
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitochondria-targeted alginate/triphenylphosphonium-grafted-chitosan for treatment of hepatocellular carcinoma
Kholoud K. Arafa a,
Mohamed A. Hamzawyb,
Shaker A. Mousac and
Ibrahim M. El-Sherbiny*a
a,
Mohamed A. Hamzawyb,
Shaker A. Mousac and
Ibrahim M. El-Sherbiny*a
aNanomedicine Research Labs, Center for Materials Science (CMS), Zewail City of Science and Technology, Ahmed Zewail Road, October Gardens, 6th of October City, 12578, Giza, Egypt. E-mail: ielsherbiny@zewailcity.edu.eg
bPharmacology & Toxicology Department, Faculty of Pharmacy, Fayoum University, Fayoum, Egypt
cPharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Rensselaer NY 12144, USA
First published on 4th August 2022
Abstract
Mitochondrial targeting of anticancer drugs can effectively eradicate chemotherapy-refractory cells through different mechanisms. This work presents the rational designing of mitochondria-targeted core–shell polymeric nanoparticles (NPs) for efficient delivery of doxorubicin (DOX) to the hepatic carcinoma mitochondria. DOX was electrostatically nano-complexed with sodium alginate (SAL) then coated with mitotropic triphenylphosphonium-grafted chitosan (TPP+-g-CS) nanoshell. Polyvinyl alcohol (PVA) was co-solubilized into the TPP+-g-CS solution to enhance the stability of the developed NPs. The optimum NPs formula is composed of TPP+-g-CS (0.05% w/v) coating a DOX-SAL core complex (0.05% w/v), with 0.2% PVA relative to CS (w/w). The optimum NPs attained an entrapment efficiency of 63.33 ± 10.18%; exhibited a spherical shape with particle size of 70–110 nm and a positive surface charge which enhances mitochondrial uptake. FTIR and DSC studies results were indicative of an efficacious poly-complexation. In vitro biological experiments proved that the developed mitotropic NPs exhibited a significantly lower IC50, effectively induced apoptotic cell death and cell cycle arrest. Moreover, the in vivo studies demonstrated an enhanced antitumor bioactivity for the mitotropic NPs along with a reduced biological toxicity profile. In conclusion, this study proposes a promising nanocarrier system for the efficient targeting of DOX to the mitochondria of hepatic tumors.
1. Introduction
Hepatic cancer has reached a daunting record for mortality rates counting for 8.3% of the total cancer deaths occurring in 2021 as reported by GLOBOCAN.1 The mainstream for liver cancer treatment protocols relays at an early stage on surgery and drug chemotherapy. Despite the clinical advances achieved by such therapies, diverse drawbacks still hamper their curative efficacy. These include for instance the disease recurrence as well as the high metastasis rates encountered post-surgical resection of tumor masses. Additionally, in case of chemotherapy, the non-selective nature of drugs used can cause severe systemic toxicity leading to hazardous health effects. Therefore, the development of targeted antitumor drug platforms with maximal therapeutic activity concomitantly with minimal toxic side effects became imperative.Recently, the application of nanomedicine in the treatment of various diseases is an expanding field of research. Advances in nanotechnology have profoundly benefited the biomedical as well as the pharmaceutical fields as it paved the road for modifying the drug's pharmacokinetic properties including solubility, bioavailability, diffusivity and release profile.2 Consequently, the deployment of such nanostructured agents can lead to the betterment of their pharmacotherapeutic efficacy through improving the drug's biodistribution and cellular uptake, reducing the dose required and guarding against excessive drug metabolism.3,4 The optimal design for the nano-scaled vehicle is principally based on the biophysical as well as biochemical properties of the loaded cargo.5 Growing attention has be drawn towards the development of targeted drug delivery systems (TDDS) as it promote the accumulation of the loaded drugs at the desired site of action and ensuring their selective spatiotemporal release hence enhancing their therapeutic efficacy while reducing any undesirable effects.6,7
Most TDDSs have relied on the cellular biochemical distinctions between cancerous cells and their normal counterparts like the overexpression of specific surface receptors or antigens hence guarantying their specific recognition, interaction and uptake within the tumor mass.8 Despite of the selective cellular deliverance, cellular-level targeting alone cannot ensure an enhanced therapeutic efficacy.9 Thus, recently, subcellular TDDSs that are directed towards certain subcellular compartments have garnered scientific interest.10 Among the various targetable intracellular organelles, we have chosen mitochondria as it significantly regulates the cellular homeostasis. Mitochondria control multiple physiological processes namely cellular respiration, metabolism, signaling, differentiation, apoptosis and intracellular Ca2+ levels.11 To date several classes of mitotropic ligands were developed for targeting purposes including lipophilic cations namely dequalinium (DQA) and triphenylphosphonium (TPP+).12,13 Chemically, TPP+ exhibits a unique structure composed of lipophilic phenyl groups as well as a phosphonium cation which endue it with specific mitochondrial accumulation.14 The discovery of TPP+ led to the formation of a plethora of mitotropic drug-conjugates as well as DDS.15,16 Interestingly TPP+ belongs to a class of salts known as quaternary phosphonium salts (QPS) which were reported to have antitumor effects against diverse cancer types including liver cancer.17,18 Moreover, TPP+ was found to preferentially amass in the tumor derived mitochondria due to their higher transmembrane potentials relative to their normal counterparts imparting it with advantageous therapeutic selectivity.19
Besides the targeting ligand, we have adopted doxorubicin (DOX) to be the model chemotherapeutic agent in this study. Intriguingly, DOX has been reported to act as a mito-active cytotoxic agent with the capability of mitochondrial accumulation aided by its specific binding to the phospholipid cardiolipin. Such DOX-mediated membrane perturbation inhibits complexes I and II deranging the electron transport chain (ETC) function. Additionally, the CI mediated reduction of the quinone moiety in the chemical structure of DOX into a reactive semiquinone free radical can derive the production of the deleterious reactive oxygen species (ROS) creating a vicious iteration of reactions known as the “redox cycling”. Alternatively, DOX can further enhance ROS formation via interacting with iron to form reactive anthracycline–iron complexes through Fenton's reactions which ultimately disrupts normal iron homeostasis. Moreover, DOX can produce another class of reactive nitrogen species (RNS) via binding to endothelial nitrogen oxide synthase (eNOS) enhancing its activity which results in peroxynitrite (ONOO−) free radical formation. Lastly, the mitochondrial DNA (mtDNA) repair enzyme, topoisomerase isoenzyme 1 is one of the main biotargets inhibited by DOX which leads to mitochondrial damage and cell death.20
Thus, in the present work, TPP+ was selected as a mitochondria-targeting moiety and was conjugated to the hydrophilic chitosan (CS) polymer (TPP+-g-CS). Then, mitochondria-targeted core–shell nanoparticles (NPs) were developed and optimized for efficient delivery of doxorubicin (DOX) to the liver mitochondria. Cationic DOX salt was electrostatically nanocomplexed with the negatively charged sodium alginate (SAL) followed by coating the obtained nanocore with the mitotropic TPP+-g-CS nanoshell. Polyvinyl alcohol (PVA) was co-solubilized into the TPP+-g-CS solution to improve the stability of the developed nanosystem. Besides, the obtained DOX-loaded core–shell NPs were optimized in such a way to have a net positive charge enhancing their delivery across the mitochondrial membrane. Next, the physicochemical, biological, morphological, and drug-encapsulating characteristics of drug-free and drug-loaded NPs were evaluated. Finally, the antitumor effect of TPP+-decorated core–shell NPs against hepatocellular carcinoma was also investigated at both in vitro and in vivo levels.
