reported several effective leaching additives, such as NaCl,9 CH3COONH4,10 NH4Cl,11 and NaC6H11O7.12 However, together with the release of Ca2+, the SO42− that is leached from CaSO4·2H2O can form a byproduct sulfate salt with the cationic component of the added basic medium. Generally, the formed sulfate salt is highly soluble in aqueous solutions and thus difficult to separate from the equally soluble additives mentioned above. Conversely, if the carbonation filtrate containing the sulfate salt and the additive is recycled directly after removing the CaCO3 precipitate, as previously reported by Chen et al.,9,11 the content of the sulfate salt increases as the filtrate cycle number increases, inhibiting the dissolution of CaSO4·2H2O due to the common ion effect. These drawbacks therefore limit the recycling and recovery of the leaching additives, which is a significant hurdle in the large-scale industrial implementation of the indirect aqueous carbonation process based on the use of BG.7
Aspartic acid (Asp) contains both carboxyl (–COOH) and amino (–NH2) groups in its molecular structure, and so it can form a soluble chelate with Ca2+ under alkaline conditions.13 Based on this complexation effect, we previously demonstrated that the use of Asp as a leaching additive can significantly improve the dissolution capability of CaSO4·2H2O in aqueous ammonia. Moreover, as an amphoteric electrolyte, the solubility of Asp in aqueous solutions is closely related to the solution pH. More specifically, the minimum solubility of Asp at 25 °C is 0.50 g per 100 g H2O at its isoelectric point (pI = 2.77),14 and this solubility increases rapidly upon increasing the solution pH. This unique feature provides a strategy to effectively separate Asp from the sulfate salt-containing carbonation filtrate by simply adjusting the pH value.
Upon reviewing previous works into leaching additives, it was found that other organic compounds containing the –COOH functionality, such as succinic acid, citrate, and EDTA acid, could enhance the degree of calcium leaching. However, because of the strong complexation capability with calcium, carbonate precipitates are not always formed during the carbonation reaction.15,16 In addition, Song et al.17 found that the addition of polyacrylic acid lowered the overall carbonation yield via CO2 emission, thus reducing the CO2 dissolution. The chelate formed by some amino acids and Ca2+ dissociates with decreasing pH, and CaCO3 precipitates are formed during the dissociation process.18 Furthermore, the –NH2 groups of amino acids can react with dissolved CO2 to promote the absorption of CO2 gas, wherein the absorption and thermal desorption reaction mechanisms are similar to those of the alkanolamines due to their comparable functional group compositions, and they are given by the following equations:19,20
Absorption reaction follows the zwitterion mechanism:
| |
 | (1) |
| | |
−OOCRNHCOO− + H2O → NH2RCOO− + HCO3− ⇌ NH3+RCOO− + CO32−
| (2) |
Thermal desorption reaction:
| |
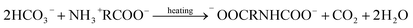 | (3) |
| |
 | (4) |
| |
 | (5) |
| |
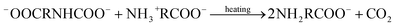 | (6) |
where R represents the side chain (R group) of each amino acid. In this context, Liu
et al.21 reported a single-step direct aqueous carbonation method for CaO and CaSiO
3, which was based on the use of sodium glycinate. This method adopted the integrated CO
2 absorption and mineralisation (IAM) technology,
22,23 which could reduce the energy consumption of glycinate regeneration through a chemical method instead of the traditional thermal method (
i.e.,
eqn (3)–(6)). According to their study, as shown in
eqn (1), the dissolved CO
2 reacts with glycinate to generate the corresponding carbamate and zwitterionic ion. The formation of CaCO
3 through carbonation reaction can greatly facilitate carbamate hydrolysis (
i.e.,
eqn (2)) to regenerate glycinate. Simultaneously, the zwitterionic ion can also be converted to glycinate under alkaline conditions. They also found that glycinate can undergo multiple CO
2 capture and regeneration cycles, thereby confirming the feasibility of amino acid regeneration through the carbonation approach. Furthermore, Ji
et al.24 recently proposed an indirect aqueous carbonation method coupled with the IAM technology, which was based on the use of protonated amino acids to dispose of alkaline solid waste (the main component being CaO). In this system, the amino acid content ranged from 1 to 2 mol L
−1, and the degree of protonation was 20–80%; however, because of the particularity of the calcium source, the only byproduct of the carbonation reaction carried out using CaO was H
2O, which differs from the generation of additional byproduct when CaSO
4·2H
2O was employed. Thus, the major drawback associated with the accumulation of sulfate salts during amino acid recycling was ignored in their report.
Thus, we herein propose an indirect aqueous carbonation method of CaSO4·2H2O with Asp as a recyclable leaching additive. Initially, we examine the dissolution capability of CaSO4·2H2O in aqueous ammonia in the presence of Asp under different ammonia contents and liquid-to-solid ratios. Subsequently, the precipitation process of CaCO3 from the leachate solution is analysed based on variations in the pH and conductivity, and a detailed transformation mechanism is described for Asp. Moreover, the feasibility of recovering Asp from the carbonation filtrate at its isoelectric point is evaluated by Fourier transform infrared (FTIR) spectroscopy. Finally, the recovery efficiency and further cycle performance of Asp based on the amount of dissolved CaSO4·2H2O and the total carbonation efficiency are examined over 10 recycling experiments.
2. Experimental
2.1 Materials
As a preliminary study, the current work ignores the influence of the BG impurity on the carbonation reaction. Therefore, analytically pure CaSO4·2H2O (Sinopharm Chemical Reagent Co., Ltd, China) was selected as the calcium source for carbonation. NH4OH (AR grade) and H2SO4 (AR grade) were purchased from Sinopharm Chemical Reagent Co., Ltd, China. Asp (levo form, biotech grade) was purchased from Macklin Biochemical Co., Ltd, China. Industry-grade CO2 with a purity of 99.9% was used for the carbonation reaction. Deionised water supplied by an ultra-pure water system was used to prepare all solutions.
2.2 Methods
2.2.1 Leaching experiments. A series of batch leaching experiments were performed in closed-glass round-bottom flasks. More specifically, Asp (9.66 g) was initially dissolved in aqueous ammonia at different volumes and ammonia contents. Subsequently, CaSO4·2H2O powder (5 g) was added to the alkaline solution and mixed well by magnetic stirring at 30 °C until the dissolution equilibrium was achieved (i.e., ∼60 min). The leaching temperature was selected to avoid the escape of gaseous NH3, and the molar ratio of Asp to CaSO4·2H2O was equal to 2.5 to ensure a slight excess of Asp in the leaching systems.13 Suspensions saturated in calcium were filtered to obtain the Ca-rich leachate solutions. The amount of dissolved CaSO4·2H2O (η) was measured by complexometric titration using an EDTA standard solution of 0.01 mol L−1. The detailed experimental parameters for the leaching process are listed in Table 1.
Table 1 Leaching experimental parameters
| No. |
Content |
L/S ratio (mL g−1) |
pHa |
| Asp (mol L−1) |
Ammonia (wt%) |
| pH values were measured under 25 °C. |
| 1 |
0.726 |
3 |
20 |
9.472 |
| 2 |
0.484 |
3 |
30 |
9.761 |
| 3 |
0.363 |
3 |
40 |
10.055 |
| 4 |
0.290 |
3 |
50 |
10.102 |
| 5 |
0.726 |
5 |
20 |
9.834 |
| 6 |
0.484 |
5 |
30 |
10.004 |
| 7 |
0.363 |
5 |
40 |
10.263 |
| 8 |
0.290 |
5 |
50 |
10.336 |
| 9 |
0.726 |
7 |
20 |
10.097 |
| 10 |
0.484 |
7 |
30 |
10.266 |
| 11 |
0.363 |
7 |
40 |
10.515 |
| 12 |
0.290 |
7 |
50 |
10.555 |
| 13 |
0.726 |
9 |
20 |
10.208 |
| 14 |
0.484 |
9 |
30 |
10.425 |
| 15 |
0.363 |
9 |
40 |
10.625 |
| 16 |
0.290 |
9 |
50 |
10.677 |
| 17 |
0.726 |
11 |
20 |
10.385 |
| 18 |
0.484 |
11 |
30 |
10.522 |
| 19 |
0.363 |
11 |
40 |
10.734 |
| 20 |
0.290 |
11 |
50 |
10.802 |
2.2.2 Carbonation reaction experiments. A five-necked glass-jacketed reactor with a 500 mL capacity was used for the gas–liquid carbonation reaction. More specifically, CaSO4·2H2O powder (15 g) was dissolved in a solution with an ammonia content of 7 wt% and a liquid-to-solid ratio of 30. This system was initially chosen considering the ideal CaSO4·2H2O leaching capability and a higher initial carbonation reaction pH, which is beneficial for CaCO3 formation. At the beginning of the reaction, CO2 gas was directly injected into the obtained Ca-rich leachate solution at a rate of 500 mL min−1 using a mass flowmeter (SLD-MFC, SENLOD, China). The stirring rate was maintained at 500 rpm using an overhead stirrer (EUROSTAR 20 digital, IKA, Germany). As the carbonation reaction is exothermic, the reaction heat was removed from the reaction system by cooling circulating water, and the reaction temperature was maintained at 30 ± 1 °C. During the 50 min reaction time, the system pH and conductivity were recorded online using a pH meter (PHSJ-6L, INESA, China) and a conductivity meter (DDSJ-319L, INESA, China), respectively. The reactive suspensions were sampled (1 mL) at predetermined time intervals and filtrated using 0.2 μm membrane filters prior to analysis of the dissolved calcium content by titration with EDTA. The amount of precipitated CaCO3 at each time interval was obtained from the difference between the determined content and the initial dissolved calcium content. At the end of the reaction, the final CaCO3 production was recovered via filtration and washing with aqueous ammonia of 7 wt%. The sample was then dried to constant weight to determine its mass with gravimetric method. The polymorphs and morphologies of the obtained CaCO3 were characterised by X-ray diffraction (XRD; Ultima IV, Rigaku, Japan, Cu Kα radiation source in a 2θ range of 5–90° at a scanning rate of 1° min−1) and scanning electron microscopy (SEM; Sigma 300, ZEISS, Germany, accelerating voltage of 5 kV). The total carbonation efficiency (γ) was calculated according to the equation, as previously described by Rahmani et al.:25| |
 | (7) |
where mCa,end (g) and mTCa,initial (g) represent the calcium amount of the final CaCO3 products and CaSO4·2H2O raw material, respectively.
