
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of new bis(1,2,4-triazolo[3,4-b][1,3,4]thiadiazines) and bis((quinoxalin-2-yl)phenoxy)alkanes as anti-breast cancer agents through dual PARP-1 and EGFR targets inhibition†
Fatma M. Thabeta,
Kamal M. Dawood *a,
Eman A. Ragaba,
Mohamed S. Nafieb and
Ashraf A. Abbas*a
*a,
Eman A. Ragaba,
Mohamed S. Nafieb and
Ashraf A. Abbas*a
aDepartment of Chemistry, Faculty of Science, Cairo University, Giza, 12613, Egypt. E-mail: ashrafabbas@cu.edu.eg; kmdawood@sci.cu.edu.eg; Fax: +202 35727556; Tel: +202 35676602
bDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, 41522, Egypt
First published on 19th August 2022
Abstract
A number of new 1,ω-bis((acetylphenoxy)acetamide)alkanes 5a–f were prepared then their bromination using NBS furnished the novel bis(2-bromoacetyl)phenoxy)acetamides 6a–f. Reaction of 6a–f with 4-amino-5-substituted-4H-1,2,4-triazole-3-thiol 7a–d and with o-phenylenediamine derivatives 9a and b afforded the corresponding bis(1,2,4-triazolo[3,4-b][1,3,4]thiadiazine) derivatives 8a–l and bis(quinoxaline) derivatives 10a–e in good yields. The cytotoxicity of the synthesized compounds as well as apoptosis induction through PARP-1 and EGFR as molecular targets was evaluated. Three compounds, 8d, 8i and 8l, exhibited much better cytotoxic activities against MDA-MB-231 than the drug Erlotinib. Interestingly, compound 8i induced apoptosis in MDA-MB-231 cells by 38-fold compared to the control arresting the cell cycle at the G2/M phase, and its treatment upregulated P53, Bax, caspase-3, caspase-8, and caspase-9 gene levels, while it downregulated the Bcl2 level. Compound 8i exhibited promising dual enzyme inhibition of PARP-1 (IC50 = 1.37 nM) compared to Olaparib (IC50 = 1.49 nM), and EGFR (IC50 = 64.65 nM) compared to Erlotinib (IC50 = 80 nM). These results agreed with the molecular docking studies that highlighted the binding disposition of compound 8i inside the PARP-1 and EGFR protein active sites. Hence, compound 8i may serve as a potential anti-breast cancer agent.
1. Introduction
Cancer is a serious public health issue that affects people all over the world. The statistics on incidence and mortality showed that it is increasing in both economically emerging and developed countries. Breast cancer is the most common malignancy in women, accounting for 23% of all cancer diagnoses and 14% of cancer deaths.1 As a result, breast cancer cells have been found to be more reliant on DNA repair pathways than normal cells, making them more vulnerable to DNA-damage response suppression.2 Anticancer research continues to be a priority, with more effective and selective chemotherapeutic anticancer drugs urgently needed.The presence of a carboxamide functionality in the composition of organic molecules was found to be essential in many clinically approved synthetic and naturally derived drugs. The carbonyl and amine groups in the carboxamide function had an interesting attention where they act as a hydrogen-bond acceptor (HBA) and a hydrogen-bond donor (HBD), respectively.3–6 It is well known that amide-based flexible pharmacophore had a significant role in enhancing the biological activity due to its hydrogen acceptor/donor (HAD) behaviour.3 A number of 1,2,4-triazole-based fused heterocycles had received a wide range of medicinal applications as appeared in some approved drugs for medical use such as Sitagliptin, Lorpiprazole, Dapiprazole, Estazolam, alprazolam and triazolam7,8 (Fig. 1). In addition, quinoxaline derivatives were among the most important class of heterocyclic compounds due to their potent therapeutic applications including anticancer, anti-HIV, antiviral, anti-inflammatory, antibacterial, antiallergic, antioxidant, … etc.9–11 Interestingly, Erdafitinib and Brimodine are quinoxaline-based commercial drugs in the market12,13 (Fig. 1). The bis-carboxamide function also constituted a main structural-unit of some commercial medicines such as Lacosamide and Batimastat (Fig. 1). In addition, some bis-amide derivatives were found to have potent anticancer activities.14–16
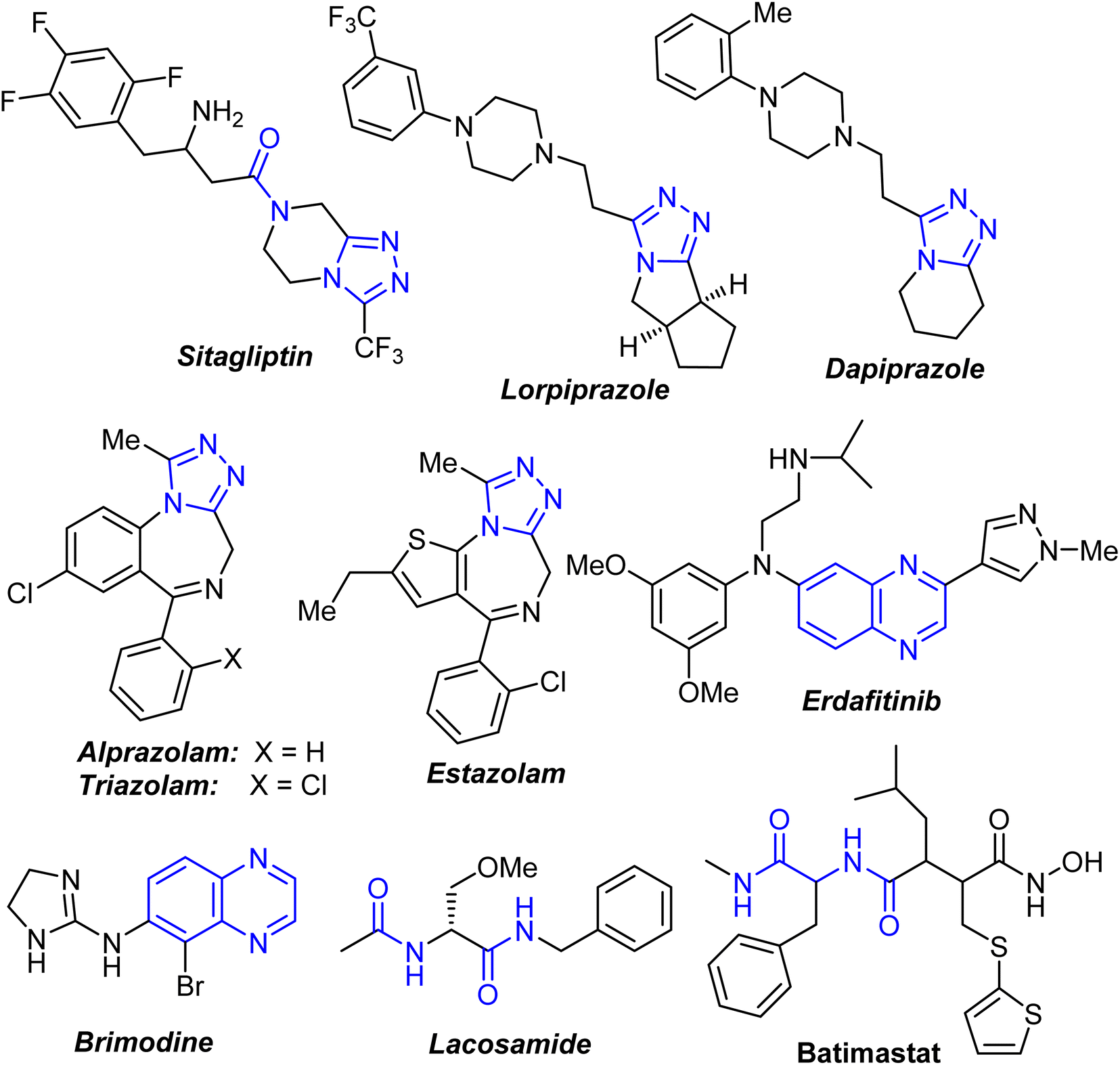 | ||
| Fig. 1 Marketed-drugs based on carboxamides, fused-triazoles and quinoxalines. | ||
The biological potencies of the fused systems: 1,2,4-triazolo[,3,4-b]thiadiazines, were broadly described in the literature.17 Some 1,2,4-triazolo[3,4-b]thiadiazine scaffolds had promising anticancer activities against several human cancer cell lines without obvious signs of toxicity.18–24 Interestingly, a list of triazole, thiadiazole and quinoxaline-based compounds with promising EGFR and PARP-1 inhibition is described in Fig. 2, hence, we thought to employ these moieties as an approach of fragment-based drug design. The 1,2,4-triazole derivative (I)25 exhibited a promising cytotoxic activity as EGFR inhibitor with IC50 value of 1.5 μM, another derivative (II)26 containing a 1,2,4-triazole moiety showed an interesting cytotoxic activity with apoptosis-induction through PARP-1 inhibition with IC50 value of 0.33 μM. The 1,2,4-triazolo[3,4-b]thiadiazine derivatives III27 and IV28 exhibited promising cytotoxic activities through EGFR and PARP-1 inhibitions with apoptosis-induction in cancer cells, respectively. Additionally, quinoxaline derivatives V29 and VI30 exhibited potent cytotoxicity against cancer cells, with inhibitory activities of EGFR (IC50 = 211.2 nM), and PARP-1 (IC50 = 71 nM).
 | ||
| Fig. 2 Examples of triazole, thiadiazine and quinoxaline-based compounds as EGFR and PARP-1 inhibitors. | ||
Poly(ADP-ribose) polymerase (PARPs) are enzymes that catalyse the transfer of ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to acceptor proteins.31 PARP-1 is the most common and well-studied member of this family, and it represents a prospective anticancer therapeutic target as it is involved in DNA repair and cell survival and death.32 Many researchers are interested in developing novel PARP-1 inhibitors, which showed promising effects in clinical studies against cancer. Epidermal growth factor receptor (EGFR) is a critical receptor that begins downstream signal transduction and causes tumor proliferation and migration, so its inhibition is a prospective cancer treatment target.33
Anticancer effects have been documented for 1,2,4-triazoles,34 quinoxalines,35 and 1,2,4-triazolo[3,4-b]thiadiazines,27,28 that all work in various ways. They work by inhibiting the enzymes including PARP-1 and EGFR, which are involved in cancer progression. For newly synthesized bioactive compounds, molecular hybridization, via combination of two or more pharmacophoric motifs in the same substrate, was recommended to enhance their biological potencies.36–38
A subtype of breast cancer known as triple-negative (TNBC) was particularly dangerous and aggressive, with only few treatment options available.39 EGFR has been found to be overexpressed in most cancers, including TNBC. For cancer treatment, the use of a PARP-1 inhibitor as a single target received considerable attention, but the potential of PARP in combination with other oncogenic targets was also accrued in recent years. Other oncogenic targets such as EGFR could be used in conjunction with PARP.40 Lin et al. 2022,41 designed a potential PARP/EGFR dual inhibitor through pharmacophore combination using Olaparib as a clinically approved drug for PRAP-1 inhibitor. Additionally, PARP-1 inhibitors showed selective sensitivity to EGFR, hence dual inhibition targets of PARP-1/EGFR were a promising therapeutic target against cancers including breast cancer.42
Analyzing the receptor ligand-binding pocket and fragment interaction with pocket amino acid side chains using fragment-based drug design (FBDD) was a critical step in developing druggable lead structures. FBDD employed computer models of the target protein to narrow the search for possible leads, and these fragments served as the starting points for “growing” the lead candidate,43,44 we thought to employ triazole, thiadiazine and quinoxaline-moieties as twin drugs against EGFR and PARP-1 proteins. It has been widely reported that functional agents combining two pharmacophoric groups in a single molecule (Twin drugs) have been effective in a wide range of medicinal chemistry applications.
During our research program aiming at synthesis of triazole-based fused heterocycles, bis(carboxamides) and bis(heterocycles) as twin drugs of significant biological applications,45–55 the reported anticancer activities of compounds employing either triazolothiadiazine or carboxamide pharmacophores inspired us to construct a series of modified novel bis-triazolothiadiazine hybrids (Fig. 3) having variable bis(carboxamide) spacers, starting from bis(2-chloroacetamide) derivatives, to investigate their potency as anticancer agents against a panel of cancer cell lines and to investigate the effective molecular target and the apoptotic cell death.
