DOI:
10.1039/D2RA03377A
(Paper)
RSC Adv., 2022,
12, 21704-21712
Intumescent flame retardancy and smoke suppression of Eucommia ulmoides gum/natural rubber blends based on synergistic g-C3N4@Fe3O4 nanocomposites
Received
31st May 2022
, Accepted 12th July 2022
First published on 5th August 2022
Abstract
A flame retardant synergist (g-C3N4@Fe3O4) was designed through an in situ co-precipitation method by using graphitized carbon nitride (g-C3N4) and Fe3O4, and its structure was characterized by Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). A series of different contents of g-C3N4@Fe3O4 and intumescent flame retardant (IFR) were simultaneously added into eucommia ulmoides gum/natural rubber (EUG/NR) blends to improve the flame retardancy and reduce the smoke release. The flame retardancy, smoke suppression performance, thermal degradation behaviors and thermal stability of EUG/NR blends were evaluated by the limiting oxygen index (LOI), UL-94, cone calorimetry test (CCT) and thermogravimetric analysis (TGA). The results showed that using g-C3N4@Fe3O4 as a flame retardant synergist, the LOI value of the 9 phr g-C3N4@Fe3O4 synergist (EUG/NR 5) blend was 29.5%, which was much higher than the 20.0% of the original composite. Moreover, the 6 phr g-C3N4@Fe3O4 synergist (EUG/NR 4) and EUG/NR 5 passed the UL-94 V-0 rating, while the pristine EUG/NR blends showed no rating. Moreover, the total heat release rate (THR), peak of heat release rate (PHRR) and total smoke production (TSP) of the EUG/NR blends in the CCT test were much lower than those of the pristine EUG/NR blends. In particular, PHRR, THR and TSP of EUG/NR 5 decreased by 37.1%, 16.2% and 18.0%, respectively. This indicated that the introduction of g-C3N4@Fe3O4 decreased the release of combustible gases. TGA results also showed that the addition of g-C3N4@Fe3O4 accelerated the thermal degradation of the EUG/NR blends and changed the thermal degradation mechanism of the EUG/NR blends, indicating the synergistic effect of g-C3N4@Fe3O4. Finally, a possible degradation mechanism of EUG/NR blend composites was proposed.
1 Introduction
Natural rubber (NR), which is recognized as the general rubber with the best comprehensive properties, has been widely used in our daily lives and industrial applications due to its excellent insulation, mechanical properties and chemical resistance.1 However, owing to the scarcity of NR resources, the production of NR is no longer sufficient to meet the expanding demand.2 Therefore, there is an urgent need to find materials that can replace NR. Eucommia ulmoides gum (EUG)3,4 has been recognized as a secondary source of NR due to its similar chemical structure. The molecular structure of EUG is trans-1,4-polyisoprene, which is the trans isomer of NR.5 Researchers have studied the influence of vulcanization on the mechanical properties of EUG/NR blends, and their results showed that the vulcanized rubber had ideal bending fatigue life, perfect wear resistance and stress softening.6 Additionally, it was needed to investigate the flame retardancy of rubber, aiming to meet quite a lot of areas with high flame retardancy requirements, such as aerospace, electrical insulating products, electrical wires and cables.7,8
Adding flame retardants into rubber can effectively improve its flame retardant property. Compared with other flame retardants, the intumescent flame retardant (IFR) plays the roles of heat insulation, oxygen insulation, smoke suppression, and prevention of dripping.9,10 In the combustion process, the IFR is decomposed by heat to form a porous carbon layer on the surface of the materials. However, low strength, easy collapse of the carbon layer and large amount of addition are the points that need to be improved in IFR. Recently, Li et al.11 found that the synergistic agent as an effective additive has a positive effect on the flame retardant properties of IFR. In our previous work,7,12 the modified graphene oxide grafted with nano-mesoporous molecular sieves MCM-41 was designed as an efficient IFR system to improve the flame retardancy performance of natural rubber. In addition, graphene oxide grafted boron nitride was developed using the silane coupling agent KH550, which was applied in composite materials to improve their flame retardancy. We found that the flame retardant synergist and IFR have an ideal synergistic effect on the thermal stability and charring properties of natural rubber.
It has been proven that most metal compounds, especially transition metal compounds (such as FeO, Fe2O3, Cu2O, and MoO3), are the most effective flame retardants and smoke suppression additives.13 Iron compounds have been known as flame-retardant synergists in halogen systems, which can improve the char morphology.14,15 Chen et al.16 investigated the synergistic effect of the intumescent flame retardant and Fe2O3 on silicone rubber (SR) composites, and the results showed that increasing the Fe2O3 loading fraction in the SR composites causes a significant increase in their fire retardancy capabilities and thermal stability, particularly the smoke suppression. Similar to Fe2O3, Fe3O4 also has a synergistic effect in combination with sepiolite when they are applied in epoxy composites, which can accelerate the thermal degradation of epoxy composites.17 Beaugendre et al.18 found that the incorporation of a low amount of iron oxide particles in the coating leads to an outstanding improvement of fire performances when such coating is applied on polycarbonate (PC). A 32% LOI value and a V-0 ranking in the UL-94 test were achieved for the pigmented system. PHRR and THR decreased by 17% and 44%, respectively, compared to pure PC.
Recently, utilized as a flame retardant for polymers, two-dimensional nanosheet materials have received attention.19,20 The nanosheet structure that acts as a physical barrier can effectively block the heat transfer and the release of smoke. Graphitized carbon nitride (g-C3N4) is a typical two-dimensional nanosheet material,21 which has the advantages of easy, fast, low-cost preparation, huge specific surface area and excellent thermal and chemical stability, compared with other layered materials.22 However, g-C3N4 is far from achieving satisfactory results when it is applied alone as a flame retardant.23,24 Fortunately, owing to the huge specific surface area of g-C3N4,25 the introduction of metal nanoparticles into the layered structure of g-C3N4 can not only exert the blocking effect of g-C3N4 but also catalyze the formation of a stable cross-linked char layer effectively, thus preventing the release of smoke and further pyrolysis of polymers.26
In this study, we presented the preparation of the g-C3N4@Fe3O4 nanocomposites by in situ co-precipitation and introduced it as a flame retardant synergist together with an IFR into the EUG/NR blend system, aiming to improve its flame retardancy. The effects of different contents of g-C3N4@Fe3O4 with the IFR on the flame retardancy and smoke suppression performance of the EUG/NR blends were studied. Furthermore, via analyzing the residual char structure and morphology of the EUG/NR blends by SEM, Raman spectrum and FTIR, the flame retardant mechanism was discussed.