2. Materials
Chitosan (MWT = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300,000, 89% deacetylated), sodium alginate (SAL), polyvinyl alcohol (PVA), (3-carboxypropyl)triphenylphosphonium bromide (TPP+Br−), N-hydroxysuccinimide (NHS), ethylcarbodiimide hydrochloride (EDC·HCl) and chloroform (CHCl3) were obtained from Alfa Aesar. Doxorubicin hydrochloride (DOX.HCl), glacial acetic acid (CH3COOH), hydrochloric acid (HCl), sodium hydroxide (NaOH) and triethylamine (TEA) were all bought from Sigma-Aldrich (St Louis, MO, USA).
000–300,000, 89% deacetylated), sodium alginate (SAL), polyvinyl alcohol (PVA), (3-carboxypropyl)triphenylphosphonium bromide (TPP+Br−), N-hydroxysuccinimide (NHS), ethylcarbodiimide hydrochloride (EDC·HCl) and chloroform (CHCl3) were obtained from Alfa Aesar. Doxorubicin hydrochloride (DOX.HCl), glacial acetic acid (CH3COOH), hydrochloric acid (HCl), sodium hydroxide (NaOH) and triethylamine (TEA) were all bought from Sigma-Aldrich (St Louis, MO, USA).
3. Methods
3.1. Preparative procedure
000 rpm, 15 min) to remove excess EDC/NHS. Afterwards, pellet was resuspended in ultra-pure water and the TPP+ solution was dropped onto the CS solution for which pH is adjusted to 4.7 using 1 N NaOH and left to stir over-night on ice (molar ratio 2:1 CS:TPP+). Afterwards, the solution pH is raised to reach 9 using 1 N NaOH leading to precipitation of the grafted CS polymer which is in-turn collected via ultra-high-speed centrifugation (20000 rpm, 20 min). The obtained pellet was washed twice with distilled water (2 cycles × 5 min) and centrifuged. Afterwards, the collected pellet was resuspended in water pH 4.7, frozen and freeze dried (brownish yellow powder).:1 v/v) resulting in complex formation. Next, the formed complex solution was mixed into the TPP+-g-CS polymer solution up on dropwise addition (ratio = 1:2 v/v) under homogenization to reach the desired final drug concentration. To increase the stability of the developed NPs, TPP+-g-CS polymer was dissolved in PVA containing aqueous solution (0.2% w/w relative to CS) and the above scheme is repeated. Thus, four formulations F1–F4 were obtained as displayed in Table 1.
| Code | TPP+-g-CS (% w/v) | SAL:DOX (w/w) |
SAL/DOX complex solution (% w/v) | PVA |
|---|---|---|---|---|
| F1 | 0.05 | 1:1 |
0.05 | Absent |
| F2 | 0.05 | 1:1 |
0.05 | Present |
| F3 | 0.1 | 1:1 |
0.1 | Absent |
| F4 | 0.1 | 1:1 |
0.1 | Present |
3.2. Structural validation of TPP+-g-CS
3.3. Evaluation of the prepared core–shell NPs
000 rpm, 30 min) at RT until complete pelleting of the NPs and separation from the rest of the solution (supernatant).The concentration of DOX in the obtained supernatant was determined spectrophotometrically. All the measurements were repeated thrice. The drug EE% was mathematically estimated as follows:
 | (1) |
000 Da; Severa) was hydrated overnight. Afterwards, the membrane was soaked in PBS for 1 h before the start of the experiment. Accurately weighed 10 mg of the dried NPs were reconstituted in PBS (2 mL) with adjusted pH. Next, the reconstituted sample was transferred into an Eppendorf tube, sealed tightly with the dialysis membrane and inverted upside-down into the receiving chamber. A 50 mL falcon tube (receptor compartment) was filled with 10 mL of PBS adjusted at specific pHs either to mimic the physiological environment (pH = 7.4) or the tumor microenvironment (TME, pH = 6.5). The entire system was continuously shaken (100 rpm, 37 °C). One mL aliquot was drawn from the receptor compartment at predetermined time points and the concentration of DOX released from the NPs was determined spectrophotometrically. The withdrawn sample volume was replenished with fresh release medium to keep the volume inside the receptor compartment constant during the entire experiment.The cumulative percentage of DOX released at a particular time (t) can be calculated using the following equation:
 | (2) |
As controls, blank NPs were used, and the plain drug solution was deployed for comparison rationale applying the same above-mentioned procedure.
3.4. In vitro biological analysis
Additionally, HepG2 cells were incubated with SAL/TPP+-g-CS NPs (as a negative control) using comparative NPs weight to their drug loaded counterparts and the rest of the experiment proceeded following similar procedures to the section above.
000 events per sample) were collected and analyzed in a FACScan flow cytometry analyzer. Excitation (λ = 488 nm) and the emission green fluorescence of AV (FL1, λ = 530 nm, x-axis) and red fluorescence of PI (FL2, λ = 575 nm, y-axis) were collected, respectively. Later, the obtained data were analyzed using the flow cytometry analysis software WinMDI 2.8.3.5. In vivo biological studies
Group (1): served as non-treated control and received normal saline only (negative control).