2.2.3 Asp recovery and recycling experiments. After the removal of CaCO3, the filtrate was evaporated at 90 °C under reduced pressure to remove any excess aqueous ammonia. After evaporation, the pH of the filtrate was close to neutral. Subsequently, a 40 wt% H2SO4 solution was added at 25 °C to further adjust the pH to the isoelectric point of Asp (i.e., 2.77). The variation in pH during addition of the H2SO4 solution was recorded, and the precipitated solid phase obtained at the isoelectric point was filtered and dried in an oven at 100 °C until reaching a constant weight (12 h). The identities of the obtained products were confirmed by means of FTIR spectrometry. In addition, the recovery efficiency of Asp (φ) was determined as follows:| |
 | (8) |
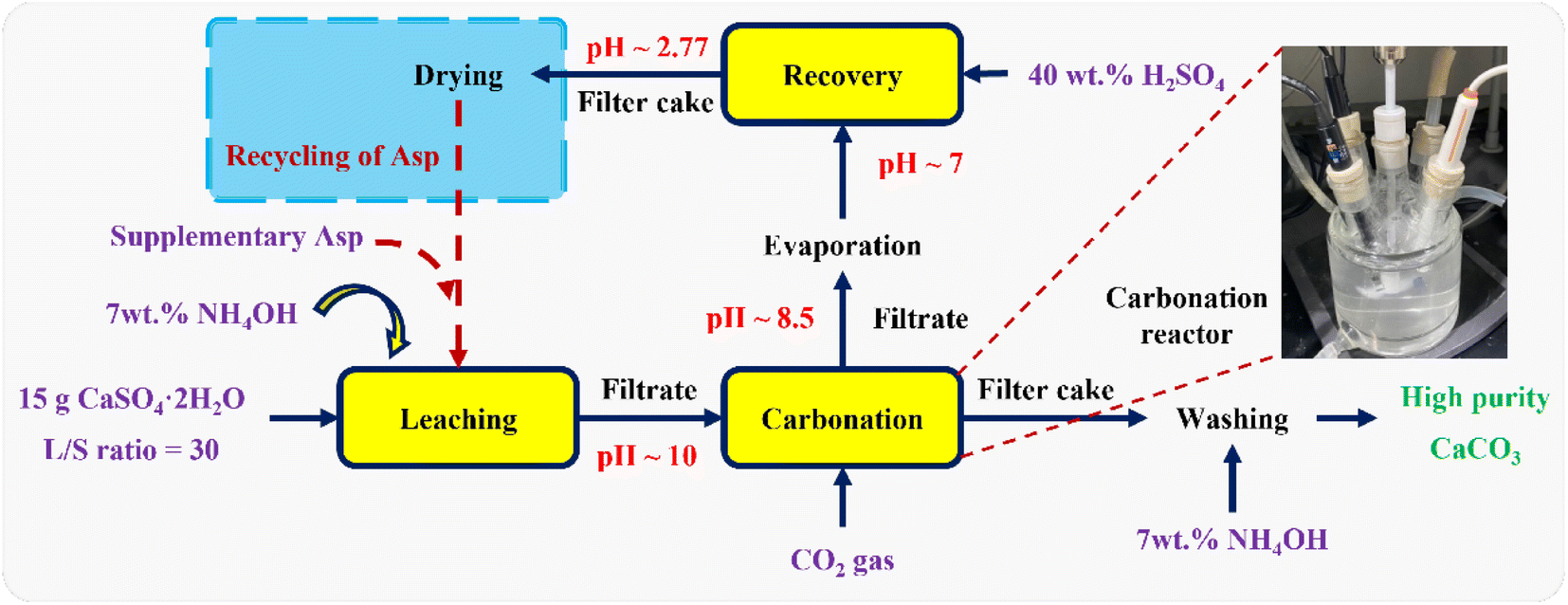
where mAsp, recovered (g) and mAsp,leaching (g) denote the amount of Asp recovered from carbonation filtrate and added during the leaching step, respectively. Finally, as outlined in Fig. 1, the Asp recycling experiments were performed after supplementing the loss of Asp with the fresh reagent to examine the cycling performance of Asp. It should be noted here that the additional fresh Asp required for each recycle was <20 wt% of the total Asp content.
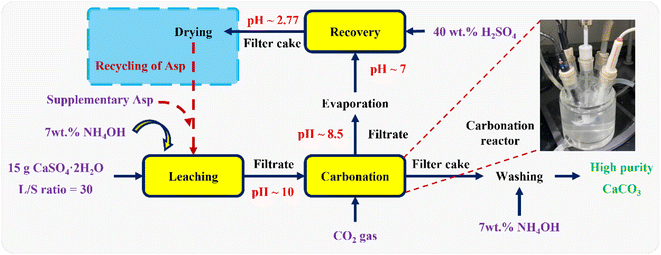 |
| | Fig. 1 Schematic outline of the multistep experimental procedure, including calcium leaching, the carbonation reaction, Asp recovery, and Asp recycling. | |
3. Results and discussion
3.1 Influence of the ammonia content and liquid-to-solid ratio on calcium leaching in the presence of Asp
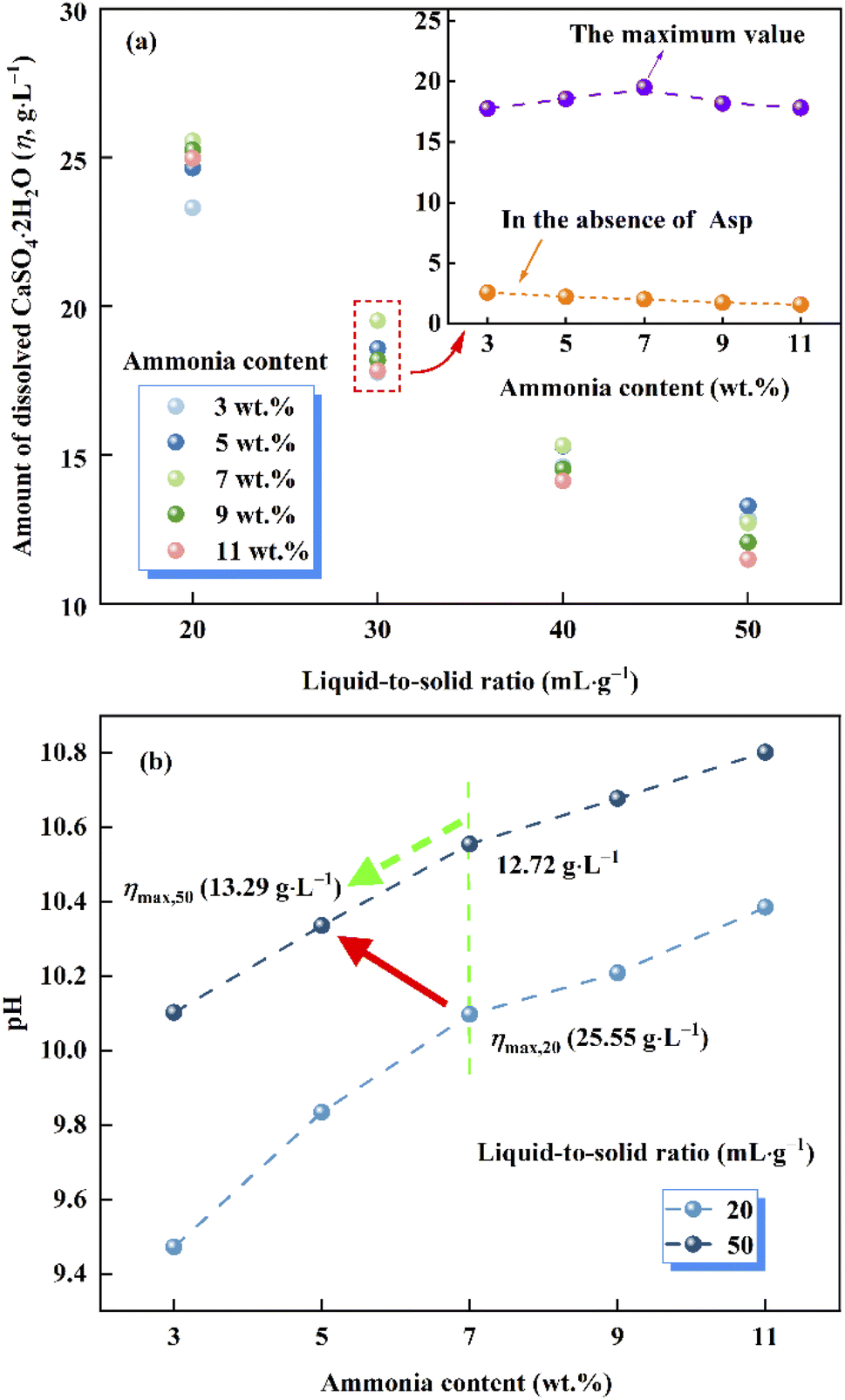
As shown in Fig. 2a, compared with the extremely low solubility of CaSO4·2H2O in aqueous ammonia, the presence of Asp resulted in a significant increase in the amount of dissolved CaSO4·2H2O (η). Furthermore, upon varying the ammonia content and liquid-to-solid ratio, η was found to exhibit different tendencies. More specifically, at a given ammonia content, the value of η gradually decreased as the liquid-to-solid ratio increased due to the increased activity of H2O, while the solubility constant of CaSO4·2H2O, which is defined as the product of the activities of Ca2+, SO42−, and H2O, remained unchanged at a given temperature.26,27 Besides, the complexing effect weakened due to the Asp content decreasing with the increase of the liquid-to-solid ratio. In contrast, the value of η increased upon increasing the ammonia content to 7%, beyond which point, the value of η decreased again. Moreover, it is worth noting that with the exception of a liquid-to-solid ratio of 50, the ammonia content corresponding to the maximum value of η remained constant at 7% (c.f., 5% for a liquid-to-solid ratio of 50) over the various liquid-to-solid ratios examined herein (see Fig. 2a).
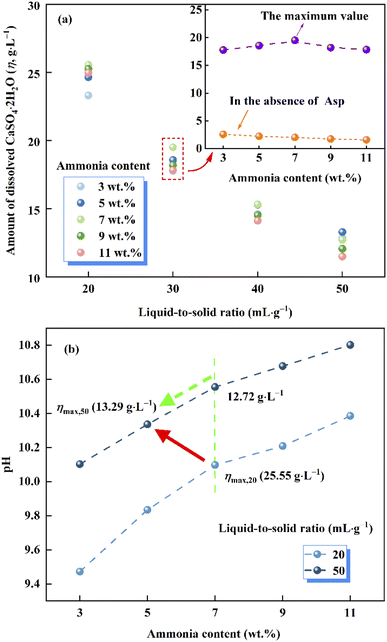 |
| | Fig. 2 (a) Plot of the amount of dissolved CaSO4·2H2O (η) vs. the liquid-to-solid ratio and the ammonia content. (b) Plot of the pH vs. the ammonia content at liquid-to-solid ratios of 20 and 50. | |