2 Materials and methods
2.1 Materials
EUG was provided by Xiangxi Old Father Biological Co., Ltd, NR was provided by Xishuangbanna Jingyang Co., Ltd, and pentaerythritol (PER), melamine (MEL), FeCl2·4H2O, FeCl2·6H2O, ammonia solution and urea were purchased from Tianjin Damao Chemical Reagent Factory. The commercial product APP was bought from Shandong Yousseo Chemical Co. Ltd. The mass ratio of APP, PER, and MEL in the IFR mixture was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. Carbon black and age inhibitor 4010 were bought from Sheng Ao Chemical Co., Ltd., Tianjin, China. Analytical grade ZnO was provided by Dalian ZnO Factory, Dalian, China. Accelerant NOBS was purchased from Tianjin No. 1 Organic Chemical Plant, Tianjin, China. Sulfur was purchased from Tong Chuang Chemical Co., Ltd., Taizhou, China.
:1:1. Carbon black and age inhibitor 4010 were bought from Sheng Ao Chemical Co., Ltd., Tianjin, China. Analytical grade ZnO was provided by Dalian ZnO Factory, Dalian, China. Accelerant NOBS was purchased from Tianjin No. 1 Organic Chemical Plant, Tianjin, China. Sulfur was purchased from Tong Chuang Chemical Co., Ltd., Taizhou, China.
2.2 Synthesis of flame retardant synergist g-C3N4@Fe3O4 nanocomposites
g-C3N4@Fe3O4 was synthesized by a two-step method, involving the pristine g-C3N4 and in situ co-precipitation synthesis of the g-C3N4@Fe3O4 nanocomposite.27,28 The schematic representation of the in situ co-precipitation of Fe3O4 nanoparticles on g-C3N4 is shown in Scheme 1. Firstly, the urea was heated directly to 550 °C in a muffle furnace at a heating rate of 2.5 °C min−1 and held 3 h to prepare g-C3N4. Then, in an open ceramic container, g-C3N4 was further heated at a heating rate of 2 °C min−1 to 500 °C for 2 h.29 Second, according to a typical procedure, the obtained g-C3N4 (140 mg) was dispersed in water with a volume of 250 ml and ultrasonicated for 3 h at room temperature to achieve the nanoscale lamellar structured g-C3N4. Then, 0.92 g FeCl2·6H2O and 0.35 g FeCl2·4H2O were weighed and added according to the molar ratio of 2:3 into the g-C3N4 suspension at 80 °C. 10 ml of ammonia solution was quickly injected into the reaction mixture, followed by stirring at 80 °C for 30 min. Finally, the reaction mixture was cooled to room temperature and washed with deionized water and anhydrous ethanol. The g-C3N4@Fe3O4 nano-composites were dried in air at 60 °C.
 |
| | Scheme 1 Schematic representation of the in situ co-precipitation of Fe3O4 nanoparticles on g-C3N4. | |
2.3 Preparation of EUG/NR blend composites
Flame retardant EUG/NR blend composites were prepared with different proportions of g-C3N4@Fe3O4, IFR and EUG/NR blends by two-roll milling. The compositions of EUG/NR blend composites are listed in Table 1. First, the NR was plasticized on a rolling mill (25 °C) evenly to wrap the roll. Then, the EUG was plasticized on the rolling mill (70 °C), and the treated NR was added to the treated EUG for mixing. Further, zinc oxide, stearic acid, carbon black, IFR, g-C3N4@Fe3O4, age inhibitor 4010, accelerant NOBS, and sulfur were added successively. After filling, roll spacing was reduced to mix the rubber with the filler well. Finally, the rubber compounds were treated into thin sheets by using a two roll calender. All samples were vulcanized at 150 °C. The optimum cure time t90 was determined using a GT-M2000-rheometer (Gao Tie Limited Co., Taiwan). All specimens were cut into vulcanized sheets at room temperature for 24 h before subsequent use.
Table 1 Compositions of EUG/NR blend composites
| Samples |
NR/phr |
EUG/phr |
IFR/phr |
g-C3N4@Fe3O4/phr |
| EUG/NR 1 |
70 |
30 |
0 |
0 |
| EUG/NR 2 |
70 |
30 |
40 |
0 |
| EUG/NR 3 |
70 |
30 |
37 |
3 |
| EUG/NR 4 |
70 |
30 |
34 |
6 |
| EUG/NR 5 |
70 |
30 |
31 |
9 |
| EUG/NR 6 |
70 |
30 |
28 |
12 |
2.4 Characterization
Fourier transform infrared spectroscopy (FTIR), with a Nicolet MNGNA-IR560 (Artisan Technology Group, Austin, TX, USA), was applied under transition mode with samples in the wavenumber range between 400 cm−1 and 4000 cm−1. X-ray photoelectron spectroscopy (XPS) measurements were performed using a Thermo Scientific K-Alpha+ spectrometer from Thermo Fisher with Al Kα X-ray radiation at 15 kV and 15 mA to determine the chemical states and the valence states of the involved elements. The X-ray diffraction (XRD) patterns of the samples were collected on a Bruker D8 Advance X-ray diffractometer (Bruker, Germany) with Cu Kα radiation (λ = 0.154 nm). Thermogravimetric analysis (TGA) was performed on an STA 449C thermal analyzer (Selb, Germany). The heating rate was set at 10 °C min−1 from room temperature to 800 °C under nitrogen at a flow rate of 80 ml min−1. The vulcanization curve of raw rubber was tested using a GTM2000-A rubber vulcanization tester produced by High-speed Rail Testing Instrument Co., Ltd. The limiting oxygen index (LOI) (JF-3 oxygen index instrument, Jiangning Analytical Instrument Co., Ltd, China) was measured at room temperature with the GB/T10707–2008 standard. The dimension of the specimens was 126 mm × 6.5 mm × 3 mm and all of the composites were tested five times. The vertical burning tests (UL-94 test, Jiangning Analysis Instrument Company, China) were performed with a CZF-3 instrument. The sizes of the samples were 130 mm × 13 mm × 3 mm. All the samples were tested multiple times to prove repeatability. The cone calorimeter tests (CCT) (FTT, UK) were performed on a Fire Testing Technology cone calorimeter, which was used to characterize the combustion behavior of samples with dimensions of 100 mm × 100 mm × 4 mm under a heat flux of 35 kW m−2 according to ISO 5660-1. The scanning electron microscopy (SEM) images of burnt samples were obtained using a scanning electron microscope JEOL JSM-6360LV (Japan Electronics Co., Tokyo, Japan). Raman spectroscopy was performed using a Raman spectrometer (Horiba Scientific LabRAM HR Evolution) at an excitation wavelength of 514 nm.