Group (2): mice were treated with of DEN (200 mg kg−1, i.p) and CCl4 (3 ml kg−1, s.c) for six consecutive weeks.
Group (3): animals were treated with SAL/TPP+-g-CS NV carrier system, which is similar in composition to (F2) two times weekly for last consecutive 4 weeks. The aim of G3 is to investigate the biological toxicity as well as the biological effect of the nanocarrier perse.
Group (4): mice were treated with DEN + CCl4, then treated with free DOX·HCl (5 mg kg−1, i.p) two times weekly for the last consecutive 4 weeks.
Group (5): mice were treated with DEN + CCl4, then treated with DOX-loaded SAL/CS NPs (F2′) in a dose equivalent to (5 mg kg−1, i.p) two times weekly for last consecutive 4 weeks.
Group (6): mice were treated with DEN + CCl4 and DOX-loaded SAL/TPP+-g-CS NPs (F2) in a dose equivalent to (2.5 mg kg−1, i.p) two times weekly for last consecutive 4 weeks.
The results obtained from G5 would confer a comparative stance to G6 about the biological toxicity profile as well as the therapeutic efficiency of DOX-loaded mitochondrial NPs versus non-targeted counterparts.
3.6. Statistical analysis
The statistical analysis was done using one-way Anova followed by Tukey's test (Sigma plot software 11.0). Statistical differences with (P ≤ 0.001) denote extreme significance, (P ≤ 0.01) denotes high significance and (P ≤ 0.05) indicates statistical significance. Data were represented as mean ± SD.4. Results and discussion
4.1. Preparation of TPP+-g-CS conjugate
The TPP+-g-CS conjugate was prepared as demonstrated in chemical synthesis Scheme 1. Initially, TPP+ carboxylic group was activated through its reaction with the coupling reagents EDC.HCl followed by NHS thus forming an amine-reactive O-acyl-isourea intermediate then the more stable amine-reactive NHS ester, respectively. Subsequently, the produced NHS ester was added to an acidified aqueous solution of CS-polymer yielding the final desired conjugate (TPP+-g-CS) connected via a stable amidic bond. The grafted polymer was collected through centrifugation followed by lyophilization (brownish yellow powder).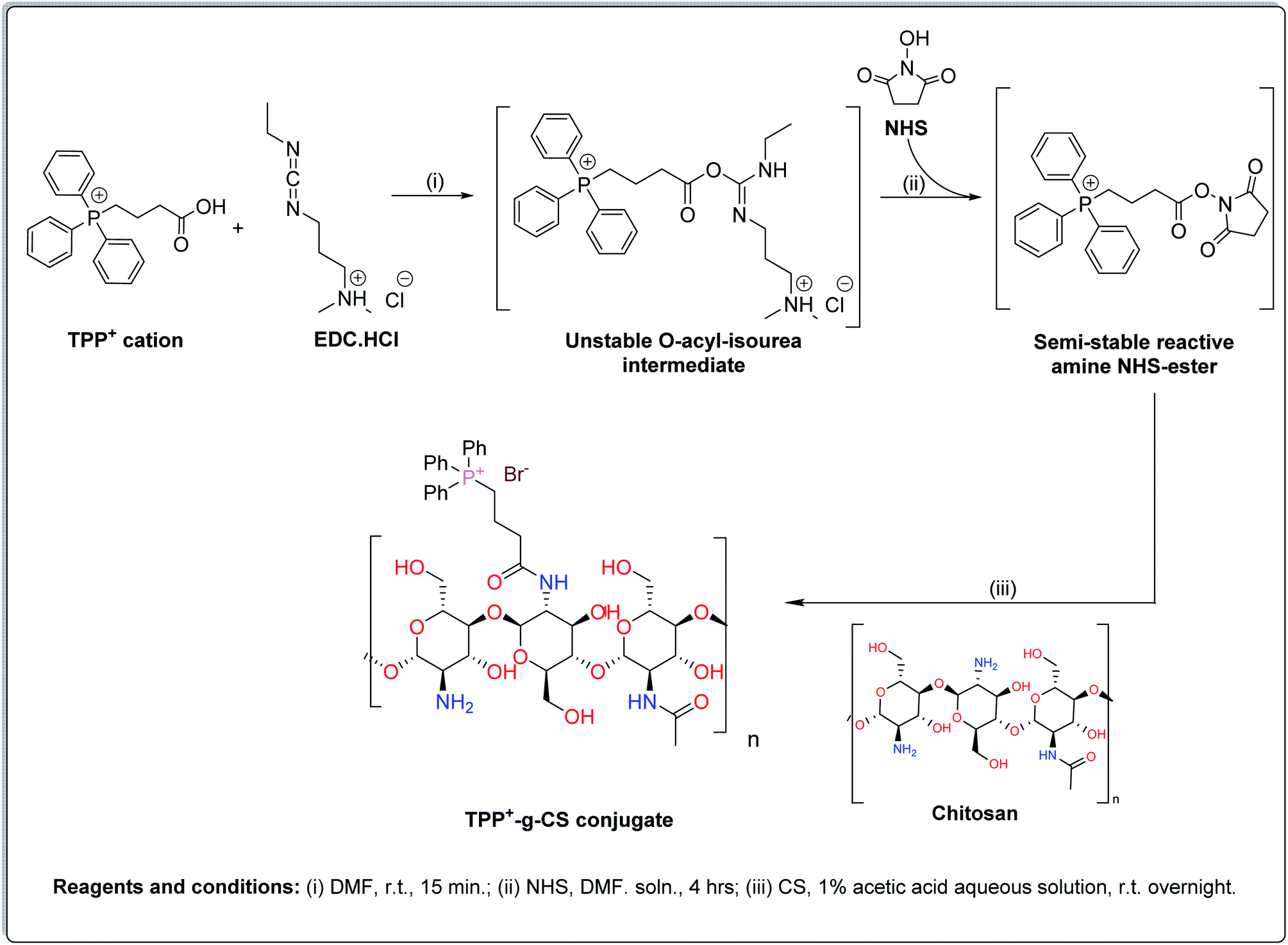 | ||
| Scheme 1 Chemical synthesis steps of TPP+-g-CS conjugate. | ||
4.2. Structural validation of TPP+-g-CS
1H NMR spectrum of TPP+-g-CS is shown in Fig. 1. Conjugation was affirmed due to the presence of signals between 7.80 and 8.57 ppm appearing at the aromatic region of the spectrum which are pertinent to the three phenyl rings of the newly introduced triphenylphosphonium groups.25,26 However, the signals of the methylene protons in TPP+ could not be distinguished clearly due to their relatively lower intensity and overlapping with the CS peaks at the aliphatic region of the spectrum. | ||
| Fig. 1 NMR chart for TPP+-g-CS polymer. | ||
FTIR spectra of TPP+-g-CS, TPP+, and CS from 500 to 4000 cm−1 are depicted in Fig. 2a. In TPP+-g-CS, there was a new absorption peak presenting at 1548 cm−1 corresponding to the absorption bands of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the phenyl rings pertinent to TPP+ moiety.25 Moreover, compared to the spectrum of CS, the appearance of additional peaks at 1640 cm−1 in the spectrum of TPP+-g-CS, demonstrating that carboxylic group of TPP+ mainly reacted with the amino groups of CS to form an amide bond.27
C of the phenyl rings pertinent to TPP+ moiety.25 Moreover, compared to the spectrum of CS, the appearance of additional peaks at 1640 cm−1 in the spectrum of TPP+-g-CS, demonstrating that carboxylic group of TPP+ mainly reacted with the amino groups of CS to form an amide bond.27
 | ||
| Fig. 2 (a) FTIR charts, (b) DSC thermograms for TPP+, CS-polymer and TPP+-g-CS conjugate. | ||
