
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of water environment in chemical degradation of a covalent organic framework tethered with quaternary ammonium for anion exchange membranes†
Siyao Qiua,
Wei Wanga,
Jibao Lu *ab and
Rong Sunab
*ab and
Rong Sunab
aShenzhen Institute of Advanced Electronic Materials, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: jibao.lu@siat.ac.cn
bShenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
First published on 1st July 2022
Abstract
The anion exchange membrane (AEM) is a main component for AEM fuel cells. Recently, a series of electrolytes based on covalent organic frameworks (COFs) functionalized with quaternary ammonium (QA) of showed extraordinary ionic conductivities thanks to the intrinsic porosity of the COF structures, which also provide a robust backbone for good mechanical strength. However, the chemical stability of the COF-based AEMs in alkaline conditions is yet to be understood. Here we systematically investigate the chemical degradation of the COF-based structures tethered with alkyl spacers by combining molecular dynamics (MD) simulations and density functional theory (DFT) calculations. We find that the water environment protects the cationic groups from chemical degradation in terms of both physical and chemical effects, which play a synergistic role. Moreover, we introduce the effective density of water as an order parameter to quantitatively characterize the level of degradation of the COF-based systems with similar design of architecture. The results provide guidance for estimation of the chemical stability of COF-based AEMs.
Polymer electrolyte fuel cells, also known as ion exchange membrane fuel cells, are a promising technology for efficient energy conversion from chemical to electrical energy.1,2 The anion exchange membrane fuel cells (AEMFC) attract tremendous attention for their high pH operating conditions, which could avoid cathode flooding and catalyst corrosion, and be low-cost electrodes.3,4 The anion exchange membrane (AEM) is constituted by polymers functionalized with cationic groups, which are used to conduct the anions from one electrode to another in terms of vehicular (standard diffusion) and Grotthuss (proton hopping) mechanisms.5 Though hydroxide anions has relatively lower diffusivity than protons in water, AEMs with comparable ionic conductivities with that of the proton exchange membranes have been fabricated by engineering the cationic groups, including the quaternary ammonium (QA),6 guanidinium,7 phosphonium,8 pyridinium,9 imidazolium,10 morpholinium,11 multi-cation side chains.12 Researchers have also introduced microphase segregation in AEMs to build highways for anion transport, such as the cross-linked AEMs13–15 and pore-filled AEMs.16,17 In particular, the cross-linking could improve the mechanical strength of the AEM to depress water swelling, which is particularly beneficial to the mechanical processability in the fuel cells.
Recently, a series of AEMs based on Covalent Organic Frameworks (COFs) tethered with QA cationic groups have been proposed and synthesized,18 which have several advantages thanks to the structure. On one hand, the high porosity of the COF structure provides channels for the ion transport. On the other hand, the structure supported by the stable covalent bonds act as a robust framework to prevent the mechanical swelling, which have been a pain point for traditional AEMs.19 Moreover, the organic backbone units offer variable joint for the functional side chains with cationic groups to bind. As a result, the COF-based AEMs exhibits remarkable hydroxide conductivities up to 212 mS cm−1 at 80 °C, which is one of the highest among the AEMs reported so far.18 To fabricate COF with functional side chains, QA-functionalized hydrazide are initially synthesis with corresponding side chains. Then COF membranes could be prepared with QA-functionalized hydrazide and 1,3,5-triformylbenzene building units via phase-transfer polymerization process.18
Besides the ionic conductivity, the chemical stability is another critical point, which determines the lifetime of the AEMs.20–22 Unfortunately, the quaternary ammonium (QA) cationic functional groups of the AEM may react with the hydroxide anions under high pH condition, which leads to a detrimental degradation of the performance and reduced lifetime of the membrane. Three possible degradation routes have been proposed in the previous works,23–25 including the loss of the entire cationic group, the demethylation and the backbone degradation. The former has been observed in simulations with the ReaxFF force field, which was specifically developed by van Duin et. al. for AEMs.23,24 The density functional theory (DFT) calculations also provide insights into the reactions mostly by characterizing the change of energy of those reactions from first principles, which has been used to study the degradation of cationic functional groups including imidazolium, pyrrolidinium, phenyltrimethylammonium and so on.26–32 It has been shown that the chemical degradation of the AEMs could be substantially influenced by the water environment.32 Thanks to the rigid framework of the COF structure, the COF-based AEMs can achieve high water uptake with relatively low swelling, which in principle facilitates a better chemical stability. However, the chemical degradation of the COF-based membranes has not yet been theoretically studied, especially for the novel outstanding COF-QA structure. Quantifying the effect of water environment on the chemical degradation and understanding the interplay between them will definitely facilitate the future design of the COF-based AEMs. To this end, we systematically investigate the chemical degradation of the COF-QA structures by combining the molecular dynamics (MD) simulations and DFT calculations. We find that the water environment protects the cationic groups from chemical degradation in terms of both physical and chemical effects, which play the role synergistically. Moreover, we introduce the effective density of water as an order parameter to quantitatively characterize the level of degradation of the COF systems.