3 Results and discussion
3.1 Characterization of g-C3N4@Fe3O4 nanocomposites
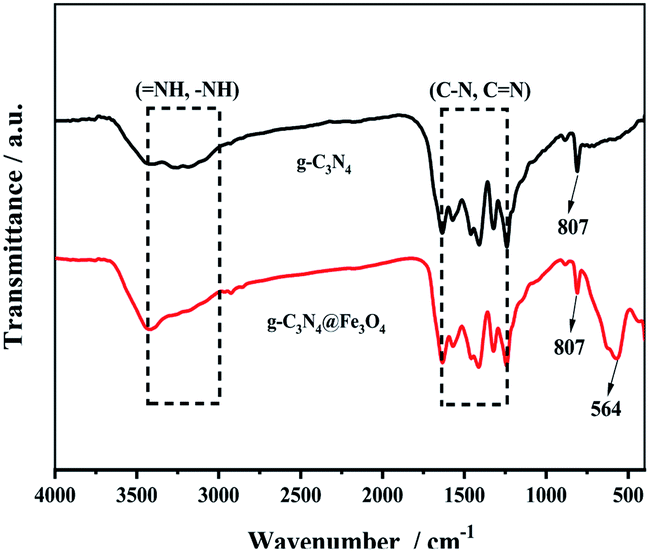
The FTIR spectra of g-C3N4 and g-C3N4@Fe3O4 are illustrated in Fig. 1. The primary (–NH) and secondary (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) N–H bonds that remained following calcination at high temperatures are responsible for the characteristic wide adsorption bands at 3430 cm−1. The stretching vibration peaks of C–N and CN of g-C3N4 as a heterocyclic compound were assigned to the bands ranging from 1241 cm−1 to 1635 cm−1.30 The vibration of the s-triazine ring is related to the peak at 807 cm−1.31 Furthermore, the broad Fe–O band at 564 cm−1 in g-C3N4@Fe3O4 is clearly visible and explained by the Fe–O absorption peak generated on the surface of carbon nitride by in situ chemical coprecipitation.32
NH) N–H bonds that remained following calcination at high temperatures are responsible for the characteristic wide adsorption bands at 3430 cm−1. The stretching vibration peaks of C–N and CN of g-C3N4 as a heterocyclic compound were assigned to the bands ranging from 1241 cm−1 to 1635 cm−1.30 The vibration of the s-triazine ring is related to the peak at 807 cm−1.31 Furthermore, the broad Fe–O band at 564 cm−1 in g-C3N4@Fe3O4 is clearly visible and explained by the Fe–O absorption peak generated on the surface of carbon nitride by in situ chemical coprecipitation.32
 |
| | Fig. 1 FTIR spectra of g-C3N4 and g-C3N4@Fe3O4 nanocomposites. | |
The full XPS spectra of g-C3N4 and g-C3N4@Fe3O4 are exhibited in Fig. 2(a). Compared with the spectrum of g-C3N4, the survey XPS spectrum of g-C3N4@Fe3O4 showed the coexistence of elements C, N, O and Fe. Fig. 2(b–e) show the high-resolution XPS spectra of C 1s, N 1s, O 1s, and Fe 2p. There are three peaks in C 1s at 284.4, 285.2 and 288.5 eV, representing sp2 C–C bonding, (CN–) group and sp3 hybrid carbon atoms. The N 1s spectrum is shown in Fig. 2(c); the three convolution peaks at 398.4, 399.1, and 400.6 eV are linked to sp2 hybridization N (C–NC), tertiary nitrogen binding to carbon atoms in the form of (N–(C)3) and the amino functional group, respectively. The O 1s peak (530.2 eV) is assigned to Fe3O4's lattice oxygen, whereas the other peak (531.9 eV) is attributed to the OH group generated by oxygen. As shown in Fig. 2(e), the XPS spectrum of Fe 2p shows four broad peaks of Fe species at about 710.4, 712.5, 717.7, and 724.5 eV. The broad peaks at 710.4 and 712.5 eV correspond to the Fe 2p3/2 Fe2+ and Fe3+ ions of Fe3O4, respectively. The broad peak at 724.7 eV is related to 2p1/2 Fe2+ and Fe3+.33 The peaks in the Fe 2p spectrum indicate the presence of Fe2+ and Fe3+ ions, which confirmed the existence of Fe3O4 particles.
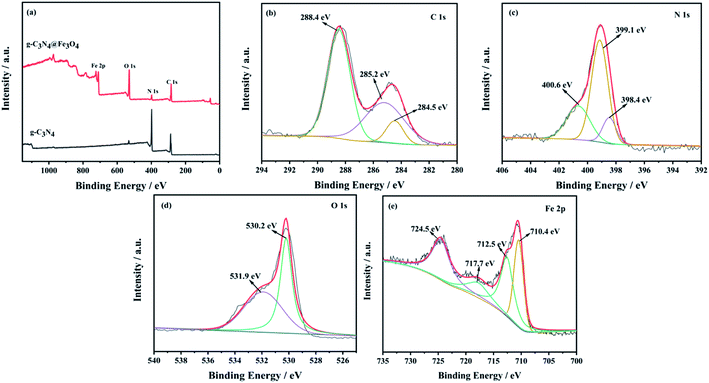 |
| | Fig. 2 (a) XPS spectra of g-C3N4 and g-C3N4@Fe3O4, (b) C 1s, (c) N 1s, (d) O 1s, and (e) Fe 2p. | |
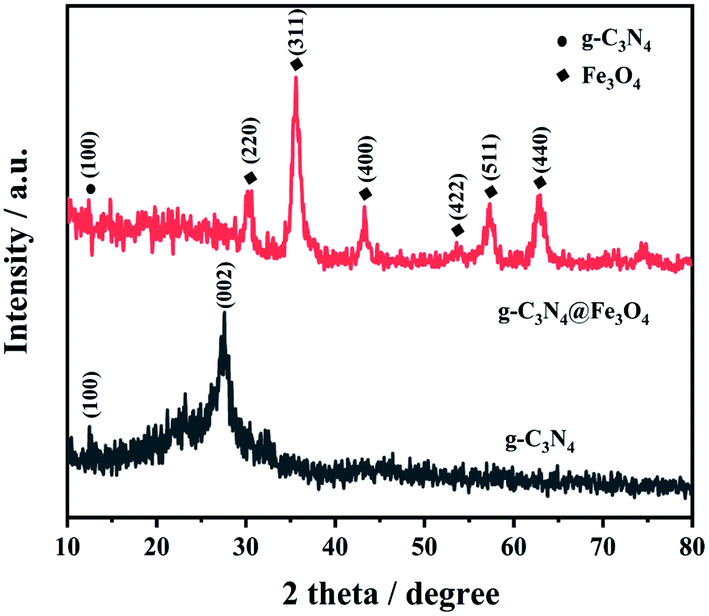
Fig. 3 shows the XRD patterns of g-C3N4@Fe3O4 composites; six characteristic peaks with reflection planes at 30.4°, 35.5°, 43.1°, 53.5°, 57.1°, and 62.8° correspond to (220), (311), (400), (422), (511), and (440), respectively, which are in accordance with the standard Fe3O4 crystal data (JCPDS 19-0629) and literature description.34 The g-C3N4@Fe3O4 displays a typical g-C3N4 characteristic peak, which corresponds to the g-C3N4 peaks (100). There are also Fe3O4 diffraction peaks in the pattern, showing that g-C3N4@Fe3O4 is a two-phase composite made up of g-C3N4 and Fe3O4. The typical diffraction peak of g-C3N4 is clearly reduced in g-C3N4@Fe3O4; this phenomenon can be explained by the distribution and effective dispersion of the majority of Fe3O4 on the surface of g-C3N4.
