
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico investigation of the role of vitamins in cancer therapy through inhibition of MCM7 oncoprotein†
Sunny Mukherjeea,
Sucharita Dasb,
Navneeth Sriramac,
Sandipan Chakraborty *d and
Mahesh Kumar Sah*a
*d and
Mahesh Kumar Sah*a
aDepartment of Biotechnology, Dr B. R. Ambedkar National Institute of Technology, Jalandhar, Punjab-144011, India. E-mail: sahmk@nitj.ac.in
bDepartment of Microbiology, University of Calcutta, 35 Ballygunge, Kolkata, 700 019, India
cDepartment of Biosciences and Bioengineering, Indian Institute of Technology, Guwahati, Assam-781039, India
dCenter for Innovation in Molecular and Pharmaceutical Sciences (CIMPS), Dr Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500046, India. E-mail: sandipanchakraborty.13@gmail.com; sandipanc@drils.org
First published on 31st October 2022
Abstract
An overabundance of MCM7 protein, a component of the minichromosome maintenance complex that normally initiates DNA replication, has been reported to cause different types of cancers with aggressive malignancy. Inhibition of MCM7 may lead to a significant reduction in cancer-associated cell proliferation. Despite such significance of MCM7 in cancer, the protein structure is yet to be resolved experimentally. This significantly halts the structure-guided ligand designing for cancer therapy targeting the MCM7. The present study aims to resolve the tertiary structure of MCM7 and repurpose the FDA-approved clinically used drugs for cancer therapy by targeting MCM7 protein. The secondary and 3D structures of MCM7 were generated using multiple bioinformatics tools, including the Self-Optimized Prediction Method with Alignment (SOPMA), SWISS-MODEL, and I-TASSER. The reliability of the modeled structure was assessed using PROCHECK. Initially, a structure-guided virtual screening was performed on the approved drug library to identify potential hits against MCM7. The detailed molecular mechanism of receptor interactions of the identified hits was evaluated using extensive molecular dynamics simulation. The results from this study reveal an intriguing discovery of the potential of ergocalciferol (vitamin D2), cholecalciferol (vitamin D3), ergosterol (precursor of vitamin D2) and menaquinone (vitamin K2) as oncoprotein inhibitors for cancer therapy via inhibition of MCM7.
1. Introduction
MCM7, a vital component of the Minichromosomal Maintenance (MCM) complex, exists as a hexameric (MCM2-7) protein.1 MCM7 is an essential constituent of the pre-replication complex, which develops at the DNA replication origin site and leads to replication fork formation and the regulation of DNA helicase activity.2 The helicase activity of the MCM7 protein subsequently regulates the longitudinal growth of eukaryotic cells.3 Thus, the helicase activity of the MCM2-7 hexameric assembly regulates the DNA replication during cell cycle stages.4,5 During the G1 phase of the cell cycle, a dimerized MCM7 complex latches onto the origin of replication of the double-stranded DNA (dsDNA).6 In normal cells, the CMG (Cdc45-Mcm2-7-GINS) complex, as depicted in Fig. 1, blocks the hexamer formation before initiation of the S phase, to stop further DNA replication.7 In cancer cells, however, MCM7 phosphorylation mediated by tyrosine kinase Lyn (p53 and p56 Lyn) spliced isoforms promote the MCM complex formation and subsequent chromatin loading, leading to unrestricted DNA replication in mutated cancer cells.8 An overabundance of the MCM7 protein has been reported to be a prognostic factor for lung, breast, colorectal, and ovarian cancer and a reliable diagnostic marker for cervical cancer and ovarian cancer.9–11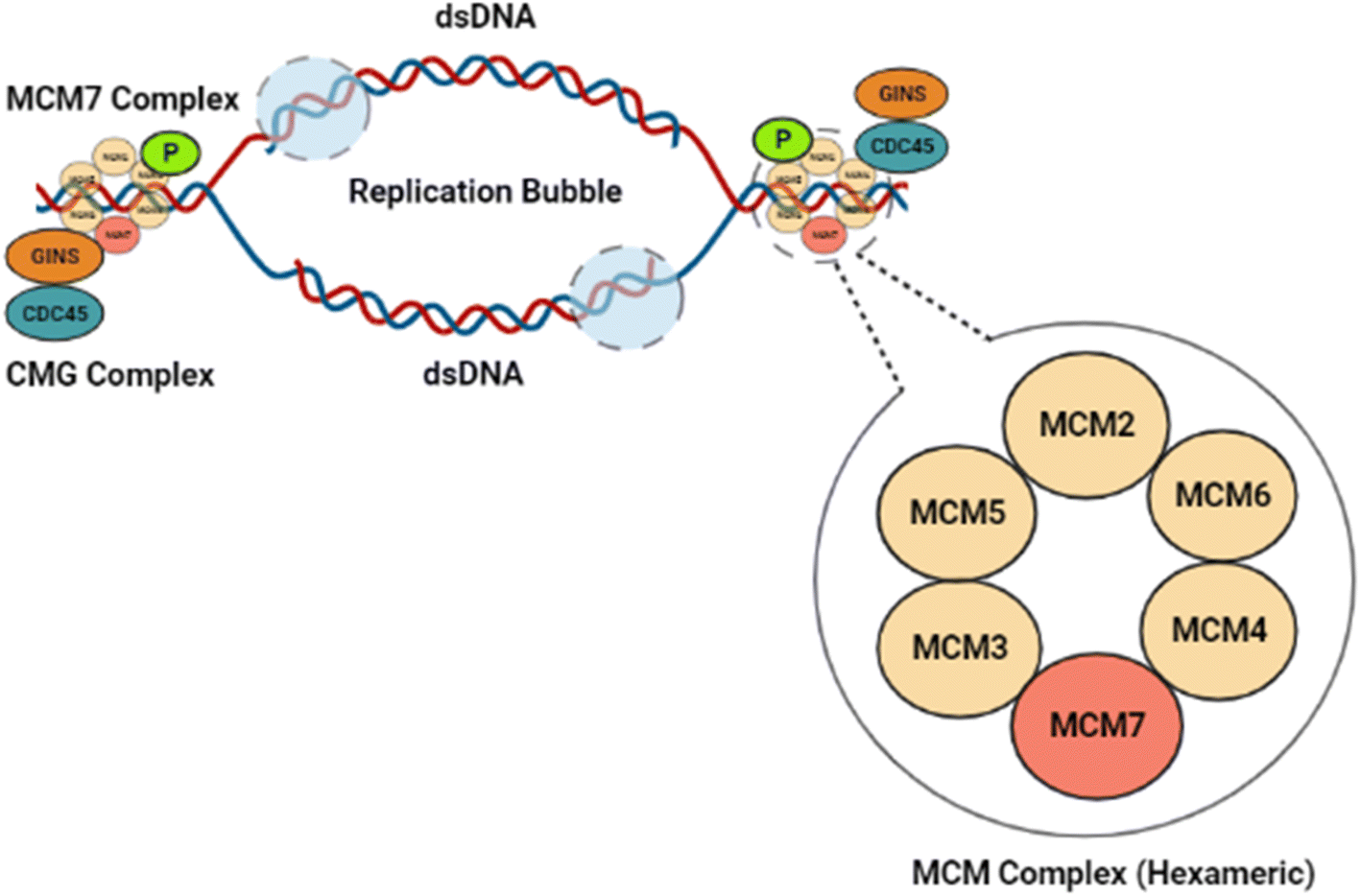 | ||
| Fig. 1 Illustration of the role of MCM7 in DNA replication. MCM7 forms a hexameric ring with the other MCM complex members that bind to DNA and assist the replication pathway. GINS and CDC45 collectively form the CMG complex that helps to catalyze the helicase activity of the complex. The origin of replication forks in the double-stranded DNA at origin, and synthesis by DNA polymerases occurs upon DNA unwinding. | ||
Cancer originating from MCM7 overexpression necessitates discovering appropriate control measures.12 In this direction, few selected small-molecule antagonists have been studied in vitro against MCM7.9,13–15 Till now, only heliquinomycin (antibiotic), ciprofloxacin (antibiotic), and simvastatin (anti-cholesterol drug) have shown positive responses against cancer cells in vitro, indicating their therapeutic potential as MCM7 inhibitors.9,13–15 Heliquinomycin has been shown to inhibit the DNA helicase activity of MCM 4–6–7 complex at an IC50 value of 2.4 μm.13 Ciprofloxacin showed inhibitory activity towards MCM 2–7 complex with an IC50 value of 632 μm, while simvastatin has demonstrated an inhibitory activity towards MCM7 at 5 μm and inhibited cell proliferation in Hep3B cells by inducing endoplasmic reticulum stress and activating autophagy signaling pathway.14,15 Even though these few studies have shown promising results in vitro, exploring potential MCM7 inhibitors with high specificity is necessary.
