DOI:
10.1039/D2RA03540E
(Review Article)
RSC Adv., 2022,
12, 22678-22694
Electrophilic halogenations of propargyl alcohols: paths to α-haloenones, β-haloenones and mixed β,β-dihaloenones†
Received
8th June 2022
, Accepted 2nd August 2022
First published on 12th August 2022
Abstract
The Meyer–Schuster rearrangement of propargyl alcohols or alkynols leading to α,β-unsaturated carbonyl compounds is well known. Yet, electrophilic halogenations of the same alkynols and their alkoxy, ester and halo derivatives are inconspicuous. This review on the halogenation reactions of propargyl alcohols and derivatives intends to give a perspective from its humble direct halogenation beginning to the present involving metal catalysis. The halogenation products of propargyl alcohols include α-fluoroenones, α-chloroenones, α-bromoenones and α-iodoenones, as well as β-haloenones and symmetrical and mixed β,β-dihaloenones. They are, in essence, tri and tetrasubstituted alkenes carrying halo-functionalization at the α- or β-carbon. This is a potential stepping stone for further construction towards challenging substituted alkenones via Pd-catalysed coupling reactions.
 Pakorn Bovonsombat | Pakorn Bovonsombat is an Associate Professor of organic chemistry at Mahidol University, Thailand. He completed his O- and A-levels at Wellington School Somerset, while his BA and MA (Chemistry) are from Vassar College, supervised by Miriam Rossi. His PhD (organic chemistry) is from New York University. Following a postdoctoral fellowship at Rockefeller University, he returned to Thailand and joined the Petroleum Authority of Thailand, then Degussa Catalyst (Thailand) Co. Ltd. In November 2003, he joined Mahidol University International College. His research interests are halogenations, alkynol rearrangements, and halogen bonds in crystal structures and organic reactions. |
 Punyanuch Sophanpanichkul | Punyanuch Sophanpanichkul received her BSc (Chemistry) from Mahidol University International College in 2015. She received her MSc (Organic Chemistry) from the University of Nevada, Reno, mentored by Dr Wesley A. Chalifoux, in 2017. She returned to Thailand and joined Kaowna Electric and Business Co., Ltd. She is currently a first-year PhD student in Pharmaceutical Sciences at the University of California, Irvine. |
 Satreerat Losuwanakul | Satreerat Losuwanakul received her BSc (Chemistry) from Mahidol University International College in 2019. After graduation, she joined Pepsi-Cola (Thai) Trading Co., Ltd as a management trainee. Currently, she is pursuing her MSc Pharmaceutical formulation and Entrepreneurship at University College London. |
1. Introduction
Propargyl alcohols or alkynols are functional group entities comprising an alcohol, be it primary, secondary, or tertiary, and an alkyne with or without substitution at the terminal carbon. The synthesis of these propargyl alcohols is straightforward, involving a 1,2-nucleophilic addition of acetylide to aldehydes or ketones. While the alcohol group can participate in typical alcohol reactions such as oxidation and dehydration, the alkynes can show specific alkyne chemistries such as 1,2-additions of hydrogen, halogens, and acids. Together, the combined alcohol-alkyne entity displays unique reactions, namely the Meyer–Schuster and the Rupe rearrangements.
Their rearrangement products, which include α,β-unsaturated aldehydes, ketones, carboxylic acids, and esters,1 are ultimately equivalent to an olefination of aldehydes and ketones. Moreover, the rearrangement of the propargyl alcohols amounts to an atom-economical route to synthesise the geometric-challenging tetrasubstituted acyclic alkenes. Reagents affecting propargyl alcohol rearrangement are crucial to account for the atom-economical purpose. They include Brønsted acid in the initial Meyer–Schuster and Rupe reactions, the precious metal complex catalysts such as Au(I) in the recent Meyer–Schuster reaction, and halogenations, either directly from halonium producing systems or via an interception (entrapment) of the reaction intermediate formed from metal catalysed propargyl alcohol rearrangement. The halogenations above, which are the subject of this article, provide a valuable path to α-vinyl and β-carbon halo-functionalization through which α-haloenones, β-haloenones and β,β-dihaloenones are the products. These haloenones are a functional synthetic template for Pd-catalysed coupling reactions,2 via vinyl halide exchanges with aryl or alkyl groups, potentially yielding valuable and precious bioactive tetrasubstituted alkenes like toremifene, afimoxifene and tamoxifen.3 Henceforth, this article will review the synthesis of α-haloenones, β-haloenones, and mixed β,β-dihaloenones from the perspective of direct halogenations and halonium ions interception of the metallated intermediates in the rearrangement of propargyl alcohols and their esters.
From the point of view of the mechanism or reaction conditions, the synthesis of α-iodoenones has its root in the Meyer–Schuster rearrangement.1 Since the Meyer–Schuster and the Rupe reactions have been periodically reviewed since 1971,1,4–6 a brief outline of the Meyer–Schuster and Rupe rearrangements before the haloenones formation discussions is sufficient to show the evolutionary development of the haloenone reactions.
Severe conditions of concentrated Brønsted acids and high temperature hampered the early Meyer–Schuster and the Rupe rearrangements of propargyl alcohols (Scheme 1, eqn (1) and (2)).7 Since then, the precious metal complexes catalysts, particularly soft electrophilic gold(I) and silver(I) alkynophiles – have been explored and used as the replacements because the conditions are milder. They show a potential towards high Z/E or E/Z selectivity, which was difficult to attain in the original acidic conditions (Scheme 1, eqn (3)). These metallic catalysts or promoters include Au(I),1b Ag(I),8 Au(I) with silver co-catalysts,9 Au(III),10 Mo–Au–Ag complexes,11 and other precious,12 and non-precious metals catalysts.13
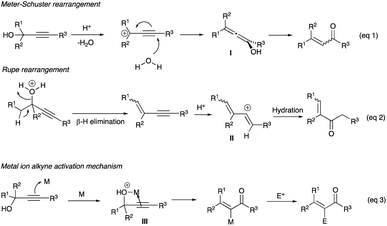 |
| | Scheme 1 Propargyl alcohol rearrangements by acid or metal activation. | |
2. α-Haloenones and α-halo-α,β-unsaturated esters formations
2.1 Direct electrophilic halogenations
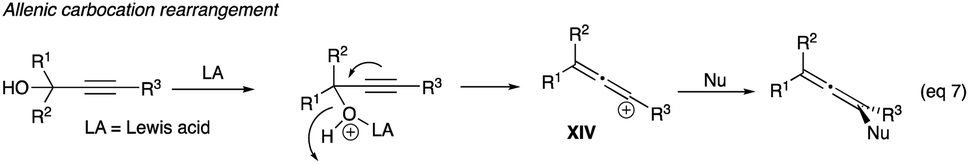
2.1.1 Direct halogenations of propargyl alcohols. A halonium ion, a soft electrophile no less, could be considered a proton replacement in the propargyl alcohol rearrangement initiation. Iodonium and bromonium ions from diverse halonium producing reagents could react directly with propargylic alcohols containing acetylenic hydrogen, alkyl or aryl (R3) via a halonium-bridged ion (IV) to give α-haloenones (eqn (4)). These are discussed in Sections 2.1.1 to 2.1.3.| |
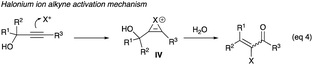 | (4) |
Examples of iodo-Meyer–Schuster rearrangement involving electrophilic iodination of linear tertiary propargyl alcohols (1a–e) to produce α-iodo-α,β-unsaturated aldehydes or α-iodoenals (2a–e) were first reported by Piancatelli and co-workers (eqn (4) and Scheme 2).14 The iodonium ions were produced in situ by oxidation of molecular iodine by pyridinium dichromate (PDC) in anhydrous CH2Cl2 containing molecular sieves. Three of the five starting propargyl alcohols contain steroidal skeletons with propargylic alcohol motifs at the 3-, 17-, and 20- positions of androstane (1c), cholestane (1d), and pregnane (1e), respectively. Among these three examples, two – 2c and 2e, showed complete selectivity for the thermodynamically stable (Z)-isomer (Scheme 2). As other rearrangements of propargyl alcohols, acetates and tosylates from direct iodination or Au(I), Bi(III), and Ag(I) catalysed reactions are examined below; the emergence of the (E)-isomer as the dominant product is elusive. These reactions show a preference for the thermodynamically stable (Z)-isomer, and in some, the preference for the (E)-isomer is increased only when the Z/E ratio approached parity. Nevertheless, the formation of kinetic (E)-isomer main product is possible, and this is discussed in Section 2.2.2.
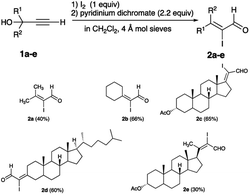 |
| | Scheme 2 Iodination of propargyl alcohols to yield α-iodoenals. | |
A decade later, Angara and McNelis, employing secondary alkynols with ethyl or a phenyl substitution (R3) at the terminal ethyne and methyl (R1 = CH3, R2 = H) at the secondary propargyl carbon (eqn (4)), reported the conversions of 3a and 3b to (Z)-α-haloenones (4a–b and 5a–b). Electrophilic iodination of 3a and 3b with N-iodosuccinimide (NIS) and catalytic hydroxy(tosyloxy)iodobenzene (HTIB) in room temperature methanol gave 4a and 4b with moderate to good yields (Scheme 3).15 With N-bromosuccinimide (NBS) as the bromonium ion source, the same alkynols furnished 5a and 5b, albeit in low yields with HTIB catalyst but more with AgBF4 replacement catalyst (Scheme 3).15
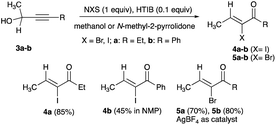 |
| | Scheme 3 Halogenation of secondary propargyl alcohols catalysed by HTIB. | |
Using HI in a 50 °C toluene solution and oxygen to produce iodonium ions, a variety of linear secondary alkynols possessing unsubstituted alkyne (eqn (4), R3 = H) and an aryl group at the propargyl carbon (6a–h) were iodinated to yield (Z)-α-iodoenals 7a–h (Scheme 4). Except for 7h, which shows an equal mix of kinetic E and thermodynamic Z isomers, all other products show high selectivity for Z-geometry.16 β-Iodoenones, a product of a 1,2-phenyl shift, were not reported, despite a phenyl ring, conducive to a 1,2-Meerwein shift, present at the propargylic carbon.