Two COF structures, COF-QA-2 and COF-QA-6, are built based on the experimental data provided in the reference work, as shown in Fig. 1.18 The former has a –(CH2)2–N(CH3)3+ side chain binding on the backbone, while the latter tethers a side chain of –(CH2)6–N(CH3)3+. For each structure, a supercell of 8 COF layers is constructed with 24 OH− ions in total (Fig. 1). The degradation of the COF structure as a function of time are simulated by using MD simulations with ReaxFF, while the degradation energy of the side chains is obtained with the DFT calculations (computational details in ESI†).
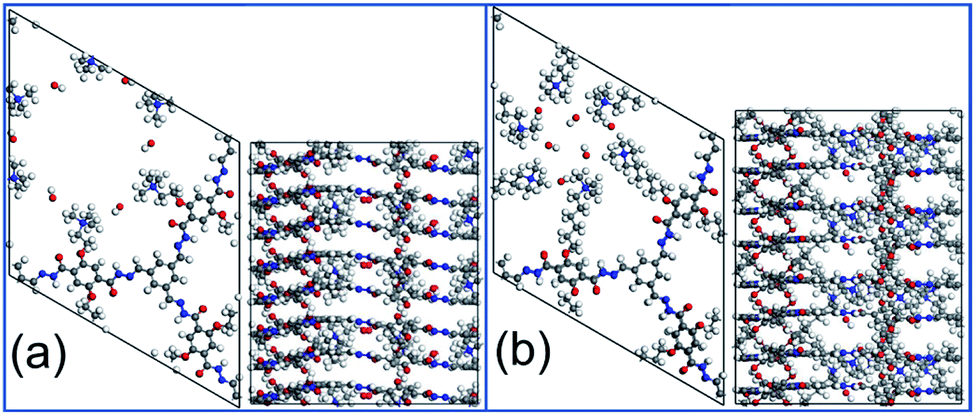 | ||
| Fig. 1 The structures of COF-QA-2 (a) and COF-QA-6 (b). Colour scheme: C (grey), O (red), H (white) and N (blue). | ||
The overall chemical degradation of the COF-QA-2 and COF-QA-6 structures are characterized at the same operating condition in terms of water fugacity. The water uptakes of the two COF structures are determined with Grand canonical Monte Carlo (GCMC) simulations at water fugacity of 101 kPa. After equilibration, the COF-QA-2 and COF-QA-6 accommodate 440 and 479 water molecules, respectively. Then, the force field is switched to ReaxFF to allow the chemical reaction to occur, in which the residual numbers of hydroxide ions as functions of time (Fig. 2) are collected. The reduction profile of the hydroxide ions reveals the levels of degradation of the quaternary ammonium in the COF-QA-2 and COF-QA-6 systems. After 800 ps simulation, the average numbers of OH− residues reach plateaus at 9.25 and 7.32 for QA-2 and QA-6, respectively, which are computed from averaging the last 200 ps simulation. Similar with the previous degradation research on AEM,23,24 no backbone degradation has been sampled with ReaxFF simulations on these two models. Fig. 2 suggests that the COF-QA-2 with ethyl side chains has the better degradation resistance than the COF-QA-6 with hexyl chains at the operating condition with the water fugacity. It is worth mentioning that, in experiments, COF-QA-2 membrane presents higher ionic conductivity than COF-QA-6, as well.18 We then try to understand the difference in chemical stability of those COF-based membranes through DFT calculation.