According to previous research, the dissolution of CaSO4·2H2O in aqueous ammonia is inhibited due to the formation of hydrogen bonds between free ammonia molecules and bound water molecules in the layered crystal structure of CaSO4·2H2O,28 thereby accounting for the later decrease in solubility at higher ammonia contents. More specifically, the observed variation in η appeared to be controlled by competition between the positive complexing effect (Asp with Ca2+) and the negative inhibitory effect (free ammonia molecules with bound water molecules); the former was dominant at relatively low ammonia contents, while the latter played a more significant role at high ammonia contents.
As shown in Fig. 2b, over a given ammonia content range, the mass of Asp remained constant, thereby indicating that the pH of the leachate solution is proportional to the liquid-to-solid ratio. More specifically, upon increasing the liquid-to-solid ratio, a higher pH led to an increase in the free ammonia molecules content, considering the shift in the dissociation equilibrium of ammonia. According to the above competitive relationship, the inhibitory effect of free ammonia molecules with bound water is enhanced, which leads to the maximum value of η moving toward the lower ammonia content.
3.2 Analysis of the CaCO3 precipitation process
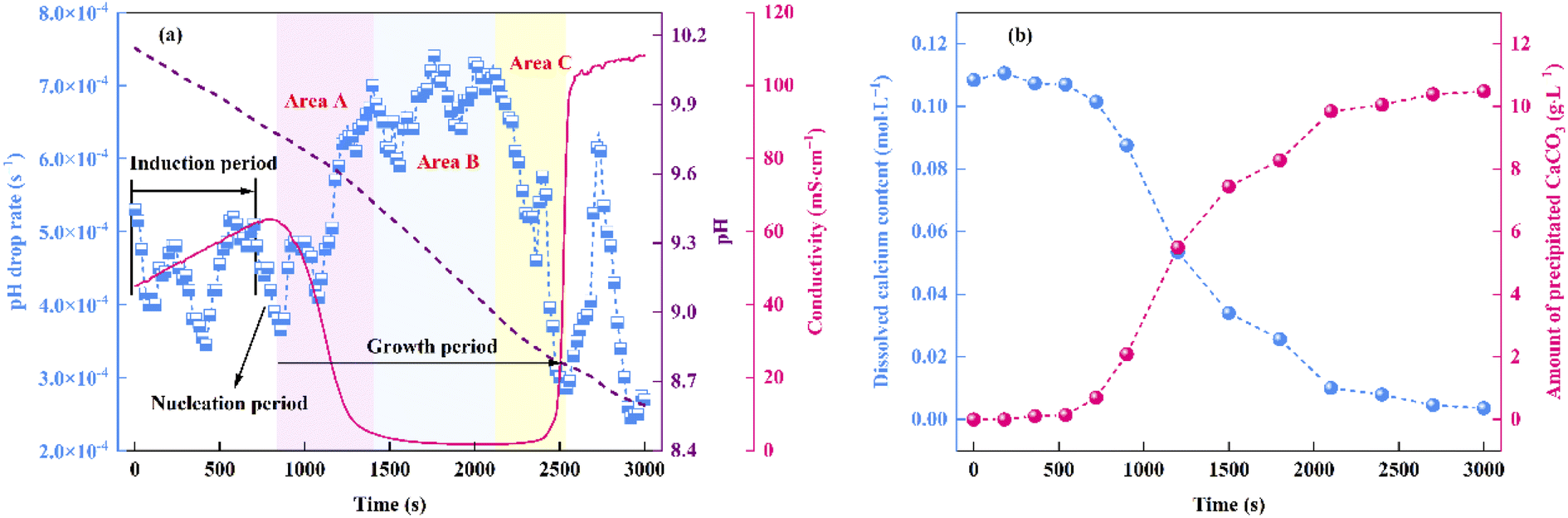
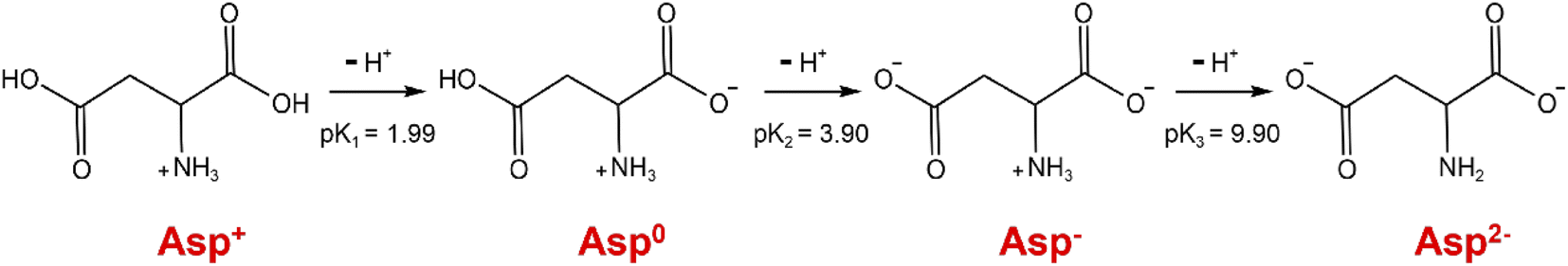
As can be seen from Fig. 3a, within the initial 420 s of the carbonation reaction, the pH drop rate showed an overall decrease, which was mainly attributed to the dissociation equilibrium shift of Asp from Asp2− to Asp− over the pH range of 10.15 to 9.97. More specifically, the dissociation of Asp upon variation in the solution pH is shown in Fig. 4. Thus, upon decreasing the solution pH, the unstable complex gradually releases Ca2+, and the electron lone pair of the Asp2− amino group can recombine with the protons (H+) generated by the chemical absorption of CO2, ultimately generating Asp−. This leads to a slight reduction in the pH drop rate. Subsequently, owing to the accumulation of free Ca2+ and CO32−, the reaction of CaCO30 ion pair formation is enhanced, which further promotes CO2 absorption.29 As a result, the pH drop rate increased during the time interval from 420 to 700 s. In addition, a crystallization induction period of ∼700 s was observed for the precipitation of CaCO3, and this was due to the above variation in the pH drop rate. Notably, the dissociation equilibrium shift of Asp from Asp2− to Asp− significantly prolongs the induction time compared to the system where Asp is absent.30 Furthermore, the conductivity was found to increase continuously during this period, and this was predominantly controlled by the chemical absorption of CO2.29 As shown in Fig. 3b, the dissolved calcium content remained relatively unchanged during the initial 700 s of the reaction, during which the reaction solution became transparent, and only a very small amount of CaCO3 precipitated. These results further confirm the existence of an extended induction period in the presence of Asp.