DSC thermograms are shown in Fig. 2b. First the CS chart demonstrated an endothermic peak at 109 °C indicating loss of hydration water as well as an exothermic signal at 236 °C indicative of the thermal decomposition of amine units in CS chains. As for the TPP+ cation thermogram, two endothermic peaks at 250 °C (sharp) and 350 °C due to its melting and decomposition, respectively. Comparing the conjugate chart to its respective precursors, the bimodal endotherm at 124 °C with a fused shoulder 236 °C confirms the proper conjugation process. The observed decrease in intensity of endothermic peak at 125 °C, may indicate a reduction in the amount of water bound to the CS polymer post-conjugation due to consumption of free amino groups. Moreover, the broad endothermic peak at 352 °C can be attributed to the decomposition of TPP+ moiety linked in the conjugate. Besides, the observed exothermic signal at 294 °C correlates to the residual unconjugated amine groups in CS backbone.
4.3. Particle size, surface charge and entrapment efficiency
Results of the particle size, surface charge and entrapment efficiency experiments are displayed in Table 2. It can be inferred from the obtained results that the presence of the PVA partially shielded the surface charge of the TPP+-g-CS rendering it less positive, this observation is similar to previous literature.28 PVA shielding of surface charge in turn affected particle size creating an observable pattern. Where, the highly positive NPs lacking PVA (F1 and F3) tend to be unstable resulting in relatively high aggregate PS and widely distributed PDI. On the contrary, PVA addition to NPs (F2 and F4) increased their stability and reduced their PS thus yielding a more homogenous population. Another pattern seen amongst PVA lacking NPs (F1 and F3), is the direct proportionality of the PS to the CS percentage used; an effect that got moderated after PVA addition. Also, the drug entrapment followed a noticeable pattern, where the EE% increased with PVA addition (F2 and F4) relative to the rest of the developed NPs (F1 and F3). The observed results might be attributed to increased NPs stability. From the obtained results of physicochemical characterization F2 formula was considered optimum and hence was selected for further studies.| Code | PS (nm) | PDI | ZP (mV) | EE% |
|---|---|---|---|---|
| F1 | 542.9 ± 6.01 | 0.99 ± 0.06 | 34.00 ± 2.40 | 22.46 ± 5.32 |
| F2 | 372 ± 4.72 | 0.39 ± 0.02 | 24.10 ± 0.71 | 63.33 ± 10.18 |
| F3 | 521 ± 7.03 | 0.67 ± 0.04 | 31.05 ± 0.21 | 23.05 ± 2.69 |
| F4 | 326 ± 2.91 | 0.44 ± 0.05 | 24.65 ± 0.78 | 50.43 ± 3.46 |
4.4. Morphological investigation (TEM images)
TEM images of the DOX-SAL/TPP+-g-CS NP (F2) demonstrated that the particles exhibited a spherical morphology with the size range (80–110 nm). The observed NPs size was lower than that measured using DLS. DLS obtained results might reflect the hydrated particle size (372 nm with PDI values 0.391), Fig. 3. As reported repeatedly, TEM imaging is considered the technique of choice to accurately assess NP's PS as it depends on direct high-resolution visualization of particles. Moreover, artifacts affecting the quality of DLS measurements including solution dynamics, NPs aggregation, presence of hydration shells by the surrounding solvent as well as Brownian motion induced fluctuation in light-scattering are well eliminated.29 Therefore, PS obtained by TEM imaging were selected over DLS. | ||
| Fig. 3 TEM image of the DOX-SAL/TPP+-g-CS NP (F2). | ||
4.5. Physicochemical characterization of NPs
The absorption spectrum for pristine DOX shows a characteristic band at 1722 cm−1 (CO bond stretching) at atom C-13. The peaks occurring at both 1575 and 1615 cm−1 (CO bond stretching and bond vibration) of the anthracene ring, respectively. Moreover, the wide band observed at 3360 cm−1 signals overlapped peaks (–OH and –NH2 groups vibration). As for DOX-SAL/CS NPs chart, the peak at 3298 cm−1 is similarly present in the parent TPP+-g-CS and the nanovoid (NV) confirming proper formation of NPs. Moreover, the disappearance of 3360 cm−1 related to the pristine DOX confirms the effective physical entrapment of the drug inside the NPs, Fig. 4a. Additionally, the occurrence of peaks at 1588 and 1405 cm−1 were observed. Conclusively, FT-IR charts indicate the electrostatic interactions between DOX, SAL and CS.