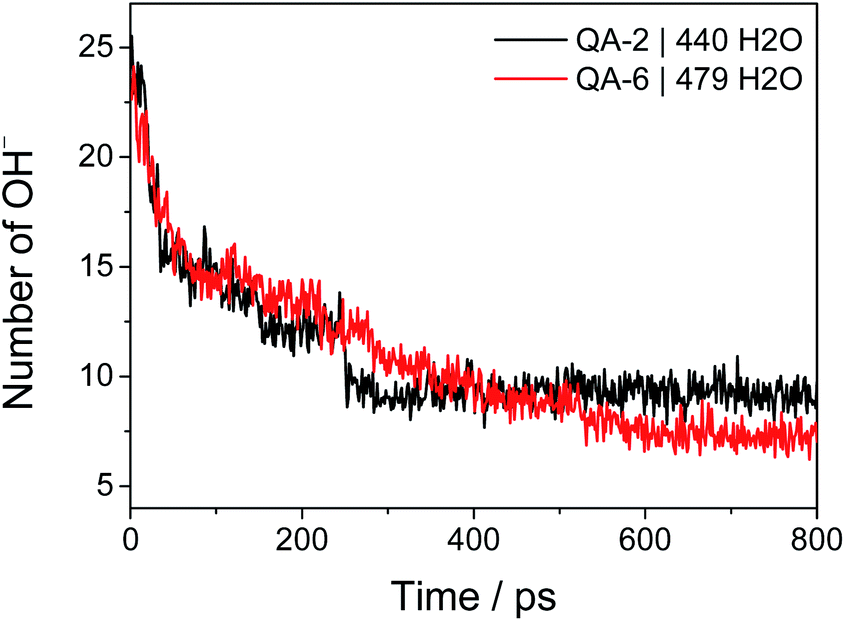 | ||
| Fig. 2 Time dependence of the residual number of OH− in the COF-QA-2 and COF-QA-6 systems with 440 and 479 water molecules, respectively. The numbers of water molecules are determined from the GCMC simulations in advance. | ||
There generally two possible reaction routes for the degradation of the side chains, the loss of the entire cationic group and the demethylation, which are described in (1) and (2), respectively:
| –R–N(CH3)3+ + OH− → –R–OH + N(CH3)3 | (1) |
| –R–N(CH3)3+ + OH− → –R–N(CH3)2 + CH3OH | (2) |
The energy differences for the two reactions, without water molecules around, are obtained from the DFT calculations (Fig. 3). In this work, we focus on the level of degradation after long relaxation time. And the level of degradation is determined the thermodynamics of the reaction. Relatively, the energy barrier mainly affects the degradation rate. Therefore, we have calculated the Gibbs free energy changes for the degradation reactions. The changes of the energy for reaction path (1) and (2) are −2.26 eV and −1.89 eV for COF-QA-2, respectively, and −2.11 eV and −1.75 eV for COF-QA-6, respectively. This indicates that the degradation prefers route (1) rather than route (2) for both the COF-QA-2 and COF-QA-6 structures. Moreover, the degradation energy of route (1) for the two structures are quite similar (−2.26 eV vs. −2.11 eV), which implies that the number of carbons on the side chain has little impact on the chemical stability of the system.