 |
| | Fig. 3 XRD patterns of g-C3N4 and g-C3N4@Fe3O4. | |
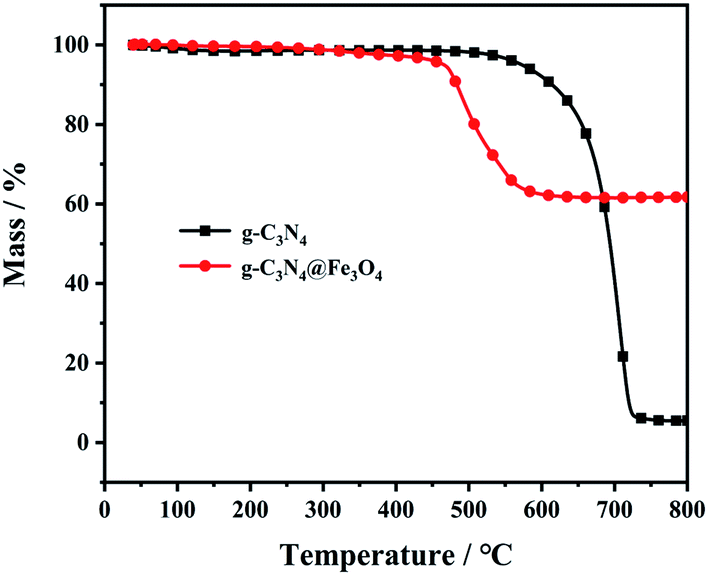
The thermal stability of g-C3N4@Fe3O4 was determined by TGA. Fig. 4 shows the TGA curves of g-C3N4 and g-C3N4@Fe3O4 under a N2 atmosphere. The materials have hardly no weight loss below 450 °C, indicating that the carbon nitride structure has high thermal stability. Compared with g-C3N4@Fe3O4, g-C3N4 is more stable at 450–580 °C without any significant weight loss. However, there is a significant thermal decomposition phenomenon at 600–700 °C. The decomposition completed above 700 °C, with 5.74% residual carbon. Compared with g-C3N4, g-C3N4@Fe3O4 shows thermal decomposition earlier than g-C3N4. This was because the loaded Fe3O4 bound water evaporates at a certain temperature and promotes the further decomposition of g-C3N4,35 which greatly reduces the weight loss. Thermal breakdown of g-C3N4@Fe3O4 is completed at 800 °C, with 62.08% residual carbon. This means that the thermal breakdown temperature of g-C3N4 loaded with Fe3O4 is lower, which contributes to the accumulation of residual carbon.
 |
| | Fig. 4 TGA curves of g-C3N4 and g-C3N4@Fe3O4. | |
3.2 Thermal stability of flame retardant EUG/NR blends
The TGA curves of the pristine EUG/NR blend and its composites with different contents of additives are depicted in Fig. 5, along with the corresponding thermal data given in Table 2. The initial degradation temperature, 20% weight loss and 50% weight loss are denoted as T10wt%, T20wt% and T50wt%, respectively. The initial degradation temperature of the EUG/NR blend added with the IFR is 320 °C; it is lower than 335 °C of the original EUG/NR blend. This is because the P–O–C bond is more unstable than the ordinary C–C bond; this reduces the initial decomposition temperature. The degraded phosphate groups contribute greatly to the formation of carbon residue, thus protecting the sample from further degradation. The addition of g-C3N4@Fe3O4 catalyzes the decomposition of the EUG/NR blends, and the amount of residual char of the EUG/NR blends increases from 22.4% to 35.4%. This is due to the unique laminar structure of g-C3N4@Fe3O4, which has a synergistic carbon formation effect. In addition, Fe3O4 forms an Fe–O–P network structure with the flame retardant, resulting in an increase in the rate of rubber carbonisation. The introduction of metal oxides also has good thermal conductivity, resulting in more uniform heat transfer in the early stages of the reaction and a better char layer, thus increasing the amount of residual char.36 This indicates that the addition of synergistic g-C3N4@Fe3O4 nanoparticles was conducive to the formation of the charring layers resulted from the degradation products of the polymer.
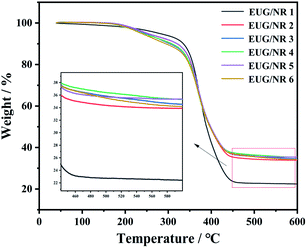 |
| | Fig. 5 TGA curves of EUG/NR blend composites. | |
Table 2 TG data of EUG/NR blend compositesa
| Samples |
T10wt%/°C |
T20wt%/°C |
T50wt%/°C |
W600/% |
| T10wt% denotes the temperature at 10% weight loss, T20wt% denotes the temperature at 20% weight loss, T50wt% denotes the temperature at 50% weight loss, and W600 denotes the weight of char residue at 600 °C. |
| EUG/NR1 |
335 |
358 |
389 |
22.4 |
| EUG/NR2 |
320 |
353 |
396 |
33.9 |
| EUG/NR3 |
304 |
350 |
397 |
34.5 |
| EUG/NR4 |
308 |
350 |
396 |
35.3 |
| EUG/NR5 |
321 |
355 |
398 |
35.4 |
| EUG/NR6 |
294 |
348 |
396 |
34.2 |
3.3 Flame retardancy
The LOI and UL-94 vertical burning tests were applied to investigate the flame retardancy properties of the composites. As shown in Table 3, pristine EUG/NR is extremely flammable, and its LOI value is only 20%, which cannot reach the required level in the UL-94 test. When IFR is added to the EUG/NR blends, the flame retardancy is significantly improved with an LOI value of 25.5%. This can be explained by the fact that when IFR is heated, the organic acid decomposed by the acid source interacts with the decomposed water molecules of the carbon source to promote the formation of a porous expanding carbon layer. This carbon layer covers the polymer surface, but it is easy to break and can form a transmission channel for gas and heat. After adding g-C3N4@Fe3O4, the LOI value of EUG/NR 2 reaches 27.8%, and the flame retardancy of the composite is further improved. The layered structure of g-C3N4 can accelerate heat transport and inhibit local heat overheating, resulting in delay of rubber combustion at the beginning of burning. Also, Fe3O4 plays a catalytic role in carbon formation. Therefore, a higher oxygen index is obtained. With the increase in the g-C3N4@Fe3O4 content, the LOI value of EUG/NR 5 reaches a maximum of 29.5%. However, the LOI value of EUG/NR 6 is reduced to 28.3%; this can be attributed to the excessive heat transfer caused by the excess of Fe3O4.