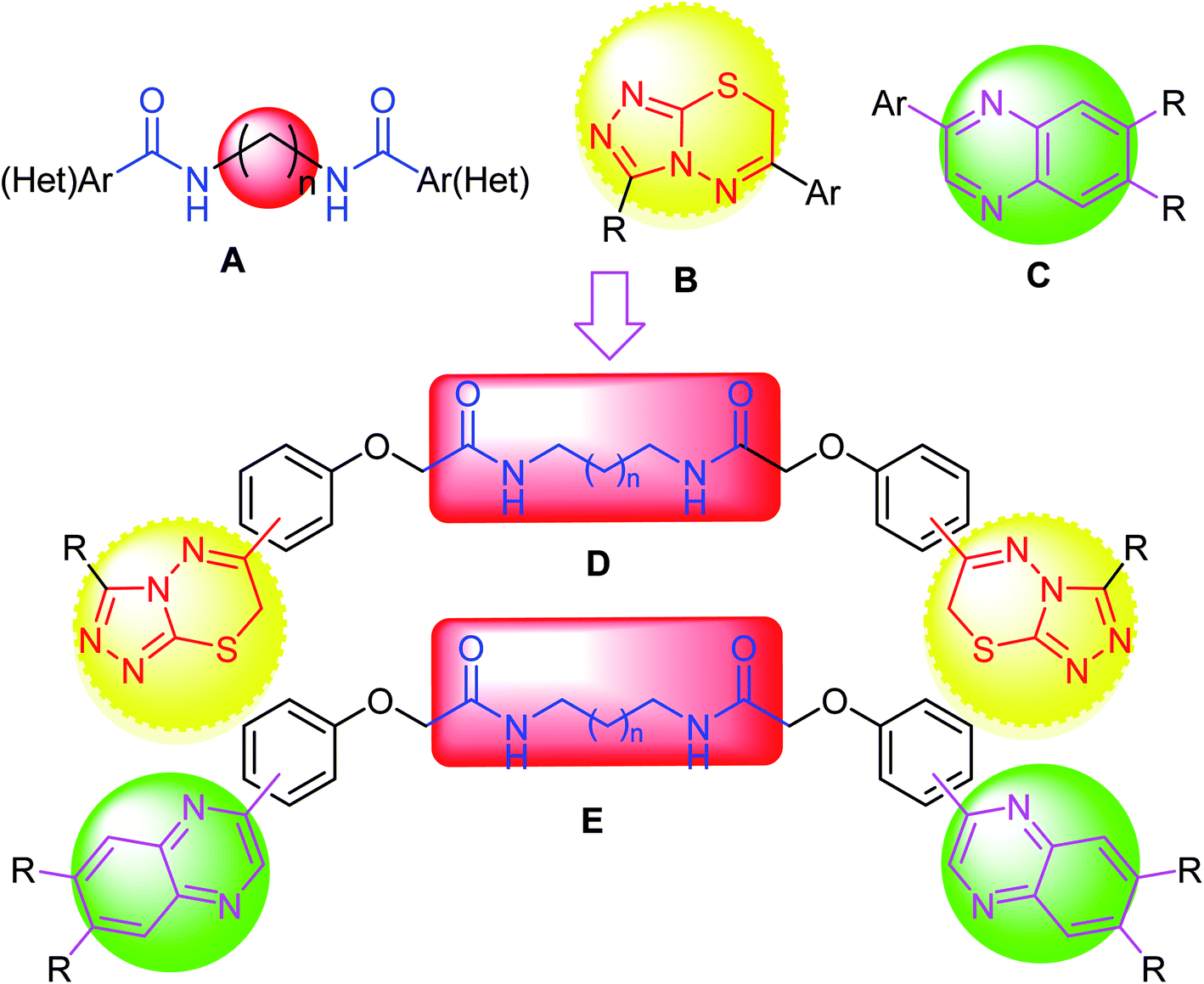 | ||
| Fig. 3 Design strategy of our target; triazolothiadiazine or quinoxaline merged bis-carboxamide pharmacophores. | ||
2. Results and discussion
2.1. Chemistry
The activated bis-(2-chloroacetamide) derivatives 3a–c were firstly prepared by conventional double N-alkylation of the readily available aliphatic 1,ω-diamino-alkanes 2a–c with the highly reactive chloroacetyl chloride 1 following a procedure reported in the literature56 (Scheme 1). The obtained bis-chloro compounds 3a–c were then used to alkylate the potassium salts of p-hydroxyacetophenone 4a and its o-isomer 4b [obtained upon treatment of hydoxyacetophenone derivatives 4a, b with methanolic potassium hydroxide solution] in boiling DMF to afford the corresponding 1,ω-bis((acetylphenoxy)acetamide)alkanes 5a–f in 60–65% yields as depicted in Scheme 1. Thus, reaction of one equivalent of N,N′-(ethane-1,2-diyl)bis(2-chloroacetamide) 3a with two equivalents of the potassium salt of p-hydroxyacetophenone 4a in boiling dimethylformamide (DMF) delivered the new bis(carboxamide); N,N′-(ethane-1,2-diyl)bis(2-(4-acetylphenoxy)acetamide) 5a in 65% yield. Structure of the new product 5a was fully confirmed from its elemental and spectral analyses (IR and 1H NMR). The 1H NMR spectrum of 5a recorded a singlet signal at 2.49 for two acetyl protons, and two singlet peaks at 3.25 and 4.55 assignable to CH2N and CH2O protons, two doublets at 7.03 and 7.90 due to the p-phenylene aromatic protons, besides a singlet peak at 8.22 ppm assignable to NH function, respectively. Moreover, the IR spectrum of 5a showed two sharp absorption peaks at 3286 and 1665 cm−1 assigned for NH and CO groups, respectively. Similarly, compounds 3b, c reacted with the potassium salt of p-hydroxyacetopheneone 4a and o-hydroxyacetopheneone 4b, under the same reaction conditions, to furnish the corresponding N,N′-(alkane)bis(2-bromoacetyl)phenoxy)acetamides 5b–f in 60–65% yields, as shown in Scheme 1. Structures of all the new bis(acetylphenoxy)acetamide)alkane derivatives 5b–f were completely confirmed from both elemental and spectral analyses (IR, 1HNMR and 13CNMR) (c.f. Experimental section). The formed bis(acetyl) derivatives 5a–g were characterized by the presence of an elongated spacer between the two carboxamide groups as well as the position of acetyl moiety on the aromatic ring.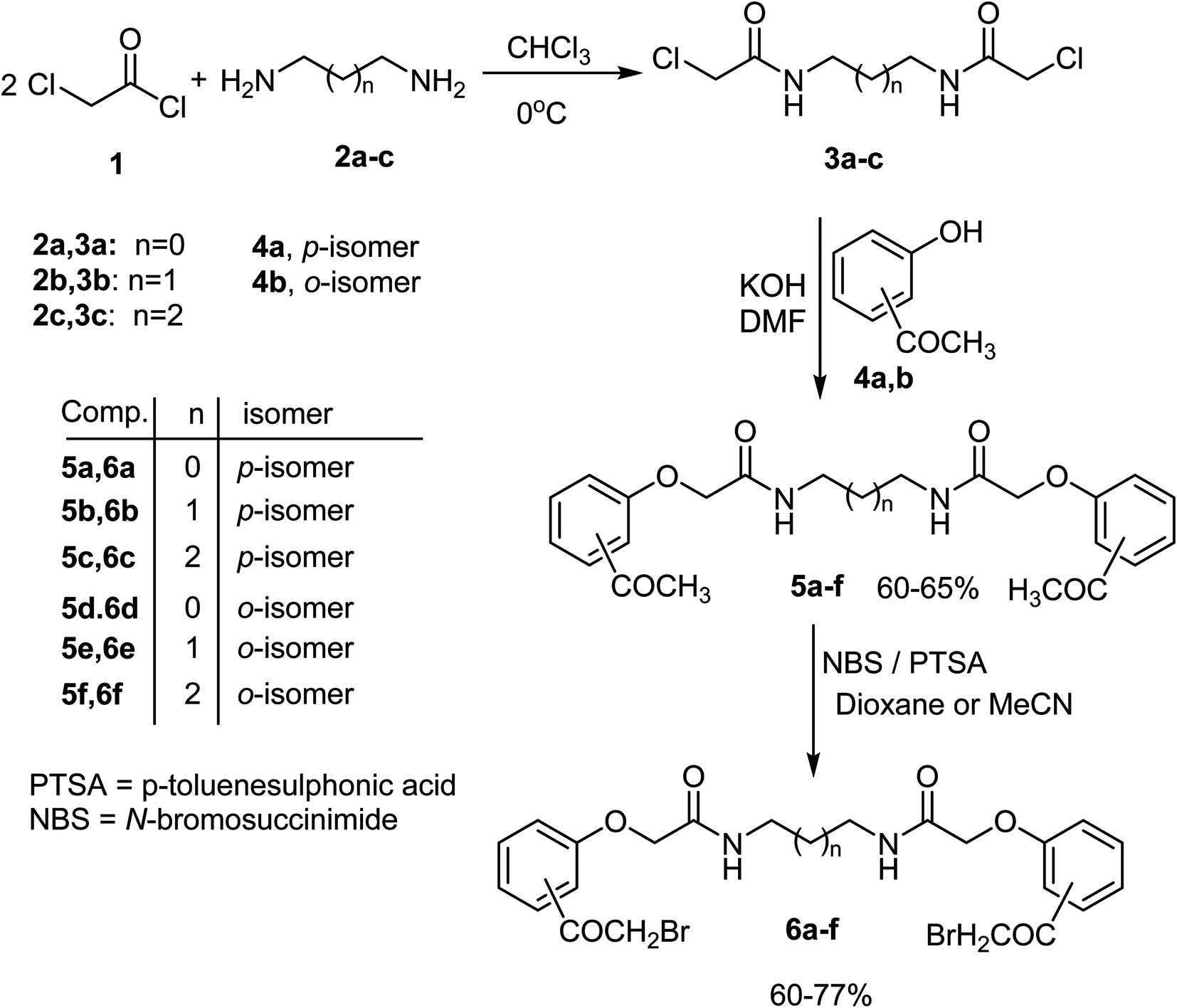 | ||
| Scheme 1 Synthesis of N,N′-(alkane)bis((2-bromoacetyl)phenoxy)acetamides 6a–f. | ||
Next, the bis-carboxamide-based bis(acetyl) derivatives 5a–f were chosen as key compounds for preparation of the hitherto unreported N,N′-(alkane)bis(2-bromoacetyl)phenoxy)acetamides 6a–f which were useful for synthesis of the target bis(heterocycles) in our project. Two strategies were attempted to synthesize the bis(2-bromoacetyl) derivatives 6a–f. The first involved conventional direct bromination of 5a with bromine in acetic acid as solvent at room temperature, however, this method led to the formation of a mixture of bis(α-bromoketone) 6a and its bis(α,α-dibromoketone) derivatives as shown by TLC and 1H NMR of the crude reaction mixture. The mixture was difficult to be separated into the corresponding pure products. In anticipation of getting an efficient and modified protocol to synthesize a pure sample of 6a, an alternative synthetic way was attempted. Thus, bromination of the model compound 5a with N-bromosuccinimide (NBS) catalyzed by p-toluenesulfonic acid (p-TsOH) in acetonitrile as solvent resulted in the formation of the desired bis(α-bromoketone) 6a as a single pure product in 60% yield as proved by the 1H NMR and TLC of the crude product (Scheme 1). 1H NMR spectrum of the pure compound 6a showed all the expected features with signals at δ 3.24–3.26 (m, 4H, 2CH2N), 4.57 (s, 4H, 2CH2O), 4.94 (s, 4H, 2CH2Br), 7.05 (d, 4H, J = 8.7 Hz, ArH's), 8.04 (d, 4H, J = 9 Hz, ArH's), and 8.26 (s, 2H, 2NH) ppm. This result stimulated us to generalize this protocol to prepare the rest bis(α-bromoketone) derivatives 6b–f. Thus, double bromination of 5b–f with NBS, under typical reaction condition, led to the formation of the desired compounds 6b–f in 66–77% yields (Scheme 2). It is noteworthy to mention here that trials to improve the reaction yield, taking compound 6b as a representative example, by switching the reaction solvent into dioxane instead of acetonitrile under the same reaction conditions failed, where compound 6b was isolated in only 37% yield.
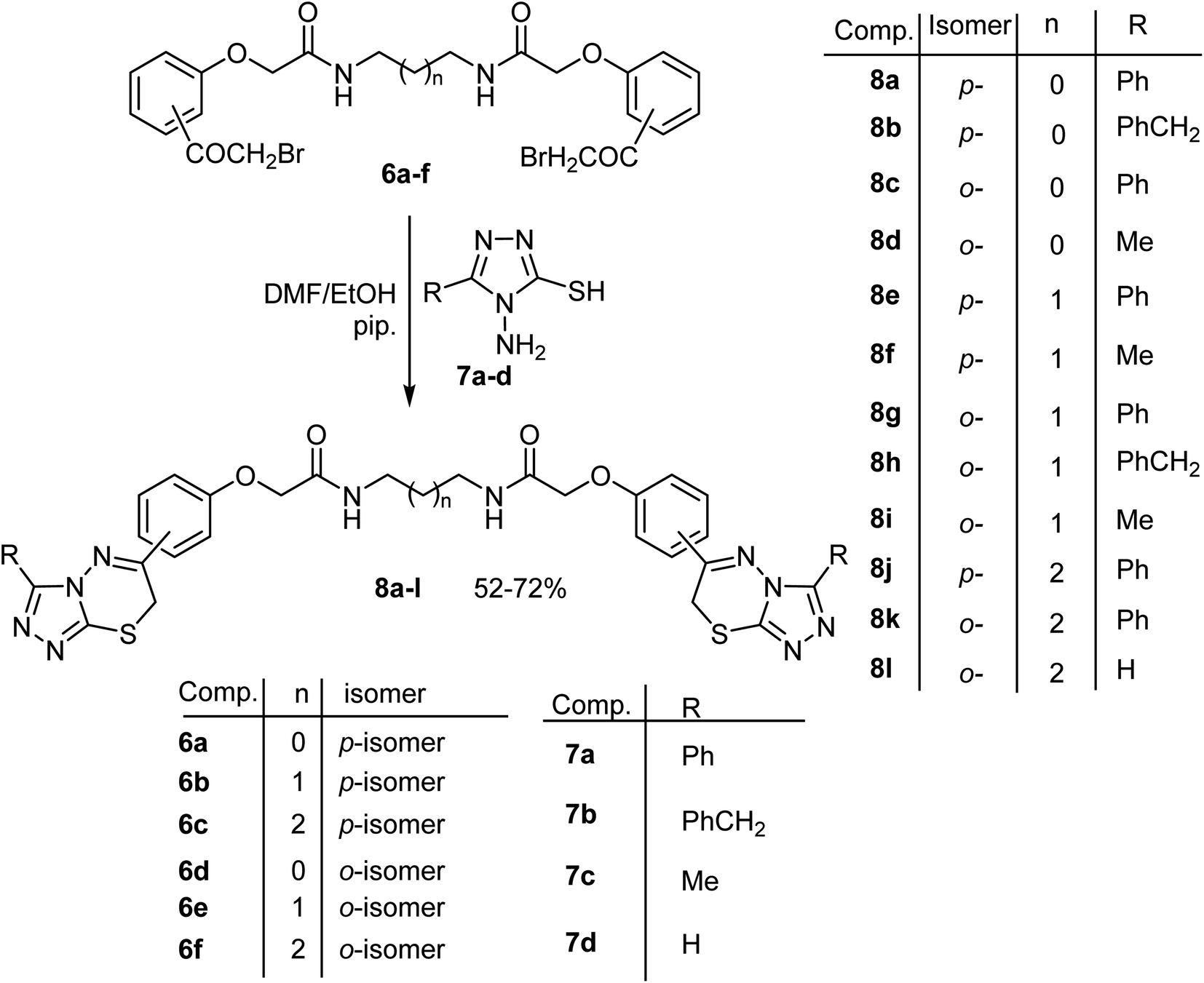 | ||
| Scheme 2 Synthesis of the bis(triazolothiadiazine) derivatives 8a–l. | ||
Next, our target was expanded to investigate the synthetic potentiality of the bis[((2-bromoacetyl)phenoxy)alkanamide] derivatives 6a–f to access the bis[((1,2,4-triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)alkanamide] derivatives 8a–l. Firstly, reaction of 6a with two equivalents of 4-amino-5-phenyl-4H-1,2,4-triazole-3-thiol (7a) in refluxing ethanol, in the presence of a catalytic amount of piperidine, for 8 h afforded a low yield (30%) of N,N′-(ethane-1,2-diyl)bis(2-(4-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)-acetamide) (8a). The low yield of 8a might be attributed to the fair solubility of the starting materials in the reaction solvent (ethanol). Repeating the same reaction in DMF/ethanol mixed solvent (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at reflux temperature, in the presence of piperidine, for 3 h furnished the desired product 8a in 62% yield. The structure of 8a was elucidated by elemental analyses, as well as its spectral data (1H NMR, MS and IR), that were in complete accordance with the proposed structure 8a (Scheme 2). The 1H NMR spectrum of 8a showed characteristic multiplet signals at δ 3.27 for CH2N integrating for four protons, two singlet signals at δ 4.36 and 4.57 corresponding to CH2S and CH2O protons, respectively, multiplet signals in the region 7.11–8.01 for the aromatic protons in addition to a singlet signal at 8.23 ppm for two NH protons. Its IR spectrum showed characteristic sharp absorption peaks for NH and CO at ν 3433 and 1666 cm−1, respectively. Finally, its mass spectrum showed a fragment at m/z 755 due to the correct molecular ion peak.