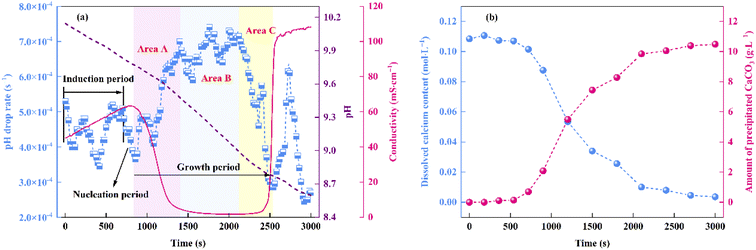 |
| | Fig. 3 Variations in the (a) pH and conductivity of the reaction system over time. (b) Contents of dissolved Ca and precipitated CaCO3 during the progress of the carbonation reaction. | |
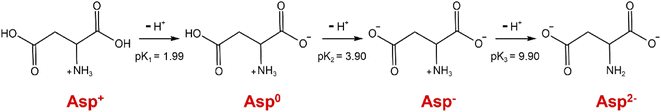 |
| | Fig. 4 Dissociation equilibrium shift of Asp. | |
Beyond a reaction time of 700 s, the pH drop rate began to decrease and nucleation occurred. During a period of ∼160 s, a large number of CaCO3 nuclei were formed through the consumption of CaCO30 ion pairs, and as a result, the rate of CaCO30 ion pair formation decreased;31 this was accompanied by a decrease in the pH drop rate. During the nucleation period, the conductivity of the reaction solution continued to increase since the total volume of the nucleus was relatively small and thus it contributed only weakly to the total ion content.31
Fig. 3a shows that the growth period began at 860 s, and this period can be divided into three stages. More specifically, area A (860–1400 s) corresponded to the rapid formation of CaCO3, wherein the rate of CO32− consumption was significantly higher than that of its formation. This led to a surge in the pH drop rate and a plunge in the conductivity. In addition, as shown in Fig. 3b, the dissolved calcium content decreased, and a large amount of CaCO3 precipitated from the liquid phase. Subsequently, area B (1400–2100 s) corresponds to the stable crystal growth stage. During this stage, the solution conductivity and pH drop rate were close to their extreme points, and a dynamic balance between the consumption and formation rates of CO32− was achieved. Finally, area C (2100–2500 s) represents the end of the growth period. Due to the minimal amount of dissolved calcium in the bulk solution at this stage (Fig. 3b), the CO32− consumption rate decreased, and reached a value lower than its formation rate. As a result, the pH drop rate decreased significantly, while the conductivity jumped to >100 ms cm−1 at the end of this stage. In the final 500 s of the carbonation reaction, the variations in the pH drop rate and the conductivity were mainly controlled by the chemical absorption of CO2. The amount of precipitated CaCO3 reached a maximum level, and the termination of the carbonation reaction was considered achieved. Finally, the total carbonation efficiency was calculated as 54.2%.
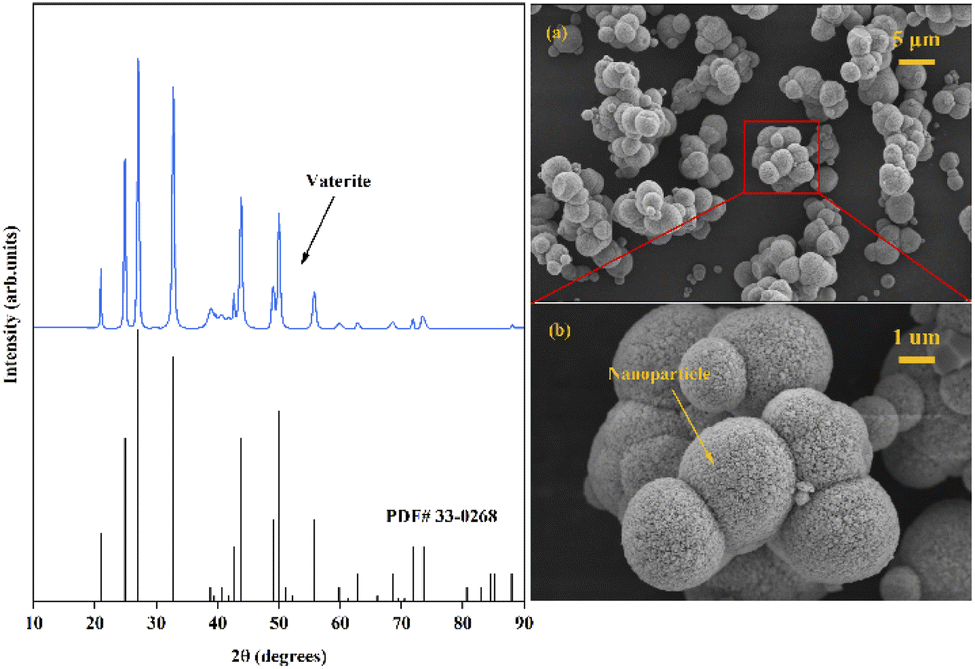
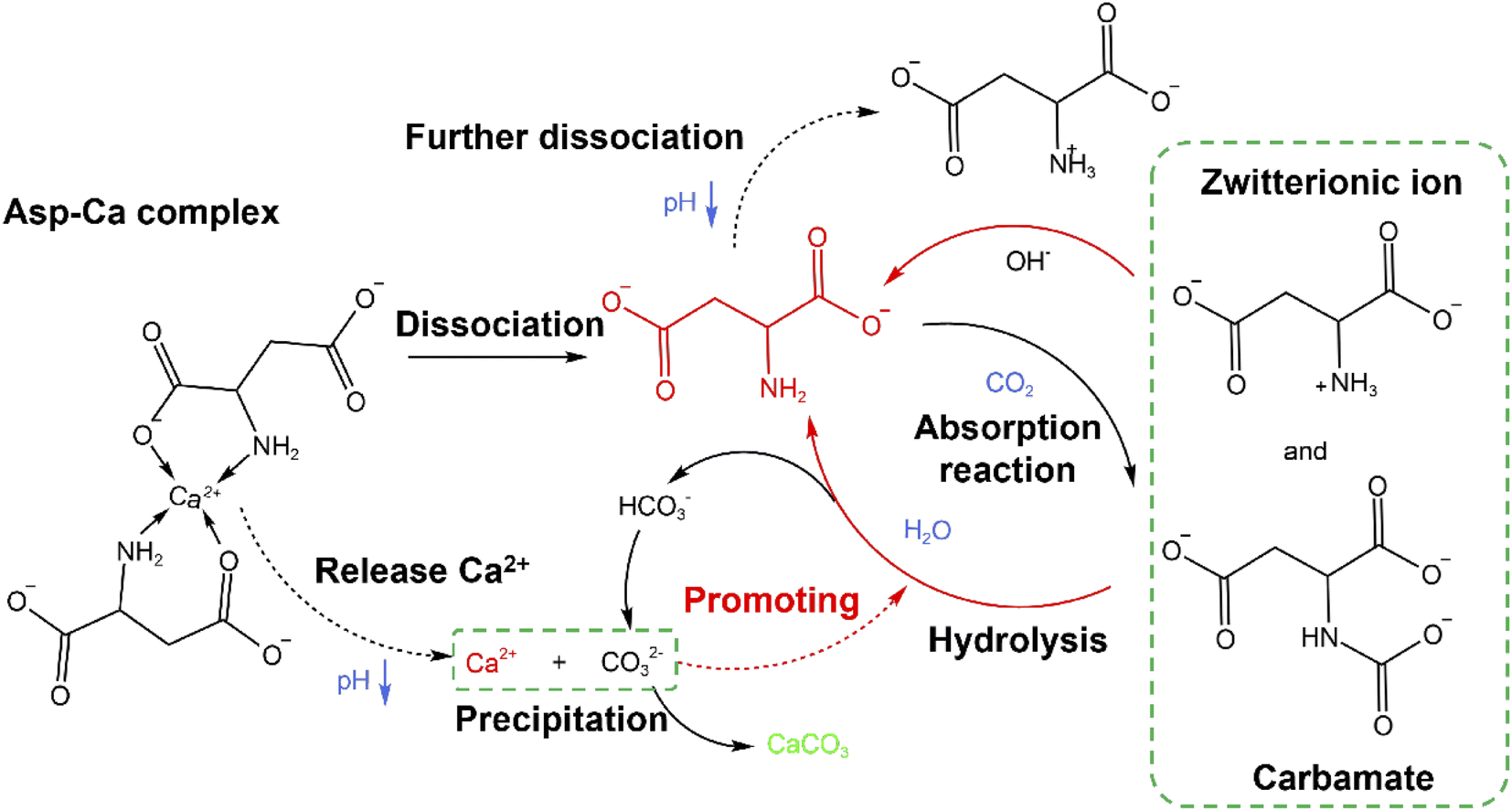
Through examination of the XRD and SEM results presented in Fig. 5, the precipitated CaCO3 products obtained in the presence of Asp were identified as spherical particles of the only vaterite phase. No characteristic peaks of calcite or aragonite phase were detected. The result was consistent with previous reported by Zheng et al.32 who proposed a glycine-mediated leaching-mineralization cycle from coal fly ash. It seems that a relatively high content of amino acid can help the formation of vaterite phase. According to Ostwald's step rule, the