Table 3 LOI and UL-94 of EUG/NR blend composites
| Samples |
LOI/% |
UL-94 |
| EUG/NR 1 |
20.0 |
No rating |
| EUG/NR 2 |
25.5 |
V-1 |
| EUG/NR 3 |
27.8 |
V-1 |
| EUG/NR 4 |
28.5 |
V-0 |
| EUG/NR 5 |
29.5 |
V-0 |
| EUG/NR 6 |
28.3 |
V-1 |
3.4 Cone calorimeter tests (CCT)
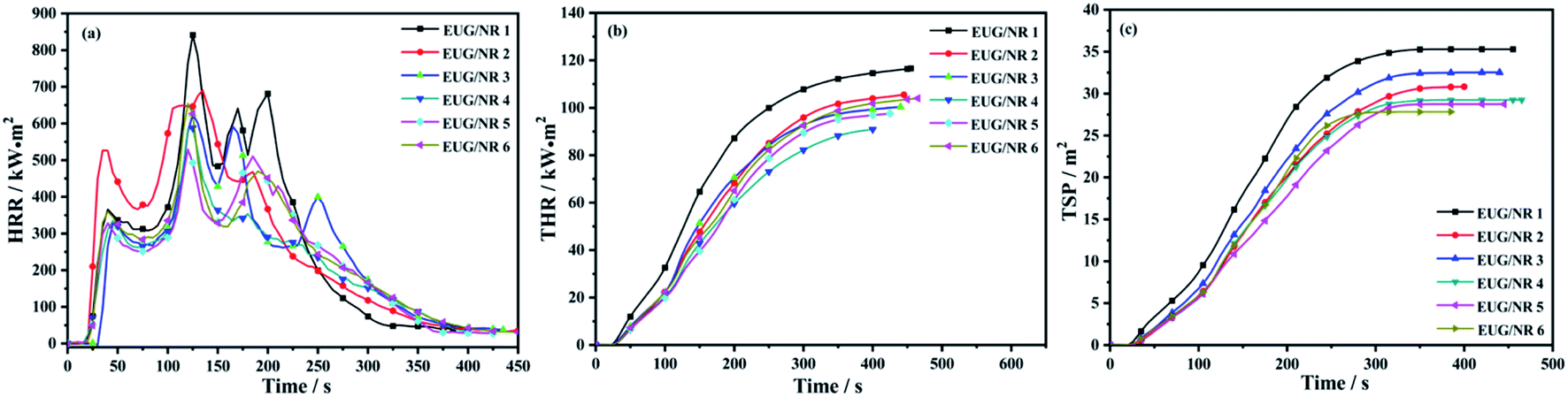
In order to further study the flame retardant and fire safety properties of the composite materials, a cone calorimetry test was carried out. The cone test is a bench scale fire test that can simulate the actual combustion of the material under the real fire conditions. The data obtained from the tests can give information on the combustion behavior of the material in fire. The heat release rate (HRR) and total heat release (THR) rate of flame retardant EUG/NR are shown in Fig. 6(a and b) and Table 4. It can be seen from Fig. 6(a and b) that EUG/NR 1 presents the highest flammability, corresponding to the largest HRR (841 kW m−2) and THR (117 MJ m−2), respectively. After adding the IFR, the PHRR and THR of EUG/NR 2 decreased by 18% and 10%, respectively. After 3 phr g-C3N4@Fe3O4 is added, the PHRR and THR of EUG/NR 3 are reduced by 26% and 15%, respectively, indicating that the addition of g-C3N4@Fe3O4 leads to the formation of an excellent synergistic flame retardant. The decrease of PHRR and THR is due to the fact that PER can produce ester polyols and acids acting as dehydrating agents. Ester polyols are carbonized to generate water molecules to reduce the ambient temperature, and the acid is esterified with PER to form a carbon layer. MEL and APP can produce the non-flammable gases NH3 and H2O, which can prevent further burning. The synergistic action makes it possible for the IFR to have high flame retardant efficiency. The decrease of ignition time (TTI) is due to the fact that APP decomposes earlier than the pristine EUG/NR blends, accompanied by the formation of small volatile molecules during the burning process, leading to the early ignition phenomenon. In addition, the total smoke production (TSP) curve of the flame retardant rubber is shown in Fig. 6(c). After adding g-C3N4@Fe3O4, the values of TSP decrease significantly. It is mainly because the Fe3O4 loaded on the surface of carbon nitride could adsorb the small combustible molecules decomposed by IFR and crosslink to form large molecules, thereby blocking their transfer to the outside through the heat flow process and forming a dense carbon layer. This carbon layer isolates the connection between the combustible gases inside the substrate and the outside; moreover, it could also form an Fe–O–P mesh structure with the decomposition products of IFR, further promoting the formation of a carbon layer and thus reducing the generation and release of smoke. This indicated that g-C3N4@Fe3O4 is a preferable flame retardant and smoke suppressant in rubber.37
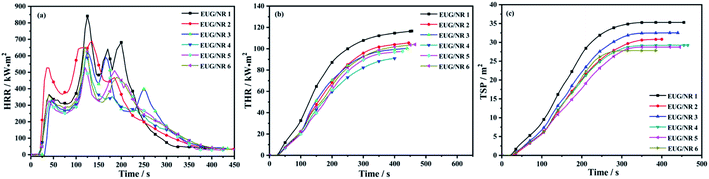 |
| | Fig. 6 (a) HRR curves of the EUG/NR blend composites; (b) THR curves of the EUG/NR blend composites; (c) TSP curves of the EUG/NR blend composites. | |
Table 4 Cone data of EUG/NR blend compositesa
| |
PHRR/kW m2 |
THR/MJ m2 |
TSP/m2 |
TTI/s |
FIGRA |
| PHRR represents peak of heat release rate, THR represents total heat release, TSP represents total smoke production, TTI means time to ignition, and FIGRA represents fire growth index. |
| EUG/NR 1 |
841 |
117 |
35.0 |
19 |
6.7 |
| EUG/NR 2 |
690 |
105 |
30.8 |
16 |
5.1 |
| EUG/NR 3 |
626 |
100 |
32.5 |
18 |
5.0 |
| EUG/NR 4 |
588 |
91 |
29.0 |
19 |
4.7 |
| EUG/NR 5 |
529 |
98 |
28.7 |
21 |
4.4 |
| EUG/NR 6 |
654 |
104 |
27.8 |
21 |
5.4 |
The fire growth rate (FIGRA) is defined as the ratio of the PHRR value to the time to PHRR, and it is created to evaluate the risk of developing fire.38 The lower the FIGRA value, the better the fire safety of materials. The values of FIGRA of EUG/NR composites are listed in Table 4. After adding IFR, it can be seen that all of the FIGRA decrease to somewhat. The FIGRA value falls as the concentration of g-C3N4@Fe3O4 increases. The FIGRA value of EUG/NR 5 is the lowest, 4.4, when the amount of g-C3N4@Fe3O4 is 9 phr. According to the above findings, g-C3N4@Fe3O4 has a good synergistic flame retardant effect.