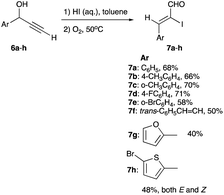 |
| | Scheme 4 Iodination of linear and aryl-substituted propargyl alcohols using HI. | |
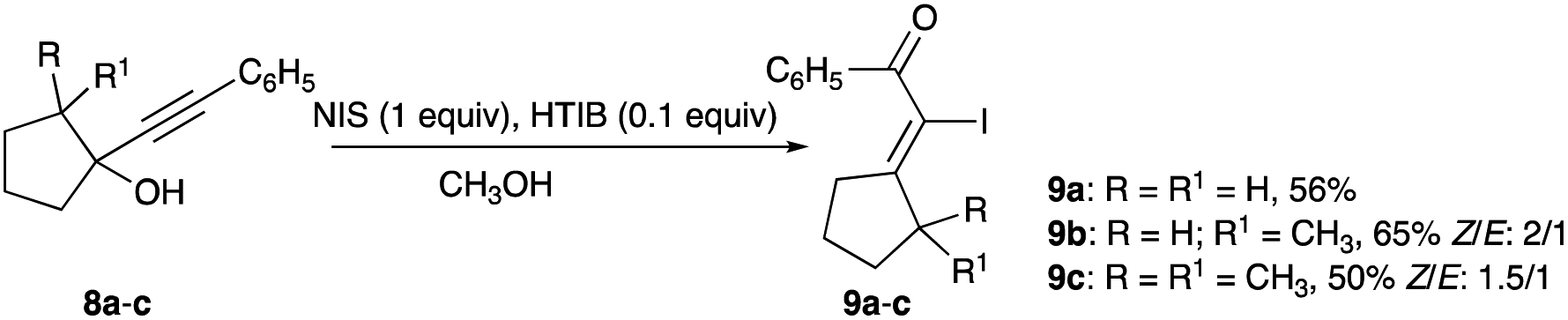
Phenylalkynylcyclopentanols containing methyl or dimethyl substitutions at C-2 of the 5-membered ring (8a–c) were iodinated with a combination of NIS and catalytic HTIB in room temperature methanol solution.17 Their rearrangements, equivalent to an α-iodophenylketone olefination of the cyclopentanone analogues, give α-iodoenones 9a–c with Z/E selectivity ranging from 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:1, showing a slight preference for the thermodynamic isomer (Scheme 5).17
:1 to 2:1, showing a slight preference for the thermodynamic isomer (Scheme 5).17
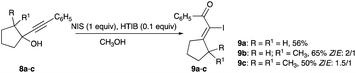 |
| | Scheme 5 Iodination of propargyl alcohols with NIS and catalytic HTIB. | |
In a different study, various tertiary alkynols with diverse arylacetylene motifs 10a–l were iodinated with an iodonium-producing combination of m-CPBA, Cl3CCO2H, and NaI to give α-iodoenones, 11a–l, in room temperature acetonitrile (Scheme 6).18 Except for the reaction of 10l and 10k yielding 11l and 11k, respectively, where the former shows an inclination for the kinetic E-isomer, presumably due to the bulky effect of the t-butyl group; and the latter shows no preference for either isomer (Scheme 6).18
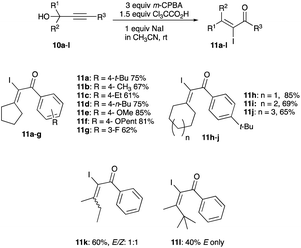 |
| | Scheme 6 Iodination of propargyl alcohols with iodide and m-CPBA. | |
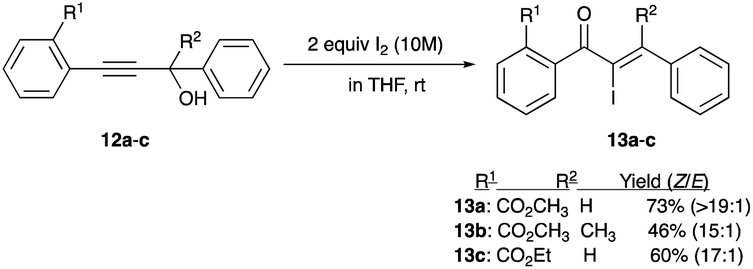
A series of phenyl propargyl alcohols 12a–c was reported to undergo rearrangements with only iodine (without Lewis acid assistance), albeit in a 10 M solution in THF, to give a variety of chalcone-like skeleton α-iodoenones 13a–c.19 A trio of examples shows an impressive thermodynamic isomer preference with the Z/E ratio ranging from 15:1 to >19:1 (Scheme 7).19
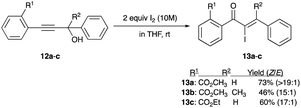 |
| | Scheme 7 Rearrangement of propargyl alcohols with 10 M iodine in THF. | |
2.1.3 Halogenations of alkoxy propargyl alcohols. Electron-enriched alkoxy propargyl alcohol is another substrate in the iodo Meyer–Schuster reaction. Its reaction with iodine or NIS in 0.25 M CH2Cl2 or toluene without a Lewis acid assistance or a Brønsted acid activation, gives α-iodo-α,β-unsaturated esters 18 with high selectivity for the thermodynamic (Z)-isomer, especially from alkoxy secondary propargyl alcohol substrates 17 (Scheme 11A and B).21 The reaction time of phenoxy secondary alkynols 17g–l compared to the ethoxy analogues 17a–f is shorter; spanning 2 to 5 hours for the former, and 10 hours for the latter (Scheme 11A and B). The Z/E selectivity of β,β-disubstituted α-iodo-α,β-unsaturated ester products 18m–q, derived from tertiary alkoxy alkynol 17m–q is less pronounced and their Z/E ratio range from 3.7:1 to 1.1:1 (Scheme 11C and D).21
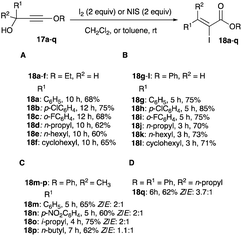 |
| | Scheme 11 (A–D) Iodination of alkoxyl propargyl alcohols. | |
2.2 Metal catalysed rearrangements
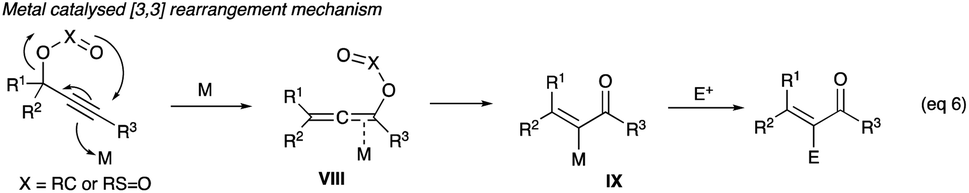
2.2.1 (Z)-Haloenones. The strategy of halogen interception (entrapment) of an allenoate VIII (eqn (6)) sees the use of the soft Lewis acid alkynophilic metal catalysts, particularly Au(I) and Au(III), Mo(VI), Ag(I) and V(V), which in the recent Meyer–Schuster reaction development are acid replacements in propargyl alcohol rearrangements, and the revival of the pursuit of α-haloenones synthesis from propargyl alcohols, acetates and tosylates methodology development. The halonium-donating reagents, which include Selectfluor, NCS, trichloroisocyanuric acid (TCICA), NBS, N-bromophthalalimide (NBP), NIS and iodine monochloride (ICl), are also typical reagents deployed in electrophilic aromatic halogenations.
2.2.1.1 With Au(PPh3)NTf2. Gold(I) complex, Au(PPh3)NTf2, first used in the Meyer–Schuster reactions to replace the severe acid conditions,1 along with halonium ion interception of the allenoate intermediate IX was shown by Zhang to be effective in converting propargyl acetates and tosylates to α-haloenones (eqn (6)), where X is carbon in acetate or sulphur in tosylate).| |
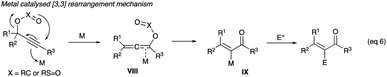 | (6) |
With two mol% of Au(PPh3)NTf2 and stoichiometric NIS in a solution of acetone and water (800:1), propargyl acetates 19a–h rearrange to α-iodoenones 20a–h with a Z/E ratio ranging from 1:1 to greater than 50:1; selected examples are illustrated in Scheme 12.22,23
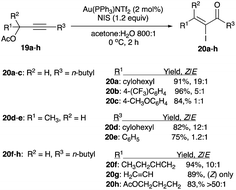 |
| | Scheme 12 Propargyl acetates iodination with gold(I) complex catalyst. | |
The same conditions of Au(I) catalyst, solvent, propargyl acetates 21a–d and NBS as an electrophilic bromine source give a high preference for the thermodynamic α-bromoenones 22a–d with a Z/E ratio spanning from 20:1 to >99:1 (Scheme 13).23 Interestingly, 22b and 22c gravitated towards the kinetic (E)-geometry and the E/Z ratio of these (E)-α-bromoenones are from 1.4:1 to 5:1 (Scheme 13).23
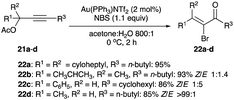 |
| | Scheme 13 Propargyl acetates bromination with Au(I) complex catalyst. | |
A proposed mechanism involving Au(PPh3)NTf2 catalysed propargyl acetate (or tosylate) rearrangement to α-haloenone is reproduced in Scheme 14. The intermediate allenoate XI, formed from an intramolecular 3,3-migration of the acetate – mediated by the soft electrophilic precious metal coordination of the alkyne (X),24,25 is crucial in defining the Z/E geometry of the product. When RL is aromatic, the α-iodoenone shows an increased tendency towards E-geometry.23 On the other hand, with RL being aliphatic, the resulting α-iodoenones show an overwhelming preference for the Z-geometry. The interception of the metal-carbon (sp2) nucleophilic site in the intermediate XIII by electrophilic iodine of NIS or bromine from NBS results in α-iodoenones and α-bromoenones (Scheme 14). Other non-halogen electrophiles such as an iminium,24 and activated acyl group,25 have been utilised to intercept related α-metallated–allenoate.
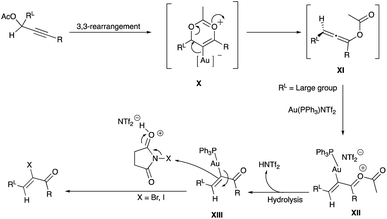 |
| | Scheme 14 Proposed mechanism of propargyl acetates halogenation with Gold(I) complex catalyst. | |
With propargyl alcohol as the substrate in the iodo Meyer–Schuster rearrangement, the olefination of the ketone or aldehyde is a two-step process, as opposed to three for a propargyl acetate or tosylate. The advantage of forgoing the third esterification step is desirable in terms of atom economy but the loss of geometric-selectivity control in the α-haloenone product, due to the absence of intramolecular 3,3-migration of acetate or tosylate, is a disadvantage. To overcome this problem with propargyl alcohol 23 usage and to convert them to α-haloenones with high Z/E selectivity as seen with propargyl acetate or tosylate substrates, Zhang employed an Au/Mo bimetallic catalyst, comprising the same catalyst, Au(PPh3)NTf2, and 1–2 mol% of MoO2(acac)2, in room temperature dichloromethane (Scheme 15).26 The α-iodoenone products 20d–e and 20i–l show a high preference for the thermodynamically stable (Z)-isomer with the Z/E ratio ranging from 6:1 to 50:1 while a Z/E ratio of 20:1 to 50:1 was observed with α-bromoenones 22d–g when NBS replaces NIS in the gold–allenoate XIII interception step (Schemes 14 and 15).26 An in situ formations of an oxomolybdenum ester, formed from a MoO2(acac)2 coordination to the alcohol, was implicated, which after a 3,3-migration of this alcohol–MoO2(acac)2 complex (analogous to the 3,3-migration of the acetate), is surmised to give an intermediate resembling VIII (eqn (6), where X is Mo).26
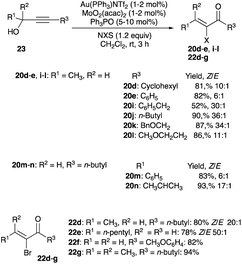 |
| | Scheme 15 Propargyl alcohols halogenation with Au/Mo bimetallic catalyst. | |
| |
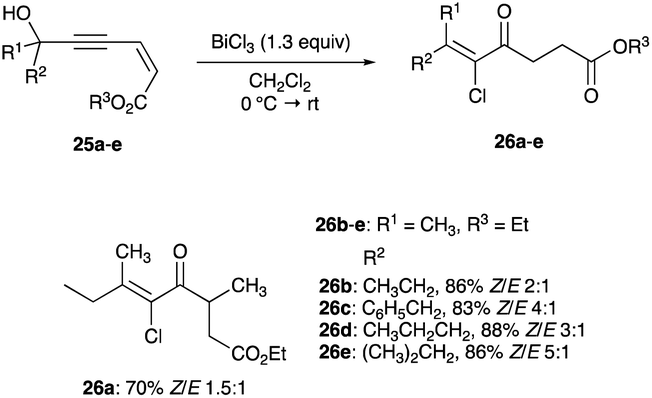
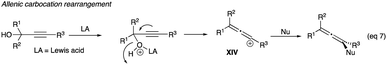 | (7) |
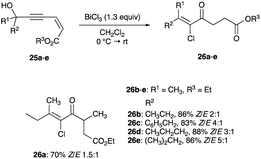 |
| | Scheme 16 Propargyl alcohols chlorination with stoichiometric BiCl3. | |
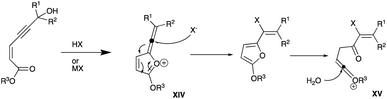 |
| | Scheme 17 Proposed mechanism of propargyl alcohol chlorination via nucleophilic addition. | |
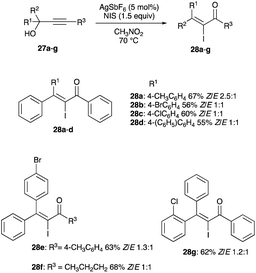 |
| | Scheme 18 Propargyl alcohols iodination with 5 mol% AgSbF6. | |
2.2.2 (E)-Haloenones from gold(I), gold(III), and vanadium(V) catalysis.