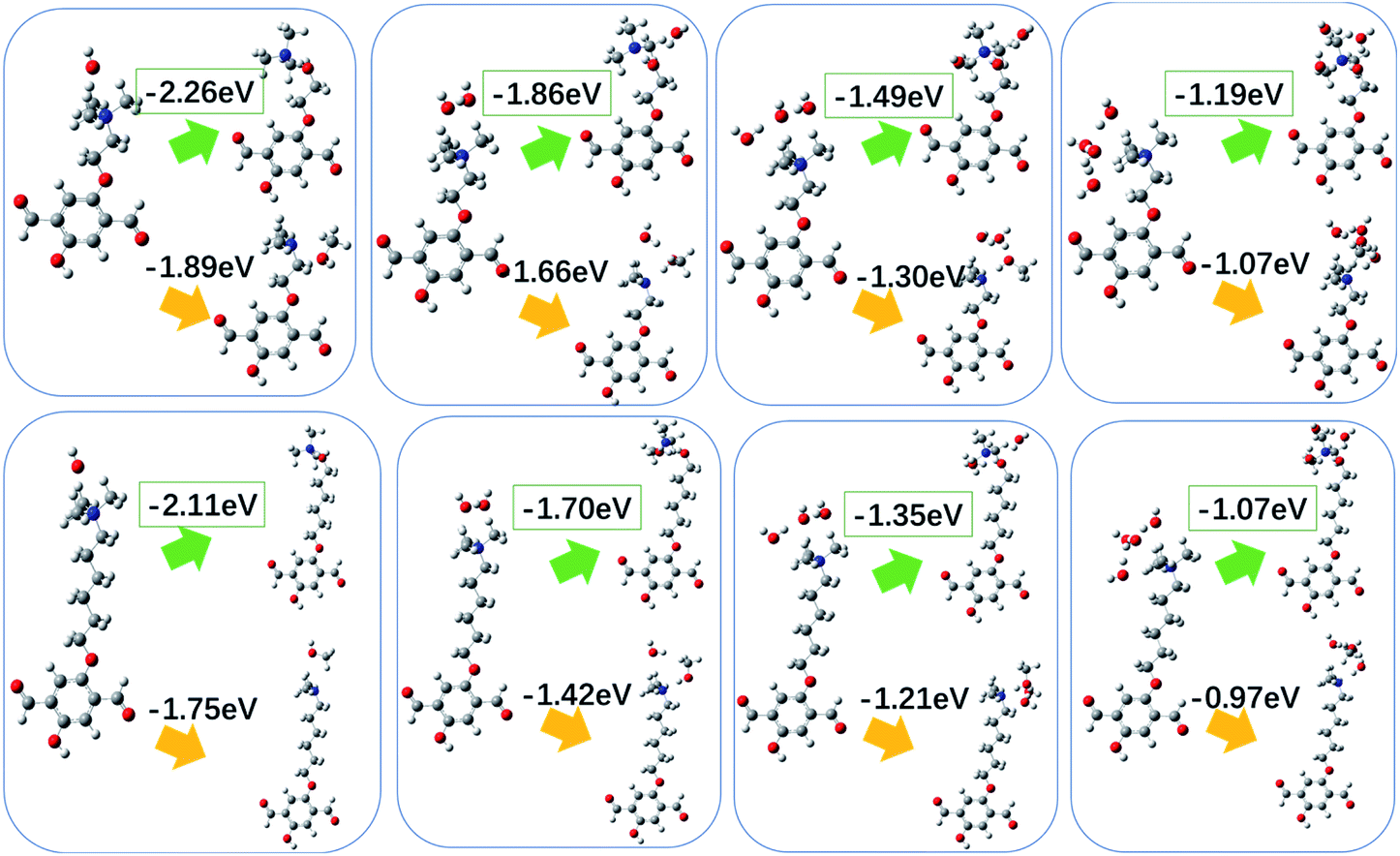 | ||
| Fig. 3 Relative Gibbs free energy difference of the chemical degradation on COF-QA-2 (up) and COF-QA-6 (down) with different numbers of surrounding water molecules, with the loss of the entire cationic group route directed by green arrow and the demethylation route by yellow. | ||
When water molecules are present around the OH− moieties, the energy difference before and after the degradation for both route (1) and (2) become less negative (Fig. 3). Taking the favoured route (1) as the example for discussion, the energy difference for the degradation of QA-2 are −2.26, −1.86, −1.49, and −1.19 with zero, one, two, and three water molecules around, respectively, which becomes less negative with increase of the number of water molecules. For QA-6, the energy differences are −2.11, −1.70. −1.35 and −1.07 eV, respectively, when zero to three water molecules are around, which shows similar trend as in QA-2. Note that the different initial configurations have been tested (see ESI†), the different energy result are very similar and would not affect the energy variation trend with the increasing of water molecules around. Considering that the first coordination shell of water molecules around the N atom of the quaternary ammonium is about 23, it is impossible to model all water molecules in the first coordination shell. To further exam the trend of energy change with increasing water molecules around, we test the reaction route (1) for QA-2 models with 4 and 5 water molecules around, and find that the energy difference follows the trend of becoming less negative (see ESI†). The result agrees well with the previous work on a different side chain of trimethylbenzyl ammonium bromide, which also suggests that the energy difference become less negative with adding water molecules around.32 The less negative energy difference for the reaction because of water molecules presenting around makes the chemical degradation less likely to happen. To understand the origin of the change of degradation energy, we calculate the Mulliken charge distribution of OH− with/without water molecules around, as shown in Table 1. We find that the more surrounding water molecules around the OH− moiety, the less negative the OH− charge distribution is. This is because the negative charge transfers from OH− to water molecules when forming hydrogen bonds between them, making the less negative OH− moiety harder to react with the side chains.