3.5 Char residue analysis
SEM was applied to investigate the microstructure of residual carbon after CCT. The SEM images of residual carbon in EUG/NR blend composites are shown in Fig. 7. It can be seen from Fig. 7(a) that the carbon layer of the EUG/NR 1 without flame retardants is prone to cracks and defects after the combustion process. The formed carbon residue structure has large pores on the surface. The cracked carbon layer enables the circulation of heat and air between the inside of the EUG/NR and the outside atmosphere, leading to a further increase of the intensity of combustion. Fig. 7(b) shows EUG/NR 2 combustion residues containing intumescent flame retardants. After the system is fully burned, a complete carbon layer is formed with a large number of bubbles. This is due to the thermal decomposition of the gas source in the intumescent flame retardant along with the entry of H2O and CO2 nonflammable gases, and moreover, the acid source catalyzes the formation of carbon. The carbon layer has expanded clearly, and its surface possesses folds and protrusions. Such a structure increases the strength of the carbon layer, which makes the carbon layer easier to maintain under high temperature conditions. It is worth noting that after a small amount of g-C3N4@Fe3O4 is added, the thickness of the formed carbon layer is increased, as can be observed in Fig. 7(c). However, the strength of the carbon layer is not enough, and a “bubble-like” carbon layer appears, implying that heat and gas are easily released to provide “fuel” for combustion under this condition. As shown in Fig. 7(d and e), when the addition amount of g-C3N4@Fe3O4 is satisfactory, a thick, strong and continuous carbon layer can be produced. The g-C3N4@Fe3O4 synergist significantly catalyzes the formation of more carbon residues, and the dense carbon layer produced by catalytic carbonization can act as a better insulation barrier, thereby reducing the flow between the “fuel” inside the EUG/NR and the outside. It can be found from Fig. 7(f) that the “bubble” carbon layer is destroyed, forming numerous irregular type carbon layers. This structure is not beneficial to the improvement of flame retardant performance.
 |
| | Fig. 7 SEM images of residual carbon in EUG/NR blend composites. (a) EUG/NR 1, (b) EUG/NR 2, (c) EUG/NR 3, (d) EUG/NR 4, (e) EUG/NR 5, and (f) EUG/NR 6. | |
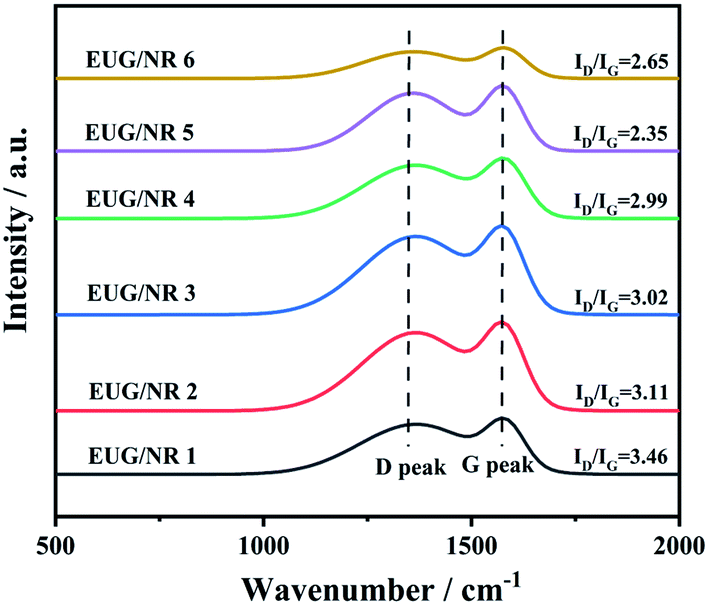
Fig. 8 shows the Raman spectra of the char remnants of EUG/NR after burning, which is used to evaluate the charring impact of the EUG/NR blends. G and D bands can be found in all of the spectra at 1585 cm−1 and 1360 cm−1, respectively.39 The graphitization degree of char is traditionally indicated by the area ratio of the D and G bands (ID/IG). In general, the lower the ID/IG value, the better the structure of the carbon layer. It is evident that the value of ID/IG follows the sequence of EUG/NR 1 (3.46) > EUG/NR 2 (3.11) > EUG/NR 3 (3.02) > EUG/NR 4 (2.99) > EUG/NR 6 (2.65) > EUG/NR 5 (2.35), indicating that addition of g-C3N4@Fe3O4 can significantly increase the graphitization level of char residues of the EUG/NR blend system. The production of compact and efficient thermally insulated residual char is aided by the high quantity of graphitized carbons. The results of Raman spectra show that the higher graphitization degree of the content of g-C3N4@Fe3O4 is 9 phr, resulting in improved thermal stability and flame retardancy of the EUG/NR blends. They also have the highest degree of thermal stability and graphite, which is consistent with TGA and cone results.
 |
| | Fig. 8 Raman spectra of the char residues of EUG/NR blend composites. | |
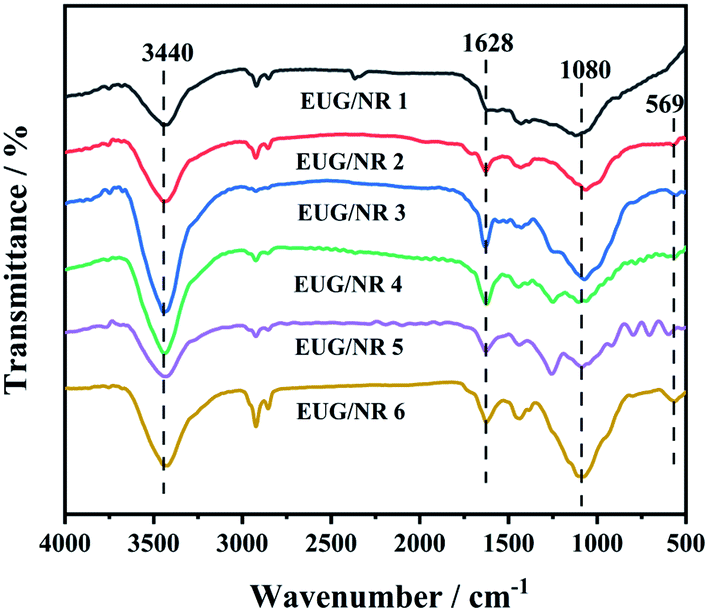
The FTIR spectra of residual carbon of EUG/NR blends after CCT are shown in Fig. 9. It can be observed that the absorption peak of all combustion products is at about 3400 cm−1, which is due to the formation of compounds with hydroxyl (–OH) during combustion. The residual carbon is composed of a polyaromatic structure, and the vibration absorption peak is at 1628 cm−1, which is due to the olefin unsaturated bond caused by insufficient combustion of the EUG/NR blend. Moreover, an evident absorption peak appearing at 1459 cm−1 can be related to CC tensile vibration. In addition, there are P–O–C characteristic absorption peaks at 1080 cm−1, corresponding to the produced phosphorous-containing carbonaceous structure.40 This finding suggests that a thermal insulator (phosphorous-containing carbonaceous layer) can effectively slow heat and mass transmission between the gaseous and condensed phases.41 At the same time, the characteristic Fe–O stretching vibration peak of Fe3O4 in g-C3N4@Fe3O4 occurred at 569 cm−1,32 showing that there is some residual Fe atom after the combustion.