Further, the current cancer therapies are non-specific, with frequent resistance to chemotherapeutic drugs and recurrence of cancer. Such impediment to cytotoxic chemotherapy with adverse clinical side effects warrants the development of novel chemotherapeutic agents.16 The recent developments in the process of drug discovery have uncovered the potential of many non-cytotoxic drugs in targeted cancer therapy.17 Even though phytochemicals as lead compounds for chemotherapy have gained widespread interest in the scientific community, significant challenges still exist. It lies in identifying, extracting, and characterizing natural phytochemicals, determining their potential target sites, and formulating effective workflows for evaluating drug potency, toxicity, and pharmacokinetic properties (PK).18 In experiments involving de novo drug design or phytochemicals, the lead compounds from virtual screening undergo ADMET (absorption, distribution, metabolism, excretion, toxicity) trials to assess their potential for adverse clinical side effects. In the case of drug repurposing using FDA-approved commercial drugs, no additional pharmacokinetics or toxicity testing is required.19 This exponentially reduces the timeline for applying these drugs to treat new diseases, with the most contemporary example being remdesivir (a drug developed for the Ebola virus) being employed to treat COVID-19.20,21
The recent advances in drug designing methods enable efficient compound screening, identify probable target sites, and repurpose drugs against a specific molecule.22,23 The application of molecular docking in virtual screening platforms aided by molecular dynamics (MD) simulations enables identifying new leads with reasonable accuracy against a particular target.24,25 A previous study by our research team discovered the role of MCM7 in promoting cancer using gene expression analysis and protein–protein interaction data.26 Therefore, the current study aims to further elucidate the investigation by decoding the complete tertiary structure of the MCM7 protein and identifying promising FDA-approved pharmaceutical drugs that can inhibit MCM7 for effective cancer therapy.
2. Materials and methods
2.1 Protein structure preparation, analysis, and validation
The amino acid sequence of MCM7 was obtained from UniProt (Uniprot ID: P33993). The secondary structure of the sequence was analyzed using the SOPMA webserver (https://www.npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). The 3D model of MCM7 was predicted from the sequence utilizing homology modeling and threading methodology using the SWISS-MODEL (https://www.swissmodel.expasy.org/)27 and I-TASSER molecular threading web server (https://www.zhanggroup.org/I-TASSER/).28,29 The 3D structure was validated with PROCHECK on the SAVES v6.0 server (https://www.saves.mbi.ucla.edu/).30,31 The pre-processing of the predicted model for molecular docking was performed in AutoDock MGLTools 1.5.6.322.2 Ligand retrieval and preparation
To search and retrieve structures of clinically approved FDA drugs common in use without side effects, the two largest known repositories for chemical structures of pharmaceutical ligands, viz. PubChem (https://www.pubchem.ncbi.nlm.nih.gov/) and DrugBank (https://www.go.drugbank.com/) were screened. Then atom types and atomic charges were assigned after merging non-polar hydrogen. The aromatic carbons were detected, and then the ‘torsion tree’ was set in AutoDock MGLTools 1.5.6.2.3 Active site identification and receptor grid generation
The identification of active site (AS) and generation of receptor grid coordinates for MCM7 was performed using the BIOVIA Discovery Studio Visualizer v21.1.0.20298 (DSV).332.4 Molecular docking
The molecular docking for each ligand was performed with the help of the Lamarckian Genetic Algorithm (LGA) in AutoDock Vina v1.2 (ref. 32 and 33) with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 energy evaluations for twenty runs. Further, the protein–ligands complex's binding interactions were analyzed using BIOVIA DSV.
000 energy evaluations for twenty runs. Further, the protein–ligands complex's binding interactions were analyzed using BIOVIA DSV.
2.5 Molecular dynamics simulations of the MCM7-ligand complexes
Conformational dynamics of the free and ligand-bound MCM7 complexes were studied using the GROMACS 2018 simulation package.34 AMBER99SB-ILDN forcefield in combination with the TIP3P water model was used. We considered five different systems: MCM7, MCM7-cholecalciferol, MCM7-ergosterol, MCM7-ergocalciferol, and MCM7-menaquinone. Parameters for all ligands were generated according to the general Amber force field (GAFF), and atomic charges for ligands were computed with the help of the AM1-BCC method using ANTECHAMBER tools. Parameters for ligands compatible with GROMACS were then generated using the ACPYPE program. Each system was solvated in a triclinic box such that at least 10 Å was the minimum distance maintained between box walls and any protein atom. The initial box dimensions of all the systems were 93.4 × 145.8 × 108 Å3. Charges in the simulated system were neutralized by the addition of six Na+ ions. The solvated systems were subjected to 500 steps of energy minimization using the steepest descent algorithm. The energy minimized systems were subjected to position restrained simulation for 1 ns in the NVT ensemble. The protein backbone atoms were restrained, but solvent molecules could move freely. Each system was equilibrated initially for 2 ns in the NVT ensemble at 298 K, followed by another 2 ns simulation in the NPT ensemble (298 K, 1 bar). The final production runs were carried out for 300 ns in the NPT ensemble. A Nose–Hoover thermostat with a coupling time constant of 0.1 ps. was used to keep the temperature fixed at 298 K. Parrinello–Rahman isotropic barostat with a coupling time constant of 2 ps was used to maintain a constant pressure of 1 bar. The particle mesh Ewald summation method was used to calculate the electrostatic interactions.The trajectories were stored at every 20 ps. The trajectories were analyzed with the trajectory analysis tools implemented in GROMACS. Principal component analysis (PCA) was carried out on a 300 ns trajectory for each system. The Cα atomic positional fluctuations retrieved from the trajectory were used to generate a mass-weighted covariance matrix. Then the essential subspace was analyzed in terms of the calculated eigenvectors. The present study's overall experimental procedure and methodology have been elucidated in a concise flowchart, as shown in Fig. S1.†
3. Result and discussions
3.1 Protein structure preparation, analysis, and validation
Secondary structure prediction is an essential step in the accurate prediction of the tertiary structure of a protein which is required for molecular docking.35 The amino acid sequence of MCM7 was extracted from UniProt in FASTA format (UniProt ID: P33993). The secondary structure of MCM7 protein was analyzed by SOPMA (Fig. S2†). The secondary structure of MCM7 is primarily helical, with 49.65% α-helix followed by random coils (32.27%) and extended strands (13.77%). It also showed the presence of 4.31% of beta-turns.The prediction of the 3D model for MCM7 started with a sequence similarity search using NCBI protein BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi),36 keeping PDB (Protein Data Bank) as the template database.37 Based on a 100% sequence identity score for the input query sequence, the PDB crystal structure 6XTX was selected for further evaluation38 (Fig. 2A). The crystal structure is of human MCM7 protein as Chain F in conjunction with the hexameric MCM–CMG complex. Therefore, chain F from the complex was isolated from the rest of the complex using BIOVIA DSV (Fig. 2B). The MCM7 protein structure contains 4 large missing sections, which were modelled using the SWISS-MODEL and the I-TASSER molecular threading webserver (Fig. 2C).27,29 The final structure thus retrieved was further processed in AutoDock MGL Tools 1.5.6 to remove water molecules and add polar hydrogen groups for molecular docking.