:1 v/v) at reflux temperature, in the presence of piperidine, for 3 h furnished the desired product 8a in 62% yield. The structure of 8a was elucidated by elemental analyses, as well as its spectral data (1H NMR, MS and IR), that were in complete accordance with the proposed structure 8a (Scheme 2). The 1H NMR spectrum of 8a showed characteristic multiplet signals at δ 3.27 for CH2N integrating for four protons, two singlet signals at δ 4.36 and 4.57 corresponding to CH2S and CH2O protons, respectively, multiplet signals in the region 7.11–8.01 for the aromatic protons in addition to a singlet signal at 8.23 ppm for two NH protons. Its IR spectrum showed characteristic sharp absorption peaks for NH and CO at ν 3433 and 1666 cm−1, respectively. Finally, its mass spectrum showed a fragment at m/z 755 due to the correct molecular ion peak.
The above reaction was generalized where treatment of the appropriate bis(α-bromoacetophenone) 6a–f, with two equivalents of the 4-aminotriazole-3-thiol derivatives 7a–d under the same reaction conditions afforded the corresponding bis(triazolothiadiazine) derivatives 8a–l in 52–72% yields as shown in Scheme 2. The structures of the obtained products 8a–l were substantiated from their elemental analyses and spectral data (1H and 13C NMR, MS and IR) as described in the experimental part. Mechanistically, formation of 8 took place via initial S-alkylation of the thiol group of aminotriazole-thiol with loss of two HBr molecules, followed by an intramolecular cyclocondensation with concurrent extrusion of two water molecules to furnish the product 8. Strong evidence for the proposed mechanism was based on the following remarks; (a) disappearance of the characteristic signals NH2 belong to amino-triazole moiety in both IR and 1H-NMR spectra of all the products 8a–l, (b) presence of new SCH2 protons, resonating at δ ∼ 4.3 ppm as singlet signal integrating for four protons, which clearly indicated the occurrence of ring closure.
Finally, our work was extended to comprise synthesis of the new functionalized bis((quinoxalin-2-yl)phenoxy)alkane derivatives 10a–e. Thus, when the bis(α-bromoketones) 6a, b, d, f were allowed to react with two equivalents of o-phenylenediamine derivatives 9a, b in absolute ethanol containing piperidine (two equiv.), at reflux temperature, gave the corresponding bis(quinoxalin-2-yl)phenoxy)alkane derivatives 10a–d in 50–70% yields as shown in Scheme 3. The structures of the isolated products were inferred from their elemental and spectral analyses as described in detail in the experimental part. For example, the spectral data (IR, 1H- and 13C-NMR and MS) of the bis(quinoxaline) derivative 10e was interpreted in detail. The mass spectrum of 10e showed the correct molecular ion peak at m/z = 668, and its IR spectrum revealed distinctive stretching absorption peaks at 3240, 1674 and 1620 cm−1 corresponding to NH, CO and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N functions, respectively. The 1H NMR of 10e showed three singlet signals at δ 1.29, 2.46, 4.59 for four groups of CH3 protons, CH2CH2NH, and OCH2 protons, respectively, multiplet signals in the range 3.19–3.21 for four protons of NHCH2CH2CH2CH2NH, in addition to a singlet peak at δ 8.93 integrated for two protons referring to quinoxaline-3-CH and all the remaining protons appeared at the expected chemical shifts with correct integral values (see experimental part). The 13C-NMR spectrum of 10e, using APT technique, showed two signals at δ 20.35 and 20.41 for two CH3 groups, three signals at 26.45, 38.35 and 67.61 for three aliphatic CH's, seven signals at 112.79, 122.52, 127.73, 127.78, 131.38, 131.54 and 144.83 for seven aromatic CH's, besides eight signals at 126.48, 139.74, 140.46, 140.69, 141.10, 150.27, and 154.85 for seven aromatic C's and 167.91 for CO group. Formation of the bis(quinoxaline) derivatives 10a–e took place via initial nucleophilic attack at the bromoacetyl moiety followed an intramolecular cyclization via consecutive elimination of two molecules of water and finally aromatization via air oxidation to afford bis(quinoxalin-2-yl)phenoxy)alkane derivatives 10a–e.
N functions, respectively. The 1H NMR of 10e showed three singlet signals at δ 1.29, 2.46, 4.59 for four groups of CH3 protons, CH2CH2NH, and OCH2 protons, respectively, multiplet signals in the range 3.19–3.21 for four protons of NHCH2CH2CH2CH2NH, in addition to a singlet peak at δ 8.93 integrated for two protons referring to quinoxaline-3-CH and all the remaining protons appeared at the expected chemical shifts with correct integral values (see experimental part). The 13C-NMR spectrum of 10e, using APT technique, showed two signals at δ 20.35 and 20.41 for two CH3 groups, three signals at 26.45, 38.35 and 67.61 for three aliphatic CH's, seven signals at 112.79, 122.52, 127.73, 127.78, 131.38, 131.54 and 144.83 for seven aromatic CH's, besides eight signals at 126.48, 139.74, 140.46, 140.69, 141.10, 150.27, and 154.85 for seven aromatic C's and 167.91 for CO group. Formation of the bis(quinoxaline) derivatives 10a–e took place via initial nucleophilic attack at the bromoacetyl moiety followed an intramolecular cyclization via consecutive elimination of two molecules of water and finally aromatization via air oxidation to afford bis(quinoxalin-2-yl)phenoxy)alkane derivatives 10a–e.
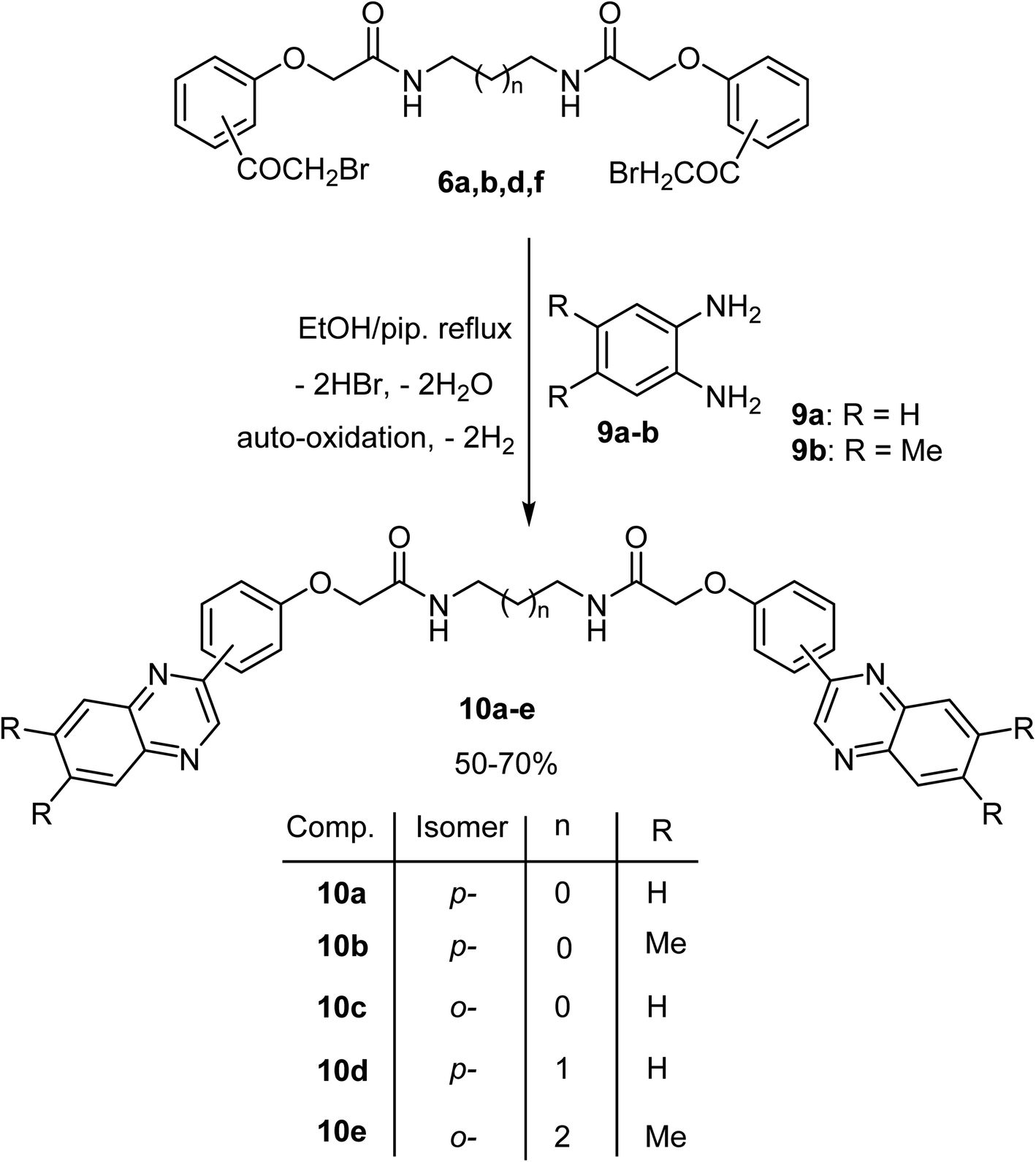 | ||
| Scheme 3 Synthesis of the bis((quinoxalin-2-yl)phenoxy)alkane derivatives 10a–e. | ||
2.2. Biology
| No. | IC50, μM | No. | IC50, μM | ||
|---|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | MCF-7 | MDA-MB-231 | ||
| a IC50 values were calculated as the average of three independent trials using dose–response curve in GraphPad prism. | |||||
| 8a | ≥50 | 31.7 | 10a | 32 | ≥50 |
| 8b | 26.1 | 29.2 | 10b | ≥50 | 22.37 |
| 8c | 16.1 | 23.3 | 10c | 7.61 | 11.01 |
| 8d | 2.14 | 0.421 | 10d | 23.21 | ≥50 |
| 8e | 12.7 | 10.9 | 10e | 31.21 | 28.1 |
| 8f | 7.12 | 3.01 | Erlotinib | 0.32 | 1.02 |
| 8g | 11.20 | 8.01 | |||
| 8h | 21.1 | ≥50 | |||
| 8i | 1.31 | 0.12 | |||
| 8j | 14.9 | 15.01 | |||
| 8k | 13.1 | 12.7 | |||
| 8l | 5.31 | 0.86 | |||
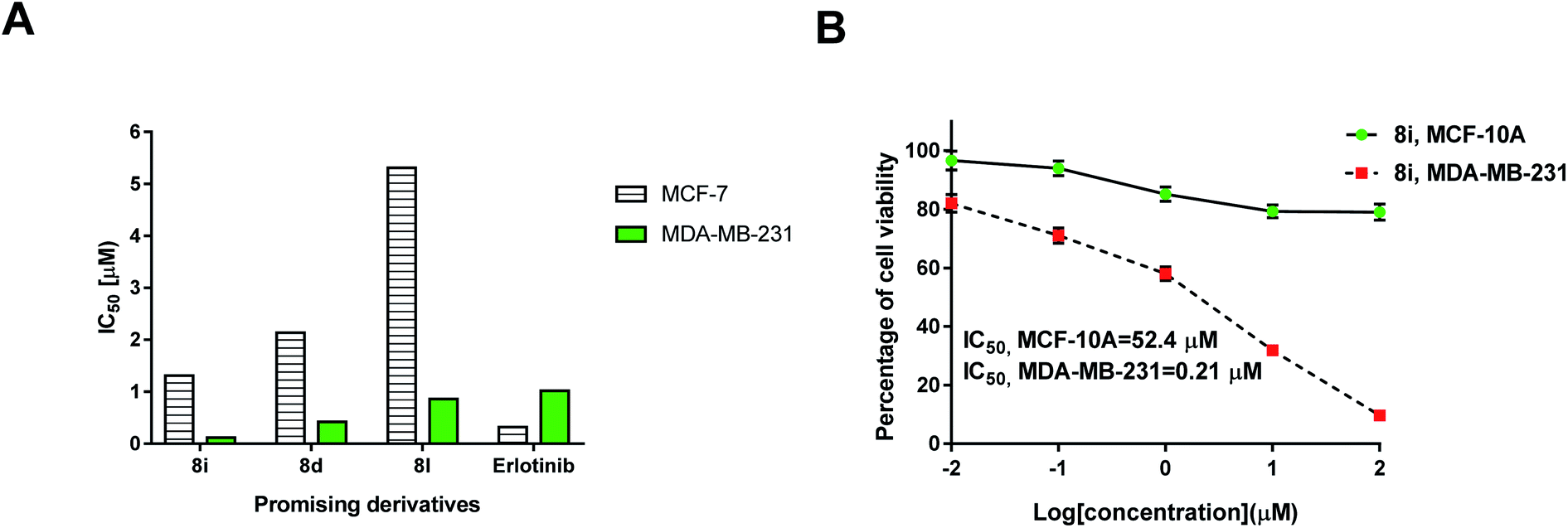 | ||
| Fig. 4 (A) IC50 (μM) values of the promising derivatives 8i, 8d, and 8l against MCF-7 and MDA-NB-231 cells compared to Erlotinib. (B) Cell viability of compound 8i against breast cancer (MDA-MB-231) and normal breast (MCF-10A) cells using MTT assay for 48 h incubation. | ||
Biological results of the current investigation revealed that there was an increase in the potency of the cytotoxicity and enzyme inhibition results of the tested bis-heterocyclic derivatives compared to the reported results of the mono-heterocyclic derivatives of triazole, thiadiazine and quinoxaline compounds.25–30 Therefore, synthesis of the current bis-fused derivatives was an added value towards the anticancer activity (Fig. 5).