 |
| | Fig. 9 FTIR spectra of char residues of EUG/NR blend composites after burning. | |
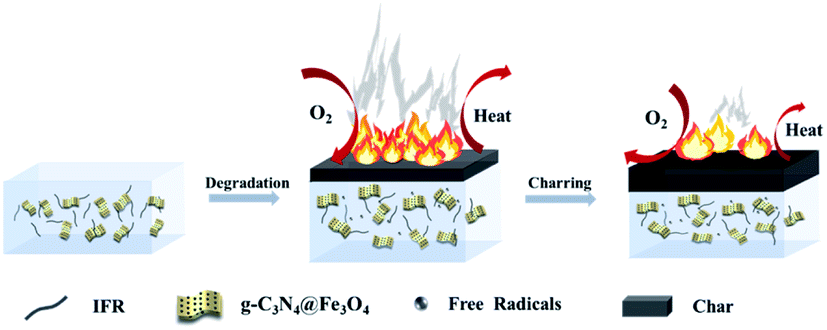
Based on the above results and analysis, the flame retardant mechanism of g-C3N4@Fe3O4 and IFR in the EUG/NR composite is proposed in Scheme 2. The IFR consists of PER, MEL and APP. PER, as a polyol, can react with the polyphosphoric acid produced by the decomposition of APP to form esters. Ester polyols are esterified to generate water molecules to reduce the ambient temperature, and acids are esterified with PER to form a carbon layer. MEL and APP can also produce non-combustible gases NH3 and H2O, which expand the carbon layers. This phenomenon enables the IFR to possess high flame retardancy efficiency. In the early stage of the test, the rubber combustion was inhibited and delayed. Meanwhile, Fe3O4 can absorb the decomposed free radicals of the flame retardant and promote the carbonization between the free radicals and part of the combustion matrix, thus improving the utilization rate of APP and preventing the combustion of unburned rubber.42
 |
| | Scheme 2 Fire retardancy mechanism of EUG/NR blend composites. | |
3.6 Mechanical properties
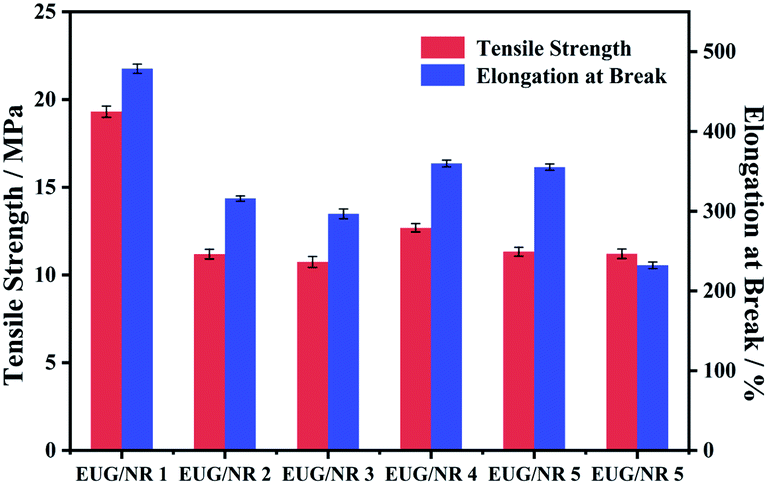
The mechanical properties of the EUG/NR blend composites are displayed in Fig. 10. As is well known, the incorporation of flame retardant additives will cause deteriorated mechanical properties.43 As can be seen from Fig. 10, the tensile strength of EUG/NR 1 is 19.31 MPa, and the elongation at break can reach 478.65%. Compared with pristine rubber, the tensile strength of EUG/NR blends with the flame retardant is significantly reduced. The tensile strength and elongation at the break of EUG/NR 2 are 11.21 MPa and 315.87%, respectively, which are 42.05% and 34.01% lower than those of pristine rubber. The reason is that rubber is an organic compound, and the addition of an inorganic filler tends to cause phase separation due to poor compatibility between inorganic substances and organic substances. The tensile strength and elongation at the break of EUG/NR 4 and EUG/NR 5 are improved. g-C3N4@Fe3O4 has a larger specific surface area; it can enhance the interface adhesion with the EUG/NR matrix. Thus, the flame retardant can be dispersed better in the rubber. Therefore, the crosslinking sites of rubber are increased and the adhesive density of rubber is also improved, implying that optimized mechanical properties of the EUG/NR blend composites can be obtained. However, once the addition of g-C3N4@Fe3O4 is too much, the mechanical properties are decreased relatively due to its easy agglomeration.
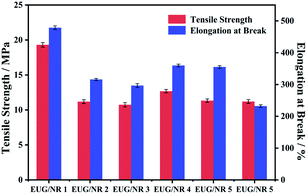 |
| | Fig. 10 Mechanical properties of EUG/NR blend Composites. | |
4 Conclusion
This work reported an in situ co-precipitation method to successfully prepare a g-C3N4@Fe3O4 flame retardant synergist, and XRD, FTIR and XPS results confirmed the successful preparation of g-C3N4@Fe3O4. The flame test results showed that the addition of g-C3N4@Fe3O4 improved the flame retardancy of EUG/NR blends. Compared to the pristine EUG/NR blends, the specific performance with the addition of 9 phr (EUG/NR 5) showed that the LOI value increased from 20.0% to 29.5%, and PHRR, THR, and TSP decreased by 37.1%, 16.2%, and 18.0%, respectively. The analyses of SEM and Raman spectra indicated that g-C3N4@Fe3O4 had a promoting effect on improving the quality of the carbon layer during the combustion process, leading to formation of a dense graphitized carbon layer with better thermal stability in the condensed phase. In short, it was hoped that the flame retardant synergist reported in this paper would provide a new direction to improve the flame retardancy of EUG/NR blends.
Author contributions
All authors contributed to the innovation. L. Z., L. Y. J. and L. H. conceived and designed the experiments; L. Z., L. Y. J. and W. N. performed the experiments; J. Y. and W. N. performed characterization and analysis of the experiments; L. Z., C. X. Y. and J. Y. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China, grant number 51973124; Liao Ning Revitalization Talents Program, grant number XLYC2005002; “Jie Bang Gua Shuai” of Science and Technology Projects of Liaoning Province in 2021, grant number 2021JH1/10400091; Shenyang Science and Technology Program-Major Key Core Technology Project, grant number 20-202-1-15.
References
- K. Ninyong, E. Wimolmala, N. Sombatsompop and K. Saenboonruang, Polym. Test., 2017, 59, 336–343 CrossRef CAS.
- N. Hayeemasae, S. Z. Salleh and H. Ismail, J. Polym. Environ., 2019, 27, 2119–2130 CrossRef CAS.
- L. Xia, S. Chen, W. Fu and G. Qiu, Polymers, 2019, 11, 508 CrossRef PubMed.
- Y. Nakazawa, T. Takeda, N. Suzuki, T. Hayashi, Y. Harada, T. Bamba and A. Kobayashi, Planta, 2013, 238, 549–560 CrossRef CAS.
- N. Rattanasom, U. Thammasiripong and K. Suchiva, J. Appl. Polym. Sci., 2005, 97, 1139–1144 CrossRef CAS.