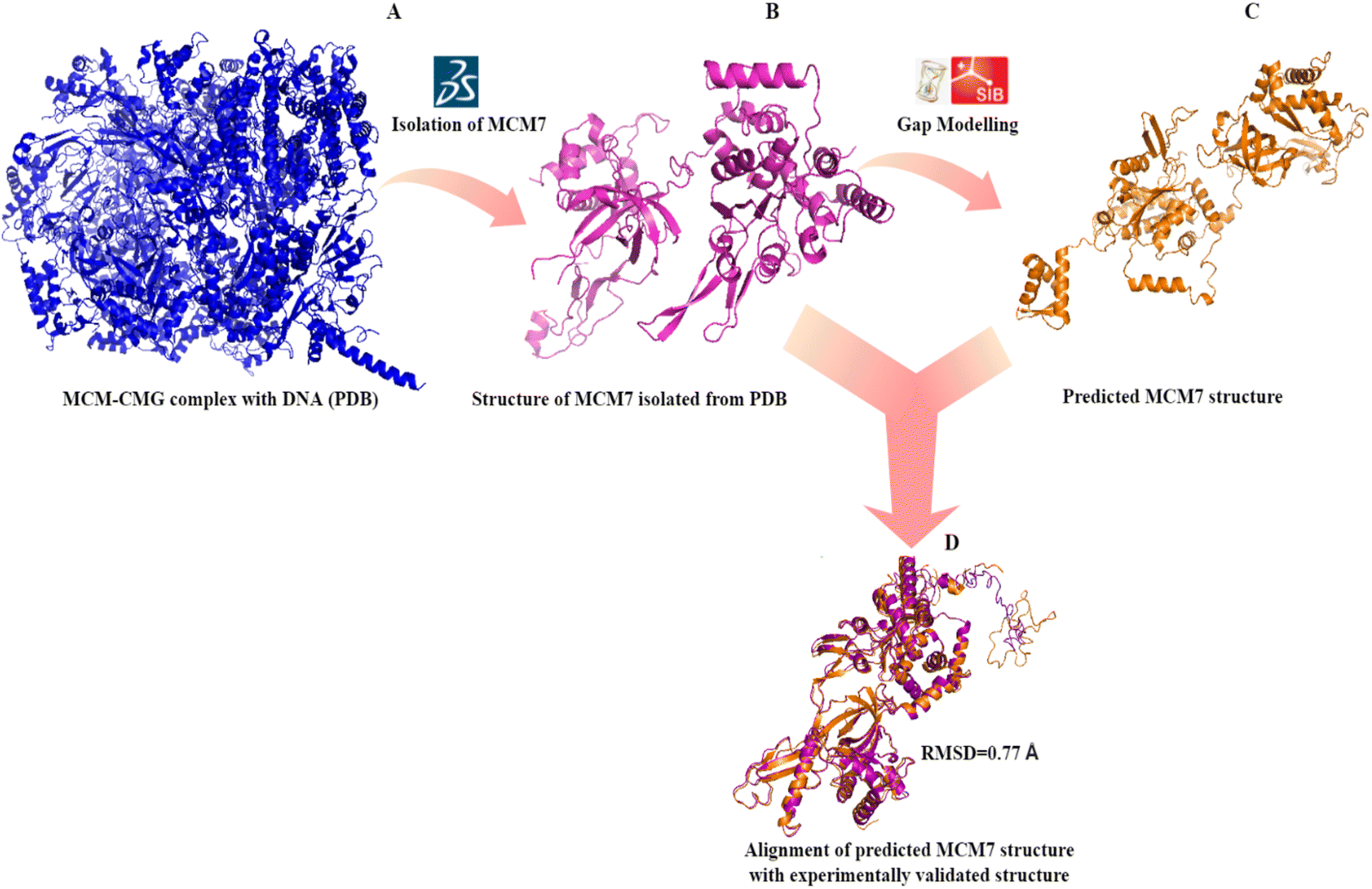 | ||
| Fig. 2 3D structure prediction of MCM7 (A) crystal structure of MCM–CMG complex (6XTX) retrieved from PDB; (B) MCM7 chain isolated from the PDB complex (6XTX: chain F); (C) complete structure prediction by I-TASSER after gap correction for unmodelled residues; (D) validation of the predicted structure by superimposition on 6XTX: chain F. | ||
The validation of the predicted MCM7 3D structure ensured the correctness of the predicted model. The 3D model showed no error that could lead to steric hindrance. The validation of the predicted MCM7 model is first performed by the superposition or alignment of the predicted model with the experimentally resolved structure obtained from the PDB, depicted in Fig. 2D. The resulting alignment or RMSD (Root Mean Square Deviation) score was 0.77 Å, significantly less than the 2 Å threshold value. The low RMSD score indicates the close resemblance of MCM7 predicted complete structure with its experimentally validated structure with the missing region.
The revalidation of the predicted model by analyzing its Ramachandran plot using PROCHECK on the SAVES V6.0 server revealed that 82% of the amino acid residues are present in the most favorable regions, 15% in the allowed regions, 1.9% in the generously allowed regions, and 1.1% in the disallowed regions. Therefore, out of a total of 719 residues, 711 residues (98.9%) were found present in the allowed regions, while only 8 residues (1.1%) were located within the disallowed regions (Fig. S3†). None of these residues present in the disallowed regions are in the modelled loop regions or in the active sites of MCM7. Thus, it cannot impart any severe error during the molecular docking process. These results confirmed the accuracy of the predicted 3D model, and thus it was selected for the molecular docking procedure. The final structure was uploaded to the PMDB database39 with the accession number PM0084198.
3.2 Ligand retrieval and preparation
PubChem and DrugBank were screened to curate a ligand library of FDA-approved drugs with minimal side effects consisting of 1100 pharmaceutical ligands. These compounds were retrieved in a 3D (.sdf) file format and converted into the pdbqt file format for further molecular docking.3.3 AS identification and receptor grid generation
The active site of an enzyme is constituted from a diverse portfolio of amino acids located within a specific region, known as the binding site, that establishes non-covalent interactions with the substrate. It allows ligand binding and promotes catalysis of biochemical reactions. The present study identified the active site of MCM7 and retrieved the coordinates of the binding site. The results revealed CYS184, CYS187, CYS206, CYS211, GLU343, ILE344, TYR345, MET369, PRO383, GLY384, VAL385, ALA386, LYS387, SER388, GLN389, ASN489, ARG514, LEU533, ARG604, LEU607 as the residues present in the active site of MCM7 that frames the ligand binding cavity (Fig. 3).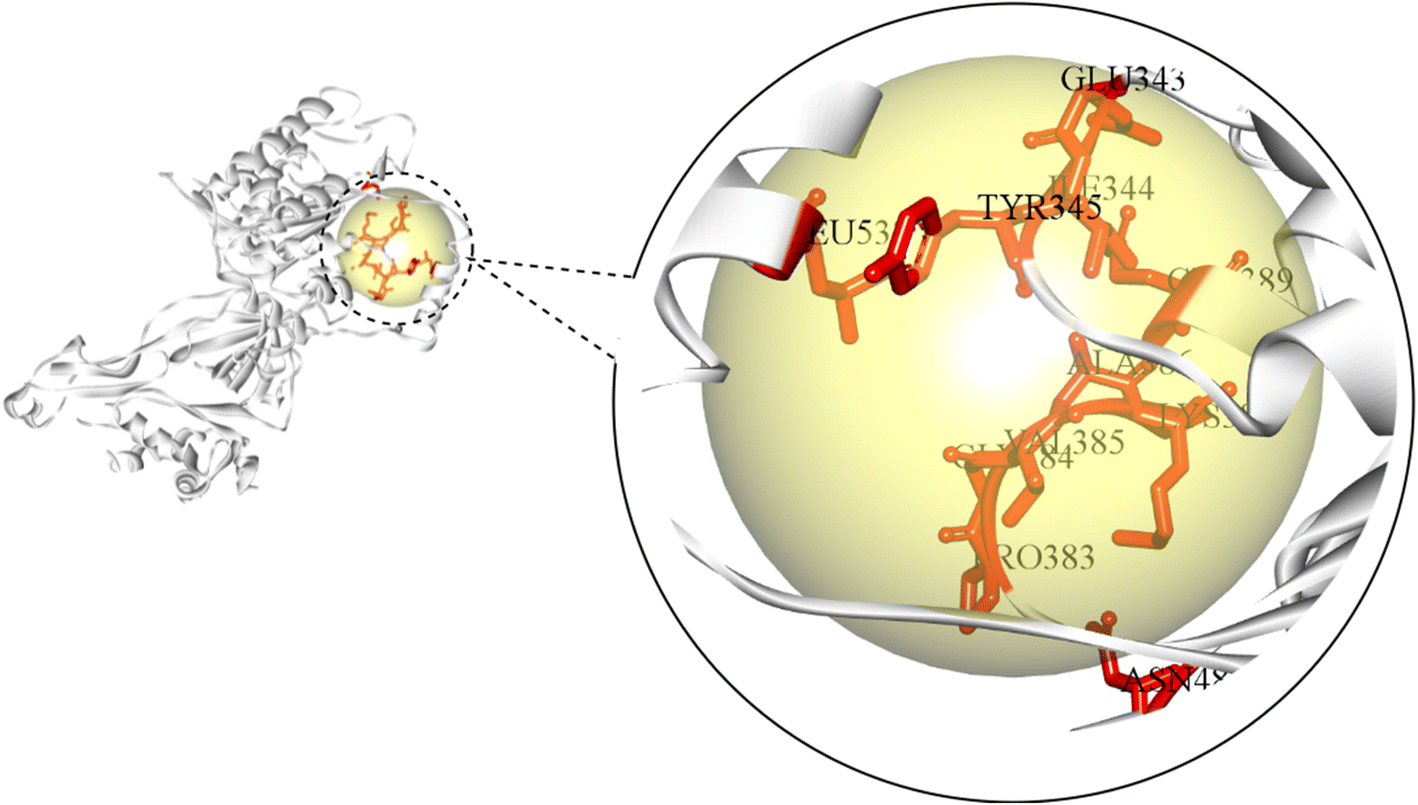 | ||
| Fig. 3 Identification of the amino acid residues constituting the active site of MCM7. | ||
A receptor grid of MCM7 was generated based on the spatial coordinates of its binding site residues. The molecular docking grid box was set to the dimensions of 60 Å × 80 Å × 60 Å with a spacing of 0.375 Å such that it covers the entire active site.