| Models | OH− charge |
|---|---|
| QA2-OH | −0.6355 |
| QA2-OH-H2O | −0.6072 |
| QA2-OH-2H2O | −0.5890 |
| QA2-OH-3H2O | −0.5575 |
Besides the abovementioned protection in terms of reaction energies, the water may provide protection for the cationic groups from a physical perspective, as found in the traditional AEMs.23,24,33,34 To study how that takes effect in the COF structure, we perform MD simulations on the COF-QA-2 model. Radial distribution function (RDF) for quantitatively analysing the number of water molecules around the OH- has been conducted in previous works.33,34 For instance, Chen et. al. performed RDF analysis with quaternary ammonium functionalized Poly (ether ether ketone) to study the alkaline stability of single quaternary and gemini quaternary membranes. The radial distribution functions (RDFs) between the hydroxide and –N(CH3)3+ moieties (Fig. 4) clearly indicate that the hydroxide ions get farther away from the ammonium as increasing the water molecules in the COF cavity. As a result, the degradation of the side chains of the COF could be inhibited by preventing the OH− from approaching the –N(CH3)3+, which superimposes a physical effect to the protection of the cations in addition to making the reaction energy difference of degradation less negative.
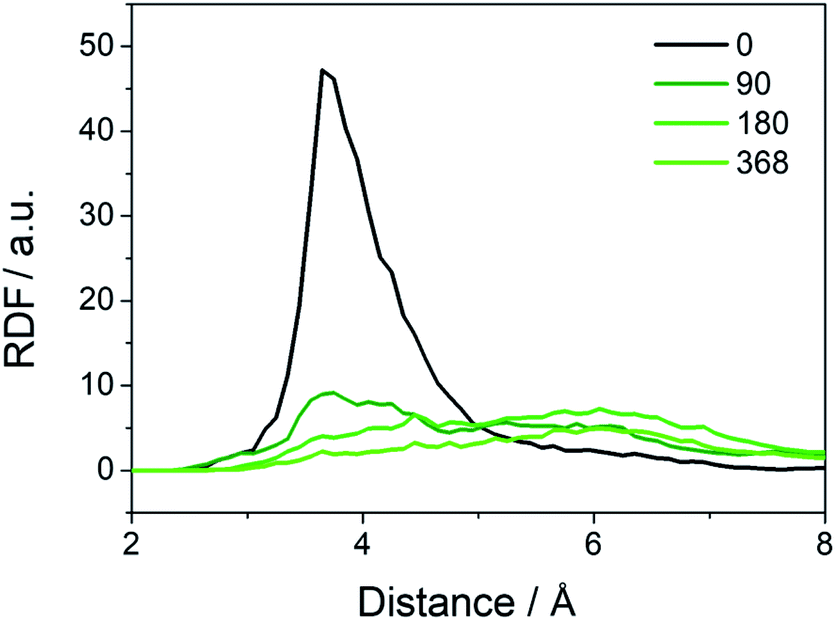 | ||
| Fig. 4 Radial distribution functions (RDFs) of the oxygens of (OH−) around the nitrogens of (-N(CH3)3+) in the COF-QA-2 system with different number of surrounding water molecules. | ||
To quantitatively characterize the degradation process, we perform ReaxFF MD simulations and monitor the variation of number of OH− residues when different number of water molecules are filled in the cavity. The initial number of OH− residues is approximately 24; after 800 ps, the numbers of OH− residues in COF-QA-2 reduce to plateaus at 0.15, 1.06, 2.22, 6.04 and 9.25, averaged over the last 200 ps, for the structures filled with water molecules of 0, 90, 180, 360 and 440, respectively (Fig. S1a†). A similar trend can be seen in COF-QA-6, with OH− residual numbers of 0.11, 0.88, 1.65, 4.43 and 7.32 in the models containing 0, 96, 192, 384 and 479 water molecules, respectively (Fig. S1b†). The increase of OH− residual with increasing number of filled water molecules evidences the important role of water environment in protecting the COF membranes from chemical degradation. The water environment is usually described by the hydration level λ, defined as the number of water molecules divided by the number of cationic ions.23,32 Fig. 5a shows the degradation of COF-QA-2 and COF-QA-6 structures as functions of λ. It can be seen