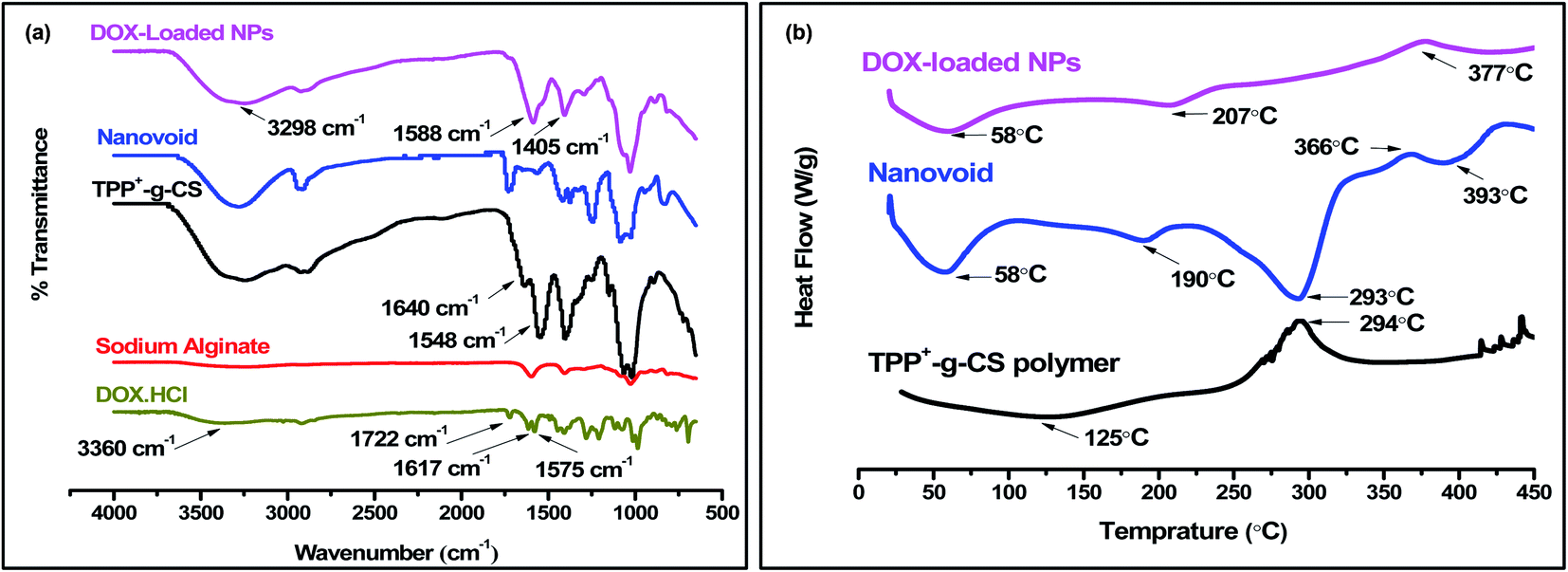 | ||
| Fig. 4 (a) FTIR charts, (b) DSC thermograms of SAL/TPP+-g-CS NPs (DOX-loaded and NVs) along with precursor pristine polymers. | ||
DSC was employed to understand the thermal behavior of the developed DOX-SAL/CS NPs and to elucidate possible interactions between the different constituting polymers along with the drug. Generally, the endothermic peaks around 100 °C are mainly associated with the dehydration of the hydrophilic molecules. The obtained thermograms demonstrate a minor lowering in the melting endotherm observed in the developed NPs relative to pristine TPP+-g-CS polymer which confirms the formation of NPs. Furthermore, comparing the thermogram of NVs to that of DOX-loaded CS NPs reflects the disappearance of the endothermic peak at 293 °օC as well as the step change at 377 °C suggesting that the electrostatic interactions occur between DOX, the carboxylic groups of SAL and the amino-groups of CS. Moreover, DOX-loaded NPs thermogram was showing a minor endothermic peak at 207 °C that might be DOX-related endothermic melting peaks indicating the proper inclusion of drug in the polymer, Fig. 4b.
4.6. In vitro drug release experiments
The prepared DOX-loaded core–shell NPs (F2) revealed a sustained release profile of the drug that lasted for 168 h, Fig. 5. Moreover, the results demonstrated that the cumulative drug release rate was significantly greater at the TME mimicking acidic environment (pH = 6.5) in comparison to physiological conditions (pH = 7.4), P < 0.05. The obtained findings suggest that there is a relative spatial selectivity in drug release within the cancerous niche. | ||
| Fig. 5 Cumulative release profiles of drug from the developed DOX-SAL/TPP+-g-CS core shell NPs in two different pHs (6.5 and 7.4).Inset is the release profile during the first day. Results presented as Mean ± SD (n = 3). | ||
To completely comprehend the kinetics of drug release, the empirical results were fitted using mathematical modeling, Table 3. Modeling outcomes showed that all the tested NPs predominantly followed the Korsmeyer-Peppas model pertinent to polymeric systems (R2 = 0.999). Where, the numerical value of the diffusional exponent (n) can predict the drug-release mechanism. Conventionally, Fickian diffusion is represented by (n ≤ 0.5) whereas, non-Fickian model can be either anomalous transport (0.5 < n < 1), case-II transport (n = 1) or super case-II transport (n > 1).30 Conclusively, the obtained release exponent n > 1 reflects a non-Fickian drug release follows super case-II transport that is controlled through polymer swelling and relaxation.