3.4 Molecular docking
AutoDock Vina was utilized for carrying out molecular docking between the selected ligands (1100) and the MCM7 binding site. The binding affinities obtained after molecular docking of these ligands were observed to be in the range of −3.4 to −9.2 kcal mol−1.After a knowledge-based screening procedure, the top four compounds were selected, viz. ergocalciferol (vitamin D2), cholecalciferol (vitamin D3), ergosterol (precursor of vitamin D2), and menaquinone (vitamin K2). These compounds satisfied (a) the criteria of having a binding affinity better than for a designated control drug; (b) and the absence of any reported lethal clinical side effects. The designated control drug was ciprofloxacin for this screening protocol due to its inhibitory activity against MCM7 previously reported in vitro.14 During the molecular docking, ciprofloxacin showed a binding affinity of −7.6 kcal mol−1. Therefore, any lead compounds needed to have a binding affinity better than −7.6 kcal mol−1 and no lethal side effects. The selected compounds and their binding affinity to MCM7 are shown in Table 1.
| Compound ID | Chemical name (common name) | 2D conformer | 3D conformer | Binding affinity (kcal mol−1) |
|---|---|---|---|---|
| PubChem CID: 5280793 | Ergocalciferol (vitamin D2) | 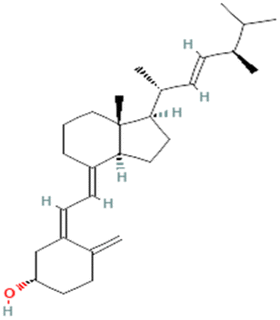 |
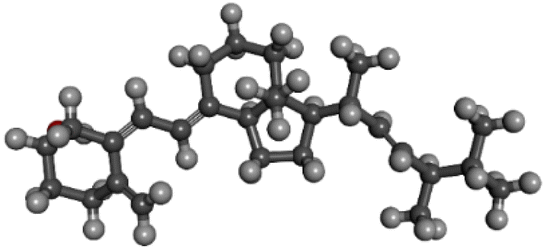 |
−9.2 |
| PubChem CID: 5280795 | Cholecalciferol (vitamin D3) | 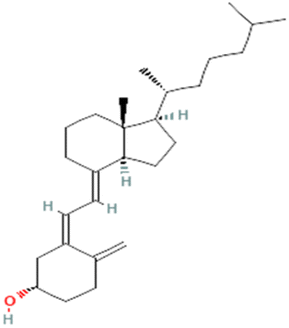 |
 |
−8.5 |
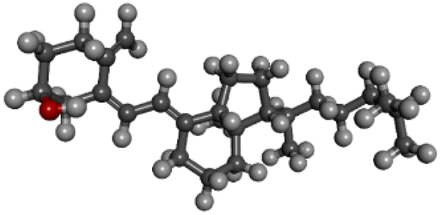
| PubChem CID: 444679 | Ergosterol (pre-vitamin D2) | 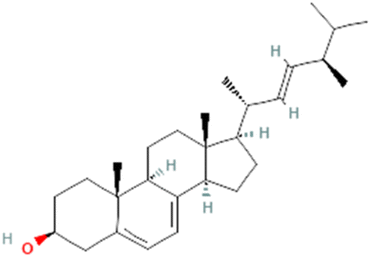 |
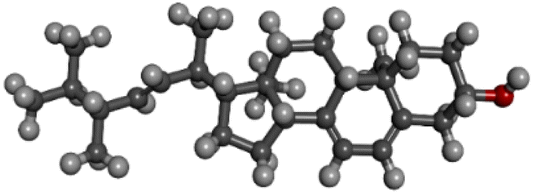 |
−8.3 |
| PubChem CID: 5287554 | Menaquinone (vitamin K2) | 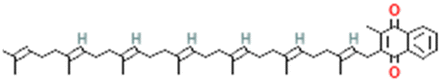 |
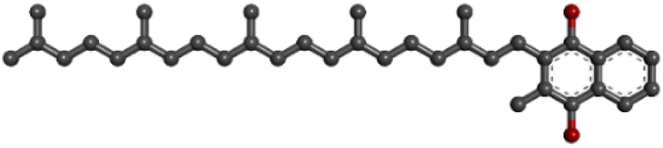 |
−8.0 |
| PubChem CID: 2764 | Ciprofloxacin (control) | 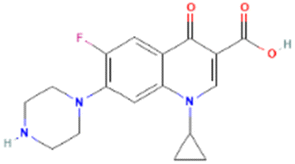 |
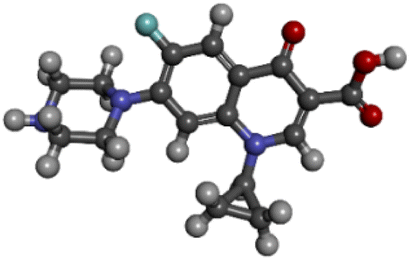 |
−7.6 |
The preliminary results from the molecular docking study direct us to an essential discovery of the potential of vitamins D (specifically D2 and D3) and K2 as oncoprotein inhibitors for cancer therapy. Although previous studies have reported the anti-proliferative action of vitamin D analogs and vitamin K2, either independently or as an adjuvant, on cancer cell lines in vitro,40–43 the molecular mechanism for the same remains unexplained.44–47 The potential target (MCM7) of vitamins to exhibit anti-cancer activity is first reported here. Thus, the findings from the present study may provide valuable insights into the role of vitamins as potential anti-cancer agents.
The analysis of protein–ligand interaction of the selected four ligands with the MCM7 was performed with BIOVIA DSV, as depicted in Fig. 4 and 5 respectively. Ergocalciferol was found to form a couple of conventional hydrogen bonds with MCM7 protein with Lys351 (6.24 Å) and Ile344 (2.83 Å). An alkyl–alkyl interaction was observed with Tyr345 (5.06 Å). Two π–alkyl interactions were also observed with Tyr539 (4.12 Å) and Pro548 (5.04 Å) (Fig. 5A and Table 2). For cholecalciferol, two conventional hydrogen bonds were observed with Lys351 (6.28 Å) and Ile344 (2.67 Å); two alkyl–alkyl interactions with Tyr539 (4.10 Å, 4.48 Å); and two pi–alkyl interactions with Pro548 (4.64 Å, 4.73 Å) were discovered (Fig. 5B and Table 2). Ergosterol formed a conventional hydrogen bond with Glu334 (2.39 Å) and three pi–alkyl interactions with Ala338 (3.87 Å), Tyr345 (4.94 Å), and Arg532 (5.32 Å) of MCM7 (Fig. 5C and Table 2). Finally, for menaquinone, three alkyl–alkyl interactions were detected with Ala338 (4.52 Å), Pro547 (5.03 Å), and Pro548 (5.05 Å), along with two π–σ interactions with Phe551 (4.07 Å, 3.69 Å) (Fig. 5D and Table 2).