that the number of OH− residues in the COF-QA-2 system is systematically higher than that in the COF-QA-6 one at each λ. To obtain a deeper insight into the role of water environment, we define effective number density of water, ρeffw, which is calculated as the hydration level λ normalized by the effective volume of the void in the COF structure. The void is estimated by multiplying the total volume of the COF model with the void ratio derived from the probability density of an helium probe inside the cavity, which is implemented in the iRASPA program.35 We find that the degree of degradation, characterized by the number of OH− residues, degenerate into a single curve as a function of ρeffw (Fig. 5b). This suggests that ρeffw, instead of λ, is the order parameter to quantitatively evaluate the chemical stability of the COF-based AEMs with the similar design of architectures. As a further example, the data points of the COF-QA-4 systems also fall into the same characteristics of the COF-QA-2 and COF-QA-6 systems, as shown in Fig. 5b. It is worth mentioning that the energy difference for the degradation reactions calculated from DFT also coincides with each other in between the COF-QA-2, COF-QA-4 (see ESI†) and COF-QA-6 systems. Interestingly, we note that the number of OH− residue is continuously growing with increasing water without evidence of approaching saturation until at the point where λ approaches the coordination number of water around the cations. This indicates that the kinetic effects of water environment on the chemical degradation may play an important role, which is not considered in this work but could be a future interest.
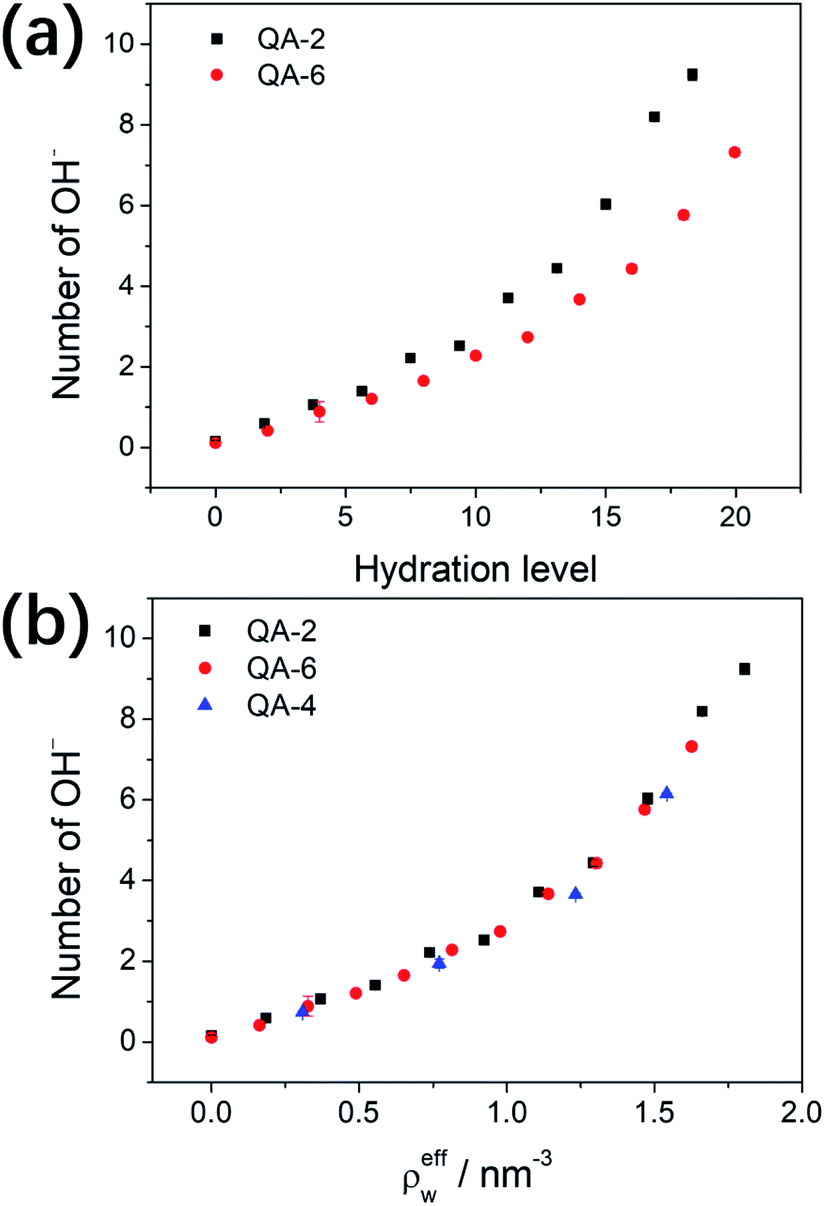 | ||
| Fig. 5 The residual number of OH− after equilibration of the degradation reaction, as functions of the hydration level λ (a) and effective number density of water ρeffw (b) in the COF-QA-2, COF-QA-4 and COF-QA-6 systems (bars denote the standard deviation, which are too small to be clearly seen). | ||
As a brief verification of the order parameter ρeffw , we take the COF-QA-6 filled with 479 water molecules in the void as a reference for the ρeffw , which corresponds to the water uptake of COF-QA-6 derived from the GCMC simulations (Fig. 2), and build a COF-QA-2 structure filled with 396 water molecules. In this case, the ρeffw in the two systems would exactly be the same, 1.626 per nm3 (see ESI† for detailed conversion of ρeffw). Then we perform MD simulations with the ReaxFF and find that the number of OH− residues converge almost to the same value, 7.05 vs. 7.32 (Fig. S2†), irrespective of the different structures of the two COFs. This demonstrates the robustness of the order parameter as an intuitive parameter to evaluate the chemical stability of the COF-based AEMs with similar design of architectures, and emphasizes the importance of water environment to the stability of the performance of the membranes.