initial formation of the metastable vaterite is followed by its transformation into the more thermodynamically stable calcite.33 However, during the growth period, organic additives such as Asp can adsorb onto the surface of the initially formed vaterite particles, which decreases the surface energy or creates a barrier to prevent the above phase transformation.13 Moreover, the presence of Asp–Ca complex reduces the content of free Ca2+ in the bulk solution. As a new Ca source, the dissociation of the complex controls the release of Ca2+, which influences the conversion of the precursor CaCO30 ion pair, and therefore benefit the formation of vaterite.13 Besides, the only vaterite phase can also obtain in excessive amount of ammonia (7 wt% in the present study).10 The SEM images show that the precipitated CaCO3 microspheres are composed of aggregates of numerous nanoparticles, which can be accounted for by considering the primary–secondary particle theory.34 In this case, Asp controls the precipitation of CaCO3 to produce stable spherical vaterite particles. Simultaneously, the Asp additive spontaneously undergoes absorption reaction and chemical regeneration, which are outlined in Fig. 6.
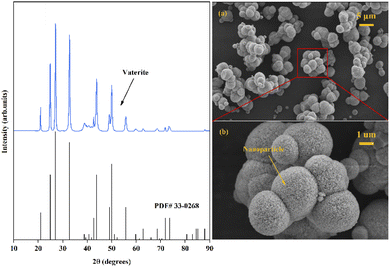 |
| | Fig. 5 XRD and SEM images ((a) scale bar 5 μm, (b) scale bar 1 μm) of the obtained CaCO3 products. | |
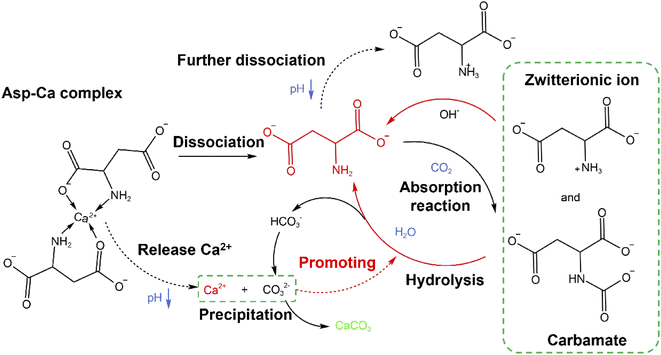 |
| | Fig. 6 The transformation mechanism of Asp during the carbonation reaction. | |
3.3 Recovery of Asp and determination of its cycling performance
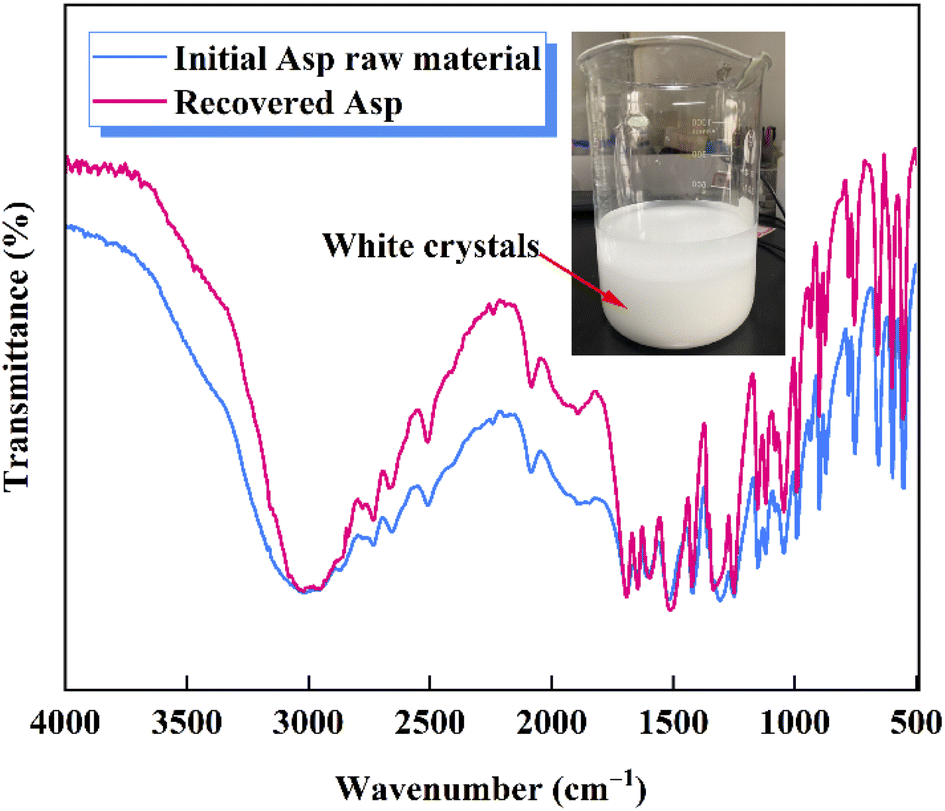
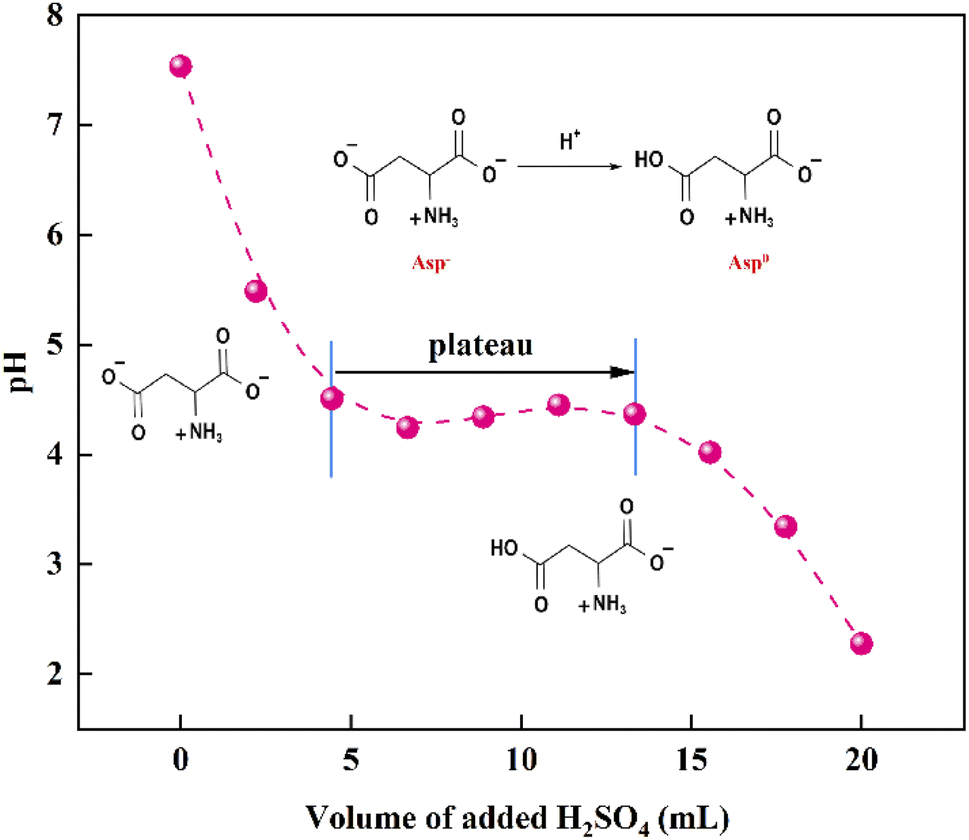
Following the removal of CaCO3, a 40 wt% H2SO4 solution was added to the evaporated filtrate to generate large quantities of white crystals (Fig. 7). As shown in the FTIR spectra, the recovered crystals were confirmed to be Asp. It should also be noted that upon the addition of H2SO4, the pH of the filtrate decreased, and so the dissociation equilibrium of Asp shifted toward the zwitterionic ion (Asp0). As shown in Fig. 8, during the initial stage of H2SO4 addition (0–4.4 mL), the pH value decreased sharply from 7.5 to 4.5, at which point Asp was present mainly in the form of Asp−. Subsequently, a plateau period was observed following the further addition of H2SO4 (4.4–13.3 mL), which leads to the combination of Asp− with the abundant H+ present in the bulk solution to generate Asp0, and so the solution pH does not change to any great extent during this addition period. In addition, owing to the substantially lower solubility of Asp0 under these conditions, high supersaturation was achieved, leading to the precipitation of large quantities of Asp from the filtrate during this plateau period. Upon the further addition of H2SO4 to a total volume of 20 mL, the pH value began to drop again until reaching the isoelectric point of Asp. As a result, the neutral Asp0 became the main form of Asp present in the bulk solution. The Asp recovery efficiency was 89.2%.