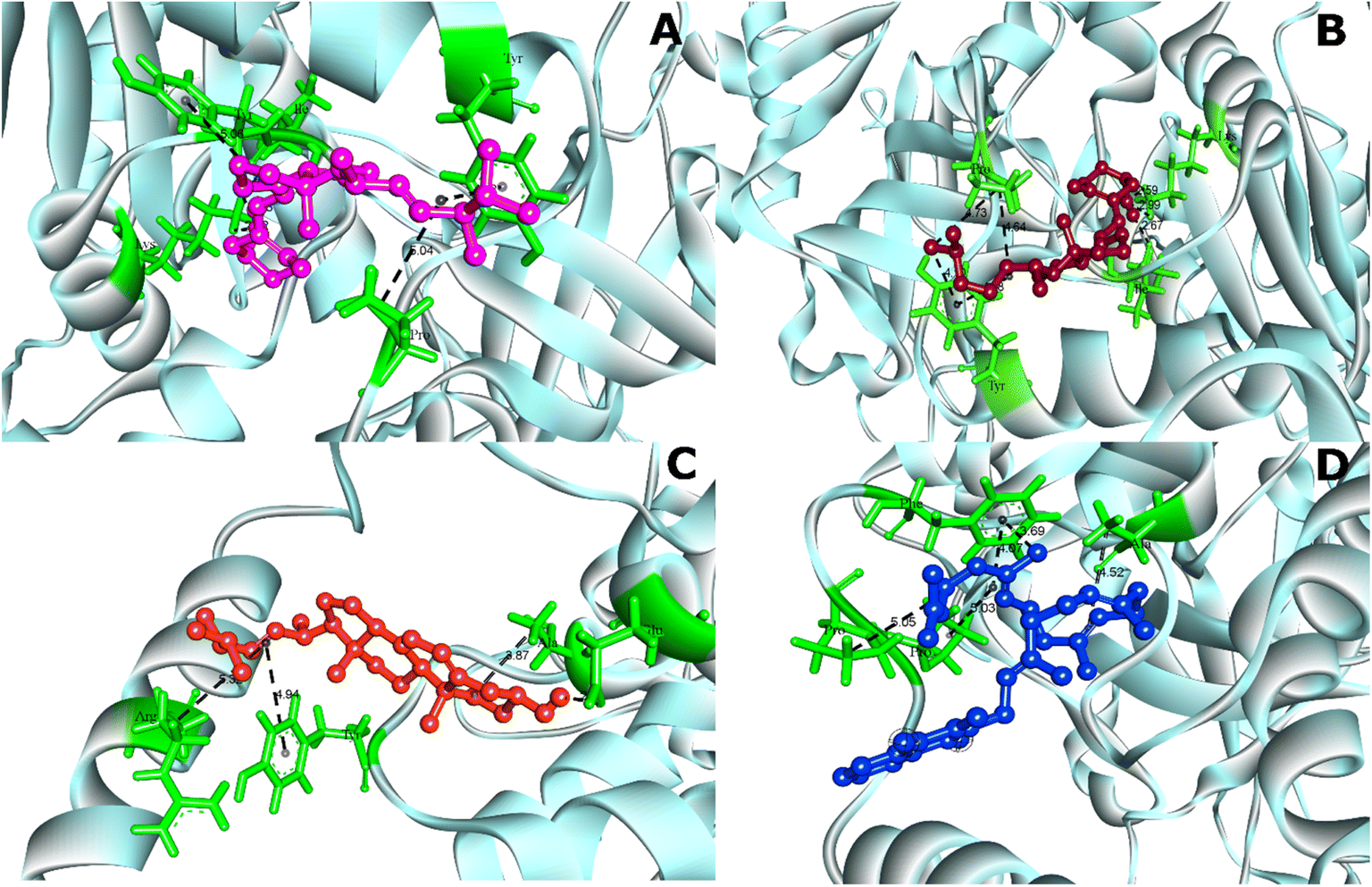 | ||
| Fig. 4 3D interaction diagrams for MCM7-ligand docked complexes. (A) MCM7-ergocalciferol; (B) MCM7-cholecalciferol; (C) MCM7-ergosterol; (D) MCM7-menaquinone. | ||
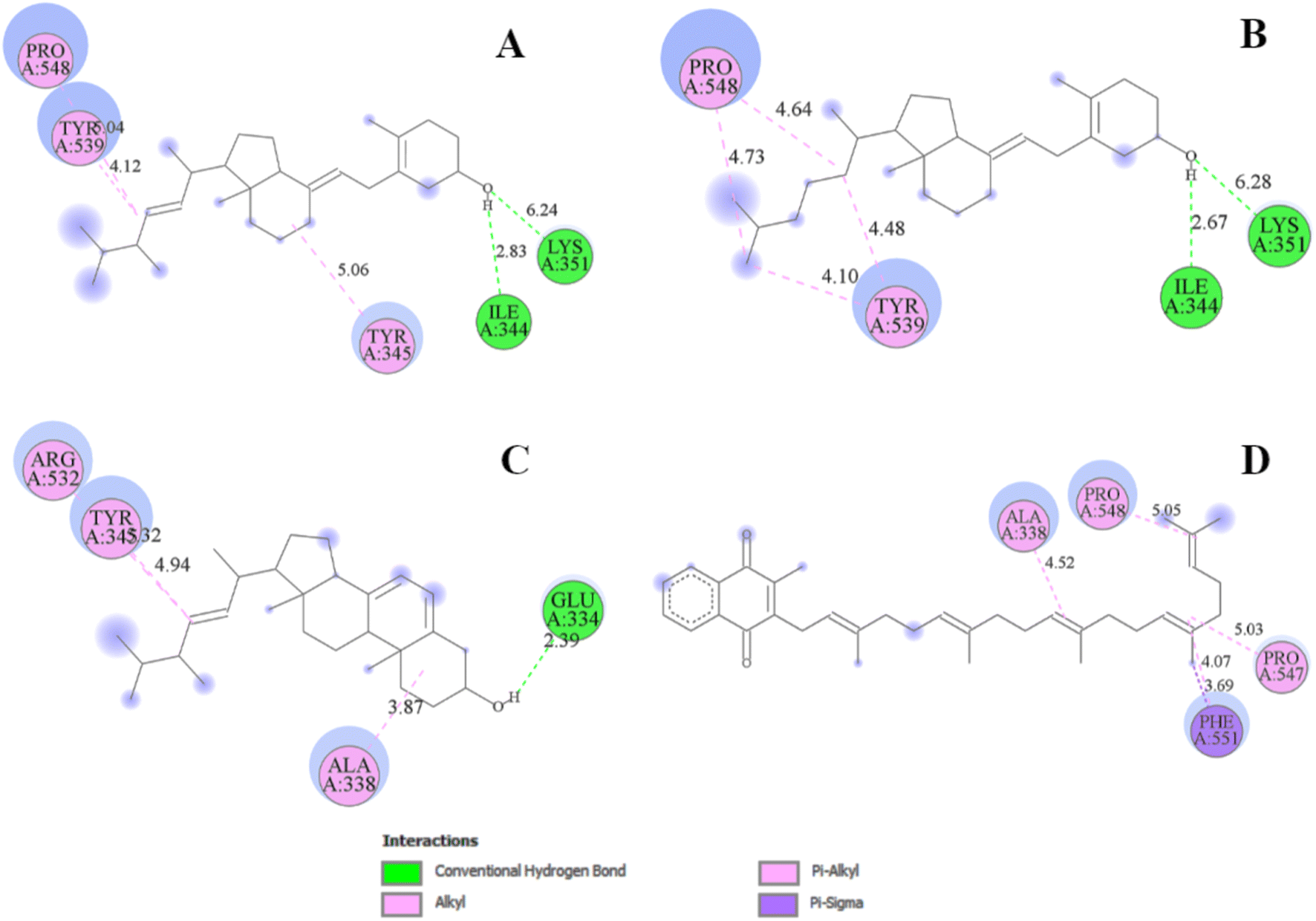 | ||
| Fig. 5 2D interaction diagrams for the protein–ligand docked complexes: (A) MCM7-ergocalciferol; (B) MCM7-cholecalciferol; (C) MCM7-ergosterol; (D) MCM7-menaquinone. | ||
| Compound name | Residues | Distance (Å) | Bond category | Bond type |
|---|---|---|---|---|
| Ergocalciferol | Ile 344 | 2.83 | Hydrogen | Conventional hydrogen bond |
| Tyr 345 | 5.06 | Alkyl | Alkyl | |
| Lys 351 | 6.24 | Hydrogen | Conventional hydrogen bond | |
| Tyr 539 | 4.12 | Pi–alkyl | Pi–alkyl | |
| Pro 548 | 5.04 | Pi–alkyl | Pi–alkyl | |
| Cholecalciferol | Ile 344 | 2.67 | Hydrogen | Conventional hydrogen bond |
| Lys 351 | 6.28 | Hydrogen | Conventional hydrogen bond | |
| Tyr 539 | 4.10, 4.48 | Alkyl | Alkyl | |
| Pro 548 | 4.64, 4.73 | Pi–alkyl | Pi–alkyl | |
| Ergosterol | Glu 334 | 2.39 | Hydrogen | Conventional hydrogen bond |
| Ala 338 | 3.87 | Alkyl | Alkyl | |
| Tyr 345 | 4.94 | Pi–alkyl | Pi–alkyl | |
| Arg 532 | 5.32 | Pi–alkyl | Pi–alkyl | |
| Menaquinone | Ala 338 | 4.52 | Alkyl | Alkyl |
| Pro 547 | 5.03 | Alkyl | Alkyl | |
| Pro 548 | 5.05 | Alkyl | Alkyl | |
| Phe 551 | 4.07, 3.69 | Pi–sigma | Pi–sigma |
3.5 MD simulations of MCM7–ligand complexes
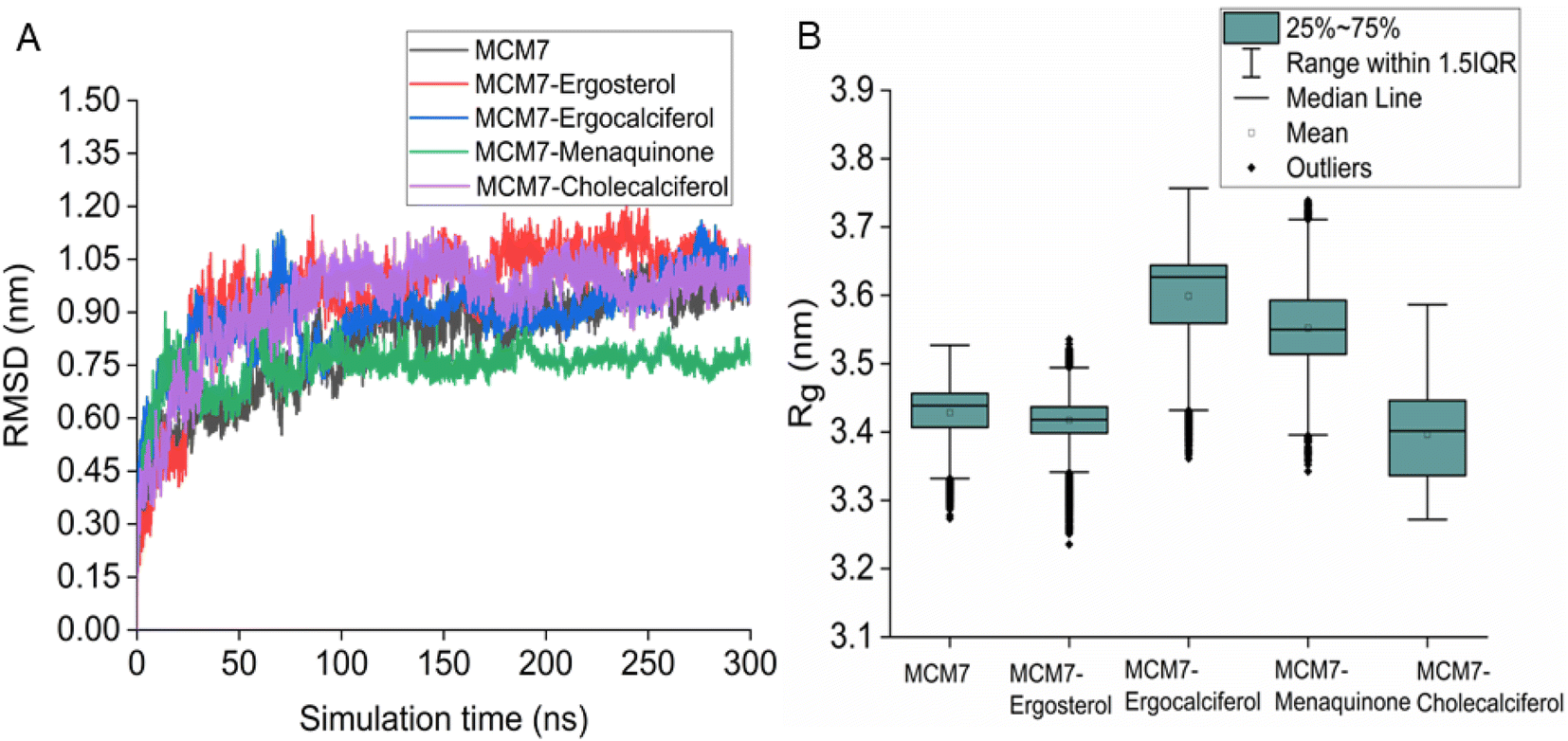 | ||
| Fig. 6 (A) Cα RMSD variation of protein and different protein–ligand complexes over the entire simulation timescale. (B) The radius of gyration of protein and different protein–ligand complexes over the entire simulation timescale. | ||
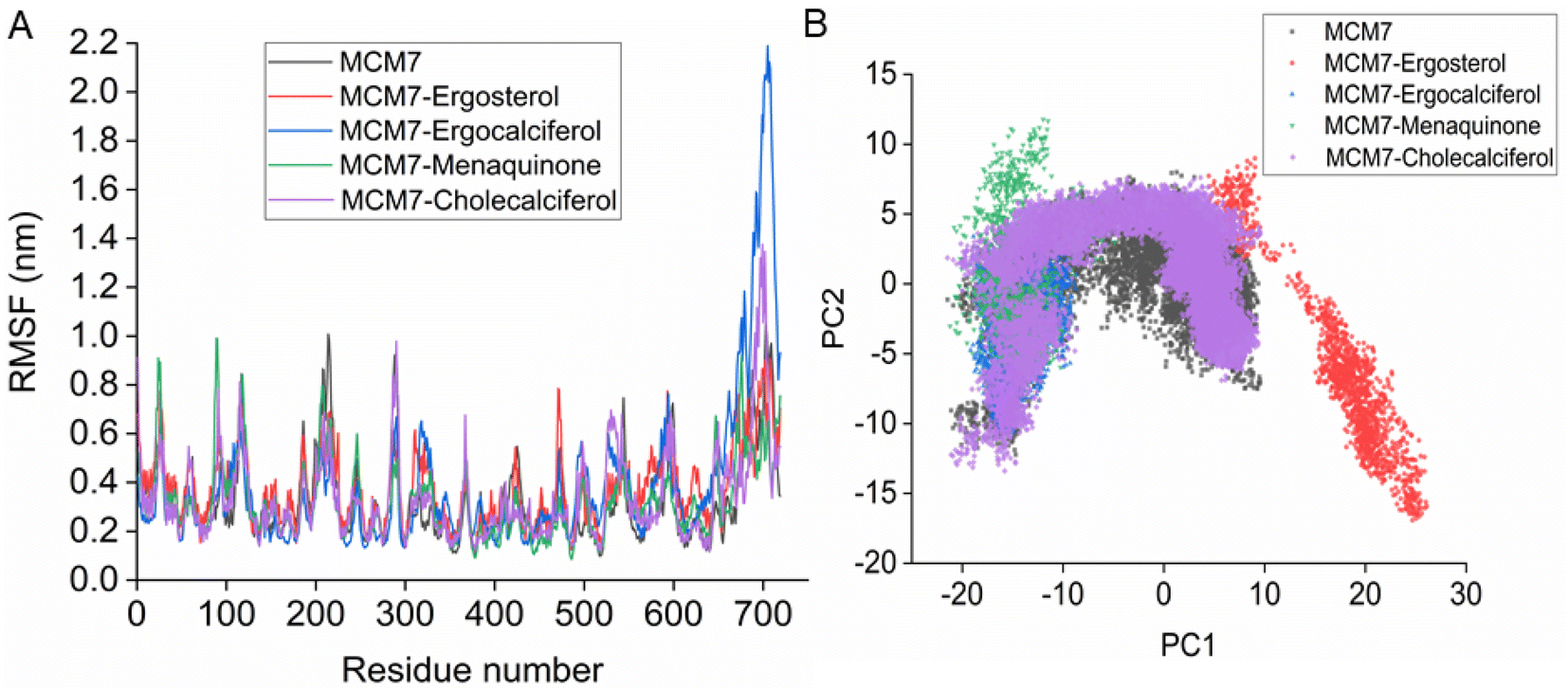 | ||
| Fig. 7 (A) RMSF plot for the protein and protein–ligand complexes derived from the MD simulation. (B) 2-D essential subspace constructed by the first two principal components (PC1 & PC2) for the protein and protein–ligand complexes obtained using results from the MD simulation. | ||
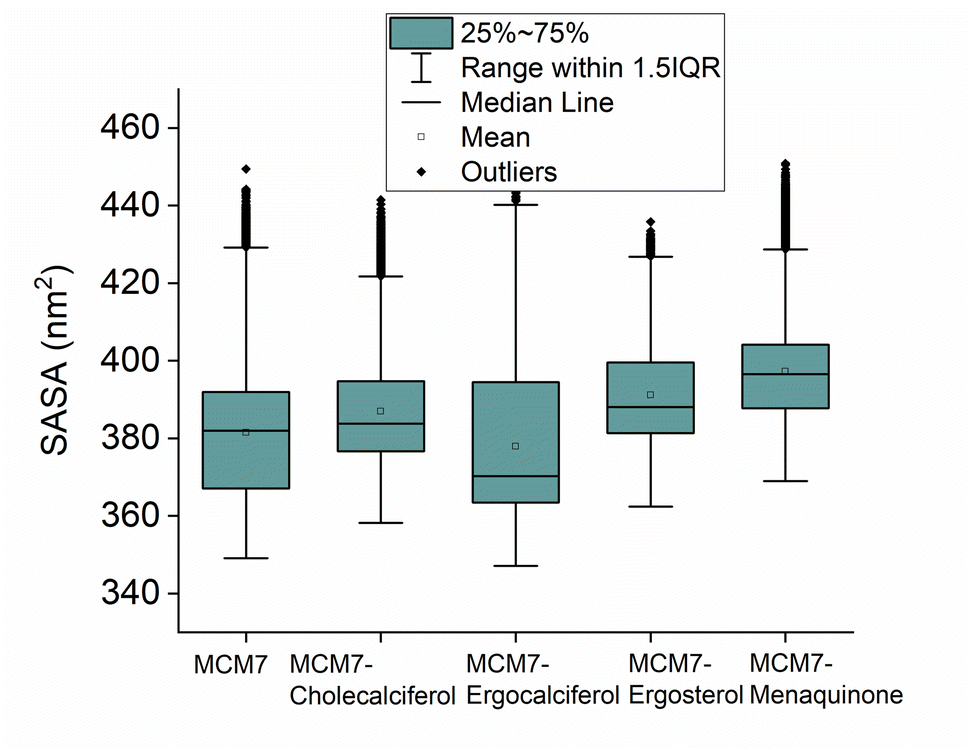 | ||
| Fig. 8 The solvent accessible surface area (SASA) distribution of the protein and different protein–ligand complexes obtained from the simulation is shown. | ||
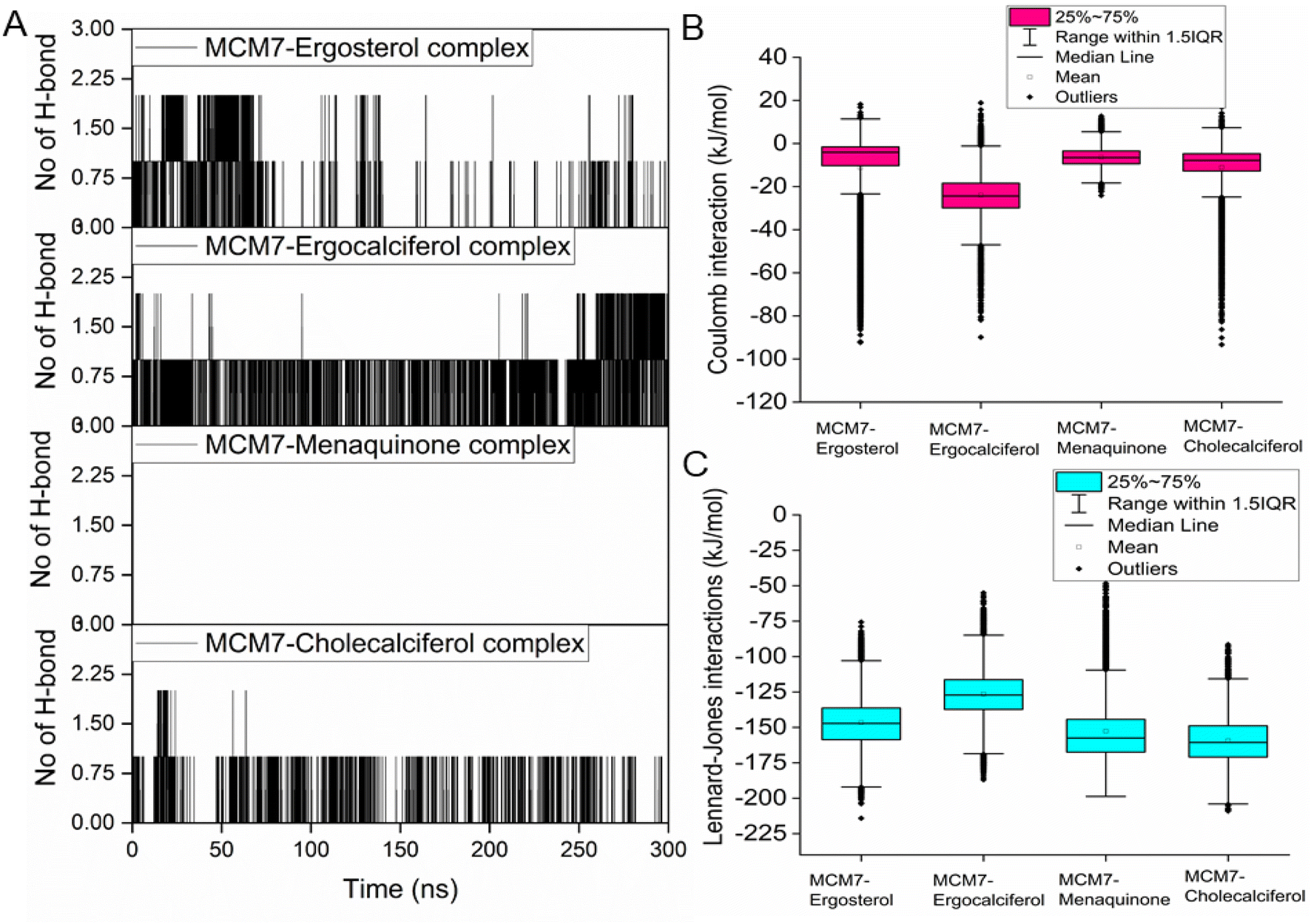 | ||
| Fig. 9 (A) Hydrogen bond interaction patterns for each of the four MCM7-ligand complexes observed from the molecular dynamics simulations. Variations of coulomb (B) and LJ (C) interaction energies between protein and each of the ligands for different MCM7–ligand complexes were obtained from the simulations. | ||
Interestingly, most of the weak interactions, π–alkyl and alkyl–alkyl, observed in all the ligand-MCM7 docked complexes are stable throughout the simulation trajectories. The average distance between the ligand atom and the side chain of the protein residue involved in each of the observed π–alkyl and alkyl–alkyl interactions obtained from the MD simulation is tabulated in Table S1 (ESI†).
4. Conclusions
Overexpression of the MCM7 gene responsible for cancer progression can be restricted with chemotherapy targeting to inhibit its action. The presented study utilized molecular docking and MD simulation to identify four compounds (vitamins) as potential drug candidates: ergocalciferol, ergosterol, cholecalciferol, and menaquinone, which can inhibit MCM7 activity. A detailed analysis has been performed to delineate the mechanism of action of the identified hits. Ergocalciferol binds with MCM7 with the highest affinity and the MCM7-ergocalciferol complex is stabilized by electrostatic interactions. The hydrogen bonding interactions are the most stable among all the four studied compounds, evident from the simulation. RMSD, Rg and SASA calculation shows that the binding of ergocalciferol induces structural alteration of the protein, MCM7. Cholecalciferol and ergosterol bind with MCM7 with comparable affinities. Both of them share a very similar moderately stable hydrogen bonding profile. Their interaction energy profiles are also very similar. The binding of both the compounds induces structural fluctuations in the MCM7, evident from the high RMSD values. RMSF indicates binding of these two compounds induces fluctuations in several loop regions. However, the overall shape and size of the