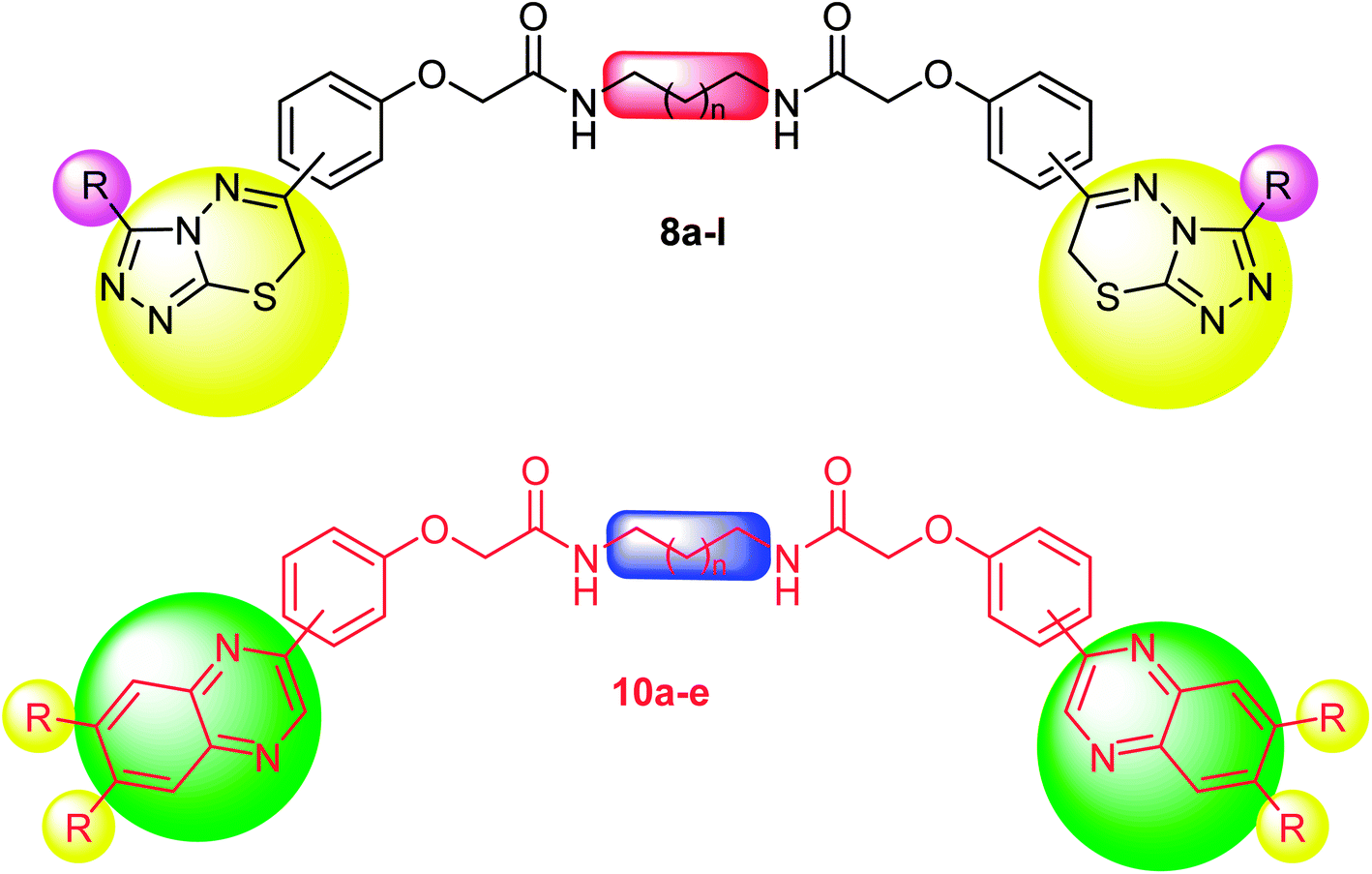 | ||
| Fig. 5 Highlighted substituents anchored on the pharmacophore with promising cytotoxic activities for compounds 8a–l and 10a–e. | ||
It is noteworthy to mention that the current synthesized derivatives 8a–l were characterized by variable alkane linkers between the two carboxamide groups from ethylene to butylene and the different substitution position of the triazolothiadiazine or quinoxaline moieties to the aromatic phenylene ring (ortho- or para-) as well as the substituents at the triazole or quinoxaline rings themselves. Such variations were necessary to optimize the potency of the anticancer activity of the synthesized products.
As illustrated in Fig. 6, the biological screening of the mentioned compounds 8a–l revealed a correlation between some compounds with comparable anticancer activity. Thus, compounds 8i, 8d and 8l were the most active inhibitors of MDA-MB-231 cells with IC50 values 0.12, 0.421 and 0.86 μM, respectively. All these compounds 8i, 8d and 8l had the triazolothiadiazinyl pharmacophore in the ortho-position of the phenylene group and had less bulky (Me or H) substituents at position 3 of the triazole scaffold. Interestingly, the most active lead compound was 8i with the shortest alkane spacer (ethylene; where n = 0) between both bis-carboxamide groups. Thus, the anticancer potency was greatly affected by the presence of a less bulky group at triazole ring, and a short-length spacer (first level activity) as shown in Fig. 6. For the second level of activity (the para-isomeric series) shown in Fig. 6, a similar correlation between the bis-carboxamide having propylene spacer and methyl substituent at position 3 of triazole scaffold (8f: IC50 = 3.01 μM), was more active than those having phenyl group at position 3 of triazole scaffold with either propylene spacer (for 8g and 8e) or butylene spacer (for 8k). Therefore, it was concluded that the most inhibitory active compounds were characterized by the following: (i) ortho-substitution was more potent than para-substitution, (ii) less bulky substituents at the triazole scaffold was more potent than others with phenyl moiety, (iii) shorter alkane spacer between the two carboxamide groups.
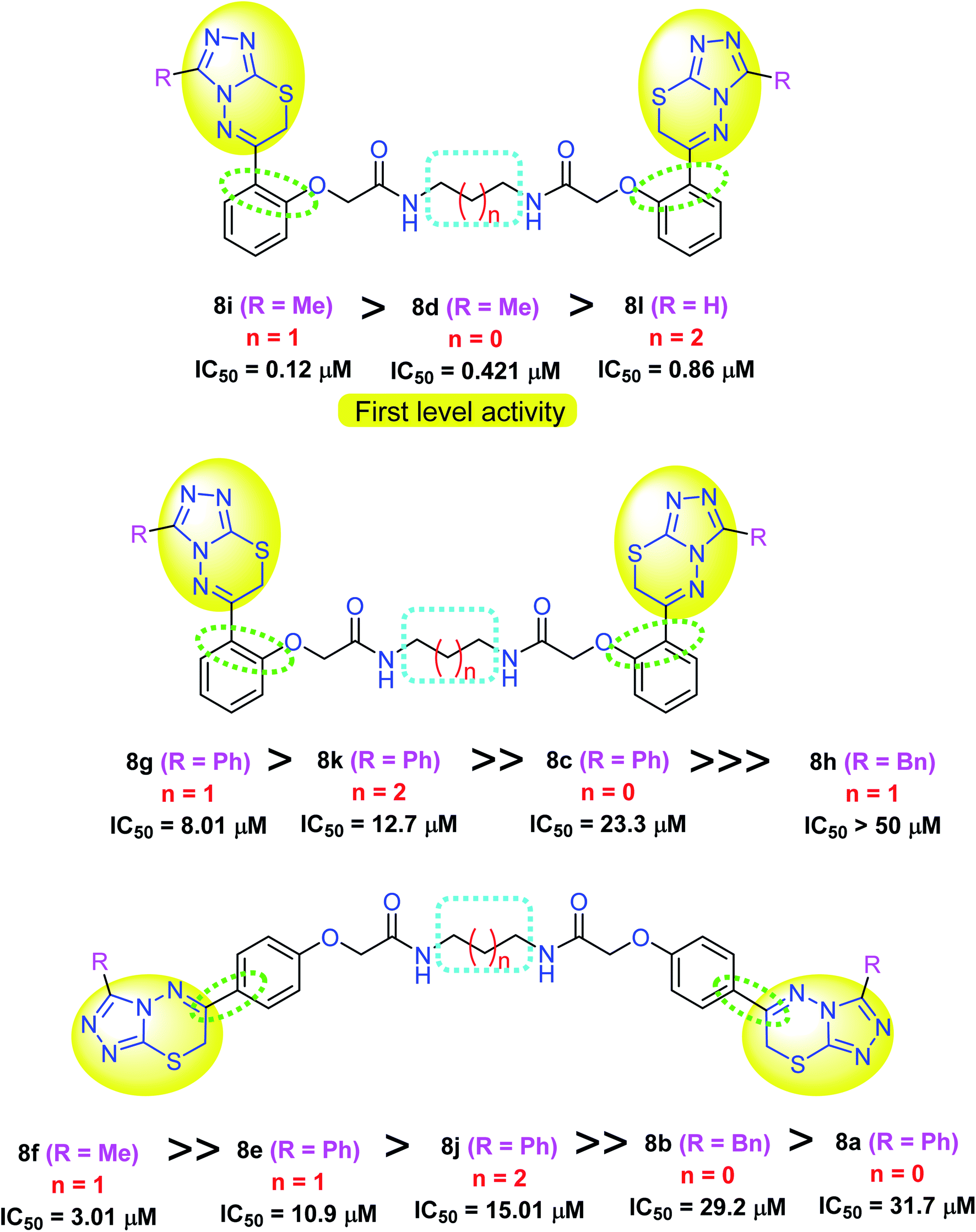 | ||
| Fig. 6 Effect of derivatization of the synthesized bis-triazolothiadiazines 8a–l on their cytotoxicity against MDA-MB-231 cells. | ||
It was noticed that, among the bis-quinoxaline series 10a–e, only three derivatives 10b, c, e exhibited moderate anticancer activity in the order 10c > 10b > 10e with IC50 values of 11.01, 22.37 and 28.1 μM, respectively, against MDA-MB-231 cells. However, the other derivatives of the bis-quinoxaline series 10a, d were totally inactive (IC50 > 50 μM).
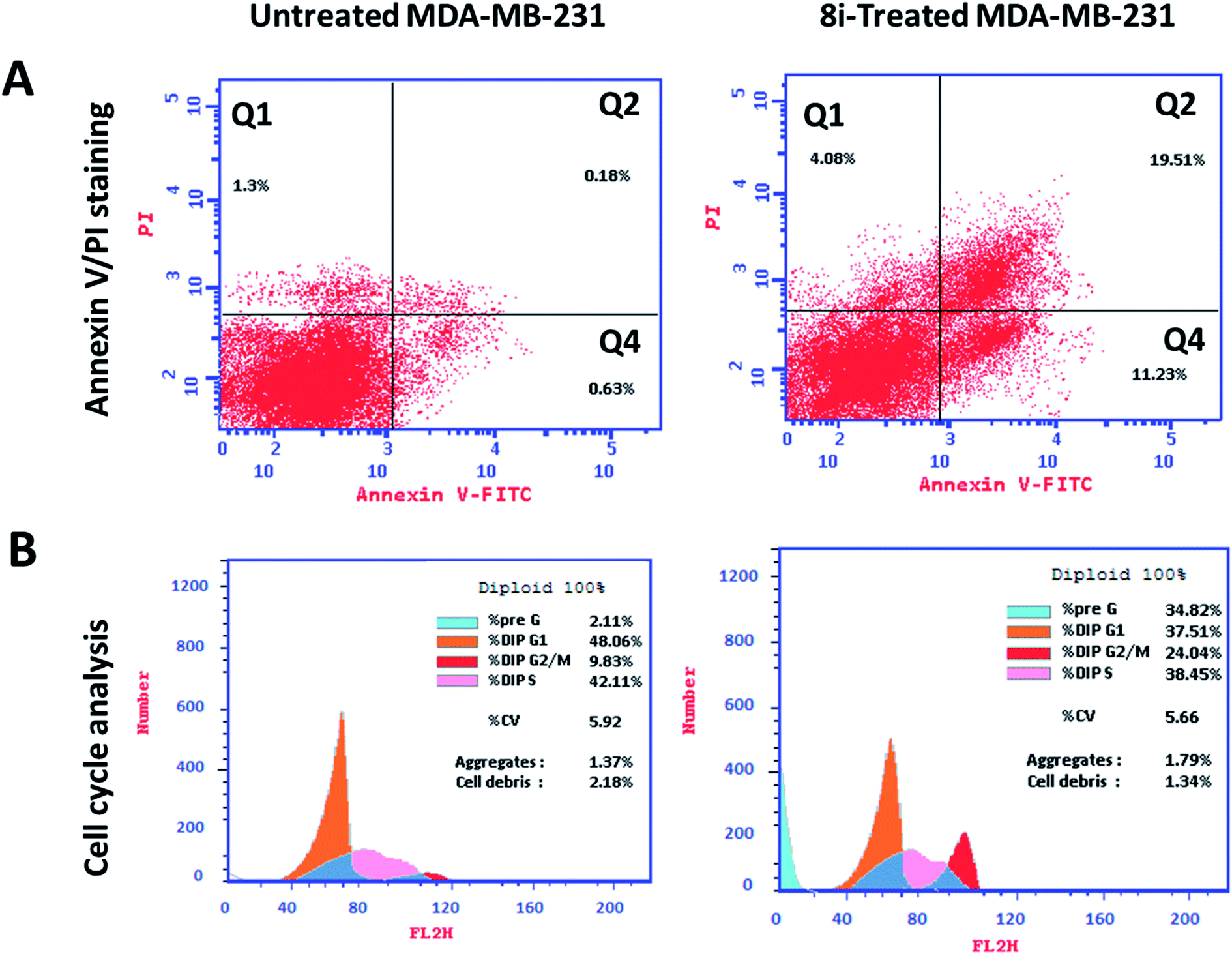 | ||
| Fig. 7 Flow cytometry analysis upper panel (A) Annexin V/PI staining for apoptosis-necrosis assessment, (Q1) necrosis, (Q2) late apoptosis, (Q4) early apoptosis. Lower panel (B) histograms of DNA content at each phase of untreated and 8i-treated MDA-MB-231 cells with IC50 value of 0.12 μM, 48 h. | ||
Investigating at which cell cycle, cells were arrested while division, MDA-MB-231 cells were tested for cell cycle analysis, which indicated the cell population at each phase in both untreated and treated cells. As seen in Fig. 7B, compound 8i significantly increased cell population at G2/M by 24.04%, compared to 9.83% for the control, so it arrested cell division at G2/M phase. Consequently, compound 8i induced apoptosis in MDA-MB-231 cells arresting the cell cycle at G2/M.
Further validation of the apoptosis-inducing activity of the tested compound 8i in MDA-MB-231 cells, the gene expression levels of apoptosis-related genes in both untreated and treated MDA-MB-231 cells were investigated through the RT-PCR. As seen in Fig. 8, compound 8i treatment increased P53 level by 4.8-fold, Bax level by 4.46-fold, caspase-3 level by 3.9-fold, caspase-8 level by 6.36-fold, caspase-9 by 2.9-fold, while the compound treatment decreased Bcl-2 level as the anti-apoptotic gene by 0.35-fold compared to the untreated control.
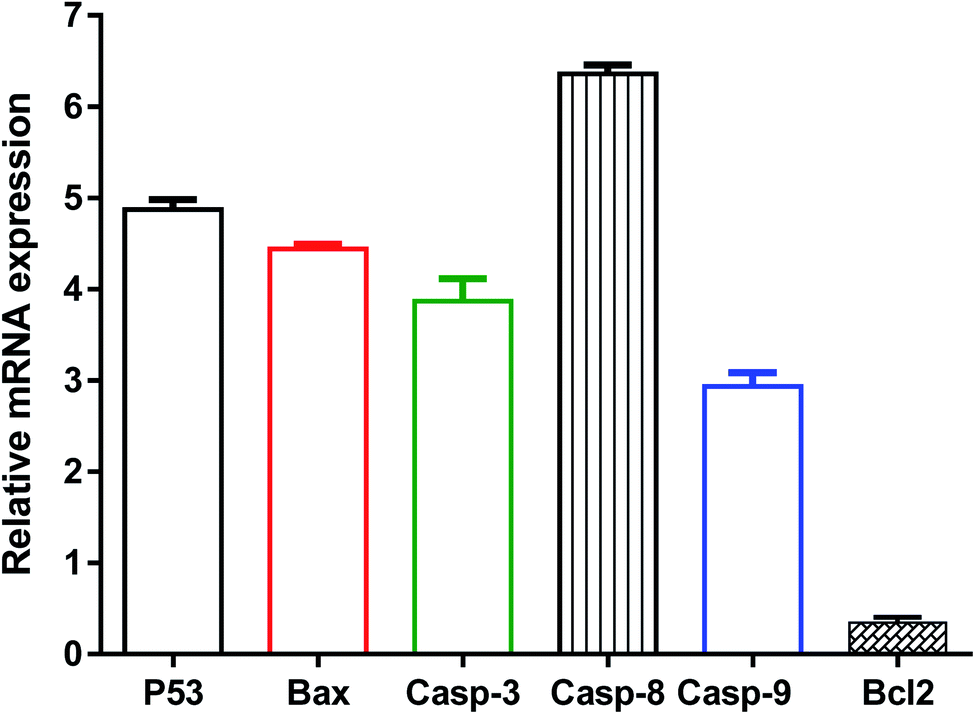 | ||
| Fig. 8 Gene expression analysis in untreated and treated MDA-MB-231 cells at the IC50 of 0.12 μM, 48 h. Values are expressed as Mean ± SD for three independent experimental runs. The housekeeping gene is β-actin. Fold-change = 2^−ΔΔCt, where ΔΔCt the difference between mean values of genes CT values in the treated and control groups. Fold of change in the untreated control = 1. | ||
The activation of caspase-3,9 and the decreased expression of the Bcl-2 gene might cause apoptosis. P53 is a tumour suppressor gene that was essential for apoptosis. The results demonstrated the intrinsic apoptotic pathway via activation of P53, Bax, caspase 3 and 9. Furthermore, in treated MDA-MB-231 cells, increased caspase-8 gene expression favours the extrinsic apoptotic pathway. As a result, our findings agreed with previous studies that used RT-PCR to highlight the apoptotic pathway by upregulating proapoptotic genes and downregulating anti-apoptotic genes.
| Target | Docking score (kcal mol−1) | 3D Interactive posea | Ligand–receptor interactions |
|---|---|---|---|
| a Binding disposition of compound 8i (Cyan-colored) and co-crystallized ligands (Yellow-colored) inside the active sites of PARP-1and EGFR proteins. Labeled amino acids are the key ones for activity. | |||
| PARP-1 | −37.09 | 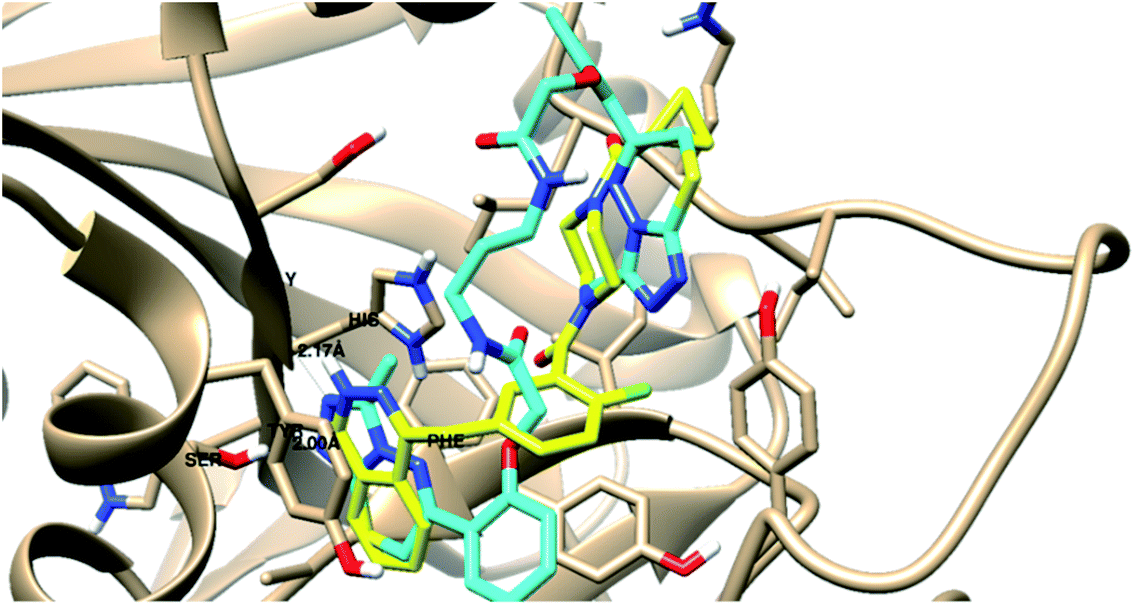 |
- 1 H-bond with Gly 863 |
| - 1 H-bond with Ser 904 | |||
| - 1 arene–cation with His 862 | |||
| - 1 arene–arene with Tyr 896 | |||
| EGFR | −29.23 | 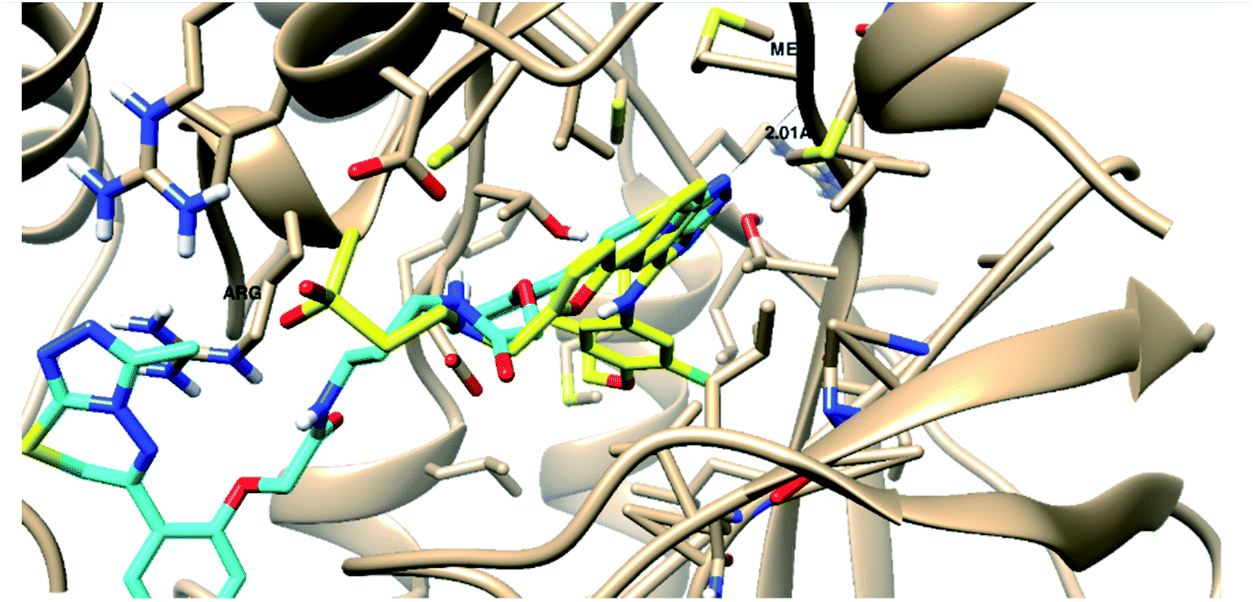 |
- 1 H-bond with Met793 |
| - 1 arene–cation with Lys 745 | |||
3. Conclusion
In this work, a synthetic protocol to a new series of bis((2-bromoacetyl)phenoxy)alkane derivatives linked by a bis-carboxaamide linker was developed and involved in the synthesis of a library of bis((triazolothiadiazinyl)phenoxy)alkane and bis((quinoxalin-2-yl)phenoxy)alkane derivatives via their reaction with 4-amino-1,2,4-triazole-3-thiol and o-phenylenediamine derivatives, respectively, in good yields. Structures of all the new compounds were established using elemental analyses and spectroscopic tools. The cytotoxicity of the synthesized compounds was tested using MTT assay, as well as apoptosis-induction through PARP-1 and EGFR as molecular targets. Compound 8i exhibited high cytotoxic activity with IC50 values of 0.12 μM and 1.31 μM against MDA-MB-231 and MCF-7 cells, respectively, compared to Erlotinib. Interestingly, compounds 8i induced apoptosis in MDA-MB-231 cells by 38-fold (30.74% compared to 0.81 for the control) arresting the cell cycle at G2/M phase, and it affected the apoptosis-related genes through RT-PCR. Additionally, compound 8i exhibited promising dual enzyme inhibition PARP-1 (IC50 = 1.37 nM) compared to Olaparib (IC50 = 1.49 nM), and EGFR with (IC50 = 64.65 nM) compared to Erlotinib (IC50 = 80 nM). Hence, this compound may serve as a potential target-oriented anti-breast cancer agent.4. Experimental
4.1. Chemistry
4.1.2.1 General procedure. 4-Hydroxyacetophenone (4a) or 2-hydroxyacetophenone (4b) (10 mmol) was added to methanolic KOH solution [prepared by dissolving 0.56 g (10 mmol) of potassium hydroxide (KOH) in 10 mL of absolute methanol], stirred for 10 min and the solvent was then removed under vacuum. The obtained potassium salt was dissolved in DMF (10 mL) and the appropriate N,N′-(alkane-1,ω-diyl)bis(2-chloroacetamide) 3a–c (5 mmol) was added. The reaction mixture was heated under reflux for 10 min during which KCl was precipitated. The solvent was then removed under vacuum and crushed ice was added to the reaction mixture. The obtained solid was collected by filtration, dried and recrystallized from EtOH/H2O to afford bis(acetylphenoxy)alkanes 5a–f.
4.1.2.2 N,N′-(Ethane-1,2-diyl)bis(2-(4-acetylphenoxy)acetamide) (5a). Obtained from 3a and 4a; colorless crystals; yield 65%, mp 176–178 °C; IR (KBr) ν 3286 (NH), 1666 (br, 2C
O) cm−1; 1H NMR (DMSO-d6) δ 2.49 (s, 6H, 2CH3CO), 3.25–3.26 (m, 4H, 2CH2N), 4.55 (s, 4H, 2CH2O), 7.03 (d, 4H, J = 9 Hz, ArH's), 7.90 (d, 4H, J = 8.7 Hz, ArH's), 8.22 (brs, 2H, 2NH) ppm. Anal. calcd for C22H24N2O6 (412.44): C, 64.07; H, 5.87; N, 6.79%. Found: C, 64.19; H, 5.90; N, 6.81%.
4.1.2.3 N,N′-(Propane-1,3-diyl)bis(2-(4-acetylphenoxy)acetamide) (5b). From 3b and 4a; colorless crystals, yield 64%, mp 134–136 °C; IR (KBr) ν 3387 (NH), 1658 (C
O) cm−1; 1H NMR (CDCl3) δ 1.73 (quintet, 2H, J = 6 Hz, CH2CH2CH2), 2.56 (s, 6H, 2CH3CO), 3.36–3.42 (m, 4H, 2CH2N), 4.56 (s, 4H, 2CH2O), 6.99 (d, 4H, J = 8.7 Hz, ArH's), 7.20 (brs, 2H, 2NH), 7.95 (d, 4H, J = 8.7 Hz, ArH's) ppm; 13C NMR (CDCl3, APT) δ 26.18 (CH3), 29.38, 35.35, 66.99, (3CH2 aliphatic), 114.26, 130.56 (2CH, ArC's) 131.25, 166.72 (2C, ArC's), 167.91, 196.54 (2CO) ppm. Anal. calcd for C23H26N2O6 (426.47): C, 64.78; H, 6.15; N, 6.57%. Found: C, 64.85; H, 6.18; N, 6.66%.
4.1.2.4 N,N′-(Butane-1,4-diyl)bis(2-(4-acetylphenoxy)acetamide) (5c). From 3c and 4a; colorless crystals; yield 62%; mp 185–187 °C; IR (KBr) ν 3387 (NH), 1728, 1627 (C
O) cm−1; 1H NMR (DMSO-d6) δ 1.41 (brs, 4H, 2CH2CH2N), 2.49 (s, 6H, 2CH3CO), 3.11–3.13 (m, 4H, 2CH2N), 4.55 (s, 4H, 2CH2O), 7.03 (d, 4H, J = 8.7 Hz, ArH's), 7.91 (d, 4H, J = 8.7 Hz, ArH's), 8.12 (t, 2H, J = 5.7 Hz, 2NH), ppm. Anal. calcd for C24H28N2O6 (440.50): C, 65.44; H, 6.41; N, 6.36%. Found: C, 65.50; H, 6.45; N, 6.39%.
4.1.2.5 N,N′-(Ethane-1,2-diyl)bis(2-(2-acetylphenoxy)acetamide) (5d). From 3a and 4b gave crude 5d; colorless crystals; yield 65%; mp 122–124 °C; IR (KBr) ν 3379 (NH), 1712, 1643 (C
O) cm−1; 1H NMR (CDCl3) δ 2.46 (s, 6H, 2CH3CO), 3.58–3.60 (m, 4H, 2CH2N), 4.52 (s, 4H, 2CH2O), 6.87 (d, 2H, J = 8.4 Hz, ArH's), 7.02 (t, 2H, J = 7.5 Hz, ArH's), 7.46 (t, 2H, J = 7.2 Hz, ArH's), 7.66 (d, 2H, J = 7.8 Hz, ArH's), 8.17 (brs, 2H, 2NH) ppm. Anal. calcd for C22H24N2O6 (412.44): C, 64.07; H, 5.87; N, 6.79%. Found: C, 64.19; H, 5.85; N, 6.90%.
4.1.2.6 N,N′-(Propane-1,3-diyl)bis(2-(2-acetylphenoxy)acetamide) (5e). From 3b and 4b; colorless crystals; yield 60%; mp 110–112 °C; IR (KBr) ν 3340 (NH), 1674, 1674 (C
O) cm−1; 1H NMR (CDCl3) δ 1.88 (quintet, 2H, J = 6.3 Hz, CH2CH2CH2), 2.61 (s, 6H, 2CH3CO), 3.39–3.45 (m, 4H, 2CH2N), 4.56 (s, 4H, 2CH2O), 6.92 (d, 2H, J = 8.1 Hz, ArH's), 7.07 (t, 2H, J = 7.8 Hz, ArH's), 7.49 (t, 2H, J = 7.8 Hz, ArH's), 7.74 (d, 2H, J = 7.5 Hz, ArH's), 8.06 (brs, 2H, 2NH) ppm. Anal. calcd for C23H26N2O6 (426.47): C, 64.78; H, 6.15; N, 6.57%. Found: C, 64.69; H, 6.18; N, 6.65%.
4.1.2.7 N,N′-(Butane-1,4-diyl)bis(2-(2-acetylphenoxy)acetamide) (5f). From 3c and 4b; colorless crystals; yield 63%, mp 158–160 °C; IR (KBr) ν 3333 (NH), 1674 (C
O) cm−1; 1H NMR (CDCl3) δ 1.68 (t, 4H, J = 2.7 Hz 2CH2CH2N), 2.58 (s, 6H, 2CH3CO), 3.39–3.41 (m, 4H, 2CH2N), 4.54 (s, 4H, 2CH2O), 6.91 (d, 2H, J = 8.7 Hz, ArH's), 7.06 (t, 2H, J = 7.5 Hz, ArH's), 7.49 (t, 2H, J = 8.4 Hz, ArH's), 7.73 (d, 2H, J = 7.8 Hz, ArH's), 7.93 (brs, 2H, 2NH), ppm; 13C NMR (CDCl3, APT) δ 26.63, 38.59, 67.66 (3CH2 aliphatic), 29.68 (CH3), 113.49, 121.33, 131.11, 134.13 (4CH, ArC's) 127.03, 156.26 (2C, ArC's), 167.85, 198.81 (2CO) ppm. Anal. calcd for C24H28N2O6 (440.50): C, 65.44; H, 6.41; N, 6.36%. Found: C, 65.55; H, 6.49; N, 6.39%.
4.1.3.1 General procedure. To a stirred solution of the appropriate 1,ω-bis((acetylphenoxy)acetamide)alkanes 5a–f (10 mmol) and p-toluenesulfonic acid monohydrate (p-TsOH) (5.7 g, 30 mmol) in acetonitrile (50 mL), was slowly added NBS (3.6 g, 20 mmol). After complete addition of NBS, the reaction mixture was refluxed with stirring for 3 h. The solvent was then removed under vacuum and the residue was left to cool to room temperature. Crushed ice (10 g) was added to the reaction mixture and the resulting mixture was stirred at 0 °C for 15 min. The solid product was collected by filtration, dried and recrystallized from the suitable solvent for each derivative to afford the corresponding bis(α-bromoketones) 6a–f.
4.1.3.2 N,N′-(Ethane-1,2-diyl)bis(2-(4-(2-bromoacetyl)phenoxy)acetamide) (6a). Crystalized from dioxane as colorless crystals, yield 60%, mp 198–200 °C; IR (KBr) ν 3371 (NH), 1751, 1650 (C
O) cm−1; 1H NMR (DMSO-d6) δ 3.24–3.26 (m, 4H, 2CH2N), 4.57 (s, 4H, 2CH2O), 4.94 (s, 4H, 2CH2Br), 7.05 (d, 4H, J = 8.7 Hz, ArH's), 8.04 (d, 4H, J = 9 Hz, ArH's), 8.26 (s, 2H, 2NH) ppm. Anal. calcd for C22H22Br 2N2O6 (570.23): C, 46.34; H, 3.89; N, 4.91%. Found: C, 46.41; H, 3.91; N, 4.93%.
4.1.3.3 N,N′-(Propane-1,3-diyl)bis(2-(4-(2-bromoacetyl)phenoxy)acetamide) (6b). Crystalized from dioxane as colorless crystals, yield 73%, mp 178–180 °C; IR (KBr) ν 3366 (NH), 1643 (C
O) cm−1; 1H NMR (DMSO-d6) δ 1.59 (quintet, 2H, J = 6.6 Hz, CH2CH2CH2), 3.10–3.17 (m, 4H, 2CH2N), 4.63 (s, 4H, 2CH2O), 5.47 (s, 4H, 2CH2Br), 7.05 (d, 4H, J = 8.7 Hz, ArH's), 8.03 (d, 4H, J = 9 Hz, ArH's), 8.21 (t, 2H, J = 5.7 Hz, 2NH) ppm. Anal. calcd for C23H24Br 2N2O6 (584.26): C, 47.28; H, 4.14; N, 4.79%. Found: C, 47.31; H, 4.19; N, 4.86%.
4.1.3.4 N,N′-(Butane-1,4-diyl)bis(2-(4-(2-bromoacetyl)phenoxy)acetamide) (6c). Crystalized from dioxane as colorless crystals, yield 70%, mp 162–164 °C; IR (KBr) ν 3294 (NH), 1643 (C
O) cm−1; 1H NMR (DMSO-d6) δ 1.42 (s, 4H, 2CH2CH2N), 3.12–3.13 (m, 4H, 2CH2N), 4.59 (s, 4H, 2CH2O), 4.82 (s, 4H, 2CH2Br), 7.07 (d, 4H, J = 9.3 Hz, ArH's), 7.98 (d, 4H, J = 8.7 Hz, ArH's), 8.15 (brs, 2H, 2NH) ppm. Anal. calcd for C24H26Br 2N2O6 (598.29): C, 48.18; H, 4.38; N, 4.68%. Found: C, 48.22; H, 4.45; N, 4.77%.
4.1.3.5 N,N′-(Ethane-1,2-diyl)bis(2-(2-(2-bromoacetyl)phenoxy)acetamide) (6d). Crystalized from ethanol/dioxane mixture as colorless crystals, yield 77%, mp 160–162 °C IR (KBr) ν 3294 (NH), 1674 (C
O) cm−1; 1H NMR (CDCl3) δ 3.57–3.58 (m, 4H, 2CH2N), 4.44 (s, 4H, 2CH2O), 4.57 (s, 4H, 2CH2Br), 6.91 (d, 2H, J = 8.4 Hz, ArH's), 7.07 (t, 2H, J = 7.5 Hz, ArH's), 7.53 (t, 2H, J = 7.2 Hz, ArH's), 7.73 (d, 2H, J = 7.5 Hz, ArH's), 7.83 (brs, 2H, 2NH) ppm. Anal. calcd for C22H22Br 2N2O6 (570.23): C, 46.34; H, 3.89; N, 4.91%. Found: C, 46.39; H, 3.95; N, 4.99%.
4.1.3.6 N,N′-(Propane-1,3-diyl)bis(2-(2-(2-bromoacetyl)phenoxy)acetamide) (6e). Crystalized from ethanol as colorless crystals, yield 73%, mp 140–142 °C; IR (KBr) ν 3286 (NH), 1666 (C
O) cm−1; 1H NMR (CDCl3) δ 1.79 (quintet, 2H, J = 6.3 Hz, CH2CH2CH2), 3.33–3.39 (m, 4H, 2CH2N), 4.56 (s, 4H, 2CH2O), 4.62 (s, 4H, 2CH2Br), 6.95 (d, 2H, J = 8.4 Hz, ArH's), 7.11 (t, 2H, J = 7.5 Hz, ArH's), 7.55 (t, 2H, J = 7.2 Hz, ArH's), 7.75 (brs, 2H, 2NH), 7.80 (d, 2H, J = 7.8 Hz, ArH's) ppm; 13C NMR (CDCl3, APT) δ 29.06, 35.45, 35.67, 67.83 (4CH2 aliphatic), 113.09, 121.84, 131.50, 135.07 (4CH, ArC's) 124.43, 156.66 (2C, ArC's), 167.97, 191.98 (2CO) ppm. Anal. calcd for C23H24Br 2N2O6 (584.26): C, 47.28; H, 4.14; N, 4.79%. Found: C, 47.10; H, 4.18; N, 4.65%.
4.1.3.7 N,N′-(Butane-1,4-diyl)bis(2-(2-(2-bromoacetyl)phenoxy)acetamide) (6f). Crystalized from ethanol as colorless crystals, yield 60%, mp 148–150 °C; IR (KBr) ν 3379 (NH), 1713 (C
O) cm−1; 1H NMR (CDCl3) δ 1.64 (s, 4H, 2CH2CH2N), 3.37–3.39 (m, 4H, 2CH2N), 4.47 (s, 4H, 2CH2O), 4.60 (s, 4H, 2CH2Br), 6.94 (d, 2H, J = 8.4 Hz, ArH's), 7.10 (t, 2H, J = 7.5 Hz, ArH's), 7.53 (brs, 2H, 2NH), 7.55 (t, 2H, J = 7.2 Hz, ArH's), 7.77 (d, 2H, J = 7.8 Hz, ArH's) ppm. Anal. calcd for C24H26Br 2N2O6 (598.29): C, 48.18; H, 4.38; N, 4.68%. Found: C, 48.28; H, 4.42; N, 4.75%.
4.1.4.1 General procedure. To a mixture of the appropriate bis(α-bromoacetophenone) derivative 6a–f (5 mmol) and the appropriate aminotriazolethiol derivative 7a–d (10 mmol) in DMF/absolute ethanol mixture (25 mL, 4
:1, v/v), piperidine (10 mmol) was added and the mixture was heated under reflux for 8 h, then left to cool to room temperature. Crushed ice was (5 g) added to the reaction mixture and chest in the frig overnight. The resulting precipitate was collected by filtration, dried and recrystallized from the suitable solvent to afford the corresponding bis(fused-heterocyclic) derivatives 8a–l in good yields.
4.1.4.2 N,N′-(Ethane-1,2-diyl)bis(2-(4-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8a). From 6a and 7a, crystallized from DMF as colorless crystals, yield 62%, mp 264–266 °C; IR (KBr) ν 3433 (NH), 1666 (C
O) cm−1; MS m/z (%): 77 (73%), 104 (100%), 118 (87%), 177 (59%), 308 (2%), 365 (1.3%), 444 (2%), 568 (3%), 668 (3.2%), 705 (2%), (%), 755 (M+ − 1, 1.5%); 1H NMR (DMSO-d6) δ 3.27 (m, 4H, 2CH2N), 4.36 (s, 4H, 2CH2S), 4.57 (s, 4H, 2CH2O), 7.11 (d, 4H, J = 8.1 Hz, ArH's), 7.55 (s, 6H, ArH's), 7.95 (d, 4H, J = 8.1 Hz, ArH's), 7.99–8.01 (m, 4H, ArH's), 8.23 (brs, 2H, 2NH) ppm. Anal. calcd for C38H32N10O4S2 (756.86): C, 60.30; H, 4.26; N, 18.51%. Found: C, 60.39; H, 4.16; N, 18.59%.
4.1.4.3 N,N′-(Ethane-1,2-diyl)bis(2-(4-(3-benzyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8b). From 6a and 7b, crystalized from DMF/ethanol as yellow crystals, yield 60%, mp 140–142 °C IR (KBr) ν 3194 (NH), 1666 (C
O) cm−1; MS m/z (%): 91 (100%), 118 (45%), 313 (0.6%), 400 (0.5%), 457 (0.5%), 501 (1%), 601 (0.5%), 643 (0.5%), 699 (0.3%), 750 (0.3%), 784 (M+, 0.2%); 1H NMR (DMSO-d6) δ 3.28 (s, 4H, 2CH2N), 4.26 (s, 4H, 2CH2S), 4.30 (s, 4H, 2CH2Ph), 4.56 (s, 4H, 2CH2O), 7.07 (d, 4H, J = 8.1 Hz, ArH's), 7.23 (m, 10H, ArH's), 7.93 (d, 4H, J = 8.4 Hz, ArH's), 8.24 (brs, 2H, 2NH) ppm. Anal. calcd for C40H36N10O4S2 (784.91): C, 61.21; H, 4.62; N, 17.85%. Found: C, 61.05; H, 4.69; N, 17.69%.
4.1.4.4 N,N′-(Ethane-1,2-diyl)bis(2-(2-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8c). From 6d and 7a, crystalized from DMF/ethanol as yellow crystals, yield 60%, mp 240–242 °C IR (KBr) ν 3209 (NH), 1681 (C
O) cm−1; MS m/z (%): 97 (61%), 149 (100%), 198 (20%), 322 (13%), 449 (13%), 593 (14%), 636 (15%), 707 (16%), 756 (M+, 17%); 1H NMR (DMSO-d6) δ 3.24 (m, 4H, 2CH2N), 4.36 (s, 4H, 2CH2S), 4.63 (s, 4H, 2CH2O), 7.07–7.12 (m, 4H, ArH's), 7.51–7.60 (m, 10H, ArH's), 7.99 (m, 4H, ArH's), 8.08 (s, 2H, 2NH) ppm; 13C NMR (DMSO-d6) δ 25.42, 38.29, 67.37, 113.18, 121.11, 121.64, 124.17, 125.96, 127.89, 128.77, 130.27, 132.95, 143.02, 151.65, 156.28, 157.26, 167.61 ppm. Anal. calcd for C38H32N10O4S2 (756.86): C, 60.30; H, 4.26; N, 18.51%. Found: C, 60.22; H, 4.14; N, 18.60%.
4.1.4.5 N,N′-(Ethane-1,2-diyl)bis(2-(2-(3-methyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8d). From 6d and 7c, crystalized from ethanol as yellow crystals, yield 61%, mp 240–242 °C; IR (KBr) ν 3263 (NH), 1674 (C
O) cm−1; MS m/z (%): 97 (67%), 115 (100%), 130 (49%), 172 (21%), 246 (24%), 313 (11%), 447 (3%), 525 (3%), 604 (4%), 632 (M+, 4%); 1H NMR (DMSO-d6) δ 2.44 (s, 6H, 2CH3), 3.23 (m, 4H, 2CH2N), 4.29 (s, 4H, 2CH2S), 4.61 (s, 4H, 2CH2O), 7.08–7.13 (m, 4H, ArH's), 7.50–7.59 (m, 4H, ArH's), 8.08 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ 9.96, 25.65, 38.21, 67.32, 105.78, 113.09, 120.69, 124.28, 130.13, 132.81, 140.46, 145.46, 156.20, 167.55 ppm. Anal. calcd for C28H28N10O4S2 (632.72): C, 53.15; H, 4.46; N, 22.14%. Found: C, 53.22; H, 4.52; N, 22.01%.
4.1.4.6 N,N′-(Propane-1,3-diyl)bis(2-(4-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8e). From 6b and 7a, crystalized from dioxane/ethanol as yellow crystals, yield 60%, mp 128–130 °C; IR (KBr) ν 3410 (NH), 1659 (C
O) cm−1; MS m/z (%): 58 (100%), 77 (59%), 104 (71%), 121 (46%), 177 (21%), 192 (59%), 256 (5%), 368 (1.5%), 650 (1.5%), 743 (1%), 770 (M+, 1%); 1H NMR (DMSO-d6) δ 1.62 (quintet, 2H, J = 6.6 Hz, CH2CH2CH2), 3.05–3.20 (m, 4H, 2CH2N), 4.39 (s, 4H, 2CH2S), 4.60 (s, 4H, 2CH2O), 7.13 (d, 4H, J = 9 Hz, ArH's), 7.53–7.60 (m, 6H, ArH's), 7.96–8.03 (m, 8H, ArH's), 8.12 (t, 2H, J = 5.7 Hz, 2NH) ppm. Anal. calcd for C39H34N10O4S2 (770.89): C, 60.76; H, 4.45; N, 18.17%. Found: C, 60.69; H, 4.49; N, 18.25%.
4.1.4.7 N,N′-(Propane-1,3-diyl)bis(2-(4-(3-methyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8f). From 6b and 7c, crystalized from ethanol as yellow crystals, yield 52%, mp 128–130 °C; IR (KBr) ν 3441 (NH), 1659 (C
O) cm−1; MS m/z (%): 56 (28%), 115 (100%), 130 (5%), 303 (0.5%), 390 (0.2%), 451 (0.2%), 482 (0.3%), 523 (0.5%), 551 (0.3%), 604 (0.3%), 646 (M+, 0.3%); 1H NMR (DMSO-d6) δ 1.61 (quintet, 2H, J = 6.6 Hz, CH2CH2CH2), 2.46 (s, 6H, 2CH3), 3.10–3.20 (m, 4H, 2CH2N), 4.32 (s, 4H, 2CH2S), 4.59 (s, 4H, 2CH2O), 7.12 (d, 4H, J = 8.7 Hz, ArH's), 7.99 (d, 4H, J = 8.7 Hz, ArH's), 8.18 (t, 2H, J = 5.4 Hz, 2NH) ppm. Anal. calcd for C29H30N10O4S2 (646.75): C, 53.86; H, 4.68; N, 21.66%. Found: C, 53.95; H, 4.75; N, 21.56%.