| Mathematical model | Equations | DOX-SAL/TPP+-g-CS (F2) | |
|---|---|---|---|
| pH = 6.5 | pH = 7.4 | ||
| Zero order | Q = k·t + Q0 | 0.734 | 0.717 |
| 1st order | Q = Q0 ek·t | 0.146 | 0.146 |
| 2nd order | (1/Q) = k·t + 1/Q0 | 0.088 | 0.088 |
| 3rd order | (1/Q)2 = k·t + 1/Q02 | 0.088 | 0.088 |
| Korsmeyer–Peppas | Q = k·tn | 0.999 | 0.999 |
| Hixson–Crowell | (1/Q)1/3 = k·t + 1/Q01/3 | 0.522 | 0.514 |
| Baker–Lonsdale | 3/2 × [1 − (1 − Q)2/3] − Q = k × t | 0.796 | 0.708 |
4.7. Biological assessments
| Formula | IC50 |
|---|---|
| SAL/TPP+-g-CS NV | N.D. |
| DOX-SAL/CS NP (F2′) | 12.94 ± 0.68 |
| DOX-SAL/TPP+-g-CS NP (F2) | 3.86 ± 0.20 |
| Plain DOX | 2.22 ± 0.12 |
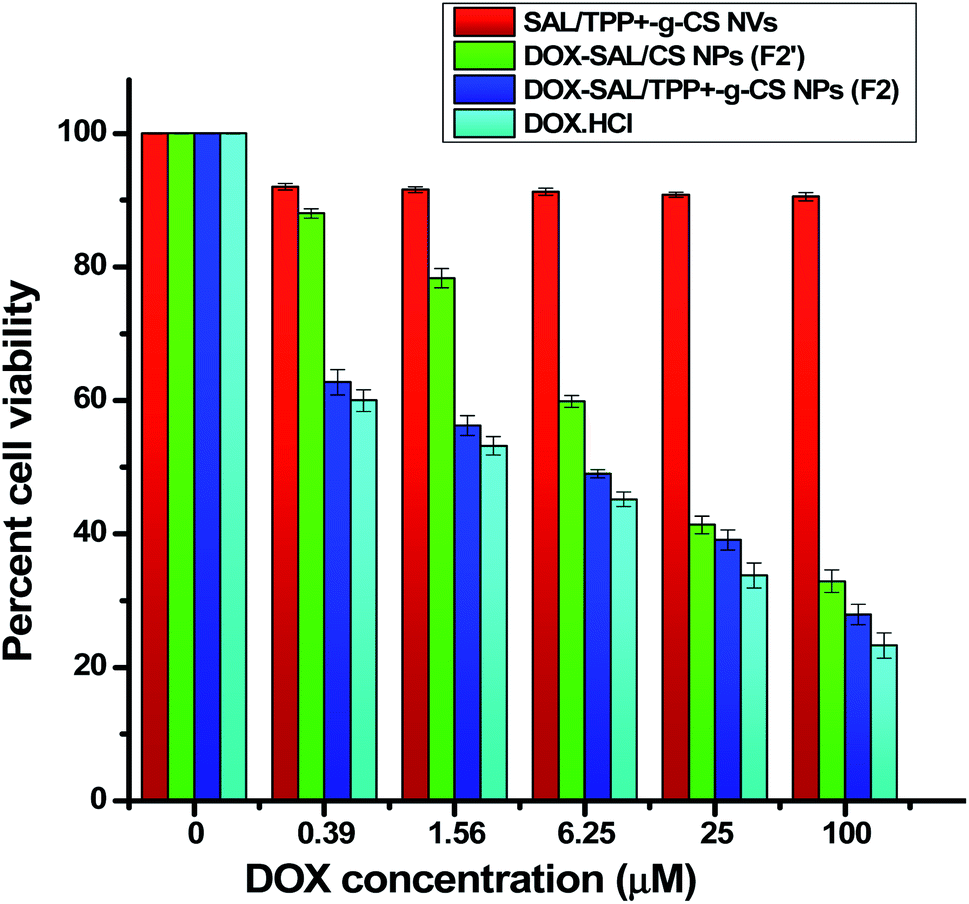 | ||
| Fig. 6 Cytotoxicity of the developed DOX-loaded NPs (IC50) relative to nanovoid and DOX using HepG2 hepatocellular carcinoma cells. Results presented as mean ± SD (n = 3). | ||
 | ||
| Fig. 7 Intracellular distribution of DOX-SAL/TPP+-g-CS NPs monitoring by fluorescence microscopy. HepG2 cells incubated with DOX-loaded TPP+-g-CS NPs (F2) for 12 h. (a) F2 NPs (DOX: 5 μM, red); (b) mitochondrial staining by mitotracker (100 nM, green); (c) merged images (a and b). Nuclear labelling by DAPI (dark blue) present in all fields. Scale bar 100 μM. | ||
| Sample | Normal | Apoptosis | Necrosis | |
|---|---|---|---|---|
| Early | Late | |||
| Non-treated Control | 98.67 ± 1.04 | 0.46 ± 0.08 | 0.26 ± 0.04 | 0.61 ± 0.05 |
| DOX-SAL/TPP+-g-CS NPs (F2) | 72.59 ± 1.36 | 3.62 ± 0.51 | 14.25 ± 0.79 | 9.54 ± 0.39 |
 | ||
| Fig. 8 Apoptosis induction cytograms for (a) non-treated control HepG2 cells; (b) DOX-SAL/TPP+-g-CS NPs (F2) treated HepG2 cells (3.86 ± 0.20 μM, 48 h). Results are presented as mean ± SD (n = 3). | ||
| Sample | %G0–G1 | %S | %G2/M | %Pre-G1 |
|---|---|---|---|---|
| Non-treated Control | 46.27 ± 1.85 | 34.02 ± 2.33 | 19.71 ± 1.88 | 1.33 ± 0.41 |
| DOX-SAL/TPP+-g-CS NPs (F2) | 42.27 ± 1.91 | 56.31 ± 1.60 | 1.42 ± 0.29 | 27.41 ± 1.72 |
 | ||
| Fig. 9 Cell cycle analysis cytograms of (a) non-treated control HepG2 cells; (b) DOX-SAL/TPP+-g-CS NPs (F2) treated HepG2 cells (3.86 ± 0.20 μM, 48 h). Results are presented as mean ± SD (n = 3). | ||
 | ||
| Fig. 10 Assessment of the antitumor effect and biological toxicity of DOX-SAL/CS core–shell NPs using hepatocellular carcinoma (HCC-bearing) in vivo animal model (a) AFP hepatic protein level, (b) ALT hepatic enzyme activity, (c) AST hepatic enzyme activity, (d) ALP hepatic enzyme activity, and (e) troponin-T cardiac protein level. Results presented as mean ± SD (n = 9), statistical significance is relative to control (aP ≤ 0.05, bP ≤ 0.01, cP ≤ 0.001, dP ≤ 0.0001). | ||
 | ||
| Fig. 11 Histopathological sections of the hepatic specimens extracted from (a) normal group, (b) HCC-bearing non-treated control group, (c) HCC-bearing group treated with SAL/TPP+-g-CS NVs, (d) HCC-bearing group treated with pristine DOX·HCl (5 mg kg−1), (e) HCC-bearing group treated with DOX-SAL/CS NPs (F2′) (5 mg kg−1) and (f) HCC-bearing group treated with DOX-SAL/TPP+-g-CS NPs (F2) (2.5 mg kg−1). (H&E stain, scale 50 μm). | ||
Post tumor induction, a dose titration study was implemented to figure out the effective lethal dose (EC50) of the free DOX.HCl (G4) as well as the DOX-loaded core–shell NPs (G5, G6). The obtained findings showed that both the free drug (G4) and non-grafted-CS NPs (F2′, G5) had similar EC50 values of (5 mg kg−1) resulting in the death of 50% of the animal test population. Intriguingly, the attained EC50 of the TPP+-g-CS NPs (F2, G6) was significantly lower than the ungrafted NPs (P < 0.001) equivalent to (2.5 mg kg−1). The last observation is reminiscent of the in vitro cytotoxicity study affirming the enhanced antiproliferative efficiency owing to the mitochondrial targeting.