protein remain unaltered, evident from similar Rg and SASA profiles of these two ligand-bound complexes and free protein obtained from MD simulation. Menaquinone, on the other hand, shows the least binding affinity among the identified hits which is accounted for its inability to form hydrogen bonds. Its binding is primarily contributed by van der Waals interactions. It forms several weak alkyl–alkyl interactions with the protein which are highly stable during the entire simulation timescale. Rg and SASA show that the binding of menaquinone to MCM7 altered the structural organization of the protein which leads to a change in functional motion of MCM7 (evident from the PCA analysis) which could lead to an altered MCM7 activity. In vitro and in vivo studies can be performed in the near future to investigate the chemotherapeutic efficacy of the selected drugs on MCM7-associated cancer progression.Author contributions
Sunny Mukherjee and Mahesh Kumar Sah designed the concept for the present study. The data analysis of molecular docking results was undertaken by Sunny Mukherjee, Navneet Sriram and Mahesh Kumar Sah, while Sucharita Das and Sandipan Chakraborty performed molecular dynamics simulations. All the authors collaboratively wrote the first draft of the manuscript, contributed to manuscript revision and finalized it before submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank MHRD (Ministry of Human Resource Development), Government of India, for the financial assistance to first author.Notes and references
- A. Samad, et al., Computational assessment of MCM2 transcriptional expression and identification of the prognostic biomarker for human breast cancer, Heliyon, 2020, 6, e05087 CrossRef.
- C. A. Nieduszynski, J. J. Blow and A. D. Donaldson, The requirement of yeast replication origins for pre-replication complex proteins is modulated by transcription, Nucleic Acids Res., 2005, 33, 2410–2420 CrossRef.
- M. A. Mou, et al., Validation of CSN1S1 transcriptional expression, promoter methylation, and prognostic power in breast cancer using independent datasets, Biochem. Biophys. Rep., 2020, 24, 100867 Search PubMed.