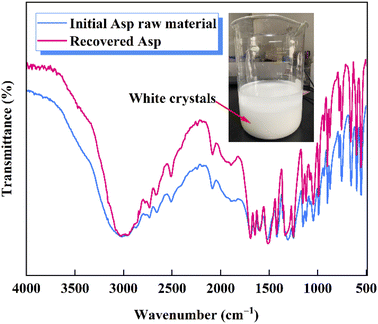 |
| | Fig. 7 FTIR spectra of the initial Asp raw material and the recovered Asp. | |
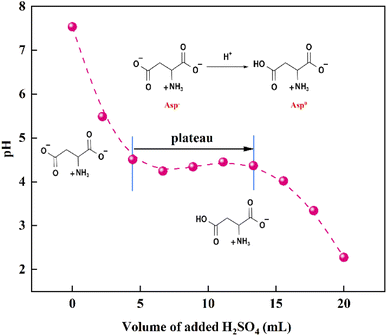 |
| | Fig. 8 Variation of the solution pH with the addition of H2SO4. | |
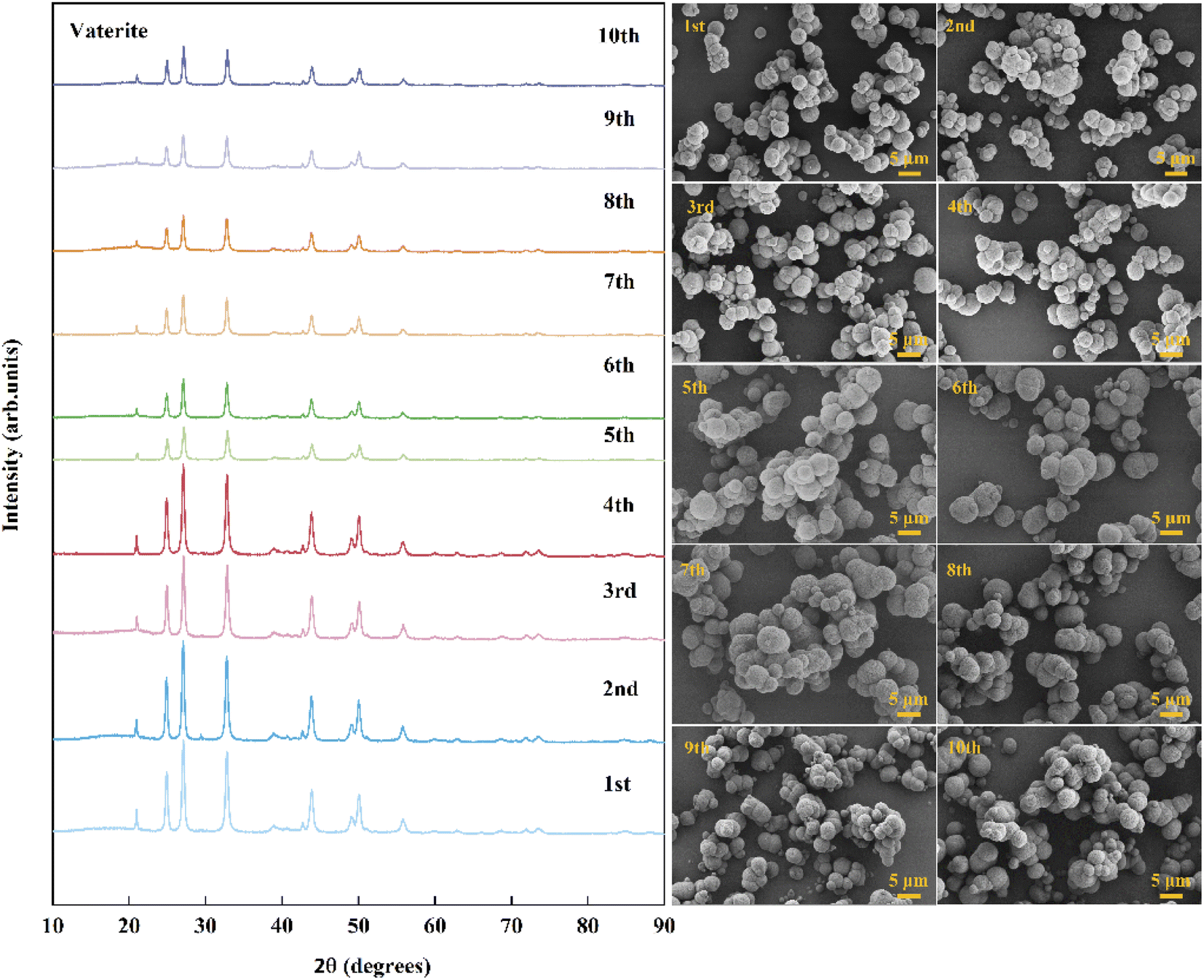
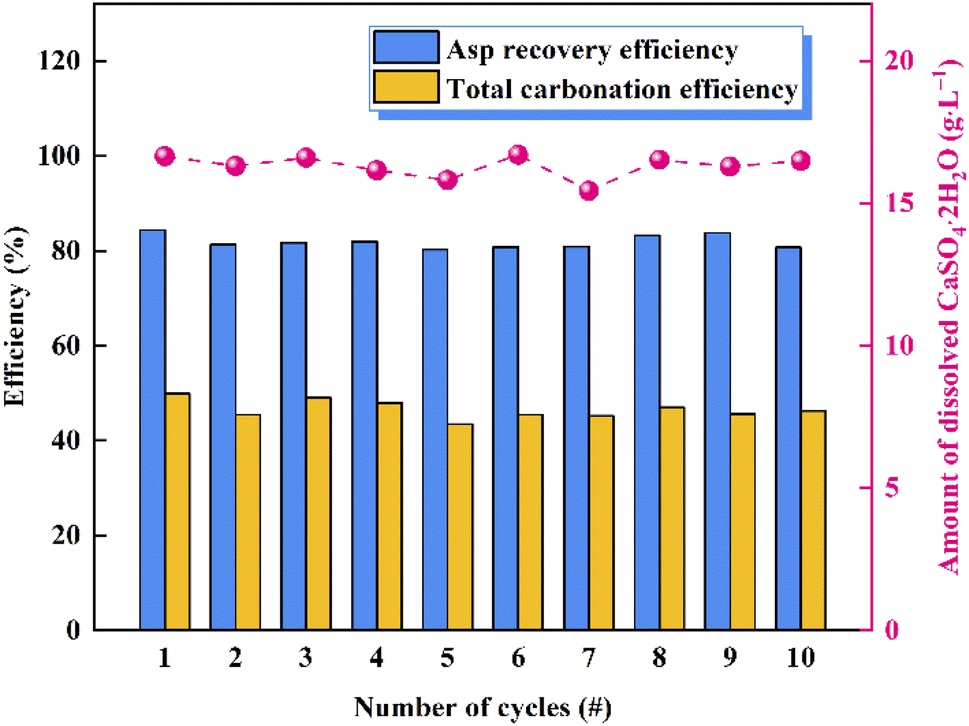
To verify the cycling performance of Asp, 10 recycling experiments were carried out under identical experimental conditions after adding fresh Asp to the recovered crystals. More specifically, each cycle involved calcium leaching, the gas–liquid carbonation reaction, and Asp recovery. As shown in Fig. 9, the Asp recovery efficiency remained relatively stable at 81.9 ± 1.4%, while the amount of dissolved CaSO4·2H2O and the total carbonation efficiency were 16.3 ± 0.4 g L−1 and 46.5 ± 1.9%, respectively. Compared with other additives, as shown in Table 2, the leaching performance of Asp was similar to that of NH4Cl, however, having higher carbonation efficiency which could be attributed to a higher liquid-to-solid ratio. Most importantly, the reaction efficiency of Asp kept an idea high level (90.5%) during the 10 recycling experiments, which was different from other inorganic additives. This indicates that the effective separation of Asp from the sulfate-rich carbonation filtrate was the key to its recycling. Finally, the precipitated CaCO3 after each cycle was examined by XRD and SEM to observe any changes taking place during these experiments. As shown in Fig. 10, the spherical vaterite particles with diameters of 2–5 μm were recovered after each stage, and overall, the XRD and SEM results were consistent, thereby further confirming the reusability of Asp.