In summary, by combining the MD simulations and DFT calculations, we find that a synergistic physical and chemical protection by the surrounding water environment inhibits the chemical degradation of the side chains of the COF-based AEMs. The MD simulations indicate that the water could prevent the OH− from physically approaching the side chains, while the DFT calculations reveal that increasing the surrounding water could make the energy difference of the degradation reaction less negative, which originates from the formation of H-bonds between OH− and water that turns the OH− charge less negative and thus the harder occurrence of electrophile-nucleophile neutralization. Moreover, we find that the effective density of water could be an order parameter to quantitatively characterize the level of degradation of the series of COF-based systems. Though a complete understanding of the degradation mechanism requires consideration of the kinetic effect of the reaction, the presented study provides a guidance for the quantitative estimation of the chemical stability of the COF-based anion exchange membranes with the similar architecture with ease.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by the National Natural Science Foundation of China (no. 52 003 289), Shenzhen Basic Research project (no. JCYJ20180302145742105), the Youth Innovation Promotion Association CAS (no. 2021363), the SIAT Innovation Program for Excellent Young Researchers (no. 201803), and Guangdong Basic and Applied Basic Research Foundation (2020A1515110978). We thank the National Supercomputer Centre in Guangzhou for computer time.Notes and references
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-i. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- Y. Wang, Y. Yang, S. Jia, X. Wang, K. Lyu, Y. Peng, H. Zheng, X. Wei, H. Ren, L. Xiao, J. Wang, D. A. Muller, H. D. Abruña, B. J. Hwang, J. Lu and L. Zhuang, Nat. Commun., 2019, 10, 1506 CrossRef PubMed.
- J. Lu, A. Barnett and V. Molinero, J. Phys. Chem. C, 2019, 123, 8717–8726 CrossRef CAS.
- D. Dong, W. Zhang, A. C. T. van Duin and D. Bedrov, J. Phys. Chem. Lett., 2018, 9, 825–829 CrossRef CAS PubMed.
- C. Chen, Y.-L. S. Tse, G. E. Lindberg, C. Knight and G. A. Voth, J. Am. Chem. Soc., 2016, 138, 991–1000 CrossRef CAS PubMed.
- M. Zhang, J. Liu, Y. Wang, L. An, M. D. Guiver and N. Li, J. Mater. Chem. A, 2015, 3, 12284–12296 RSC.