- H. Nishitani and Z. Lygerou, Control of DNA replication licensing in a cell cycle, Genes Cells, 2002, 7, 523–534 CrossRef.
- F. Rahman, et al., A multi-omics approach to reveal the key evidence of GDF10 as a novel therapeutic biomarker for breast cancer, Inform. Med. Unlocked, 2020, 21, 100463 CrossRef.
- S. Tognetti, A. Riera and C. Speck, Switch on the engine: how the eukaryotic replicative helicase MCM2–7 becomes activated, Chromosoma, 2015, 124, 13–26 CrossRef.
- X. Xu, J.-T. Wang, M. Li and Y. Liu, TIMELESS Suppresses the Accumulation of Aberrant CDC45·MCM2-7·GINS Replicative Helicase Complexes on Human Chromatin, J. Biol. Chem., 2016, 291, 22544–22558 CrossRef PubMed.
- T.-H. Huang, et al., Epidermal growth factor receptor potentiates MCM7-mediated DNA replication through tyrosine phosphorylation of Lyn kinase in human cancers, Cancer Cell, 2013, 23, 796–810 CrossRef.
- Z. Liang, et al., Simvastatin suppresses the DNA replication licensing factor MCM7 and inhibits the growth of tamoxifen-resistant breast cancer cells, Sci. Rep., 2017, 7, 41776 CrossRef PubMed.
- J. H. Rafi, et al., High expression of bone morphogenetic protein 1 (BMP1) is associated with a poor survival rate in human gastric cancer, a dataset approaches, Genomics, 2021, 113, 1141–1154 CrossRef.
- A. Tabassum, et al., Transporter associated with antigen processing 1 (TAP1) expression and prognostic analysis in breast, lung, liver, and ovarian cancer, J. Mol. Med., 2021, 99, 1293–1309 CrossRef.
- K. A. Honeycutt, et al., Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis, Oncogene, 2006, 25, 4027–4032 CrossRef PubMed.
- Y. Ishimi, et al., Effect of heliquinomycin on the activity of human minichromosome maintenance 4/6/7 helicase, FEBS J., 2009, 276, 3382–3391 CrossRef.
- N. Simon, et al., Ciprofloxacin is an inhibitor of the Mcm2-7 replicative helicase, Biosci. Rep., 2013, 33, e00072 CrossRef PubMed.
- G. Toyokawa, et al., Minichromosome Maintenance Protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer, Mol. Cancer, 2011, 10, 65 CrossRef.
- R. B. Mokhtari, et al., Combination therapy in combating cancer, Oncotarget, 2017, 8, 38022–38043 CrossRef PubMed.
- B. Dhakal, et al., The Antianginal Drug Perhexiline Displays Cytotoxicity against Colorectal Cancer Cells In Vitro: A Potential for Drug Repurposing, Cancers, 2022, 14, 1043 CrossRef CAS PubMed.
- J. Fang, C. Liu, Q. Wang, P. Lin and F. Cheng, silico polypharmacology of natural products, Briefings Bioinf., 2018, 19, 1153–1171 CAS.
- Z. Y. Low, I. A. Farouk and S. K. Lal, Drug Repositioning: New Approaches and Future Prospects for Life-Debilitating Diseases and the COVID-19 Pandemic Outbreak, Viruses, 2020, 12, 1058 CrossRef.
- R. T. Eastman, et al., Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19, ACS Cent. Sci., 2020, 6, 672–683 CrossRef PubMed.
- F.-J. Meyer-Almes, Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: Virtual screening and structure based drug design, Comput. Biol. Chem., 2020, 88, 107351 CrossRef PubMed.
- F. Ahammad, T. R. Tengku Abd Rashid, M. Mohamed, S. Tanbin and F. A. Ahmad Fuad, Contemporary Strategies and Current Trends in Designing Antiviral Drugs against Dengue Fever via Targeting Host-Based Approaches, Microorganisms, 2019, 7, 296 CrossRef PubMed.
- A. Raafat, S. Mowafy, S. M. Abouseri, M. A. Fouad and N. A. Farag, Lead generation of cysteine based mesenchymal epithelial transition (c-Met) kinase inhibitors: Using structure-based scaffold hopping, 3D-QSAR pharmacophore modeling, virtual screening, molecular docking, and molecular dynamics simulation, Comput. Biol. Med., 2022, 105526, DOI:10.1016/j.compbiomed.2022.105526.
- S. Mahmud, et al., Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2, Briefings Bioinf., 2021, 22, 1402–1414 CrossRef CAS.
- F. A. D. M. Opo, et al., Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein, Sci. Rep., 2021, 11, 4049 CrossRef CAS.
- N. Sriram, S. Mukherjee and M. K. Sah, Gene expression profiling and protein–protein interaction analysis reveals the dynamic role of MCM7 in Alzheimer's disorder and breast cancer, 3 Biotech, 2022, 12, 146 CrossRef.
- A. Waterhouse, et al., SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS.
- S. Paramashivam and K. N. Dhiraviam, Computational insights into the identification of a potent matrix metalloproteinase inhibitor from Indigofera aspalathoides to control cancer metastasis, 3 Biotech, 2021, 11, 206 CrossRef.
- J. Yang, et al., The I-TASSER Suite: protein structure and function prediction, Nat. Methods, 2015, 12, 7–8 CrossRef CAS PubMed.
- C. Geourjon and G. Deléage, SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments, Bioinformatics, 1995, 11, 681–684 CrossRef CAS PubMed.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, PRO CHECK: a program to check the stereochemical quality of protein structures, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- O. Trott and A. J. Olson, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 Search PubMed.
- M. Manne, et al., Cordifolioside: potent inhibitor against Mpro of SARS-CoV-2 and immunomodulatory through human TGF-β and TNF-α, 3 Biotech, 2021, 11, 136 CrossRef PubMed.
- M. J. Abraham, et al., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- P. Sneha and C. George Priya Doss, Chapter Seven - Molecular Dynamics: New Frontier in Personalized Medicine, in Advances in Protein Chemistry and Structural Biology, ed. R. Donev, Academic Press, 2016, vol. 102, pp. 181–224 Search PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, Basic local alignment search tool, J. Mol. Biol., 1990, 215, 403–410 CrossRef PubMed.
- S. K. Burley, et al., RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences, Nucleic Acids Res., 2021, 49, D437–D451 CrossRef CAS.
- N. J. Rzechorzek, S. W. Hardwick, V. A. Jatikusumo, D. Y. Chirgadze and L. Pellegrini, CryoEM structures of human CMG–ATPγS–DNA and CMG–AND-1 complexes, Nucleic Acids Res., 2020, 48, 6980–6995 CrossRef CAS.
- T. Castrignanò, P. D. De Meo, D. Cozzetto, I. G. Talamo and A. Tramontano, The PMDB Protein Model Database, Nucleic Acids Res., 2006, 34, D306–D309 CrossRef.
- A. Khriesha, Y. Bustanji, R. Abu Farha, R. Al-Abbasi and B. Abu-Irmaileh, Evaluation of the potential anticancer activity of different vitamin D metabolites on colorectal and breast cancer cell lines, Horm. Mol. Biol. Clin. Invest., 2021, 42, 3–9 CrossRef CAS PubMed.
- A. Murray, et al., Vitamin D receptor as a target for breast cancer therapy, Endocr.-Relat. Cancer, 2017, 24, 181–195 CAS.
- G. Wu, R. S. Fan, W. Li, T. C. Ko and M. G. Brattain, Modulation of cell cycle control by vitamin D3 and its analogue, EB1089, in human breast cancer cells, Oncogene, 1997, 15, 1555–1563 CrossRef CAS PubMed.
- F. Xv, J. Chen, L. Duan and S. Li, Research progress on the anticancer effects of vitamin K2, Oncol. Lett., 2018, 15, 8926–8934 Search PubMed.
- C. K. Chakraborti, Vitamin D as a promising anticancer agent, Indian J. Pharmacol., 2011, 43, 113–120 CrossRef CAS.
- E. Haddur, A. B. Ozkaya, H. Ak and H. H. Aydin, The effect of calcitriol on endoplasmic reticulum stress response, Biochem. Cell Biol., 2015, 93, 268–271 CrossRef CAS.
- B. Saracligil, B. Ozturk, A. Unlu, S. Abusoglu and G. Tekin, The effect of vitamin D on MCF-7 breast cancer cell metabolism, Bratisl. Lek. Listy, 2017, 118, 101–106 CAS.
- M. P. Sheeley, C. Andolino, V. A. Kiesel and D. Teegarden, Vitamin D regulation of energy metabolism in cancer, Br. J. Pharmacol., 2022, 179, 2890–2905 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra03703c |
| This journal is © The Royal Society of Chemistry 2022 |