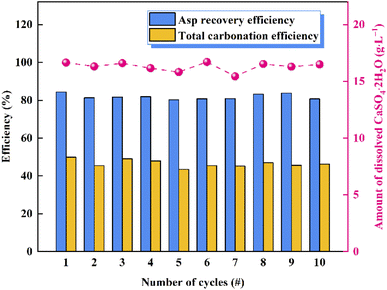 |
| | Fig. 9 Results of the 10 recycling experiments to confirm the reusability of Asp. | |
Table 2 Comparison of leaching, carbonation, and cycle performance in this study and previous studies
| Additives |
CaOa (g) |
η (g L−1) |
γ (%) |
λ (%) |
The mass of raw material was calculated based on pure CaO. λ: the reaction efficiency (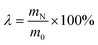 ), where m0 presents the mass of CaCO3 when the additive was first used, and mN denotes that under different cycle indices.11 c: additive content, mol L−1; LS: liquid-to-solid ratio, mL g−1; t: temperature, °C; Q: CO2 flow rate, mL min−1; θ: carbonation reaction time, min. ), where m0 presents the mass of CaCO3 when the additive was first used, and mN denotes that under different cycle indices.11 c: additive content, mol L−1; LS: liquid-to-solid ratio, mL g−1; t: temperature, °C; Q: CO2 flow rate, mL min−1; θ: carbonation reaction time, min. |
| NaCl9 |
5.92 |
9.0 (c = 3, LS = 50, t = 30) |
47.6 (Q = 80, t = 30, θ = 60) |
40.3 (after 5 cycles) |
| CH3COONH410 |
5.92 |
89.3 (c = 6, LS = 10, t = 80) |
98.3 (Q = 100, t = 30, θ = 90) |
— |
| NH4Cl11 |
5.92 |
18.7 (c = 2, LS = 10, t = 60) |
20.2 (Q = 50, t = 30, θ = 60) |
42.6 (after 10 cycles) |
| Asp (this study) |
4.87 |
19.5 (c = 0.484, LS = 30, t = 30) |
54.2 (Q = 500, t = 30, θ = 50) |
90.5 (after 10 cycles) |
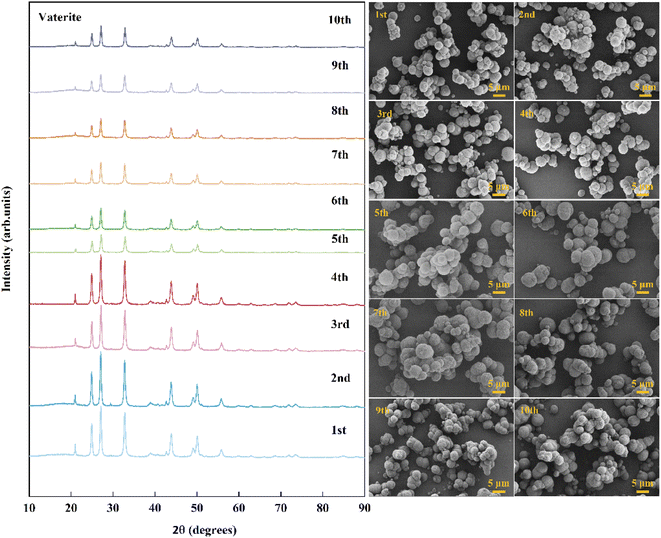 |
| | Fig. 10 XRD patterns and SEM images (scale bar 5 μm) of CaCO3 recorded during the cycling. | |
4. Conclusions
In conclusion, we proposed an indirect aqueous carbonation method of CaSO4·2H2O using aspartic acid (Asp) as recyclable additive. Our results indicated that the multiple roles of Asp were found as Ca2+ leaching agent, CO2 absorbent, and CaCO3 polymorph regulator in both the calcium leaching and subsequent gas–liquid carbonation reaction steps. More specifically, the dissolution capability of CaSO4·2H2O in aqueous ammonia was found to improve significantly through complexation between Asp and Ca2+. Furthermore, the Asp2− reacts with dissolved CO2 and was then regenerated by means of the integrated CO2 absorption and mineralisation (IAM) technique. The induction period during the carbonation reaction was extended owing to the dissociation equilibrium shift of Asp from Asp2− to Asp−, which eventually controlled the conversion of the precursor CaCO30 ion pair. As the final carbonation products, stable spherical vaterite particles were found. Based on the solubility characteristics of Asp upon variation in the pH, the regenerated Asp was successfully recovered from the sulfate-rich carbonation filtrate at its isoelectric point, with a recovery efficiency of >80% being achieved, and the reusability of the recycled Asp was further confirmed by recycling experiments. We believe that our study makes a significant contribution to the recycling and recovery of the leaching additives in the indirect aqueous carbonation process of CaSO4·2H2O. Although the present study demonstrated the feasibility of Asp recovery and recycling, the recovery efficiency of Asp must be further improved to reduce the cost of this technology. Therefore, current work in our group is focusing on the thermodynamics and kinetics of Asp crystallisation from a solution containing a sulfate salt. Moreover, the recovery method of ammonia from the final filtrate after Asp separation and the comprehensive mechanism study of the carbonation reaction system in the presence of Asp are other important directions for future work.
Author contributions
Yuan Gong: conceptualization, formal analysis, investigation, methodology, writing – original draft, writing – review & editing. Xuechen Zhu: formal analysis, validation, investigation, writing – review & editing. Zhuo Yang: validation, writing – review & editing. Xin Zhang: validation. Chunlei Li: conceptualization, supervision, project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the support of the Natural Science Foundation of Gansu Province, China (Grant No. 21JR7RA226).
References
- S. P. Veetil and M. Hitch, Int. J. Environ. Sci. Technol., 2020, 17, 4359–4380 CrossRef CAS.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- A. M. Rashad, J. Cleaner Prod., 2017, 166, 732–743 CrossRef CAS.
- W. Liu, L. Teng, S. Rohani, Z. Qin, B. Zhao, C. C. Xu, S. Ren, Q. Liu and B. Liang, Chem. Eng. J., 2021, 416, 129093 CrossRef CAS.
- N. H. Koralegedara, P. X. Pinto, D. D. Dionysiou and S. R. Al-Abed, J. Environ. Manage., 2019, 251, 109572 CrossRef CAS PubMed.
- J. Zhou, Z. Sheng, T. Li, Z. Shu, Y. Chen and Y. Wang, Ceram. Int., 2016, 42, 7237–7245 CrossRef CAS.
- B. Wang, Z. Pan, H. Cheng, Z. Zhang and F. Cheng, J. Cleaner Prod., 2021, 302, 126930 CrossRef CAS.
- O. Rahmani, M. Tyrer and R. Junin, RSC Adv., 2014, 4, 45548–45557 RSC.
- Q. Chen, W. Ding, H. Sun, T. Peng and G. Ma, Energy, 2020, 206, 118148 CrossRef CAS.
- W. Ding, Q. Chen, H. Sun and T. Peng, J. CO2 Util., 2019, 34, 507–515 CrossRef CAS.
- Q. Chen, W. Ding, H. Sun, T. Peng and G. Ma, ACS Sustainable Chem. Eng., 2020, 8, 11649–11657 CrossRef CAS.
- B. Yang, M. Yang, B. Wang, X. Fang and Q. Wan, Mater. Res. Express, 2019, 6, 45042 CrossRef.
- J. Luo, F. Kong and X. Ma, Cryst. Growth Des., 2016, 16, 728–736 CrossRef CAS.
- X. Yu, Handbook of MSG industry, China Light Industry Press Ltd, Beijing, 2009 Search PubMed.
- B. Bonfils, C. Julcour-Lebigue, F. Guyot, F. Bodénan, P. Chiquet and F. Bourgeois, Int. J. Greenhouse Gas Control, 2012, 9, 334–346 CrossRef CAS.
- R. M. Santos, Y. W. Chiang, J. Elsen and T. Van Gerven, Hydrometallurgy, 2014, 147–148, 90–94 CrossRef CAS.
- K. Song, W. Kim, S. Park, J.-H. Bang, C. W. Jeon and J.-W. Ahn, Chem. Eng. J., 2016, 301, 51–57 CrossRef CAS.
- X. Liu, B. Wang, Z. Zhang, Z. Pan, H. Cheng and F. Cheng, Environ. Chem. Lett., 2022, 20, 2261–2269 CrossRef CAS.
- Y. Li, X. Duan, W. Song, L. Ma and J. Jow, Chem. Eng. J., 2021, 405, 126938 CrossRef CAS.
- V. Sang Sefidi and P. Luis, Ind. Eng. Chem. Res., 2019, 58, 20181–20194 CrossRef CAS.
- M. Liu and G. Gadikota, Fuel, 2020, 275, 117887 CrossRef CAS.
- L. Ji, H. Yu, K. Li, B. Yu, M. Grigore, Q. Yang, X. Wang, Z. Chen, M. Zeng and S. Zhao, Appl. Energy, 2018, 225, 356–366 CrossRef CAS.
- Z. Yan, Y. Wang, H. Yue, C. Liu, S. Zhong, K. Ma, W. Liao, S. Tang and B. Liang, ACS Sustainable Chem. Eng., 2021, 9, 8238–8248 CrossRef CAS.
- L. Ji, X. Zheng, S. Yan and Q. He, CN. Pat., 113443628A, 2021 Search PubMed.
- O. Rahmani, J. CO2 Util., 2020, 35, 265–271 CrossRef CAS.
- I. Hammas, K. Horchani-Naifer and M. Férid, Int. J. Miner. Process., 2013, 123, 87–93 CrossRef CAS.
- J. Ge, J. Zhu, S. Xia and S. Liu, CIESC J., 2018, 69, 2829–2837 CAS.
- L. Wu, J. Li, J. Zhu, Y. Gong and J. Ge, CIESC J., 2020, 71, 3575–3584 CAS.
- M. Vučak, J. Perić, A. Žmikić and M. N. Pons, Chem. Eng. J., 2002, 87, 171–179 CrossRef.
- K. Song, Y.-N. Jang, W. Kim, M. G. Lee, D. Shin, J.-H. Bang, C. W. Jeon and S. C. Chae, Chem. Eng. J., 2012, 213, 251–258 CrossRef CAS.
- Y. Gong, L. Wu, J. Li and J. Zhu, J. Cryst. Growth, 2019, 522, 128–138 CrossRef CAS.
- X. Zheng, J. Liu, Y. Wei, K. Li, H. Yu, X. Wang, L. Ji and S. Yan, Chem. Eng. J., 2022, 440, 135900 CrossRef CAS.
- C. C. Yec and H. C. Zeng, J. Mater. Chem. A, 2014, 2, 4843–4851 RSC.
- Y. Lai, L. Chen, W. Bao, Y. Ren, Y. Gao, Y. Yin and Y. Zhao, Cryst. Growth Des., 2015, 15, 1194–1200 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Xuechen Zhua,
Zhuo Yanga,
Xin Zhanga and
Chunlei Li*ab
ab,
Xuechen Zhua,
Zhuo Yanga,
Xin Zhanga and
Chunlei Li*ab
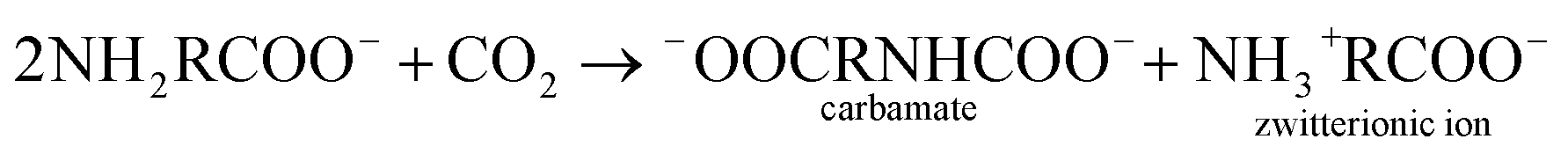
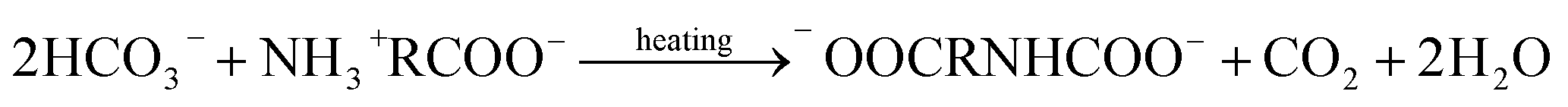

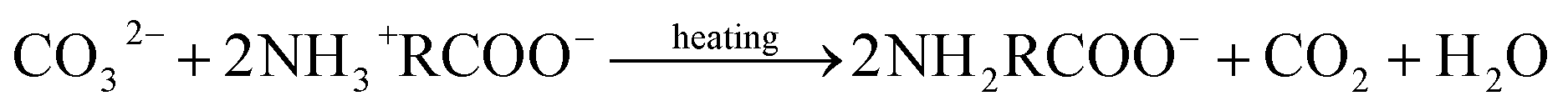
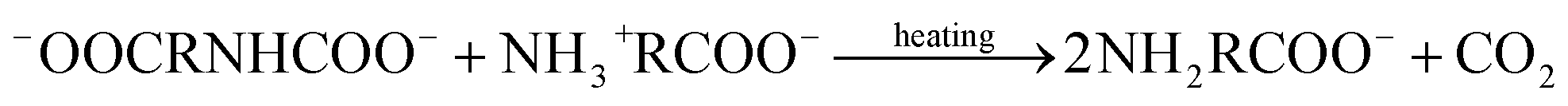
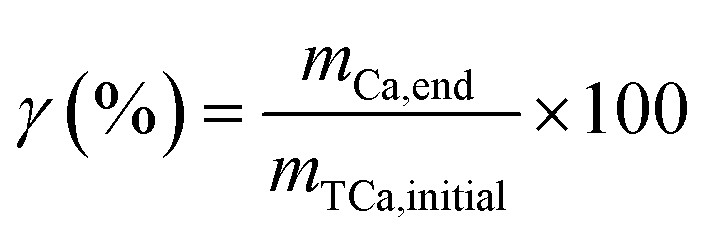
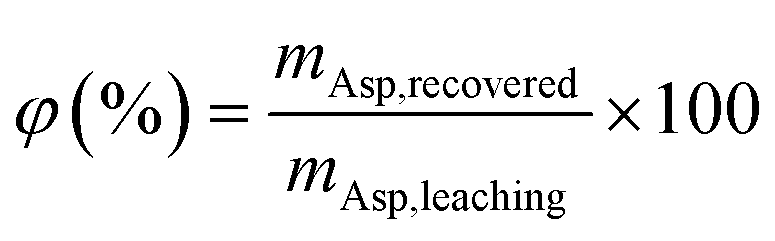
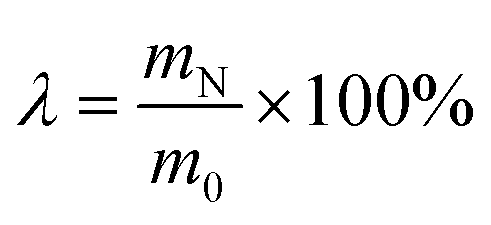 ), where m0 presents the mass of CaCO3 when the additive was first used, and mN denotes that under different cycle indices.11 c: additive content, mol L−1; LS: liquid-to-solid ratio, mL g−1; t: temperature, °C; Q: CO2 flow rate, mL min−1; θ: carbonation reaction time, min.
), where m0 presents the mass of CaCO3 when the additive was first used, and mN denotes that under different cycle indices.11 c: additive content, mol L−1; LS: liquid-to-solid ratio, mL g−1; t: temperature, °C; Q: CO2 flow rate, mL min−1; θ: carbonation reaction time, min.