The evaluation of alanine aminotransferase (ALT) enzyme levels in the serum was investigated to assess the extent of hepatic injury, Fig. 10b.36 Findings showed that HCC induction procedure in mice caused extreme hepatotoxicity accompanied with significant increase in serum ALT levels relative to normal mice (P < 0.0001). This hepatotoxic effect was reversed upon treatment with both F2′ and F2 core–shell NPs. The restoration of normal liver functioning is evident through rendering ALT protein levels similar to the normal serum value. Unlike the DOX-loaded NPs, the treatment with drug free NVs were not able to reverse the HCC pertinent functional aberrations (P < 0.0001). This last finding proves that NVs do not have any antiproliferative activity by itself. Despite, being therapeutically active free DOX was not as efficient as the NPs in restoring the normal ALT levels (P < 0.01).
Likewise, to ALT, enhanced levels of aspartate aminotransferase (AST) protein in the blood is well correlated to hepatic diseases.37 A similar pattern was observed for AST serum levels to that of ALT before and after treatment with NPs, Fig. 10c. Additionally, alanine aminotransferase (ALP) serum level, which is used as an HCC prognostic factor, was assessed.38 The results showed that trend of change in ALP serum level was resembling that of ALT and AST, Fig. 10d. Interestingly, the grafted-CS NPs (F2) were significantly efficient in restoring ALP levels to normal, which is significantly better when compared to their non-grafted counterparts (F2′) (P < 0.01). Our findings relevant to the reduction of AST and ALP serum levels post-treatment by the developed NPs (F2 and F2′) confirm that they are more therapeutically effective when compared to free DOX.
Beyond their antiproliferative capacity, the developed NPs biological toxicity profile was evaluated. The biological toxicity of the drug is an important aspect of our study as it is well reported that DOX induces severe cardiotoxicity.39,40 To this end, the cardiotoxicity biomarker troponin-T serum level was measured, Fig. 10e.41 As shown by the results, treatment by free DOX (G4) caused an extremely significant cardiotoxicity as compared to normal control (G1) (P < 0.0001). Compared to the free drug, treatment with the developed NPs (G5, G6) conferred a reduced biological toxicity profile significantly lowering the troponin-T levels (P < 0.0001). Taking the dose difference into account, treatment with the mitochondria targeted NPs (G6) was evidently safer than non-targeted NPs (G5) (P < 0.01), attaining similar protein levels to the normal control.
Taken together, the biochemical serological testing as well as the histological examination indicated the superiority of mitochondrial targeting (G6) in effectively inhibiting HCC progression at a significantly lower dose. Additionally, mitotropic NPs (F2) attained a biologically safe profile with minimized hepatotoxicity to almost null cardiotoxicity.
5. Conclusion
This study reports the development of mitotropic chitosan/sodium alginate-based core–shell nanocarriers (DOX-SAL/TPP+-g-CS NPs) for the treatment of hepatic cancer. The optimum NPs were found to exhibit favorable physicochemical characteristics mainly enhanced structural stability due to the incorporation of PVA; reflected by the enhanced drug EE% and homogenous particle size distribution. The other factor contributing to the high EE% is the electrostatic interaction between the cationic DOX with the polyanionic sodium alginate polymer. Afterwards, the mitochondria targeting was ensured through the synthesized TPP+-g-CS polymer used as an outer coating shell. Release profiles were also evaluated and were shown to be pH dependent. Biologically, the DOX-SAL/TPP+-g-CS NPs were found to be exclusively localized in the mitochondria, potentiating their antiproliferative activity. In vitro studies proved that the synthesized core–shell NPs were found to cause apoptotic cell-death and S-phase cell-cycle arrest. Finally, the in vivo studies proved that the designed NPs were biocompatible as reflected by the reduced cardiotoxicity and the therapeutic efficacy as presented by the retrieval of normal hepatic function and structure. Conclusively, the prepared NP system deems promising due to its enhanced therapeutic selectivity towards cancerous cells nominating it for wider application against various cancer types.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The principal investigator and the authors would like to thank the Academy of Scientific Research and Technology (ASRT), Egypt for funding this project (JESOR-Fund).References
- H. Sung, et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2021, 71(3), 209–249 CrossRef PubMed.
- J. K. Patra, et al., Nano based drug delivery systems: recent developments and future prospects, J. Nanobiotechnol., 2018, 16(1), 71 CrossRef PubMed.
- A. Z. Mirza and F. A. Siddiqui, Nanomedicine and drug delivery: a mini review, Int. Nano Lett., 2014, 4(1), 94 CrossRef.
- A. V. Kabanov, et al., Pluronic® block copolymers: novel functional molecules for gene therapy, Adv. Drug Delivery Rev., 2002, 54(2), 223–233 CrossRef CAS PubMed.
- R. Watkins, et al., Natural product-based nanomedicine: recent advances and issues, Int. J. Nanomed., 2015, 10, 6055 CAS.
- S. V. Boddapati, et al., Mitochondriotropic liposomes, J. Liposome Res., 2005, 15(1–2), 49–58 CrossRef CAS PubMed.
- Y. Yamada and H. Harashima, Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases, Adv. Drug Delivery Rev., 2008, 60(13–14), 1439–1462 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, Odyssey of a cancer nanoparticle: from injection site to site of action, Nano today, 2012, 7(6), 606–618 CrossRef CAS PubMed.
- L. Rajendran, H.-J. Knölker and K. Simons, Subcellular targeting strategies for drug design and delivery, Nat. Rev. Drug Discovery, 2010, 9(1), 29–42 CrossRef CAS PubMed.
- M. Mossalam, A. S. Dixon and C. S. Lim, Controlling subcellular delivery to optimize therapeutic effect, Ther. Delivery, 2010, 1(1), 169–193 CrossRef CAS PubMed.
- R. A. Smith, R. C. Hartley and M. P. Murphy, Mitochondria-targeted small molecule therapeutics and probes, Antioxid. Redox Signaling, 2011, 15(12), 3021–3038 CrossRef CAS PubMed.
- Z.-P. Chen, et al., Mitochondria-targeted drug delivery system for cancer treatment, J. Drug Targeting, 2016, 24(6), 492–502 CrossRef CAS PubMed.