2.2.2.1 With Au(I)-1,2,3-triazole. A high E/Z ratio, in which the kinetic (E)-isomer becomes the dominant product, is elusive. Direct iodination or Au(I), Bi(III) and Ag(I) catalysis show a preference for the thermodynamically stable (Z)-isomer in the rearrangement of propargyl alcohols, acetates and tosylates to α-iodoenones. However, the catalyst Au(I)–1,2,3-triazole (TriA–Au), reported by Shi and co-workers, showed that one mol% of TriA–Au in cold CH3NO2 (0 °C) converted secondary propargyl acetates 29a–i to (E)-α-iodoenones 30a–i with a E/Z selectivity, ranging from a low 1.3:1 to a high >20:1 (Scheme 19).29
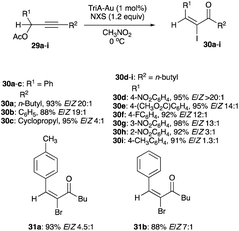 |
| | Scheme 19 Halogenation of propargyl acetates with TriA–Au catalyst. | |
For (E)-α-bromoenones 31a–b, using the same reaction conditions except with NBS instead of NIS, the E/Z ratio was from 4.5:1 to 7:1 (Scheme 19).29 Even though the geometry of Au(I) and its ligand to the sp2 carbon are linear, a high E/Z selectivity was observed, and intermediates XVII and XVIII are thought to be responsible for the resulting (E)-isomer (Scheme 20).29 The electrophilic addition of the halogen from N-halosuccinimide is surmised to be anti to the R group in XVII and XVIII, to avoid the alternate sterically-imposing syn-approach (relative to R), which would take the halogen reagent close to the large and sterically-imposing RL (Scheme 20).29
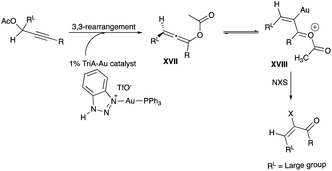 |
| | Scheme 20 Proposed mechanism of propargyl acetate rearrangement with TriA–Au catalyst. | |
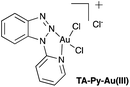 |
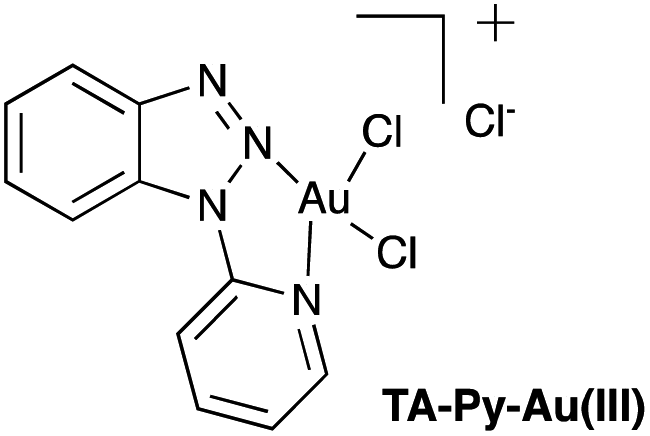
| | Fig. 1 Pyridyltriazole Au(III) complex, (TA-Py–Au(III)). | |
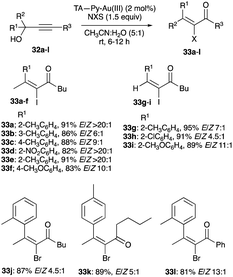 |
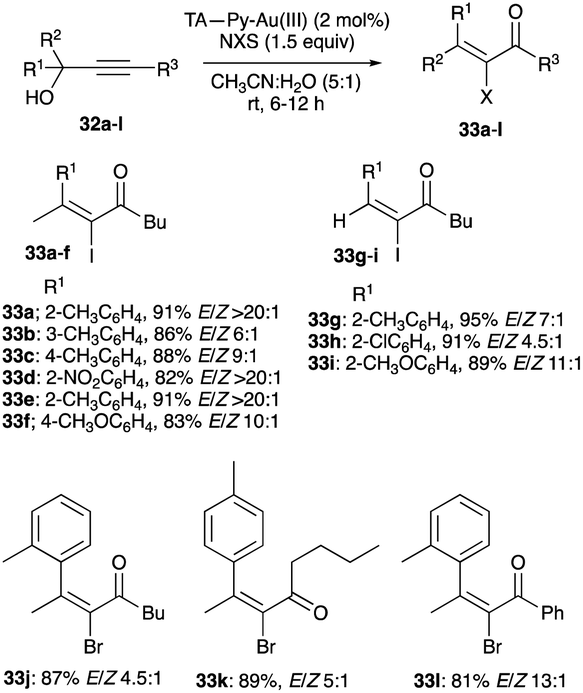
| | Scheme 21 TA-Py–Au(III) catalysed propargyl alcohols rearrangement with NIS and NBS interception. | |
 |
| | Scheme 22 TA-Py–Au(III) catalysed rearrangement of propargyl alcohols with chlorination interception. | |
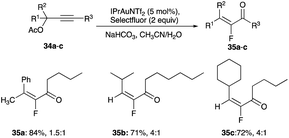 |
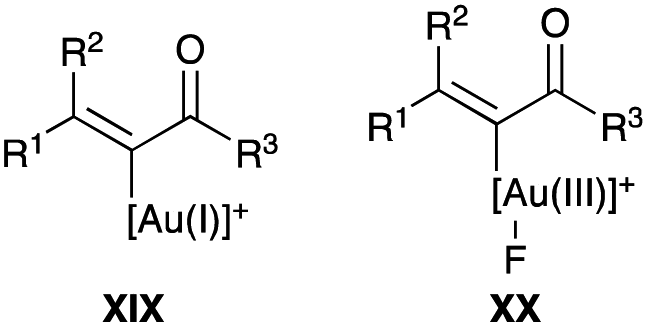
| | Scheme 23 IPrAuNTf2 catalysed propargyl acetates rearrangement with fluorination interception. | |
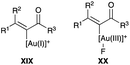 |
| | Fig. 2 (E)-α-Fluoroenone formation intermediates. | |
To that end, at five mol% of OV(OSi(p-ClC6H4)3)3, this non-precious vanadium catalyst has been shown by Trost and Tracy to be effective in converting propargyl alcohols to α-haloenones with high E/Z selectivity.32 With MgO serving as a base in acetonitrile maintained at 65 °C and NCS as a chloronium source intercept of the allenolate, the vanadium complex catalyst converts both secondary and tertiary propargyl alcohols to (E)-α-chloroenones 37a–i with E/Z ratio ranging from 11:1 to >20:1 (Scheme 24).32
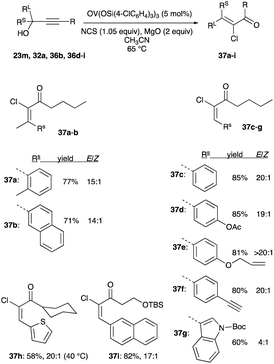 |
| | Scheme 24 Chlorination of propargyl alcohols with 5 mol% OV(OSi(p-ClC6H4)3)3. | |
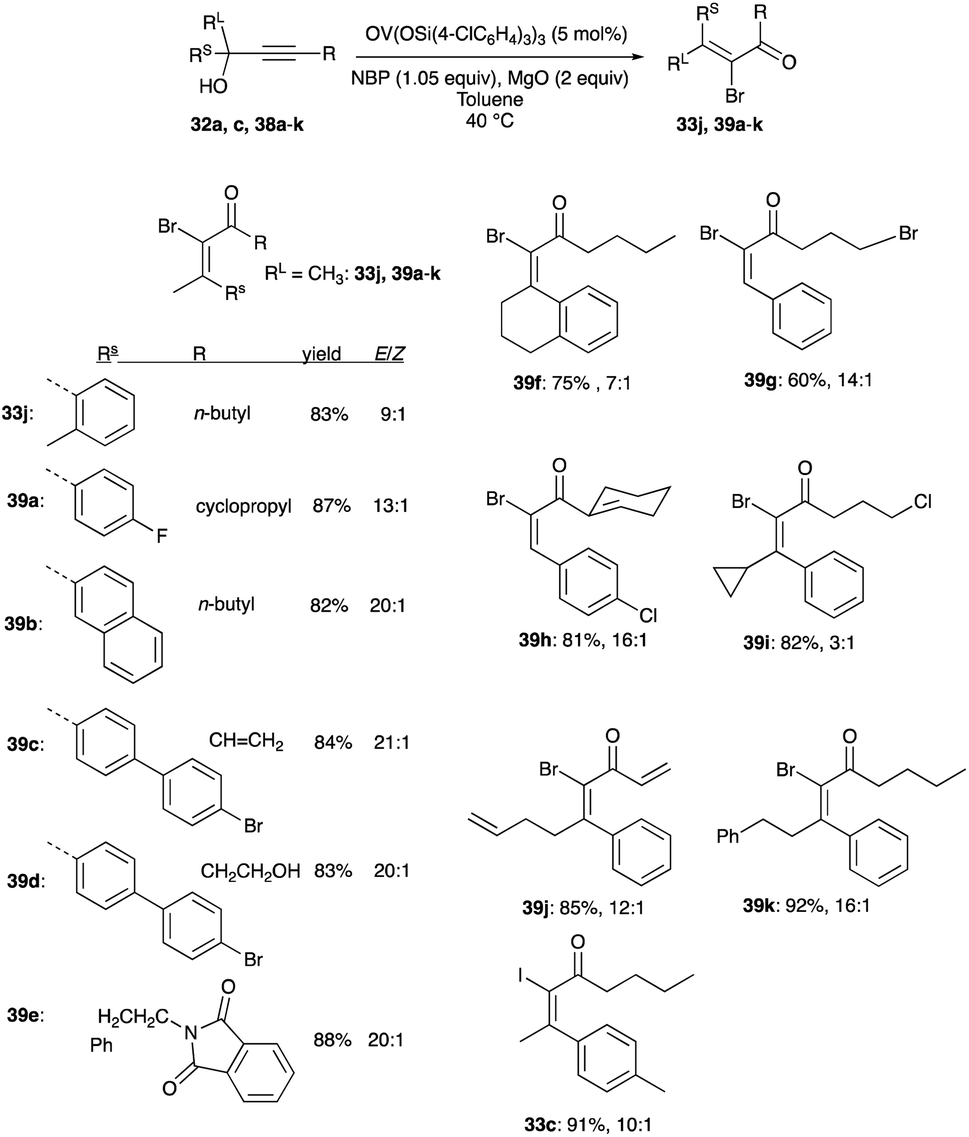
The same catalyst and loading are useful in the rearrangement of propargyl alcohols to (E)-α-bromoenones. But unlike the chlorination reactions, acetonitrile is replaced by toluene with a lower reaction temperature of 40 °C, and NBS is replaced by NBP as the bromonium ion source for the interception step. Both secondary and tertiary propargylic alcohols undergo rearrangements to (E)-α-bromoenones 33j and 39a–k with yields of 60 to 91% and an E/Z selectivity spanning 3:1 to 21:1 (Scheme 25).32 An (E)-α-iodoenone 33c was synthesised using the bromination conditions with NIS instead of NBP. The E/Z selectivity of this product is 10:1 (Scheme 25).32
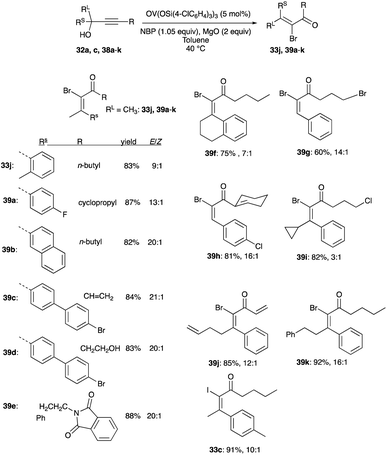 |
| | Scheme 25 Bromination of propargyl alcohols with 5 mol% OV(OSi(p-ClC6H4)3)3. | |