4.1.4.8 N,N′-(Propane-1,3-diyl)bis(2-(2-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8g). From 6e and 7a, crystalized from DMF as pale yellow crystals, yield 52%, mp 270–272 °C; IR (KBr) ν 3225 (NH), 1682 (C
O) cm−1; MS m/z (%): 77 (71%), 91 (64%), 104 (100%), 118 (67%), 177 (69%), 192 (20%), 308 (14%), 495 (3%), 598 (3%), 732 (3%), 770 (M+, 3%); 1H NMR (DMSO-d6) δ 1.57 (quintet, 2H, J = 6.6 Hz, CH2CH2CH2), 3.10–3.16 (m, 4H, 2CH2N), 4.38 (s, 4H, 2CH2S), 4.60 (s, 4H, 2CH2O), 7.09–7.14 (m, 4H, ArH's), 7.51–7.61 (m, 10H, ArH's), 7.94–8.06 (m, 6H, 4ArH's & 2NH) ppm. Anal. calcd for C39H34N10O4S2 (770.89): C, 60.76; H, 4.45; N, 18.17%. Found: C, 60.79; H, 4.55; N, 18.02%.
4.1.4.9 N,N′-(Propane-1,3-diyl)bis(2-(2-(3-benzyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8h). From 6e and 7b, crystalized from ethanol as yellow crystals, yield 62%, mp 202–204 °C; IR (KBr) ν 3287 (NH), 1682 (C
O) cm−1; MS m/z (%): 77 (21%), 91 (100%), 191 (48%), 275 (7%), 415 (3%), 567 (3%), 674 (3%), 733 (3%), 798 (M+, 2%); 1H NMR (CDCl3) δ 1.56 (brs, 2H, CH2CH2CH2), 3.20–3.22 (m, 4H, 2CH2N), 3.99 (s, 4H, 2CH2S), 4.26 (s, 4H, 2CH2Ph), 4.29 (s, 4H, 2CH2O), 6.96–7.54 (m, 18H, ArH's), 7.56 (t, 2H, J = 7.2 Hz, 2NH) ppm. Anal. calcd for C41H38N10O4S2 (798.94): C, 61.64; H, 4.79; N, 17.53%. Found: C, 61.51; H, 4.85; N, 17.59%.
4.1.4.10 N,N′-(Propane-1,3-diyl)bis(2-(2-(3-methyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8i). From 6e and 7c, crystalized from methanol as yellow crystals, yield 72%, mp 178–180 °C; IR (KBr) ν 3433 (NH), 1682 (C
O) cm−1; MS m/z (%): 97 (31%), 115 (100%), 130 (75%), 172 (36%), 246 (12%), 304 (7%), 444 (4%), 465 (2%), 588 (5%), 646 (M+, 3%); 1H NMR (CDCl3) δ 1.62 (brs, 2H, CH2CH2CH2), 2.50 (s, 6H, 2CH3), 3.20–3.35 (m, 4H, 2CH2N), 4.07 (s, 4H, 2CH2S), 4.49 (s, 4H, 2CH2O), 7.03 (d, 2H, J = 8.4 Hz, ArH's), 7.15 (t, 2H, J = 7.5 Hz, ArH's), 7.49–7.58 (m, 6H, 4ArH's & 2NH) ppm. Anal. calcd for C29H30N10O4S2 (646.75): C, 53.86; H, 4.68; N, 21.66%. Found: C, 53.92; H, 4.53; N, 21.82%.
4.1.4.11 N,N′-(Butane-1,4-diyl)bis(2-(4-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8j). From 6c and 7a, which was crystalized from DMF as yellow crystals, yield 72%, mp 260–262 °C; IR (KBr) ν 3280 (NH), 1659 (C
O) cm−1; MS m/z (%): 77 (41%), 104 (59%), 118 (69%), 177 (100%), 375 (1%), 527 (2%), 620 (1.2%), 785 (M+ + 1, %); 1H NMR (DMSO-d6) δ 1.43 (brs, 4H, CH2CH2N), 3.15 (s, 4H, 2CH2NH), 4.39 (s, 4H, 2CH2S), 4.58 (s, 4H, 2CH2O), 7.12 (d, 4H, J = 8.7 Hz, ArH's), 7.55–7.57 (m, 6H, ArH's), 7.95–8.03 (m, 8H, ArH's), 8.14 (t, 2H, J = 5.4 Hz, 2NH) ppm. Anal. calcd for C40H36N10O4S2 (784.91): C, 61.21; H, 4.62; N, 17.85%. Found: C, 61.17; H, 4.72; N, 17.77%.
4.1.4.12 N,N′-(Butane-1,4-diyl)bis(2-(2-(3-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)acetamide) (8k). From 6f and 7a, crystalized from DMF as yellow crystals, yield 65%, mp 262–264 °C; IR (KBr) ν 3371 (NH), 1682 (C
O) cm−1; MS m/z (%): 77 (62%), 118 (84%), 177 (100%), 308 (4%), 338 (2%), 442 (1.2%), 560 (1.5%), 618 (1.5%), 715 (2%), 784 (M+, 1.2%); 1H NMR (DMSO-d6) δ 1.40 (brs, 4H, CH2CH2N), 3.10–3.12 (m, 4H, 2CH2N), 4.39 (s, 4H, 2CH2S), 4.67 (s, 4H, 2CH2O), 7.08–7.13 (m, 6H, ArH's), 7.52–7.61 (m, 10H, ArH's), 7.98–8.01 (m, 4H, 2ArH's & 2NH) ppm; 13C NMR (DMSO-d6) δ 25.43, 26.50, 38.10, 67.48 (aliphatic C's), 113.19, 121.62, 124.18, 125.96, 127.89, 128.76, 130.25, 132.92, 142.99, 151.65, 156.38, 157.23, 167.09 (CO) ppm. Anal. calcd for C40H36N10O4S2 (784.91): C, 61.21; H, 4.62; N, 17.85%. Found: C, 61.15; H, 4.66; N, 17.78%.
4.1.4.13 N,N′-(Butane-1,4-diyl)bis(2-(2-(7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)phenoxy)-acetamide) (8l). From 6f and 7d, crystalized from acetic acid/ethanol as yellow crystals, yield 72%, mp 132–134 °C; IR (KBr) ν 3240 (NH), 1674 (C
O) cm−1; MS m/z (%): 101 (100%), 322 (22%), 363 (1.2%), 435 (0.2%), 483 (0.2%), 527 (0.2%), 606 (0.23%), 633 (M+ + 1, 0.2%); 1H NMR (DMSO-d6) δ 1.40 (s, 4H, CH2CH2N), 3.13 (m, 4H, 2CH2N), 4.37 (s, 4H, 2CH2S), 4.65 (s, 4H, 2CH2O), 7.08–7.12 (m, 4H, ArH's), 7.51–7.58 (m, 4H, ArH's), 8.01 (t, 2H, J = 5.4 Hz, 2NH), 9.14 (s, 2H, triazole-3H) ppm; 13C NMR (DMSO-d6) δ 26.37, 26.53, 38.13, 67.46 (aliphatic C's), 113.18, 120.90, 121.56, 124.07, 130.07, 132.95, 143.11, 156.34, 157.12, 167.13 (CO) ppm. Anal. calcd for C28H28N10O4S2 (632.72): C, 53.15; H, 4.46; N, 22.14%. Found: C, 53.27; H, 4.54; N, 22.24%.
4.1.5.1 General procedure. To a mixture of the appropriate bis(α-bromoacetophenone) derivative 6a, b, d and o-phenylenediamine derivatives 9a, b (2 mmol) in ethanol (25 mL), piperidine (0.19 mL, 2 mmol) was added. The reaction mixture was heated under reflux for 3 h. The solvent was then removed under vacuum, and the solid product was collected by filtration, dried and recrystallized from ethanol to afford the corresponding bis-quinoxaline derivatives 10a–e.
4.1.5.2 N,N′-(Ethane-1,2-diyl)bis(2-(4-(quinoxalin-2-yl)phenoxy)acetamide) (10a). From 6a and 9a, crystalized from dioxane as yellow crystals, yield 55%, mp 250–252 °C; IR (KBr) ν 3433 (NH), 1643 (C
O), 1620 (CN) cm−1; MS m/z (%): 102 (77%), 235 (100%), 293 (54%), 350 (17%), 526 (8%), 584 (M+, 38%); 1H NMR (DMSO-d6) δ 3.30 (m, 4H, 2CH2N), 4.58 (s, 4H, 2CH2O), 7.15 (d, 4H, J = 8.7 Hz, ArH's), 7.73–7.84 (m, 4H, ArH's), 8.04 (d, 4H, J = 8.7 Hz, ArH's), 8.24–8.27 (m, 6H, 4ArH's & 2NH), 9.47 (s, 2H, quinoxaline-3-H) ppm; 13C NMR (CDCl3, APT) δ 39.13, 67.49 (2CH2), 126.37, 140.84, 141.78, 151.47, 154.92, 168.81 (6C's), 112.85, 122.59, 128.88, 129.82, 130.43, 131.62, 146.13 (7CH's) ppm. Anal. calcd for C34H28N6O4 (584.64): C, 69.85; H, 4.83; N, 14.38%. Found: C, 69.74; H, 4.95; N, 14.26%.
4.1.5.3 N,N′-(Ethane-1,2-diyl)bis(2-(4-(6,7-dimethylquinoxalin-2-yl)phenoxy)acetamide) (10b). From 6a and 9b, crystalized from DMF as yellow crystals, yield 60%, mp 254–256 °C IR (KBr) ν 3356 (NH), 1643 (C
O), 1620 (CN) cm−1; MS m/z (%): 103 (57%), 233 (58%), 250 (100%), 263 (89%), 320 (39%), 378 (17%), 515 (4%), 612 (3%), 640 (M+, 89%); 1H NMR (DMSO-d6) δ 2.42 (s, 12H, 4CH3), 3.33 (m, 4H, 2CH2N), 4.55 (s, 4H, 2CH2O), 7.09 (d, 4H, J = 8.7 Hz, ArH's), 7.77 (s, 4H, 2ArH's & 2NH), 8.16 (m, 6H, ArH's), 9.29 (s, 2H, quinoxaline-3H) ppm. Anal. calcd for C38H36N6O4 (640.74): C, 71.23; H, 5.66; N, 13.12%. Found: C, 71.12; H, 5.70; N, 13.01%.
4.1.5.4 N,N′-(Ethane-1,2-diyl)bis(2-(2-(quinoxalin-2-yl)phenoxy)acetamide) (10c). From 6d and 9a, crystalized from ethanol as brown crystals, yield 50%, mp 210–212 °C; IR (KBr) ν 3341 (NH), 1666 (C
O), 1600 (CN) cm−1; MS m/z (%): 102 (24%), 207 (21%), 235 (100%), 292 (24%), 349 (9%), 554 (10%), 584 (M+, 43%); 1H NMR (CDCl3) δ 3.43 (m, 4H, 2CH2N), 4.46 (s, 4H, 2CH2O), 6.94 (d, 2H, J = 8.1 Hz, ArH's), 7.17 (t, 2H, J = 7.5 Hz, ArH's), 7.44 (t, 2H, J = 6.9 Hz, ArH's), 7.68–7.76 (m, 8H, 6ArH's & 2NH), 7.99–8.02 (m, 4H, ArH's), 9.07 (s, 2H, quinoxaline-3H) ppm. Anal. calcd for C34H28N6O4 (584.64): C, 69.85; H, 4.83; N, 14.38%. Found: C, 69.77; H, 4.77; N, 14.25%.
4.1.5.5 N,N′-(Propane-1,3-diyl)bis(2-(4-(quinoxalin-2-yl)phenoxy)acetamide) (10d). From 6a and 9a, crystalized from ethanol as brown crystals, yield 66%, mp 184–186 °C; IR (KBr) ν 3418 (NH), 1659 (C
O), 1610 (CN) cm−1; MS m/z (%): 98 (69%), 121 (19%), 205 (6%), 235 (100%), 281 (14%), 364 (30%), 512 (5%), 598 (M+, 17%); 1H NMR (DMSO-d6) δ 1.65 (quintet, 2H, J = 6.6 Hz, CH2CH2CH2), 3.10–3.25 (m, 4H, 2CH2N), 4.61 (s, 4H, 2CH2O), 7.17 (d, 4H, J = 8.1 Hz, ArH's), 7.80 (m, 4H, ArH's), 8.06 (d, 4H, J = 7.8 Hz, ArH's), 8.21 (brs, 2H, 2NH), 8.30 (d, 4H, J = 8.4 Hz, ArH's), 9.50 (s, 2H, quinoxaline-3H) ppm. Anal. calcd for C35H30N6O4 (598.66): C, 70.22; H, 5.05; N, 14.04%. Found: C, 70.29; H, 4.94; N, 13.89%.
4.1.5.6 N,N′-(Butane-1,4-diyl)bis(2-(2-(6,7-dimethylquinoxalin-2-yl)phenoxy)acetamide) (10e). From 6f and 9b, crystalized from DMF/ethanol as yellow crystals, yield 70%, mp 210–212 °C; IR (KBr) ν 3240 (NH), 1674 (C
O), 1620 (CN) cm−1; MS m/z (%): 103 (17%), 235 (21%), 249 (19%), 263 (100%), 405 (6%), 638 (3%), 668 (M+, 32%); 1H NMR (CDCl3) δ 1.29 (s, 4H, 2 CH2CH2N), 2.46 (s, 12H, 4CH3), 3.19–3.21 (m, 4H, 2 NHCH2CH2), 4.59 (s, 4H, 2CH2O), 7.02 (d, 2H, J = 8.1 Hz, ArH's), 7.17 (t, 2H, J = 7.5 Hz, ArH's), 7.35 (s, 2H, 2NH), 7.43 (t, 2H, J = 7.5 Hz, ArH's), 7.65–7.75 (m, 6H, ArH's), 8.93 (s, 2H, quinoxaline-3H) ppm; 13C NMR (CDCl3, APT) δ 20.35, 20.41 (2CH3), 26.45, 38.35, 67.61 (3CH2's, aliphatic), 112.79, 122.52, 127.73, 127.78, 131.38, 131.54, 144.83 (7CH's), 126.48, 139.74, 140.46, 140.69, 141.10, 150.27, 154.85, 167.91 (8C's) ppm. Anal. calcd for C40H40N6O4 (668.80): C, 71.84; H, 6.03; N, 12.57%. Found: C, 71.79; H, 6.22; N, 12.45%.