- Z. Wang, et al., Nanopreparations for mitochondria targeting drug delivery system: current strategies and future prospective, Asian J. Pharm. Sci., 2017, 12(6), 498–508 CrossRef PubMed.
- P. G. Finichiu, et al., Mitochondrial accumulation of a lipophilic cation conjugated to an ionisable group depends on membrane potential, pH gradient and pK a: implications for the design of mitochondrial probes and therapies, J. Bioenerg. Biomembr., 2013, 45(1–2), 165–173 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics, Proc. Natl. Acad. Sci., 2012, 109(40), 16288–16293 CrossRef CAS PubMed.
- D. Y. Cho, et al., Triphenylphosphonium-conjugated poly (ε-caprolactone)-based self-assembled nanostructures as nanosized drugs and drug delivery carriers for mitochondria-targeting synergistic anticancer drug delivery, Adv. Funct. Mater., 2015, 25(34), 5479–5491 CrossRef CAS.
- V. Kumar and S. V. Malhotra, Study on the potential anti-cancer activity of phosphonium and ammonium-based ionic liquids, Bioorg. Med. Chem. Lett., 2009, 19(16), 4643–4646 CrossRef CAS PubMed.
- B. Bachowska, et al., High Cytotoxic Activity of Phosphonium Salts and Their Complementary Selectivity towards HeLa and K562 Cancer Cells: Identification of Tri-n-butyl-n-hexadecylphosphonium bromide as a Highly Potent Anti-HeLa Phosphonium Salt, ChemistryOpen, 2012, 1(1), 33–38 CrossRef CAS PubMed.
- J. Shi, et al., Cancer nanomedicine: progress, challenges and opportunities, Nat. Rev. Cancer, 2017, 17(1), 20 CrossRef CAS PubMed.
- S. Gorini, et al., Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib, Oxid. Med. Cell. Longevity, 2018, 7582730 Search PubMed.
- H. Huang, et al., Antitumor activity and antitumor mechanism of triphenylphosphonium chitosan against liver carcinoma, J. Mater. Res., 2018, 33(17), 2586–2597 CrossRef CAS.
- N. P. Katuwavila, et al., Chitosan-alginate nanoparticle system efficiently delivers doxorubicin to MCF-7 cells, J. Nanomater., 2016, 736–747 Search PubMed.
- A. Hefnawy, I. A. Khalil and I. M. El-Sherbiny, Facile development of nanocomplex-in-nanoparticles for enhanced loading and selective delivery of doxorubicin to brain, Nanomedicine, 2017, 12(24), 2737–2761 CrossRef CAS PubMed.
- H. K. Shaikh, R. Kshirsagar and S. Patil, Mathematical models for drug release characterization: a review, World J. Pharm. Pharm. Sci., 2015, 4, 324–338 CAS.
- L. Wang, et al., Novel water soluble phosphonium chitosan derivatives: synthesis, characterization and cytotoxicity studies, Int. J. Biol. Macromol., 2011, 48(2), 375–380 CrossRef CAS PubMed.
- T. A. Theodossiou, et al., Mitochondrial delivery of doxorubicin by triphenylphosphonium-functionalized hyperbranched nanocarriers results in rapid and severe cytotoxicity, Pharm. Res., 2013, 30(11), 2832–2842 CrossRef CAS PubMed.
- J. Hou, et al., Triphenyl phosphine-functionalized chitosan nanoparticles enhanced antitumor efficiency through targeted delivery of doxorubicin to mitochondria, Nanoscale Res. Lett., 2017, 12(1), 1–9 CrossRef CAS PubMed.
- H. Shagholani, S. M. Ghoreishi and M. Mousazadeh, Improvement of interaction between PVA and chitosan via magnetite nanoparticles for drug delivery application, Int. J. Biol. Macromol., 2015, 78, 130–136 CrossRef CAS PubMed.
- A. Kim, et al., Validation of Size Estimation of Nanoparticle Tracking Analysis on Polydisperse Macromolecule Assembly, Sci. Rep., 2019, 9(1), 2639 CrossRef PubMed.
- M. P. Paarakh, et al., Release kinetics–concepts and applications, Int. J. Pharm. Res. Technol., 2018, 8(1), 12–20 Search PubMed.
- M. C. Drinane and V. H. Shah, Alcoholic hepatitis: diagnosis and prognosis, Clinical Liver Disease, 2013, 2(2), 80 CrossRef PubMed.
- R. Tolba, et al., Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice, Lab. Anim., 2015, 49(1 Suppl), 59–69 CrossRef CAS PubMed.
- L. W. Weber, M. Boll and A. Stampfl, Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model, Crit. Rev. Toxicol., 2003, 33(2), 105–136 CrossRef CAS PubMed.
- T. Sakurai, S. Maeda and M. K. Lufen Chang, INAUGURAL ARTICLE by a Recently Elected Academy Member: Loss of hepatic NF-κB activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(28), 10544 CrossRef CAS PubMed.
- F. Farinati, et al., Diagnostic and prognostic role of α-fetoprotein in hepatocellular carcinoma: both or neither?, Am. J. Gastroenterol., 2006, 101(3), 524–532 CrossRef CAS PubMed.
- W. R. Kim, et al., Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease, Hepatology, 2008, 47(4), 1363–1370 CrossRef CAS PubMed.
- A. Memon, et al., A Modified Protocol of Diethylnitrosamine Administration in Mice to Model Hepatocellular Carcinoma, Int. J. Mol. Sci., 2020, 21(15), 5461 CrossRef CAS PubMed.
- S. Sarkar, et al., Pharmaceutical efficacy of harmalol in inhibiting hepatocellular carcinoma, J. Pharm. Sci., 2020, 6(1), 1–18 Search PubMed.
- Y.-W. Zhang, et al., Cardiomyocyte death in doxorubicin-induced cardiotoxicity, Arch. Immunol. Ther. Exp., 2009, 57(6), 435–445 CrossRef CAS PubMed.
- B. Kalyanaraman, Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree?, Redox Biol., 2020, 29, 101394 CrossRef CAS PubMed.
- E. H. Herman, et al., Use of cardiac troponin T levels as an indicator of doxorubicin-induced cardiotoxicity, Cancer Res., 1998, 58(2), 195–197 CAS.
| This journal is © The Royal Society of Chemistry 2022 |