The proposed interactive role of OV(OSi(p-ClC6H4)3)3 in the propargyl alcohol rearrangement and the subsequent electrophilic halogenation trapping to produce (E)-α-haloenone is reproduced in Scheme 26.32
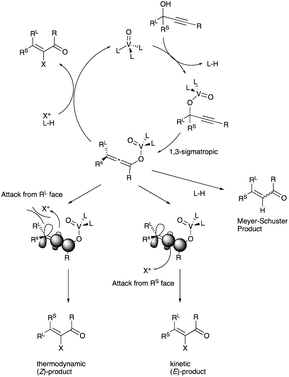 |
| | Scheme 26 Proposed interactive role of OV(OSi(p-ClC6H4)3)3 in propargyl alcohol rearrangement. | |
3. β-Haloenones
3.1 Propargyl alcohols rearrangements
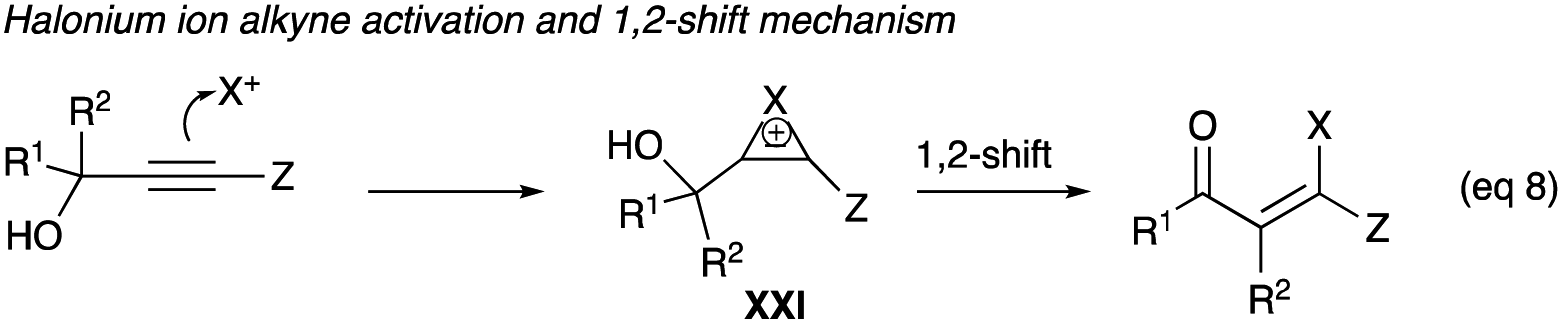
β-Haloenones formation is a possibility when either R1 or R2 or both of the propargyl alcohol is an aryl group, even if Z is an alkyl or an aryl group (eqn (8)). The first example of β-iodoenones was reported by McNelis and co-workers, in which 4-iodo-2-phenylbut-3-yn-2-ol (40c) and 2-phenylbut-3-yn-2-ol (40b) underwent an iodonium ion-induced 1,2-phenyl shift rearrangement.33 With stoichiometric amounts of iodine and iodic acid in refluxing methanol, 41c was obtained in 75% yield from the iodination of 40c (Scheme 27).33 The same reaction condition also shows the shift of the phenyl group of 40b to (Z)-41b in 60% yield. The (E)-isomer was not observed. With 2,4-diphenyl-3-butyn-2-ol (40a) as a substrate, the same conditions of iodine and iodic acid in methanol gave 41a after an overnight stirring at room temperature. The diphenyl propargyl alcohol did not produce an α-iodoenone, which is possible with a phenyl group at the terminal alkyne carbon. Rather, it underwent a 1,2-phenyl shift.| |
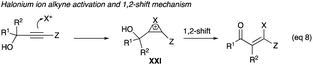 | (8) |
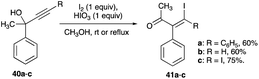 |
| | Scheme 27 Iodonium ion-induced 1,2-phenyl shift rearrangement of propargyl alcohol. | |
Several years (thirty-four years) elapsed before another study on β-haloenone formation from propargyl alcohols was reported.32,33 In the report by Trost and Tracy, 32c, induced by the bromonium ion from NBS, underwent a Wagner–Meerwein 1,2-phenyl shift (a cationic [1,2]-sigmatropic rearrangement) via 42 to give β-bromoenones 43 (Schemes 28 and 30).32
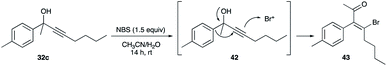 |
| | Scheme 28 Bromonium ion-induced 1,2-phenyl shift of aromatic ring at the propargyl carbon. | |
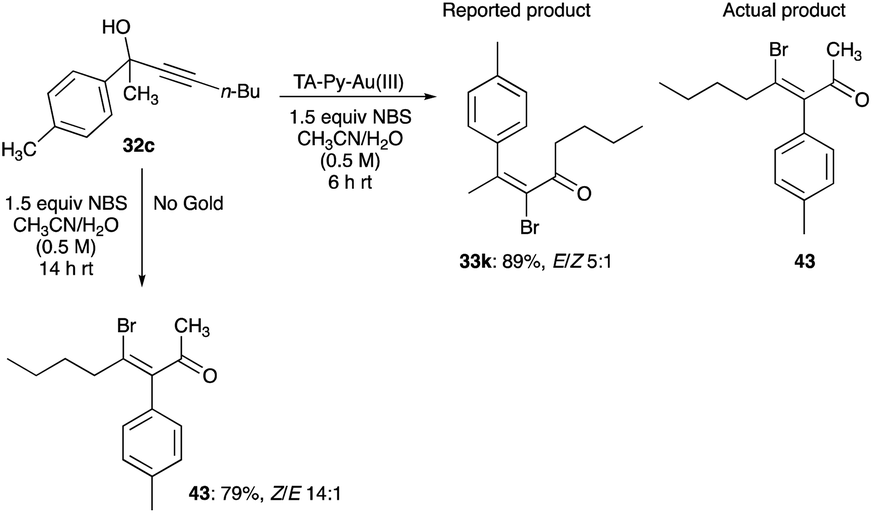
The aryl shifts show regiospecificity; in that the initial aromatic substituent pattern in the propargyl alcohols is retained in the β-haloenone products as exemplified by the retention of aryl ring substituents pattern of 4-methyl, 2-methyl, 4-bromo, 4-methoxy and 2-naphthalenyl of the original propargyl alcohols (Scheme 30).32 Their regiospecific 1,2-aryl shifts are as expected with the prediction of the cationic [1,2]-sigmatropic shift or the Wagner–Meerwein rearrangement. Curiously, it was from an erroneous structure assigned to the product of TA-Py–Au(III) complex catalysed rearrangement of 32c,30 that led Trost and Tracy to probe the halogenation rearrangement of propargyl alcohols to β-haloenones by reinvestigating the reaction of 32c and its product (Scheme 29).32
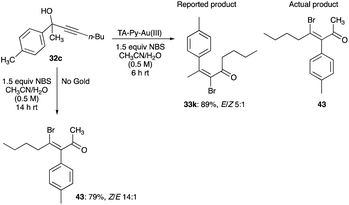 |
| | Scheme 29 Halogenated rearrangement of propargyl alcohols to β-haloenones.30,32 | |
Instead of obtaining the reported product, 33k, the substrate 32c had undergone a 1,2-phenyl shift to give 43, despite catalytic TA-Py–Au(III) and 1.5 equivalent of NBS in 0.5 M solution of acetonitrile and water.30,32 The 1,2-shift of the toluene group is likely caused by bromination of the alkyne, leading to the bromonium bridged ion and subsequent anti-1,2 aryl group shift. Intriguingly, this suggests, to account for the production of bromonium ion from NBS, that TA-Py–Au(III) catalyst is behaving as a Lewis acid and its mode of action is an activation of the bromine of NBS through its coordination with the oxygen of the imide of NBS.
Compared to the pure acetonitrile used in the McNelis reactions, hexafluoroisopropanol (HFIP) admixed with water in a ratio of 5:1 for bromination (with NBS), and iodination (with NIS) gives a more potent electrophilic halogenations of alkynols 32c and 44a–h to β-bromoenone 45a–g and β-iodoenone 45i, in terms of good to excellent yields and exemplary Z/E selectivity, that ranges from 10:1 to over 20:1 (Scheme 30).32 For chlorination of propargyl alcohol, which has one example, trichloroisocyanuric acid (TCICA) is used instead of the unreactive NCS. Chlorination with TCICA is more effective in an acetonitrile-water solution (5:1, at 0.17 M), and the β-chloroenone product 45h shows a Z/E selectivity of 5:1 (Scheme 30).32
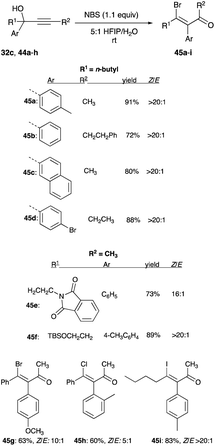 |
| | Scheme 30 Halogenated rearrangement of propargyl alcohol in 5:1 HFIP/H20. | |
Water addition to organic solvents such as HFIP and acetonitrile to promote β-bromo and β-chloroenone formations is not unprecedented. The findings of Trost and Tracy corroborate with the earlier report of Herault and McNelis who had shown that water in acetonitrile promotes the 1,2-phenyl shift in the β-haloenone formation.34 Different solvent–water mixtures were studied by Herault and McNelis to shed light on the competitive nature of the iodo-Meyer–Schuster, enyne production from the dehydration of alkynols, and the 1,2-phenyl shift rearrangements and from their study, water shows a positive influence on the course of propargylic alcohols reaction to β-iodoenones 41b, 46a, and 46b formations by favouring the phenyl shift over the other pathways (Scheme 31).34
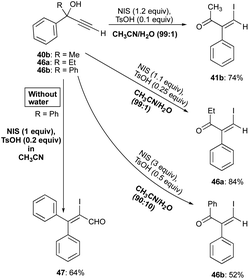 |
| | Scheme 31 Water influences on propargyl alcohols pathway to β-haloenones formation. | |
3.2 Propargyl alkoxide rearrangements
An alternative substrate to propargyl alcohol for the formation of β-iodoenone is propargyl alkoxide, made in situ from a one-pot reaction of three components; an aromatic alkyne, cyclic or linear ketone, and t-butoxide functioning as a base. Examples of alkyl shifts of the tertiary propargyl alkoxide, whose outcome is equivalent to insertion at the α-carbon of the ketone substrate of a β-aryl–β-iodovinyl motif, are illustrated in Scheme 32A (for cycloalkanones 48a–b) and Scheme 32B (for linear ketones 49a–b). For cycloalkanones, the reaction results in a ring expansion while homologation is the end result for the linear ketones. The proposed mechanism involving an iodonium bridged ion, as a result of the addition of alkynophilic iodine monochloride and a 1,2-alkyl shift to yield β-iodoenone, is reproduced in Scheme 33.35
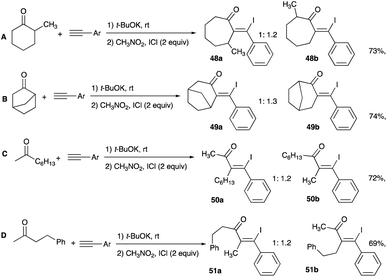 |
| | Scheme 32 (A–D) β-Iodoenones formation from alkyl shifts of the tertiary propargyl alkoxide. | |
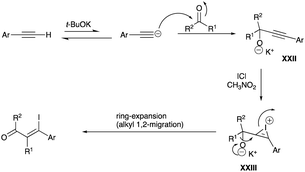 |
| | Scheme 33 Proposed mechanism of propargyl alkoxide rearrangement. | |
Recently, a computational NMR via machine learning-augmented DFT study of some unusual reactions which include iodination-rearrangements of propargyl alkoxide emerged. It casts doubt on some of the reported ring expanded products.36 In the study of Novitskiy and Kutateladze, the NMR deviations between experimental reports and those of AI-assisted DFT calculations have led to the revisions of the six to seven and seven to eight-membered ring expansions to the retention of the ring size of the original alkoxide substrate 52 and 54 and a revised structure of the product to an α-iodoenone 53 and 55 (Scheme 34A). These structural revisions also extend to the ring expanded products of cyclic propargyl alcohols rearrangements induced by iodine and catalytic amounts of polymer-immobilised iodine(III) reagents.37 Their structure revisions are shown in Scheme 34B. In Section 4.1.4, the impact of the machine learning-augmented DFT (DU8ML) study will be discussed on the structure revisions of the ring expanded product of camphor alkynol rearrangements.
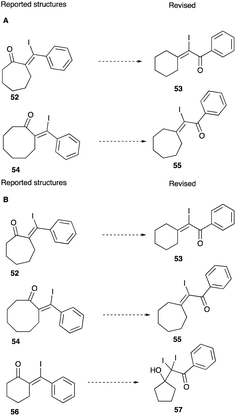 |
| | Scheme 34 (A and B) Revised structures of ring expanded products based on DU8ML analysis. | |
4. β,β-Dihaloenones
4.1 (Z)-Mixed β,β-dihaloenone
The implication of eqn (8) extends to the formation of β,β-dihaloenones, where its formation becomes possible if Z is a halogen and R1 or R2, or both are aryl groups. The first report of mixed β,β-dihaloenones comes from McNelis, who before had reported β-iodoenone and symmetrical β,β-dihaloenone formations from iodonium ion-induced propargyl alcohol rearrangements.33,38 The formation of β,β-dihaloenones and mixed dihaloenones were made more facile by new combinations of reagents developed to produce halonium species, particularly iodonium ion, at room temperature effective in both protic and aprotic solvents, which allow the difficult iodination of alkyne with electronegative terminal bromine to be possible.39,40 These reagent combinations include NIS or NBS with catalytic amounts of HTIB or p-toluenesulfonic acid (TsOH),41 and molecular iodine, bromine or anionic halides with stoichiometric amounts of HTIB.42,43
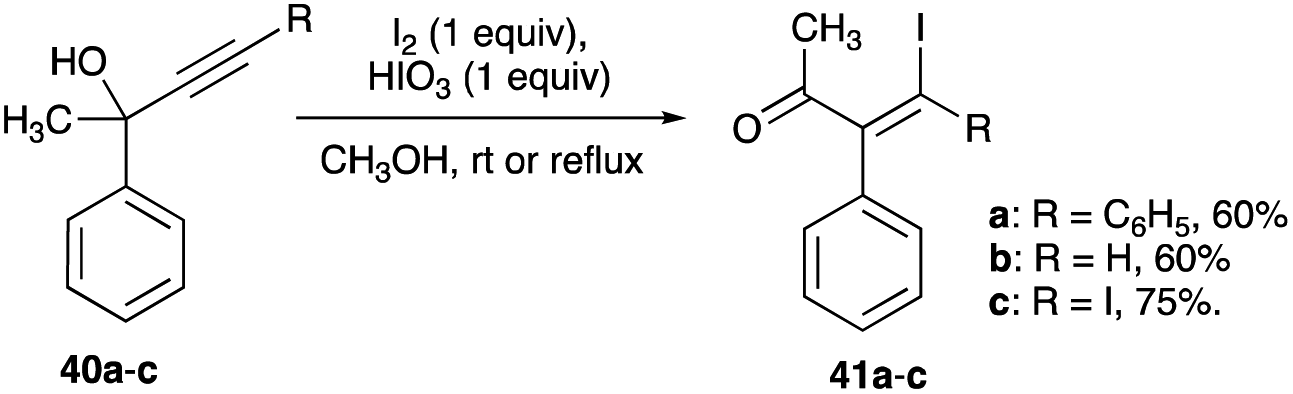
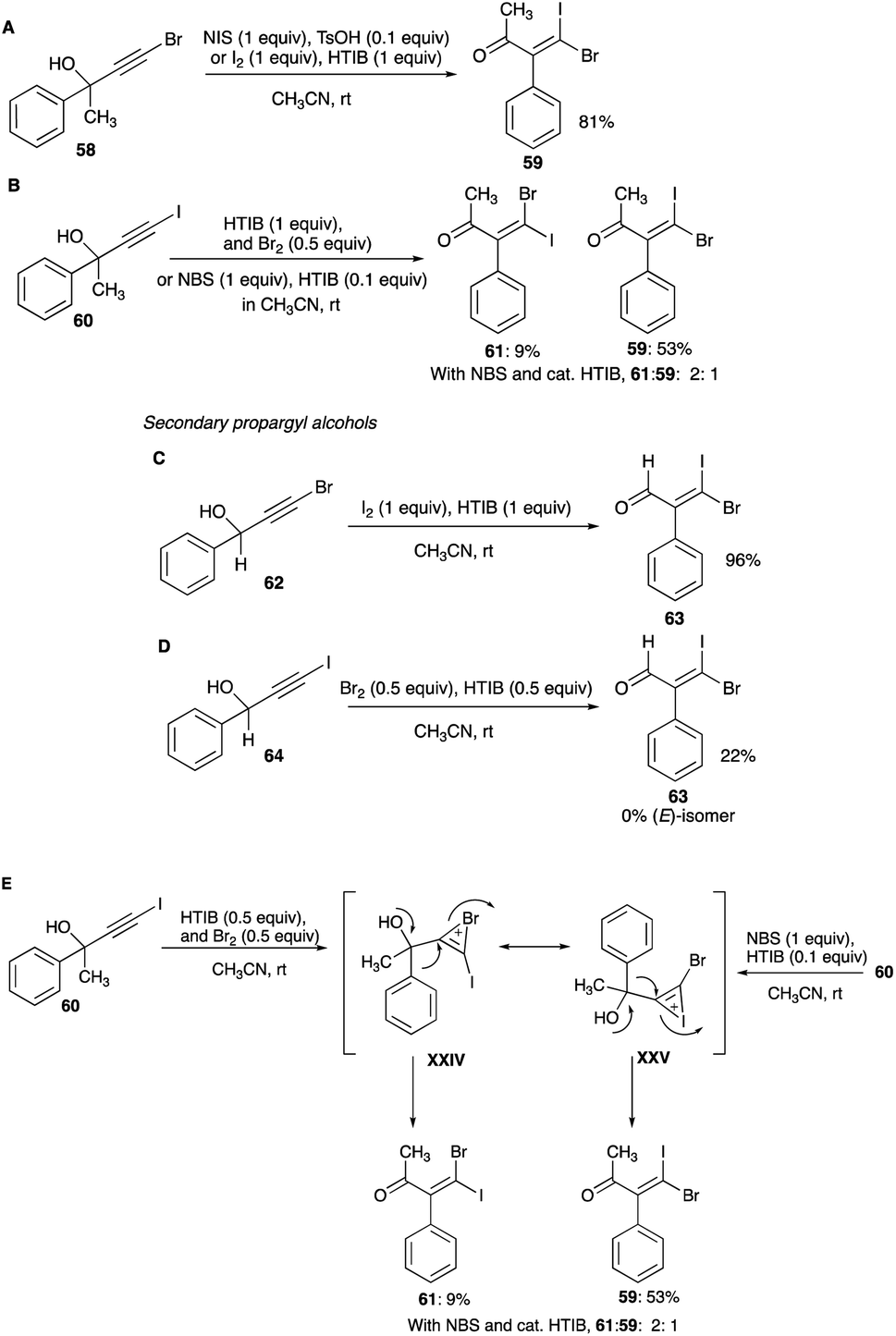
4.1.1 Linear secondary and tertiary propargylic alcohols. The first report of a mixed β,β-dihaloenones was from iodination of brominated tertiary propargyl alcohol, 4-bromo-2-phenyl-3-butyn-2-ol. A reaction of this 58 with HTIB and molecular iodine combination or NIS and catalytic amounts of TsOH in room temperature acetonitrile gave 59 with greater than 90% selectivity and 90% conversion (Scheme 35A).38 The (E)-isomer of 59 was not detected in the reaction mixture. The resulting 1,2-phenyl shift appears to be encouraged by the presence of terminal bromine, which likely curtails any open vinyl cation at the terminal carbon (eqn (8)). But the bromination of 60 with NBS and catalytic amounts of HTIB did not furnish the expected (E)-isomer 61 as the sole product of the expected bromonium bridged ion. The unexpected (Z)-isomer 59 found in the product mixture (with a Z/E composition of 1:2) suggests a presence of an iodonium bridged ion intermediate as well (Scheme 35B).38 The same reaction with more potent bromine and stoichiometric HTIB combination in acetonitrile, gave a 90% conversion to the β,β-dihaloenone products 61 and 59 with an increased Z/E ratio of 5.9:1.38 The unexpected (Z)-isomer in these reactions was a revelation. Clearly, an interconversion of the bromonium bridged ion to the iodonium bridged ion had occurred, favouring the larger iodonium bridged ion over the smaller bromonium ion. The relative stability between these two haloniums bridged ions is still today unanswered.
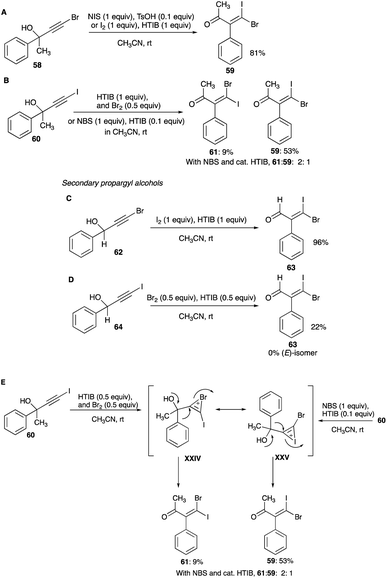 |
| | Scheme 35 (A–E) Mixed β,β-dihaloenones formation from linear secondary and tertiary propargyl alcohols. | |
Bromination of the iodo analogue of a secondary alkynol, 64, furnished none of the expected (E)-isomer of 63. The (Z)-isomer was the only product of the phenyl shift, obtained with 22% selectivity along with 27% of 3,3-diiodo-2-phenylpropenal (Scheme 35D).42 On the other hand, iodination of the same secondary alkynol but with terminal bromine, 62, with HTIB and iodine or NIS and catalytic amounts of TsOH gives only (Z)-isomer 63 with high conversion and selectivity (Scheme 35C).42
4.1.2 Ring expansion reaction of propargylic alcohols. A ring expansion of a five-membered ring propargyl alcohol to a six-membered ring β,β-dihaloenone is shown by an iodonium ion-induced ring expansion of a phenanthrene ring containing a tertiary brominated alkynol motif 65 (Scheme 36A).44 This ring expansion is the result of 1,2-aryl shift of one of the aryl rings with HTIB and iodine or NIS and catalytic TsOH which provide the activating iodonium ions. The resulting (Z)-product shows a remarkable identical mass spectrum pattern to the previous (Z)-isomer examples of other phenyl-shifted secondary and tertiary brominated alkynols rearrangements to mixed β,β-dihaloenones 66.38,42,44,45
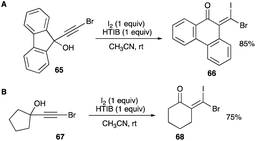 |
| | Scheme 36 (A and B) β,β-Dihaloenones formation via ring expansion of propargyl alcohols. | |
Prior to the phenanthrene ring expansion, ring expansion involving an alkyl shift was observed in the electrophilic halogenation of 67 (Scheme 36B). Iodination of the terminally brominated propargyl alcohol was achieved by a potent iodonium producing system of HTIB and iodine. As with the phenyl shifts of previous examples of terminally brominated alkynols, the electron-withdrawing bromine at the terminal alkyne carbon renders the vinyl cation at this site the least likely to form while a vinyl cation at the internal alkyne carbon is more than likely, allowing a 1,2-alkyl shift and the resulting ring expansion of 68.45
4.1.3 Migration aptitude in the ring expansion. A re-examination of the ring expanded products of C-2 substituted 1-bromoethynylcyclopentanol by Herault and McNelis revealed the reported alkyl shifts of this brominated propargyl alcohol did not follow the norms of the observed migratory aptitude of the cationic rearrangement.46 A re-evaluation of the ring expansion of 69 showed that the actual product was 71, as a result of the shift of the lesser substituted carbon (C-5), and not the earlier reported 70 (Scheme 37A).45,46 Iodonium ion-induced ring expansion of cis-72 and trans-73 were studied by Herault and McNelis to elucidate the migratory aptitude between the secondary (CH2 at C-5) of 74 and the tertiary centres (C-2) of 75 (Scheme 37B).46 For trans-72, the secondary carbon (C-5) of 74 shifted in preference to the tertiary C-2 carbon (Schemes 37B and 38A).46 In the curtailed migration of the tertiary carbon, the transition state 76A is additionally strained by the vinyl bromine atom impinging onto the axial methyl during the positioning of the iodonium bridged ion anti to the tertiary carbon, a prerequisite for the shift (Scheme 38A). On the other hand, the methylene carbon (C-5) shift, also needing to be anti to the iodonium bridged ion for the shift, has its vinyl bromine pointing away from the methyl of the tertiary carbon which poses no steric strain in the transition state.46
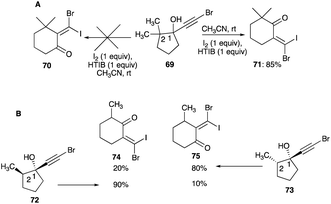 |
| | Scheme 37 (A and B) Alkyl shift of brominated propargyl alcohol in ring expansion. | |
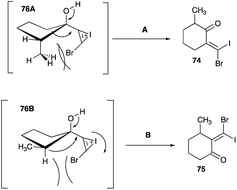 |
| | Scheme 38 (A and B) Plausible paths of propargyl alcohol alkyl shift in ring expansion. | |
For the cis-73, the alkyl shifts follow the norms of the Pinacol and other cationic rearrangements, in that the tertiary centre 75 is preferred over secondary 74 (Scheme 38 path B).46 In the rearrangement of cis-76B, the vinyl bromine of both conformations of the iodonium bridged ion – one being anti to the tertiary C-2 carbon and the other to C-5 methylene carbon – impose no steric strain to the axial hydrogen or the equatorial methyl at C-2 nor to the axial hydrogen at C-5. Both C-2 and C-5 migrations have none of the additional steric impediment and the only difference between C-2 and C-5 is the migratory aptitude between tertiary and secondary carbons (Scheme 38B).46
4.1.4 Ring expansion of camphor, norcamphor and homoadamantane systems. Rings of bicyclic 1R-(+)-camphor and norcamphor, and the tricyclic homoadamantanone can undergo ring expansion reactions to form seven-membered rings of bicyclic and tricyclic analogues of β,β-dihaloenones. Iodination of 77b with HTIB and iodine in acetonitrile at room temperature yielded a sole product 78 of 60% yield, whose spectral and analytical data were consistent with the structure of 2-[(Z)-bromoiodomethylidene]-1,8,8-trimethylbicyclo[3.2.1]octan-3-one with the Z-configuration of the halogens and the shift of the quaternary carbon (C-1) consistent with the finding of Herault and McNelis (Scheme 39A).46,47
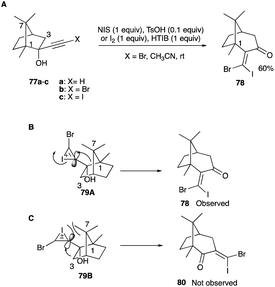 |
| | Scheme 39 (A–C) Ring expansion of bicyclic camphor to β,β-dihaloenones. | |
In the ring expansion of the bromo propargyl alcohol of 1R-(+)-camphor 77b, the quaternary carbon shifted in preference over the secondary carbon (C-3) alternative and this is in line with the expected norms of well-known cationic rearrangements (Scheme 39A). However, a closer examination of the alignments of the iodonium bridged ion between the tertiary C-1 and the secondary C-3 carbons reveals a possible steric strain for the secondary carbon shift path. For the latter, the anti-alignment of the iodonium bridge to the secondary C-3 carbon points the iodonium towards the methyl at C-1, and critically, it is also perilously close to the methyl at the C-7 bridgehead (Scheme 39C).46 On the other hand, the shift of the quaternary C-1 centre shift has none of the steric drawbacks with the neighbouring C-3 centre nor with the methyl at the C-7 bridgehead in its anti-alignment of iodonium bridged ion to C-1 prior to the shift (Scheme 39B).46
However, in lieu of the recent report of a machine learning-augmented DFT study,36 the reported structure of the alkynol of 1R-camphor and its ring expansion products might be in doubt.
Shown in Scheme 40, the revised structure of the originally reported endo alkyne in the camphor alkynol is exo (Scheme 40A), leading to 82 as the ring expanded product and not the reported 78. The implication of the alkyl shift is evident. With the alkyne unit as exo, the shift of the secondary carbon (C3) is preferred over C1 tertiary carbon, as was reported originally. But here lies the serendipity of this recent finding, the revised structure of camphor alkynol rearrangement lends further support to the thesis of Herualt and McNelis.46 A closer examination of the alkyl shift of the exo halo alkyne unit shows that the shift of the C3 secondary carbon is preferred due to the iodonium-bridged ion's orientation while having to be anti to C3 centre, is pointing away from the ring and thus inherits no strain. While the shift of the C1 tertiary centre will orient the iodonium-bridged ion anti to C1 but its vinyl bromide would point towards the methyl of C1, leading to additional strain (Scheme 40B).
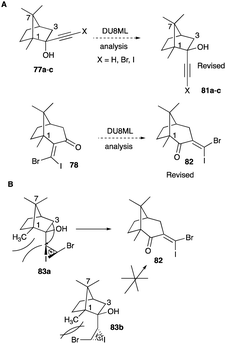 |
| | Scheme 40 (A and B) Revised structure. | |
The ring expansion reaction of norcamphor 84, unlike camphor, is not as compliant in furnishing a single ring-expanded product. Instead, this reaction yields two products 86 and 87, with a 6:4 composition, which reveals further insight into the nature of alkyl shifts in the ring expansion. These two products are not geometric isomers but they are from different neighbouring carbons to the bromoethynyl unit shifting (Scheme 41).48 With the bridgeheads at C-1 and C-7 in norcamphor lacking methyl groups, no interference on the iodonium-bridged ion or from the vinyl bromine atom is possible. The iodonium-bridge forming across the triple bond is devoid of any infringement from the hydrogen at C-7 and C-1 bridgeheads, allowing it to be anti-periplanar to both C-1 and C-3. The shifts of C-1 and C-3, without additional steric strains in the transition states 85A and 85B, reflect the norms of the migratory aptitude order (Scheme 41A and B).48
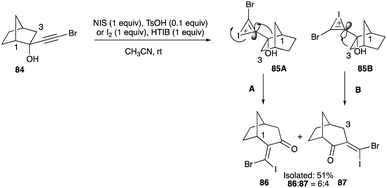 |
| | Scheme 41 (A and B) Ring expansion reaction of norcamphor to β,β-dihaloenones. | |
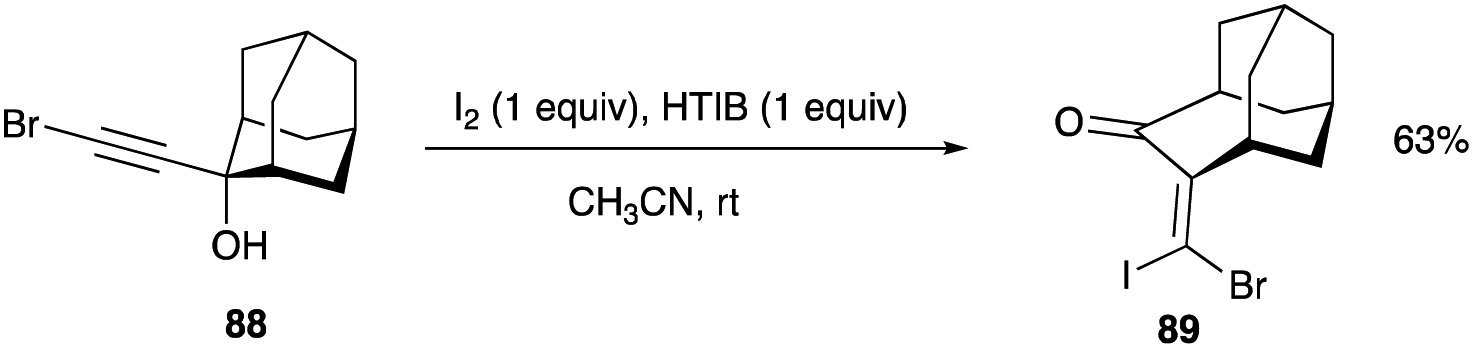
Propargyl alcohol of the tricyclic homoadamantanone system, 88, can also undergo a ring expansion with the combination of HTIB and iodine in acetonitrile at room temperature. The sole product of 89, obtained in a 63% yield, shows an expansion of one of the rings in the tricyclic system to a seven-membered ring (Scheme 42).49 Its spectral analytical data and its Z-isomeric assignment are consistent with the previous data of other (Z)-β,β-bromoiodoenone.38,42,44
 |
| | Scheme 42 Ring expansion reaction of tricyclic homoadamantanone system to β,β-dihaloenone. | |
4.2 (E)-Mixed β,β-dihaloenones
The only example of (E)-β,β-dihaloenones comes from the ring expansion reaction of 90, reported by Marchand and co-workers (Scheme 43).50 The reported X-ray structure of this ring expansion product shows the (E)-geometry 91, contrary to the previously reported (Z)-geometry of McNelis rearrangement products, and unequivocally points to which carbon adjacent to the brominated ethynyl motif has shifted.38,42,44,47–49 The ring expansion reaction of Marchand could be a particular case of the McNelis rearrangement, in which an isomerisation of the halonium bridged-ion was involved, resulting in a bromonium bridged ion in place prior to the methylidene migration and a five-membered to a six-membered ring expansion. The isomerisation of the initial iodonium bridged-ion to the bromonium one is not understood, as well as the impact on the methylidene shift when fused to four and five-membered rings. Indeed, further study is needed to understand the factors governing this (E)-isomer path and the (Z)-isomer non-path.
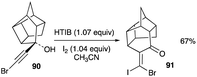 |
| | Scheme 43 (E)-β,β-Dihaloenone formation from ring expansion reaction of exo-8-(bromoethynyl)–endo-8-hydroxypentacyclo[5.4.1.02,6.03,10.05,9]undecane. | |
4.3 Symmetrical β,β-dihaloenones
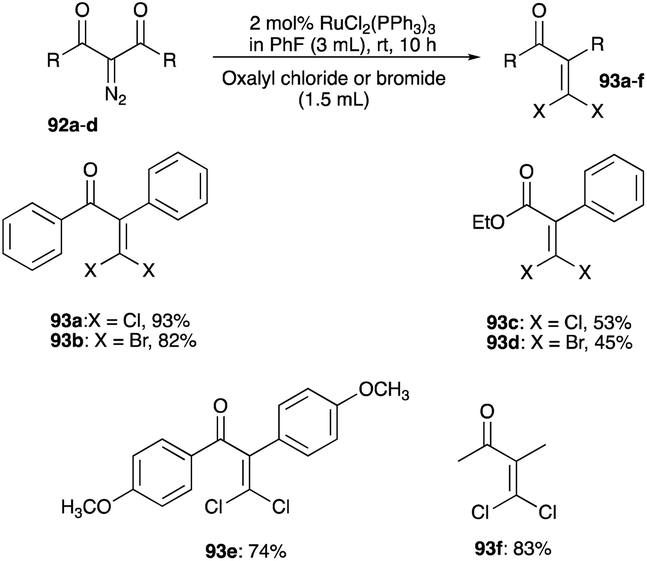
The McNelis rearrangement of bromo-propargyl alcohols by iodination forms mixed β,β-bromoiodoenones. This rearrangement can be adapted by brominating or iodinating the bromo and iodo analogues of propargyl alcohol, respectively to symmetrical β,β-dibromoenone or β,β-diiodoenone. However, the absence of symmetrical dichloroenone using the McNelis rearrangement route is documented and sheds light on a lack of effective chlorination when using NCS with a Brønsted acid catalyst or HTIB. A convenient synthesis of symmetrical β,β-dichloroenone as well as β,β-dibromoenone has been reported by Lee et al. and this entails a conversion of diazodicarbonyls 92a–d to symmetrical β,β-dihaloenones 93a–f using RuCl2(PPh3)3 catalyst with oxalyl chloride or bromide as the source of chloride or bromide (Scheme 44).51 The same group, using AuFeAg hybrid nanoparticles, later demonstrated the effectiveness of this alternative in the synthesis of α,β-dichlorenones 93e and β,β-dichloroenones 93f from diazocarbonyl substrates (Scheme 44).52,53
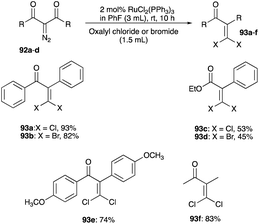 |
| | Scheme 44 Synthesis of symmetrical β,β-dihaloenones. | |
4.4 Iodination of halo propargyl alcohol furanoses
Iodination of the haloalkynols of xylofuranose and glucofuranose gives a different reaction path with NIS and catalytic TsOH or HTIB and iodine. The expected ring expansion, observed in an iodonium ion-induced rearrangement of the haloalkynols of cyclopentanones, camphor, norcamphor, adamantanone and fluorenone systems, did not occur.44,45,47,49 Instead, the haloalkynols of the two furanoses participated in a 5-exo-dig cyclisation or a haloetherification reaction (Schemes 45 and 46).54,55 3-Haloethynyl-1,2-O-isopropylidene-5-O-methyl-α-D-pentofuranose 95a–b, converted from a series of reactions to the haloalkynol adduct from commercially available 1,2-O-isopropylidene-α-D-xylofuranose 94, was iodinated at room temperature with either NIS and catalytic TsOH combination or HTIB and iodine mixture in 5% water–solution and dry acetonitrile, respectively, to give a haloetherification product in high yields. Although both the haloetherification and the ring expansion (alkyl shift) are intramolecular, it appears that the intramolecular cyclisation of the primary methyl ether with the vinyl cation at the internal ethyne carbon, produced by alkyne iodination, is preferred over the C-4 alkyl shifts of the 5-membered ring (Scheme 45).54
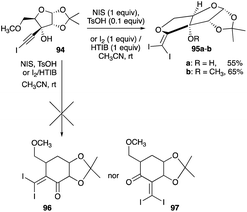 |
| | Scheme 45 Haloetherification of haloalkynols furanose. | |
 |
| | Scheme 46 Haloetherification–cyclisation reaction of glucofuranose. | |
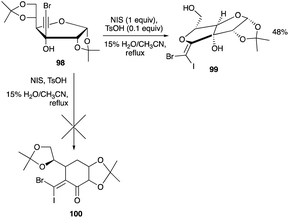
Another example of iodonium-induced rearrangement of alkynol derivatives of sugars involves the bromoalkynol adduct of 98. To encourage the alkyl shift, the iodination was conducted in a 15% water–acetonitrile solvent mixture.53 The anticipated ring-expanded product 100 involving C-4 carbon shift in the glucofuranose was again not observed. Rather, the glucofuranose undergoes a 5-exo-dig haloetherification–cyclisation reaction (Scheme 46).55 As with the haloetherification of the haloalkynols of xylofuranose, this haloetherification occurs with the loss of the acetonide group to yield the vinyl ether product 99.
5. Conclusion
Electrophilic halogenations of propargyl alcohols and analogues give α-haloenones, β-haloenones, and mixed β,β-dihaloenones. Depending on the type of alkynylphilic electrophiles and reaction conditions, these haloenones can be synthesised with varying degrees of Z/E selectivities from rearrangements of propargyl alcohols and ester, alkoxy and halo analogues. The fledgling direct halogenations of propargyl alcohols afford α-iodoenals, α-iodoenones, and α-bromoenones with low Z/E selectivities, but in propargyl acetate and tosylate instead of the alcohols, remarkable Z/E selectivity enhancements were observed. As with precious metals used in the Meyer–Schuster rearrangement, gold catalysts, among other precious metal catalysts, are useful in the synthesis of high Z/E selective α-iodoenones and α-bromoenones from propargyl acetates, and propargyl alcohols, albeit with MoO2(acac)2 co-catalyst to provide an in situ ester to curtail the loss of Z/E selectivity. For the challenging kinetic (E)-haloenones, conversions of propargyl acetates and alcohols to α-fluoroenones, α-chloroenones, α-bromoenones and α-iodoenones with high E/Z ratios have been demonstrated by gold(I), gold(III) and vanadium catalysts.
β-Haloenones, except for β-chloroenone – yet to be reported, are formed from 1,2-aryl and 1,2-alkyl shifts during halogenations of the alkyne of propargyl alcohols. Studies have shown that hexafluoroisopropanol gives superior results than acetonitrile, and water as co-solvent favours the 1,2-shift pathway over other competing dehydration to enyne and halo-Meyer–Schuster paths. A 1,2-phenyl shift from the propargyl carbon of terminal halo propargyl alcohols affords (Z)-mixed β,β-dihaloenones, and for β,β-diiodoenone from iodinations of iodo alkynols. Symmetrical β,β-dihaloenones such as β,β-dichloroenone and β,β-dibromoenone can be synthesised from diazodicarbonyls using Ru catalyst and oxalyl chloride or bromide as the source of chloride or bromide.
Halogenations of the haloalkynols of xylofuranose and glucofuranose did not give the anticipated ring expanded pyranose products; instead, the haloalkynol undergoes a haloetherification reaction via 5-exo-dig cyclisation.
These α-haloenones, β-haloenones, and mixed β,β-dihaloenones are, in essence, tri and tetrasubstituted alkenes with synthetically useful halo-functionalization at the α or β carbon. Yet the recent study of a computational NMR via machine learning-augmented DFT, shows evidence that there are structural interpretation errors of some unusual iodination rearrangements of propargyl alkoxide and bicyclic propargyl alcohols. Hence, further study is highly recommended as they serve as potential templates for the construction, via Pd-catalysed coupling reactions, of challenging substituted alkenes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to Mahidol University International College (MUIC) of Mahidol University for the continuing support.
Notes and references
-
(a) S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS;
(b) D. A. Engel and G. B. Dudley, Org. Biomol. Chem., 2009, 7, 4149–4158 RSC.
-
(a) R. F. Heck, Org. React., 1982, 27, 345–390 CAS;
(b) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef;
(c) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS;
(d) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS PubMed;
(e) G.-P. Lu, K. R. Voigtritter, C. Cai and B. H. Lipshutz, J. Org. Chem., 2012, 77, 3700–3703 CrossRef CAS PubMed.
-
(a) W. J. Gradishar and V. C. Jordan, J. Clin. Oncol., 1997, 15, 840–852 CrossRef CAS;
(b) R. Mansel, A. Goyal, E.-L. Nestour, V. Masini-Etévé and K. O'Connel, Breast Cancer Res. Treat., 2007, 106, 389–397 CrossRef CAS;
(c) V. C. Jordan, Nat. Rev. Drug Discovery, 2003, 2, 205–213 CrossRef CAS PubMed.
-
(a) B. Alcaide and P. Almendros, Acc. Chem. Res., 2014, 47, 939–952 CrossRef CAS PubMed;
(b) Y. Zhu, L. Sun, P. Lu and Y. Wang, ACS Catal., 2014, 4, 1911–1925 CrossRef CAS.
- N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2750–2752 CrossRef PubMed.
- D. Roy, P. Tharra and B. Baire, Asian J. Org. Chem., 2018, 7, 1015–1032 CrossRef CAS.
-
(a) G. F. Hennion and F. P. Kupiecki, J. Org. Chem., 1953, 18, 1601–1609 CrossRef CAS;
(b) D. Szemenyei, D. Steichen and J. E. Byrd, J. Mol. Catal., 1977, 2, 105–117 CrossRef CAS.
- For Ag(I) catalysis, see
(a) Y. Shigemasa, H. Oikawa, S. Ohrai, H. Sashiwa and H. Saimoto, Bull. Chem. Soc. Jpn., 1992, 65, 2594–2598 CrossRef CAS;
(b) M. Egi, M. Umemura, T. Kawai and S. Akai, Angew. Chem., Int. Ed., 2011, 50, 12197–12200 CrossRef CAS;
(c) J. García-Álvarez, J. Díez, C. Vidal and C. Vicent, Inorg. Chem., 2013, 52, 6533–6542 CrossRef.
- For Au(I) with Ag co-catalyst, see
(a) N. Marion, P. Carlqvist, R. Gealageas, P. de Frémont, F. Maseras and S. P. Nolan, Chem.–Eur. J., 2007, 13, 6437–6451 CrossRef CAS PubMed;
(b) S. I. Lee, J. Y. Baek, S. H. Sim and Y. K. Chung, Synthesis, 2007, 2107–2114 CAS.
- For Au(III) catalysis, see, D. A. Engel and G. B. Dudley, Org. Lett., 2006, 8, 4027–4029 CrossRef CAS PubMed.
- For Mo–Au(I)–Ag(I) complex, see, M. Egi, Y. Yamaguchi, N. Fujiwara and S. Akai, Org. Lett., 2008, 10, 1867–1870 CrossRef CAS PubMed.
- For other metal catalysis, see
(a) D. Ma and X. Lu, Tetrahedron Lett., 1989, 30, 2109–2112 CrossRef CAS;
(b) X. Lu, J. Ji, D. Ma and W. Shen, J. Org. Chem., 1991, 56, 5774–5778 CrossRef CAS;
(c) B. M. Trost and R. C. Livingston, J. Am. Chem. Soc., 1995, 117, 9586–9587 CrossRef CAS;
(d) M. K. E. Saïah and R. Pellicciari, Tetrahedron Lett., 1995, 36, 4497–4500 CrossRef;
(e) M. Picquet, C. Bruneau and P. H. Dixneuf, Chem. Commun., 1997, 1201–1202 RSC;
(f) M. Picquet, A. Fernández, C. Bruneau and P. H. Dixneuf, Eur. J. Org. Chem., 2000, 2361–2366 CrossRef CAS;
(g) T. Suzuki, M. Tokunaga and Y. Wakatsuki, Tetrahedron Lett., 2002, 43, 7531–7533 CrossRef CAS;
(h) V. Cadierno, J. Díez, S. E. Garcia-Garrido and J. Gimeno, Chem. Commun., 2004, 2716–2717 RSC;
(i) K. Tanaka and T. Shoji, Org. Lett., 2005, 7, 3561–3563 CrossRef CAS PubMed;
(j) V. Cadierno, S. E. García-Garrido and J. Gimeno, Adv. Synth. Catal., 2006, 348, 101–110 CrossRef CAS;
(k) V. Cadierno, J. Francos and J. Gimeno, Tetrahedron Lett., 2009, 50, 4773–4776 CrossRef CAS;
(l) M. Stefanoni, M. Luparia, A. Porta, G. Zanoni and G. Vidari, Chem.–Eur. J., 2009, 15, 3940–3944 CrossRef CAS PubMed;
(m) B. M. Trost and X. Luan, J. Am. Chem. Soc., 2011, 133, 1706–1709 CrossRef CAS PubMed;
(n) J. García- Álvarez, J. Díez, J. Gimeno and C. M. Seifried, Chem. Commun., 2011, 47, 6470–6472 RSC;
(o) A. Antiñolo, F. Carrillo-Hermosilla, V. Cadierno, J. García-Álvarez and A. Otero, ChemCatChem, 2012, 4, 123–128 CrossRef;
(p) J. Park, J. Yun, J. Kim, D.-J. Jang, C. Ho Park and K. Lee, Synth. Commun., 2014, 44, 1924–1929 CrossRef CAS;
(q) L. Yang and Q. Zeng, Synthesis, 2017, 49, 3149–3156 CrossRef CAS.
- For non-precious metal catalysis, see
(a) J. S. Yadav, V. Prahlad and B. Muralidhar, Synth. Commun., 1997, 27, 3415–3418 CrossRef CAS;
(b) M. Nishizawa, H. Hirakawa, Y. Nakagawa, H. Yamamoto, K. Namba and H. Imagawa, Org. Lett., 2007, 9, 5577–5580 CrossRef CAS PubMed;
(c) S. Puri, M. H. Babu and M. S. Reddy, Org. Biomol. Chem., 2016, 14, 7001–7009 RSC.
- R. Antonioletti, M. D'Auria, G. Piancatelli and A. Scettri, Tetrahedron Lett., 1981, 22, 1041–1042 CrossRef CAS.
- G. J. Angara and E. McNelis, Tetrahedron Lett., 1991, 32, 2099–2100 CrossRef CAS.
- S. Chen and J. Wang, J. Org. Chem., 2007, 72, 4993–4996 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Tetrahedron Lett., 1993, 34, 8205–8208 CrossRef CAS.
- W. J. Morań and A. Rodríguez, Org. Biomol. Chem., 2012, 10, 8590–8592 RSC.
- H.-T. Zhu, M.-J. Fan, D.-S. Yang, X.-L. Wang, S. Ke, C.-Y. Zhang and Z.-H. Guan, Org. Chem. Front., 2015, 2, 506–509 RSC.
- T. Suárez-Rodríguez, Á. L. Suárez-Sobrino and A. Ballesteros, J. Org. Chem., 2018, 83, 12575–12583S CrossRef.
- S. Puri, N. Thirupathi and M. S. Reddy, Org. Lett., 2014, 16, 5246–5249 CrossRef CAS PubMed.
- M. Yu, G. Zhang and L. Zhang, Org. Lett., 2007, 9, 2147–2150 CrossRef CAS PubMed.
- M. Yu, G. Zhang and L. Zhang, Tetrahedron, 2009, 65, 1846–1855 CrossRef CAS.
- L. Zhang, J. Am. Chem. Soc., 2005, 127, 16804–16805 CrossRef CAS PubMed.
- S. Wang and L. Zhang, J. Am. Chem. Soc., 2006, 128, 8414–8415 CrossRef CAS.
- L. Ye and L. Zhang, Org. Lett., 2009, 11, 3646–3649 CrossRef CAS PubMed.
- P. Tharra and B. Baire, Chem.–Eur. J., 2017, 23, 2014–2017 CrossRef CAS.
- N. Naveen, G. Ramesh and R. Balamurugan, ChemistrySelect, 2019, 4, 13610–13614 CrossRef CAS.
- D. Wang, X. Ye and X. Shi, Org. Lett., 2010, 12, 2088–2091 CrossRef CAS.
- Y. Yang, W. Hu, X. Ye, D. Wang and X. Shi, Adv. Synth. Catal., 2016, 358, 2583–2588 CrossRef CAS.
- T. de Haro and C. Nevado, Chem. Commun., 2011, 47, 248–249 RSC.
- B. M. Trost and J. S. Tracy, ACS Catal., 2019, 9, 1584–1594 CrossRef CAS.
- J. J. Janas, E. T. Asirvatham and E. McNelis, Tetrahedron Lett., 1985, 26, 1967–1968 CrossRef CAS.
- X. Herault and E. McNelis, New J. Chem., 1997, 3, 377–382 Search PubMed.
- K. Mahesh Kumar, T. Khan, A. I. Almansour, N. Arumugam and S. Yaragorla, Tetrahedron Lett., 2020, 61, 152374 CrossRef CAS.
- I. M. Novitskiy and A. G. Kutateladze, J. Org. Chem., 2022, 87, 8589–8598 CrossRef CAS.
- J.-M. Chen and X. Huang, Synthesis, 2004, 2459–2462 CAS.
- G. J. Angara, P. Bovonsombat and E. McNelis, Tetrahedron Lett., 1992, 33, 2285–2288 CrossRef CAS.
- P. Bovonsombat, G. J. Angara and E. McNelis, Synlett, 1992, 131–132 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Synthesis, 1993, 237–241 CrossRef CAS.
- For a review on HTIB or Koser’s reagent and the applications of hypervalent iodine compound in organic synthesis, see
(a) G. F. Koser, C-Heteroatom-Bond Forming Reactions, in Hypervalent Iodine Chemistry, Topics in Current Chemistry, ed. T. Wirth, Springer, Berlin, Heidelberg, 2003, vol. 224 Search PubMed;
(b) G. F. Koser, Aldrichimica Acta, 2001, 34, 89–102 CAS;
(c) A. Varvoglis, Tetrahedron, 1997, 53, 1179–1255 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Tetrahedron Lett., 1992, 33, 7705–7708 CrossRef CAS.
- P. Bovonsombat, E. Djuardi and E. McNelis, Tetrahedron Lett., 1994, 35, 2841–2844 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Tetrahedron Lett., 1994, 35, 6431–6432 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Tetrahedron, 1993, 49, 1525–1534 CrossRef CAS.
- X. Herault and E. McNelis, Tetrahedron, 1996, 52, 10267–10278 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Tetrahedron Lett., 1993, 34, 4277–4280 CrossRef CAS.
- E. Djuardi, P. Bovonsombat and E. McNelis, Tetrahedron, 1994, 50, 11793–11802 CrossRef CAS.
- P. Bovonsombat and E. McNelis, Synth. Commun., 1995, 25, 1223–1229 CrossRef CAS.
- A. P. Marchand, D. Rajagopal, A. Burritt, S. G. Bott, W. H. Watson and D. Sun, Tetrahedron, 1995, 51, 11673–11680 CrossRef CAS.
- K. B. Somai Magar and Y. R. Lee, Adv. Synth. Catal., 2014, 356, 3422–3432 CrossRef CAS.
- K. Mishra, N. Basavegowda and Y. R. Lee, Appl. Catal., A, 2015, 506, 180–187 CrossRef CAS.
- For a review on synthesis of symmetrical and mixed gem-dihalovinyl systems, see, G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef CAS PubMed.
- E. Djuardi and E. McNelis, Tetrahedron Lett., 1999, 40, 7193–7196 CrossRef CAS.
- M. Blandino and E. McNelis, Org. Lett., 2002, 4, 3387–3390 CrossRef CAS.
Footnote |
| † This article is dedicated to the late Professor Edward J. McNelis and Dr Geetha J. Angara. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Punyanuch Sophanpanichkul
*a,
Punyanuch Sophanpanichkul