- J. Wang, S. Li and S. Zhang, Macromol, 2010, 43, 3890–3896 CrossRef CAS.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS PubMed.
- A. Vöge, V. Deimede and J. K. Kallitsis, RSC Adv., 2014, 4, 45040–45049 RSC.
- E. Abouzari-Lotf, M. V. Jacob, H. Ghassemi, M. Zakeri, M. M. Nasef, Y. Abdolahi, A. Abbasi and A. Ahmad, Sci. Rep., 2021, 11, 3764 CrossRef CAS PubMed.
- L. Gao, C. Lu, S. Ma, X. Yan, X. Jiang, X. Wu and G. He, Int. J. Hydrogen Energy, 2020, 45, 29681–29689 CrossRef CAS.
- L. Li, J. Zhang, T. Jiang, X. Sun, Y. Li, X. Li, S. Yang, S. Lu, H. Wei and Y. Ding, ACS Appl. Energy Mater., 2020, 3, 6268–6279 CrossRef CAS.
- N. Li, L. Wang and M. Hickner, Chem. Commun., 2014, 50, 4092–4095 RSC.
- W. Chen, M. Mandal, G. Huang, X. Wu, G. He and P. A. Kohl, ACS Appl. Energy Mater., 2019, 2, 2458–2468 CrossRef CAS.
- N. A. Qaisrani, L. Ma, M. Hussain, J. Liu, L. Li, R. Zhou, Y. Jia, F. Zhang and G. He, ACS Appl. Mater. Interfaces, 2020, 12, 3510–3521 CrossRef CAS PubMed.
- M.-S. Lee, T. Kim, S.-H. Park, C.-S. Kim and Y.-W. Choi, J. Mater. Chem., 2012, 22, 13928–13931 RSC.
- D. S. Hwang, T. A. Sherazi, J. Y. Sohn, Y. C. Noh, C. H. Park, M. D. Guiver and Y. M. Lee, J. Membr. Sci., 2015, 495, 206–215 CrossRef CAS.
- X. He, Y. Yang, H. Wu, G. He, Z. Xu, Y. Kong, L. Cao, B. Shi, Z. Zhang and C. Tongsh, Adv. Mater., 2020, 32, 2001284 CrossRef CAS PubMed.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438–449 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490 CrossRef CAS PubMed.
- F. Schönberger, J. Kerres, H. Dilger and E. Roduner, Phys. Chem. Chem. Phys., 2009, 11, 5782–5795 RSC.
- W. Zhang and A. V. Duin, J. Phys. Chem. C, 2015, 119, 27727–27736 CrossRef CAS.
- W. Zhang, D. Dong, D. Bedrov and A. C. T. van Duin, J. Mater. Chem. A, 2019, 7, 5442–5452 RSC.
- S. A. Nuñez, C. Capparelli and M. A. Hickner, Chem. Mater., 2016, 28, 2589–2598 CrossRef.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- M. Kumar and S. J. Paddison, J. Mater. Res., 2012, 27, 1982–1991 CrossRef CAS.
- H. Long and B. Pivovar, J. Phys. Chem. C, 2014, 118, 9880–9888 CrossRef CAS.
- T. Xiang and H. Si, Comput. Theor. Chem., 2015, 1065, 12–17 CrossRef CAS.
- W. Wang, S. Wang, X. Xie, Y. lv and V. Ramani, Int. J. Hydrogen Energy, 2014, 39, 14355–14361 CrossRef CAS.
- S. Castañeda Ramírez and R. E. Ribadeneira Paz, ECS Trans., 2014, 64, 1233–1240 CrossRef.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- S. Chen, H. Wang, J. Zhang, S. Lu and Y. Xiang, J. Membr. Sci., 2020, 605, 118105 CrossRef CAS.
- D. R. Dekel, S. Willdorf, U. Ash, M. Amar, S. Pusara, S. Dhara, S. Srebnik and C. E. Diesendruck, J. Power Sources, 2018, 375, 351–360 CrossRef CAS.
- D. Dubbeldam, S. Calero and T. Vlugt, Mol. Simul., 2018, 44, 1–24 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods, effective density of water, chemical degradation under different water environment and at same density of water. See https://doi.org/10.1039/d2ra03449b |
| This journal is © The Royal Society of Chemistry 2022 |