4.2. Biological assays
The biological activity of the synthesized molecules including cytotoxicity against breast cancer cell lines (MDA-MB-231 cells), enzymes' assay of the most active compounds, Annexin V-FITC/Propidium iodide dual staining assay for measuring the apoptotic effect of compound 8i were done at Center of Excellence in tissue culture in the National research institute and Suez Canal University, Egypt.4.3. Cytotoxicity
Breast cancer cell lines (MCF-7 and MDA-MB-231), and the normal breast cell lines (MCF-10A) were collected from National Cancer Institute in Cairo and grown in “RPMI-1640 medium L-glutamine. The cells were grown in 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. All samples were cultured at 37 degrees Celsius in a 5% CO2 gas. On the second day, the cells were grown in triplicate on a 96-well plate at a density of 5 × 104 cells and incubated with the investigated compounds at concentrations; 0.01, 0.1, 1, 10, and 100 μM. MTT solution was used to determine the cell viability. The plate was cultured for 3 hours. An ELISA microplate reader was used to measure the absorbance. The viability was determined by comparing it to the control group, and the IC50 values were recorded.58,594.4. Enzymatic targeting
To assess the inhibitory potency of compound 8i against the PARP-1 and EGFR target proteins. Research kits of PARP-1 (Bioscience, Cat no. # 80580, CA, USA) and EGFR (ADP-GloTM kinase assay, Cat no. V9261, Promega, USA), were employed. The percentage inhibition of autophosphorylation by substances was estimated using the following equation: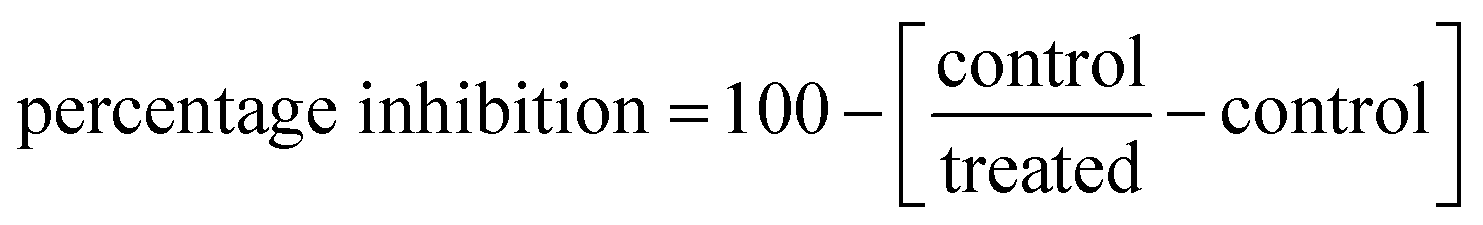 .60,61
.60,61
4.5. Apoptosis investigation
MDA-MB-231 cells were incubated overnight in 6-well culture plates (3–5 × 105 cells per well) and then treated with the IC50 values of compound 8i for 48 hours. After that, the cells were incubated in a 100 μL solution of Annexin binding buffer “25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH at pH 7.4” in the dark for 30 minutes with “Annexin V-FITC solution (1:100) and propidium iodide (PI) at a concentration equivalent to 10 μg mL−1”. The labeled cells were then extracted using the Cytoflex FACS machine. CytExpert software was used to analyze the data.62,63
4.6. Molecular docking studies
Molecular modeling studies were carried out using Chimera-UCSF and AutoDock Vina on Linux-based systems at the laboratory of Drug Design and Discovery, Suez Canal University. Proteins and compounds structures were prepared and optimized using Maestro, then binding sites inside proteins were determined using grid-box dimensions around the co-crystallized ligands. The investigated compounds were docked against the protein structures of PARP-1 (PDB = 5DS3) EGFR (PDB = 1XKK) using AutoDock Vina software following routine work.64 Vina was used to improve protein and ligand structures and to favor them energetically. Binding activities interpreted molecular docking results in terms of binding energy and ligand–receptor interactions. The visualization was then done with Chimera.Conflicts of interest
The authors declare that there is no conflict-of-interest text.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209 CrossRef PubMed.
- P. A. Jeggo and M. Löbrich, Biochem. J., 2015, 471, 1 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290 CrossRef CAS.
- R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed.
- A. Johansson, P. Kollman, S. Rothenberg and J. McKelvey, J. Am. Chem. Soc., 1974, 96, 3794 CrossRef CAS.
- W. Sneader, in Comprehensive Medicinal Chemistry, ed. C. Hansch, P. G. Sammnes and J. B. Taylor, Pergamon, London, 1990, vol. 1, p. 65 Search PubMed.
- R. Aggarwal and G. Sumran, Eur. J. Med. Chem., 2020, 205, 112652 CrossRef CAS PubMed.
- X. Jiang, K. Wu, R. Bai, P. Zhang and Y. Zhang, Eur. J. Med. Chem., 2022, 229, 114085 CrossRef CAS.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, H. A. Mahdy and H. Elkady, Bioorg. Chem., 2021, 110, 104807 CrossRef CAS PubMed.
- L. Fabian, M. Taverna Porro, N. Gomez, M. Salvatori, G. Turk, D. Estrin and A. Moglioni, Eur. J. Med. Chem., 2020, 188, 111987 CrossRef CAS.
- X. Lu, H. Chen, A. V. Patterson, J. B. Smaill and K. Ding, J. Med. Chem., 2019, 62, 2905 CrossRef CAS PubMed.
- S. Dai, Z. Zhou, Z. Chen, G. Xu and Y. Chen, Cells, 2019, 8, 614 CrossRef CAS.
- M. S. Ayoup, M. A. Fouad, H. Abdel-Hamid, E.-S. Ramadan, M. M. Abu-Serie, A. Noby and M. Teleb, Eur. J. Med. Chem., 2020, 186, 111875 CrossRef CAS PubMed.
- M. P. Gajewski, H. Beall, M. Schnieder, S. M. Stranahan, M. D. Mosher, K. C. Rider and N. R. Natale, Bioorg. Med. Chem. Lett., 2009, 19, 4067 CrossRef CAS.
- M. I. El-Gamal, M. S. Abdel-Maksoud, M. M. G. El-Din, K. H. Yoo, D. Baek and C. H. Oh, Arch. Pharm., 2014, 347, 635 CrossRef CAS.
- I. Khan, A. Ibrar and N. Abbas, Eur. J. Med. Chem., 2013, 63, 854 CrossRef CAS PubMed.
- A.-M. M. E. Omar, O. M. AboulWafa, M. E. Amr and M. S. El-Shoukrofy, Bioorg. Chem., 2021, 109, 104752 CrossRef CAS.
- W. Ma, P. Chen, X. Huo, Y. Ma, Y. Li, P. Diao, F. Yang, S. Zheng, M. Hu, W. You and P. Zhao, Eur. J. Med. Chem., 2020, 208, 112847 CrossRef CAS PubMed.
- Z. Liu, B. Lang, M. Gao, X. Chang, Q. Guan, Q. Xu, D. Wu, Z. Li, D. Zuo, W. Zhang and Y. Wu, J. Cell. Biochem., 2020, 121, 2184 CrossRef CAS PubMed.
- A. U. Çevik, B. N. Sağlık, D. Osmaniye, S. Levent, B. K. Çavuşoğlu, A. B. Karaduman, Y. Özkay and Z. A. Kaplancıklı, Arch. Pharm., 2020, 353, e2000008 Search PubMed.
- F. Yang, X.-E. Jian, L. Chen, Y.-F. Ma, Y.-X. Liu, W.-W. You and P.-L. Zhao, New J. Chem., 2021, 45, 21869 RSC.
- I. Khan, S. Zaib, A. Ibrar, N. H. Rama, J. Simpson and J. Iqbal, Eur. J. Med. Chem., 2014, 78, 167 CrossRef CAS.
- I. Khan, A. Ibrar, S. Zaib, S. Ahmad, N. Furtmann, S. Hameed, J. Simpson, J. Bajorath and J. Iqbal, Bioorg. Med. Chem., 2014, 22, 6163 CrossRef CAS PubMed.
- H. A. M. El-Sherief, B. G. M. Youssif, S. N. A. Bukhari, A. H. Abdelazeem, M. Abdel-Aziz and H. M. Abdel-Rahman, Eur. J. Med. Chem., 2018, 156, 774 CrossRef CAS PubMed.
- A. T. A. Boraei, P. K. Singh, M. Sechi and S. Satta, Eur. J. Med. Chem., 2019, 182, 111621 CrossRef CAS.
- A. T. A. Boraei, H. A. Ghabbour, M. S. Gomaa, E. S. H. El Ashry and A. Barakat, Molecules, 2019, 24, 4471 CrossRef PubMed.
- R. Aggarwal, M. Hooda, P. Kumar and G. Sumran, Top. Curr. Chem., 2022, 380, 10 CrossRef CAS.
- M. Kumar, G. Joshi, S. Arora, T. Singh, S. Biswas, N. Sharma, Z. R. Bhat, K. Tikoo, S. Singh and R. Kumar, Molecules, 2021, 26, 1490 CrossRef CAS.
- A. Iwashita, K. Hattori, H. Yamamoto, J. Ishida, Y. Kido, K. Kamijo, K. Murano, H. Miyake, T. Kinoshita, M. Warizaya, M. Ohkubo, N. Matsuoka and S. Mutoh, FEBS Lett., 2005, 579, 1389–1393 CrossRef CAS PubMed.
- G. Papeo, P. Orsini, N. R. Avanzi, D. Borghi, E. Casale, M. Ciomei, A. Cirla, V. Desperati, D. Donati, E. R. Felder, A. Galvani, M. Guanci, A. Isacchi, H. Posteri, S. Rainoldi, F. Riccardi-Sirtori, A. Scolaro and A. Montagnoli, ACS Med. Chem. Lett., 2019, 10, 534 CrossRef CAS PubMed.
- Y.-H. Kang, M.-J. Yi, M.-J. Kim, M.-T. Park, S. Bae, C.-M. Kang, C.-K. Cho, I.-C. Park, M.-J. Park, C. H. Rhee, S.-I. Hong, H. Y. Chung, Y.-S. Lee and S.-J. Lee, Cancer Res., 2004, 64, 8960 CrossRef CAS PubMed.
- P. Wee and Z. Wang, Cancers, 2017, 9, 52 CrossRef.
- M. M. Alam, Arch. Pharm., 2022, 355, 2100158 CrossRef CAS PubMed.
- A. C. Pinheiro, T. C. M. Nogueira and M. V. N. de Souza, Anti-Cancer Agents Med. Chem., 2016, 16, 1339 CrossRef CAS PubMed.
- A. Powelczyk, K. S. Kasprzak, D. Olender and L. Zaprutko, Int. J. Bio. Sci., 2018, 19, 1104 Search PubMed.
- N. Keru, P. Singh, N. Koorbanally, R. Raj and V. Kumar, Eur. J. Med. Chem., 2017, 142, 179 CrossRef PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422 CrossRef CAS PubMed.
- R. Dent, M. Trudeau, K. I. Pritchard, W. M. Hanna, H. K. Kahn, C. A. Sawka, L. A. Lickley, E. Rawlinson, P. Sun and S. A. Narod, Clin. Cancer Res., 2007, 13, 4429–4434 CrossRef PubMed.
- E. K. Lee and U. A. Matulonis, Cancers, 2020, 12, 2054 CrossRef CAS PubMed.
- S. Lin, X. Zhang, Z. Yu, X. Huang, J. Xu, Y. Liu and L. Wu, Bioorg. Med. Chem., 2022, 61, 116739 CrossRef CAS PubMed.
- W. K. A. Yung, S. Wu, F. Gao, S. Zheng, J. Ding, C. Zhang, X. Li, R. Ezhilarasan, N. Feng, J. F. De Groot, E. P. Sulman, T. Heffernan and D. Koul, J. Clin. Oncol., 2019, 37, 2047 Search PubMed.
- J.-M. Contreras and J.-J. Bourguignon, in The Practice of Medicinal Chemistry, ed. C. G. Wermuth, Academic Press, London, 2nd edn, 2003, pp. 251–273 Search PubMed.
- M. J. Wasko, K. A. Pellegrene, J. D. Madura and C. K. Surratt, Front. Neurol., 2015, 6, 197 Search PubMed.
- A. A. Abbas, M. H. Abdellattif and K. M. Dawood, Expert Opin. Ther. Pat., 2022, 32, 63 CrossRef CAS PubMed.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed and M. S. Nafie, Anti-Cancer Agents Med. Chem., 2022 DOI:10.2174/1871520622666220615140239.
- K. M. Dawood and A. A. Abbas, ChemistrySelect, 2021, 6, 279–305 CrossRef CAS.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed, M. H. Abdellattif, M. S. Nafie and M. K. Hassan, Front Org. Chem., 2021, 9, 694870 CrossRef CAS PubMed.
- K. M. Dawood and A. A. Abbas, Expert Opin. Ther. Pat., 2020, 30, 695–714 CrossRef CAS PubMed.
- K. M. Dawood and H. K. A. Abu-Deif, Chem. Pharm. Bull., 2014, 62, 439 CrossRef CAS PubMed.
- B. Hegazi, H. A. Mohamed, K. M. Dawood and F. A. R. Badria, Chem. Pharm. Bull., 2010, 58, 479 CrossRef CAS PubMed.
- K. M. Dawood, J. Heterocycl. Chem., 2019, 56, 1701 CrossRef CAS.
- K. M. Dawood, A. M. Farag and H. A. Abdel-Aziz, Heteroatom Chem, 2005, 16, 621 CrossRef CAS.
- H. Abdel-Gawad, H. A. Mohamed, K. M. Dawood and F. A. R. Badria, Chem. Pharm. Bull., 2010, 58, 1529 CrossRef CAS PubMed.
- H. K. Mahmoud, A. A. Abbas and S. M. Gomha, Polycycl. Aromat. Compd., 2021, 41, 2029 CrossRef CAS.
- R. M. Kassab, F. S. Khalil and A. A. Abbas, Polycycl. Aromat. Compd., 2022, 42(5), 2751–2766 CrossRef CAS.
- M. S. Nafie, S. M. Kishk, S. Mahgoub and A. M. Amer, Chem. Biol. Drug Des., 2022, 99, 547 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- M. S. Nafie, N. H. Elghazawy, S. M. Owf, K. Arafa, M. A. Abdel-Rahman and R. K. Arafa, Chem.-Biol. Interact., 2022, 351, 109753 CrossRef CAS PubMed.
- H. S. A. ElZahabi, M. S. Nafie, D. Osman, N. H. Elghazawy, D. H. Soliman, A. A. H. EL-Helby and R. K. Arafa, Eur. J. Med. Chem., 2021, 222, 113609 CrossRef CAS PubMed.
- S. K. Ramadan, E. Z. Elrazaz, K. A. M. Abouzid and A. M. El-Naggar, RSC Adv., 2020, 10, 29475 RSC.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Bioorg. Med. Chem., 2020, 28, 115828 CrossRef CAS PubMed.
- M. S. Nafie and A. T. A. Boraei, Bioorg. Chem., 2022, 122, 105708 CrossRef CAS PubMed.
- M. S. Nafie, M. A. Tantawy and G. A. Elmgeed, Steroids, 2019, 152, 108485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03549a |
| This journal is © The Royal Society of Chemistry 2022 |