- X. Qi, J. Zhang, L. Zhang and D. Yue, J. Mater. Sci., 2020, 55, 4940–4951 CrossRef CAS.
- N. Wang, H. Liu, J. Zhang, M. Zhang, Q. Fang and D. Wang, Arab. J. Chem., 2020, 13, 6274–6284 CrossRef CAS.
- Y. Pan, J. Zhan, H. Pan, W. Wang, H. Ge, L. Song and Y. Hu, RSC Adv., 2015, 5, 67878–67885 RSC.
- S. De Juan, J. Zhang, P. Acuña, S. Nie, Z. Liu, W. Zhang, M. Luisa Puertas, A. Esteban-Cubillo, J. Santarén and D. Y. Wang, Fire Mater., 2019, 43, 961–970 CrossRef CAS.
- N. Wang, F. Yang, J. Zhang and Q. Fang, Chem. J. Chinese. Univ., 2019, 40, 385–392 CAS.
- Y. Li, B. Li, J. Dai, H. Jia and S. Gao, Polym. Degrad. Stab., 2008, 93, 9–16 CrossRef CAS.
- N. Wang, M. Zhang, P. Kang, J. Zhang, Q. Fang and W. Li, Composites. Materials, 2018, 11, 1005–1017 Search PubMed.
- B. Li, Polym. Degrad. Stab., 2002, 78, 349–356 CrossRef CAS.
- N. Wang, H. Teng, X. Zhang, J. Zhang, L. Li, J. Zhang and Q. Fang, Polymers, 2019, 11, 1677–1690 CrossRef CAS PubMed.
- E. D. Weil and N. G. Patel, Polym. Degrad. Stab., 2003, 82, 291–296 CrossRef CAS.
- X. Chen, M. Li, J. Zhuo, C. Ma and C. Jiao, J. Therm. Anal. Calorim., 2016, 123, 439–448 CrossRef CAS.
- Y. Liu, Q. H. Kong, X. M. Zhao, P. Zhu, J. Zhao, A. Esteban-Cubillo, J. Santarén and D. Y. Wang, Polym. Adv. Technol., 2017, 28, 971–978 CrossRef CAS.
- A. Beaugendre, S. Saidi, S. Degoutin, S. Bellayer, C. Pierlot, S. Duquesne, M. Casetta and M. Jimenez, RSC Adv., 2017, 7, 40682–40694 RSC.
- X. Cao, X. Chi, X. Deng, T. Liu, B. Yu, B. Wang, A. C. Y. Yuen, W. Wu and R. K. Y. Li, Polym. Adv. Technol., 2020, 31, 1661–1670 CrossRef CAS.
- Y. Shi, L. Fu, X. Chen, J. Guo, F. Yang, J. Wang, Y. Zheng and Y. Hu, Nanomaterials, 2017, 7, 259 CrossRef PubMed.
- X. Dong and F. Cheng, J. Mater. Chem. A, 2015, 3, 23642–23652 RSC.
- F. Guo, M. Li, H. Ren, X. Huang, K. Shu, W. Shi and C. Lu, Sep. Purif. Technol., 2019, 228, 115770 CrossRef CAS.
- Y. Shi, B. Yu, L. Duan, Z. Gui, B. Wang, Y. Hu and R. K. K. Yuen, J. Hazard. Mater., 2017, 332, 87–96 CrossRef CAS PubMed.
- Y. Ma, Y. Yang, C. Lu, D. Xiao, S. Wu and Y. Liu, Polym. Compos., 2018, 39, 1928–1938 CrossRef.
- Y. Q. Shi, S. H. Jiang, K. Q. Zhou, C. L. Bao, B. Yu, X. D. Qian, B. B. Wang, N. N. Hong, P. Y. Wen, Z. Gui, Y. Hu and K. K. Yuen Richard, ACS Appl. Mater. Interfaces, 2014, 6, 429–437 CrossRef CAS PubMed.
- Q. Bi, D. Yao, G. Z. Yin, J. You, X. Q. Liu, N. Wang and D. Y. Wang, React. Funct. Polym., 2020, 155, 104690 CrossRef CAS.
- S. Wang, F. Zhang, L. Yi, R. Huan and N. Kang, J. Nanopart. Res., 2020, 22, 148 CrossRef CAS.
- J. Wang, Z. Luo, Y. Song, X. Zheng, L. Qu, J. Qian, Y. Wu, X. Wu and Z. Wu, Chemosphere, 2019, 221, 554–562 CrossRef CAS.
- X. Jia, R. Dai, Y. Sun, H. Song and X. Wu, J. Mater. Sci.: Mater. Electron., 2016, 27, 3791–3798 CrossRef CAS.
- J. Y. Hu, K. Tian and H. Jiang, Chemosphere, 2016, 148, 34–40 CrossRef CAS PubMed.
- N. Tian, H. W. Huang, S. B. Wang, T. R. Zhang, X. Du and Y. H. Zhang, Appl. Catal., B, 2020, 267, 118697 CrossRef CAS.
- Z. Zhang and J. Kong, J. Hazard. Mater., 2011, 193, 325–329 CrossRef CAS PubMed.
- X. Liu, Q. Hu, Z. Fang, Q. Wu and Q. Xie, Langmuir, 2009, 25, 7244–7248 CrossRef CAS PubMed.
- A. Habibi-Yangjeh, M. Mousavi and K. Nakata, J. Photochem. Photobiol., A, 2019, 368, 120–136 CrossRef CAS.
- P. J. Wang, D. J Liao, X. P. Hu, N. Pan, W. X. Li, D. Y. Wang and Y. Yao, Polym. Degrad. Stab., 2019, 159, 153–162 CrossRef CAS.
- E. Gallo, B. Schartel, D. Acierno and P. Russo, Eur. Polym. J., 2011, 47, 1390–1401 CrossRef CAS.
- J. Wang, Y. Cao, Z. Wang, H. Zhou, Y. Zhou, X. Zheng, X. He and C. Wang, Appl. Surf. Sci., 2022, 577, 151648 CrossRef CAS.
- D. Yao, G. Yin, Q. Bi, X. Yin, N. Wang and D. Y. Wang, Polymers, 2020, 12, 2107 CrossRef CAS.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- M. J. Chen, C. R. Chen, Y. Tan, J. Q. Huang, X. L. Wang, L. Chen and Y. Z. Wang, Ind. Eng. Chem. Res., 2014, 53, 1160–1171 CrossRef CAS.
- Z. B. Shao, C. Deng, Y. Tan, M. J. Chen, L. Chen and Y. Z. Wang, Polym. Degrad. Stab., 2014, 106, 88–96 CrossRef CAS.
- E. N. Kalali, X. Wang and D. Y. Wang, Ind. Eng. Chem. Res., 2016, 55, 6634–6642 CrossRef CAS.
- L. Zhang, S. Chen, Y. T. Pan, S. Zhang, S. Nie, P. Wei, X. Zhang, R. Wang and D. Y. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9272–9280 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab