
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycosylation of luteolin in hydrophilic organic solvents and structure–antioxidant relationships of luteolin glycosides†
Tingting Xua,
Chen Wangb,
Sijie Jiangb,
Tingting Yangb and
Xueming Wu *b
*b
aSchool of Medicine & Holistic Integrative Medicine, Nanjing University of Chinese Medicine, Nanjing, 210023, China
bState Key Laboratory Cultivation Base for TCM Quality and Efficacy, School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, 210023, China. E-mail: xuemingwu@njucm.edu.cn
First published on 22nd June 2022
Abstract
An effective approach was developed to biotransform luteolin glycosides in hydrophilic organic solvents. Bacillus cereus A46 cells showed high activity and stability in 5–20% (v/v) DMSO with 90–98% conversion rates of luteolin glycosides. Five glycosides of luteolin 7-O-β-glucoside, luteolin 4′-O-β-glucoside, luteolin 3′-O-β-glucoside, luteolin 7,3′-di-O-β-glucoside and luteolin 7,4′-di-O-β-glucoside were obtained. The addition of DMSO greatly promoted the solubility of luteolin and further regulated the formation of the main products from five luteolin glycosides to luteolin 7-O-β-glucoside (931.2 μM). Fourteen flavonoids and anthraquinones were used as tentative substrates. Glycosylation positions were located at the C-7, C-3′ or C4′ hydroxyl groups of flavonoids and C-5 hydroxyl group of anthraquinones. The 3′,4′-dihydroxy arrangement played the key role for the antioxidant activity of luteolin.
1. Introduction
Luteolin (3′,4′,5,7-tetrahydroxyflavone) is a natural flavonoid widely found in plants. It is reported to have anti-inflammatory,1 antioxidant,2 anti-tumor,3 anti-tuberculosis immunity,4 and neuroprotective properties.5 Although luteolin itself exhibits various biological activities with valuable medicinal applications, several drawbacks have been reported including poor aqueous solubility and low bioavailability.6,7 Glycosylation serves to enhance the solubility and bioactivity of flavonoid aglycones. The biosynthesis of flavonoid-O-glycosides in plants is mainly mediated by glycosyltransferases.8 Some luteolin O-glycosides have been isolated from various plants, including luteolin 3′-O-glucoside, luteolin 4′-O-glucoside,9 luteolin 7-O-glucoside, luteolin 7,4′-di-O-glucoside,10,11 luteolin-7,3′-di-O-glucoside.12 Among of them, luteolin 4′-O-glucoside and 7-O-glucoside have been confirmed to be cytotoxic activity against NCI-H929, U266, and OPM2 cell lines.13 However, further bioactivity study of luteolin glucosides is hampered by its low in planta abundance.9–12 Thus, chemical synthesis or biotransformation approach supplies potential alternatives to obtain sufficient luteolin glucosides.Compared with chemical glycosylation, which relies on a heavy dependence on protecting-group mechanisms that introduce many additional steps, biotransformation provides a direct regioselective glycosylation method to construct glycosidic bonds.14 For instance, glycosyltransferase (XcGT-2) from Xanthomonas campestris was reported regiospecifically glycosylated the 3′-hydroxyl group of luteolin.15 UDP-glycosyltransferase (RUGT-5) from rice was recorded glycosylated the 7- and 4′-hydroxyl groups of luteolin.16 However, enzyme reaction in aqueous phase is not efficient with lower yields due to the solubility issue of the substrate. The conversion rate of luteolin 7-O-glycoside by UDP-glycosyltransferase (BcGT-1) from Bacillus cereus was only 40.9%.17 Furthermore, the availability of glycosyltransferase has been constrained by need to provide activated sugar as donor for the glycosylation.15–17
Fortunately, enzymatic glycosylation in organic solvents using directly oligosaccharides as donors supply industrially attractive advantages, such as improving the solubility of hydrophobic substrates, reversing of the thermodynamic equilibrium of hydrolysis reactions, and lowing cost process.18,19 Herein, we investigated the glycosylation of luteolin in hydrophilic organic solvents using the biotransformation approach, whereas five luteolin glycosides were enzymatically biosynthesized. Furthermore, the structure–antioxidant relationships of luteolin glycosides were also studied.
2. Materials and methods
2.1 Materials and regents
Flavonoids and emodin were purchased from Zelang (Jiangsu, China). 2,2-Diphenyl-1-picrylhydrazyl (DPPH) was obtained from Sigma (St. Louis, MO, USA). Physcion, catenarin, erythroglaucin, skyrin, emodin bianthrone and emodin-catenarin bianthrone were isolated from our laboratory.20,21 Methanol used for HPLC analysis was of chromatographic grade from Sinopharm (Shanghai, China). All other chemicals were of analytical grade and purchased from Sunshine (Nanjing, China).2.2 Strain and culture condition
Bacillus cereus A46 was isolated by the previous methods and stored as 15% glycerol stocks at −80 °C.18 Bacterial identification was based on the 16S rDNA sequence, and identified as Bacillus cereus. The strain A46 was incubated in liquid culture medium containing (g L−1): sucrose 20.0, peptone 10.0, KH2PO4 2.0, CaCl2 0.6, adjusted to pH 7.5. Cultivation was conducted at 30 °C, 200 rpm. After 12 h incubation, the cultured cells were harvested by centrifugation at 8500 × g and 4 °C for 20 min.2.3 Glycosylation of luteolin
The glycosylation reactions (360 mL) were conducted in 1/15 M Na2HPO4/KH2PO4 buffer (pH 6.47) containing 2% (w/v) harvested wet cells, 20% (w/v) sucrose, 970 μM luteolin and 20% (v/v) dimethyl sulfoxide (DMSO). The reaction mixture was conducted at 30 °C with shaking at 200 rpm for 48 h.2.4 Analysis, isolation and identification of products
In general, products were analyzed by reversed-phase liquid chromatography on Agilent 1260 HPLC, using a XBridge C18 column (5 μm, 4.6 × 250 mm). The HPLC analysis method was performed using the following gradient: 0.0–13.0 min, 10–100% B; 13.0–20.0 min, 100% B; 20.0–20.1 min, 100–10% B; 20.1–23.0 min, equilibration with 10% B. The mobile phase consisted of solvents A (water) and B (methanol). The column was operated with a constant flow rate of 0.8 mL min−1 at 30 °C and a sample injection volume of 20 μL. The liquid chromatography hyphened with the high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) was run on an Agilent 6546 Accurate-Mass Q-TOF LC/MS instrument with a Poroshell 120 EC-C18 column (2.7 μm, 4.5 × 50 mm). The LC-HR/MS analysis of products was performed using the “A + B” mobile phase as specified below: 0.0–13.0 min, 10–100% B. The column was operated with a constant flow rate of 0.3 mL min−1 at 30 °C. Glycosylation products were purified by semipreparative HPLC on an Agilent Technologies 1260 Infinity LC System (Agilent, California, USA) by an Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm) at a flow rate of 2.0 mL min−1. For structure elucidation, NMR spectra were recorded on a Bruker AV-500 NMR spectrometer (Bruker, Fällanden, Switzerland). Samples were dissolved in DMSO-d6 at room temperature with tetramethylsilane (Me4Si) as the chemical shift reference.2.5 Evaluation of antioxidant activity
The effects of luteolin and its five glycosides on the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical were estimated.19 Briefly, 150 μL of various concentration samples (5, 10, 20, 40, 80 and 160 μM) were mixed with 150 μL of 150 μM DPPH solution. After being incubated at 37 °C for 30 min, the absorbance of the mixture was measured at 517 nm. The free radical scavenging activity of the reaction solution was calculated as a percentage of DPPH discolouration using the following equation: radical scavenging activity (%) = (1 − Asample/Acontrol) × 100, where Asample is the absorbance of the solution when the sample has been added at a particular level, and Acontrol is the absorbance of the DPPH solution. The results were expressed as mean values ± standard deviations (n = 8).3. Results and discussion
3.1 Biotransformation and identification of luteolin glycosides
Bacillus cereus A46 was used to biotransformation of the luteolin glycosides. The glycosylation reaction mixture of this strain showed five product peaks (compounds 2–6) with reference to the luteolin (1) (Fig. 1A), which were further analysed by LC-HR/MS. The HR-ESI mass analysis of the reaction mixture showed the ion peaks 2, 3 and 4 [M + H]+ at m/z 449.1167, 449.1168 and 449.1170, correspondingly (calculated for C21H21O11, 449.1084) (Fig. 1B). Compared with luteolin (1), the compound 2, 3 or 4 has an additional formula C6H10O5, suggesting that these compounds could be the luteolin monosaccharide derivatives. The comparison analysis between the 1H-NMR data of luteolin (1) and compound 2 revealed that the high field peak δ 4.87 (d, J = 7.2 Hz) (δ 4.88 (d, J = 7.3 Hz) for 3, δ 5.08 (d, J = 7.4 Hz) for 4) was the anomeric sugar proton signal (Table 1 and Fig. S1–S4†). The resonance of H-1′′ at δ 4.87, 4.88 or 5.08 as a double with J1,2 > 7.0 Hz indicates that compound 2, 3, or 4 is the sugar β-anomer. The 1H-NMR spectra of compounds 2–4 showed similarities to those of previous reported compounds luteolin 3′-O-β-glucoside, luteolin 4′-O-β-glucoside and luteolin 7-O-β-glucoside.10,22,23 Therefore, the chemical structure of compound 2 was elucidated as luteolin 3′-O-β-glucoside,22,23 3 was elucidated as luteolin 4′-O-β-glucoside23 and 4 was elucidated as luteolin 7-O-β-glucoside (Fig. 1C).10,22 Furthermore, the mass spectrum of compounds 5 and 6 showed the ion peaks [M + H]+ at m/z 611.1726 and 611.1728, correspondingly (calculated for C27H31O16, 611.1612) (Fig. 1B). Compared with luteolin, the compound 5 or 6 has an additional formula C12H20O10, suggesting that compounds 5 and 6 may be the luteolin disaccharide derivatives. The 1H-NMR spectra of compound 5 showed the presence of two anomeric sugar protons at δ 5.01 (d, 7.6 Hz) and δ 5.08 (d, 7.5 Hz) (Table 1, Fig. S5 and S6†). The 1H-NMR spectra of compound 5 showed similarities to those of the known compounds luteolin di-O-glucoside.24 Hence, compound 5 was established as luteolin 7,3′-di-O-β-glucoside.24 Moreover, two anomeric sugar protons appear at δ 4.89 (d, 7.3 Hz) and δ 5.09 (d, 7.4 Hz) for compound 6 (Table 1), which was elucidated as luteolin 7,4′-di-O-β-glucoside (Fig. 1C).23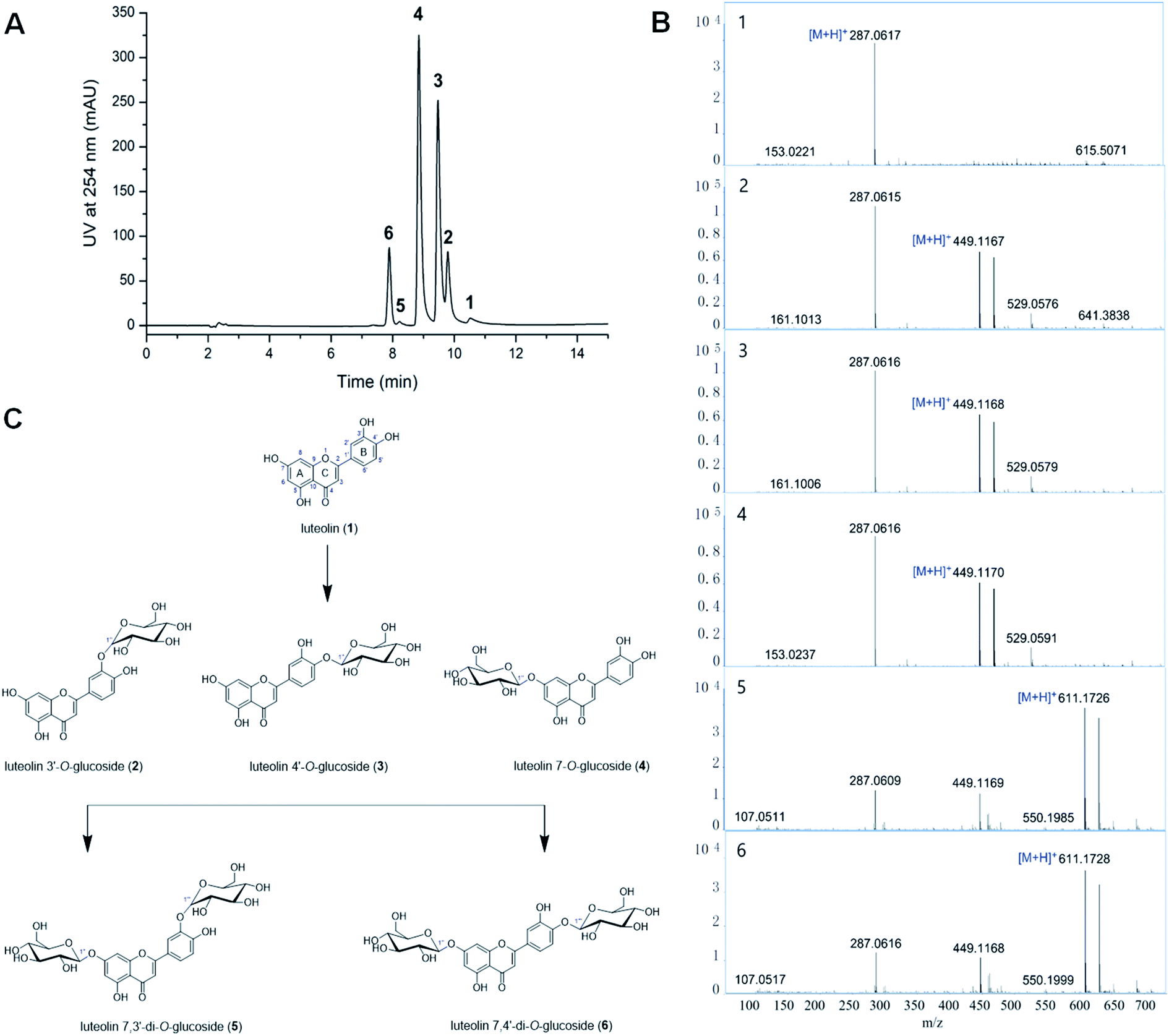 | ||
| Fig. 1 Biotransformation of luteolin glycosides by Bacillus cereus A46. (A) HPLC profiling of biotransformation for luteolin glycosides by A46 cells. (B) The LC-HR/MS analysis of the reaction products. (C) The chemical structures of luteolin and luteolin glycosides. | ||
| Position | δH (J, Hz) | |||||
|---|---|---|---|---|---|---|
| Luteolin (1) | Luteolin 3′-O-β-glucoside (2) | Luteolin 4′-O-β-glucoside (3) | Luteolin 7-O-β-glucoside (4) | Luteolin 7,3′-di-O-β-glucoside (5) | Luteolin 7,4′-di-O-β-glucoside (6) | |
| 3 | 6.67 (s) | 6.77 (s) | 6.82 (s) | 6.74 (s) | 6.79 (s) | 6.92 (s) |
| 5-OH | 12.97 (s) | 12.98 (s) | 12.91 (s) | 13.07 (s) | 12.96 (s) | 12.96 (s) |
| 6 | 6.19 (s) | 6.16 (s) | 6.19 (d, J = 1.9 Hz) | 6.44 (d, J = 2.1 Hz) | 6.44 (d, J = 2.1 Hz) | 6.45 (d, J = 2.0 Hz) |
| 7-OH | 10.81 (s) | |||||
| 8 | 6.44 (s) | 6.50 (s) | 6.49 (d, J = 1.9 Hz) | 6.79 (d, J = 1.9 Hz) | 6.75 (s) | 6.86 (d, J = 2.0 Hz) |
| 2′ | 7.40 (d, J = 2.6 Hz) | 7.63 (m) | 7.49 (d, J = 2.1 Hz) | 7.43 (m) | 7.42 (d, J = 1.7 Hz) | 7.54 (d, J = 2.0 Hz) |
| 3′-OH | 9.47 (s) | |||||
| 4′-OH | 9.92 (s) | |||||
| 5′ | 6.89 (d, J = 8.2 Hz) | 6.93 (d, J =8.5 Hz) | 7.24 (d, J = 8.6 Hz) | 6.89 (d, J = 8.2 Hz) | 6.90 (d, J = 8.3 Hz) | 7.26 (d, J = 8.6 Hz) |
| 6′ | 7.42 (m) | 7.76 (m) | 7.51 (dd, J = 2.2, 8.5 Hz) | 7.44 (dd, J = 2.0, 8.4 Hz) | 7.44 (dd, J = 2.1, 8.3 Hz) | 7.56 (dd, J = 2.1, 8.6 Hz) |
| 1′′ | 4.87 (d, J = 7.2 Hz) | 4.88 (d, J = 7.3 Hz) | 5.08 (d, J = 7.4 Hz) | 5.08 (d, J = 7.5 Hz) | 5.09 (d, J = 7.4 Hz) | |
| 1′′′ | 5.01 (d, J = 7.6 Hz) | 4.89 (d, J = 7.3 Hz) | ||||
3.2 Efficient glycosylation in hydrophilic organic solvents
The hydrophilic solvents such as methanol, ethanol, 2-propanol, DMSO and DMF were employed to promote the solubility of hydrophobic acceptors. The solubility of luteolin in this reaction system was improved about 4.0 times than in aqueous solution. Strikingly, highly efficient glycosylation of luteolin was obtained in the hydrophilic solvents by Bacillus cereus A46 (Fig. 2). The A46 cell shows high activity and stability in 20% (v/v) DMSO and DMF with 90–96% conversion rates of luteolin glycosides, however methanol and 2-propanol have solvent denaturing effect for A46 cells resulting less 80% conversion rates (Fig. 2A).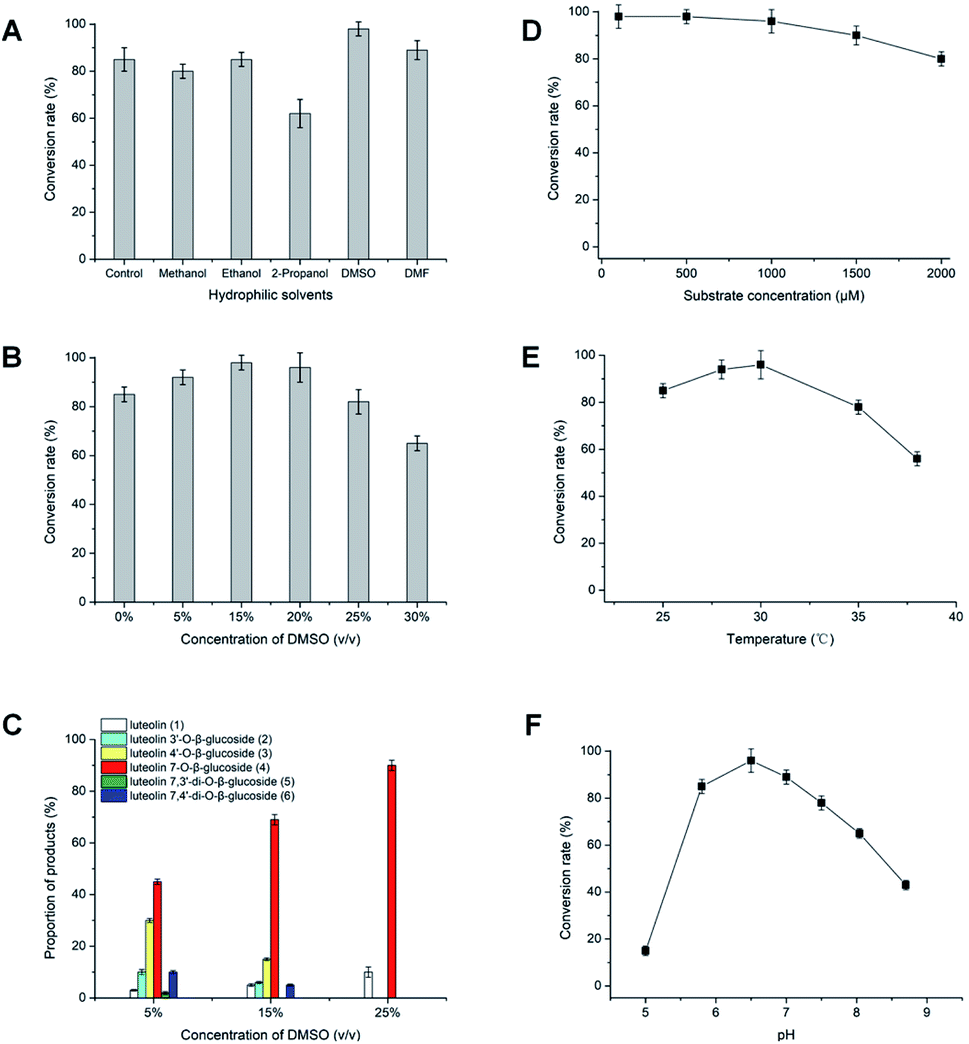 | ||
| Fig. 2 Effects of multi-factors on the glycosylation of luteolin catalyzed by Bacillus cereus A46. | ||
Significantly, the participation of DMSO not only obviously increased the solubility of luteolin, but also displayed the regulation for the products of luteolin glycosylation. The main products changed from five luteolin glucosides (luteolin 7-O-β-glucoside, luteolin 4′-O-β-glucoside, luteolin 3′-O-β-glucoside, luteolin 7,3′-di-O-β-glucoside and luteolin 7,4′-di-O-β-glucoside) to luteolin 7-O-β-glucoside with increasing DMSO concertation (Fig. 2C). This regulation of the formation of main products depends the balance between the concentration of organic solvents and the catalytic activity of A46 cell. As shown in Fig. 2B and C, A46 cell displays highly catalytic activity in 5–15% (v/v) DMSO with 90–98% conversion rates and multi-glucoside products. However, the activity and stability of A46 cell began to reduce in 25–30% (v/v) DMSO with less 80% conversion rate. Luteolin 7-O-β-glucoside was the first glucoside products, followed by luteolin 3′-O-β-glucoside, luteolin 4′-O-β-glucoside and luteolin 7,4′ or 3′-di-O-β-glucoside catalyzed by A46 cell (Fig. 1C and 2C). The multi-glucoside products changed to luteolin 7-O-β-glucoside in 5–25% (v/v) DMSO mainly due to the reduction of catalytic activity. Furthermore, the optimization of luteolin glycosylation process was conducted shown in Fig. 2D–F. The efficient glycosylation was achieved in 30 °C, pH 6.47 and 970 μM substrate concentration.
Efficiently production of the luteolin glucosides was attained in 20% (v/v) DMSO solvent system, which gave a final conversion rate of 96%. This resulted in 931.2 μM (417.2 mg L−1) of luteolin 7-O-β-glucoside. The substrate concentration (970 μM) of luteolin in 20% (v/v) DMSO solvent was over tenfold than other reports, such as glycosylation of luteolin (30.0 μM) with glycosyltransferase (XcGT-2) from X. campestris15 and trans-glycosylation of luteolin (70.0 μM) by UDP-glycosyltransferase (RUGT-5 and BcGT-1).16,17 The high concentration and simple products of luteolin glycosides (mainly luteolin 7-O-β-glucoside) have a great facility for separating and achieving products in large-scale process, which supply convenience for further bioactivity study of luteolin glucosides.
3.3 Antioxidant activities of luteolin glycosides
The role of 3′-OH and 4′-OH groups in the B ring on DPPH radical scavenging potential of luteolin glycosides was studied. As shown in Table 2, luteolin (IC50 20.2 μM) and luteolin 7-O-β-glucoside (IC50 21.2 μM) showed higher antioxidant activities in comparison with luteolin 3′ or 4′-mono glucosides and 7,3′ or 7,4′-diglucosides (IC50 > 100 μM). Both luteolin and luteolin-7-O-β-glucoside contain a 3′,4′- dihydroxy structure in the B ring. The results showed clear differences of antioxidant activities between these luteolin glucosides and suggest that the B ring 3′,4′-dihydroxy group may be particularly important to their activities in this system. These structure–activity relationships are consistent with the observations by Tournaire25 and Nagai.26 Who both demonstrated that the presence of the O-dihydroxy group in the B ring is essential for 1O2 quenching in chemical systems.25,26 Clearly, the presence of the 3′,4′-dihydroxy arrangement in this biological system is also important for the superior capacity of luteolin on DPPH radical scavenging activities, and this appears to relate to the formation of a more stable ortho-hydroxyl phenoxyl radical and an intramolecular hydrogen bond in the process of oxidation27 or increase the rate of H-atom transfer to radicals.28| Compounds | Free OH | IC50 (μM) |
|---|---|---|
| Luteolin | 5, 7, 3′, 4′ | 20.2 |
| Luteolin 3′-O-β-glucoside | 5, 7, 4′ | >100 |
| Luteolin 4′-O-β-glucoside | 5, 7, 3′ | >100 |
| Luteolin 7-O-β-glucoside | 5, 3′, 4′ | 21.2 |
| Luteolin 7,3′-di-O-β-glucoside | 5, 4′ | >100 |
| Luteolin 7,4′-di-O-β-glucoside | 5, 3′ | >100 |
3.4 Substrate specificity for glycosylation by Bacillus cereus A46
The specificity for glucosyl acceptor catalyzed by A46 cell was investigated using flavonoids and anthraquinones. All seven flavonoids contain a 7-OH group in the A ring or 3′-OH/4′-OH group in the B ring (Fig. 3 and S7–S12†), could be glycosylated by A46 cells. The results showed broad substrate specificity of B. cereus A46, which made us interested in studying other structural types of substrates. Seven anthraquinone derivatives were used as substrate, A46 cells were found to convert emodin, catenarin, skyrin, emodin bianthrone and emodin-catenarin bianthrone to glycosylated forms (Fig. 3 and S13–S27†). When the 5-OH group of anthraquinones was replaced by a methyl group, such as physcion and erythroglaucin, these anthraquinones could not be glycosylated by A46 cells. The results showed that B. cereus A46 cells have exquisite regioselectivity for the hydroxyl group at C-5 of anthraquinones. The glycosylation catalysed by B. cereus A46 with broad substrate specificity will beneficial to exploit the new candidate pharmaceuticals.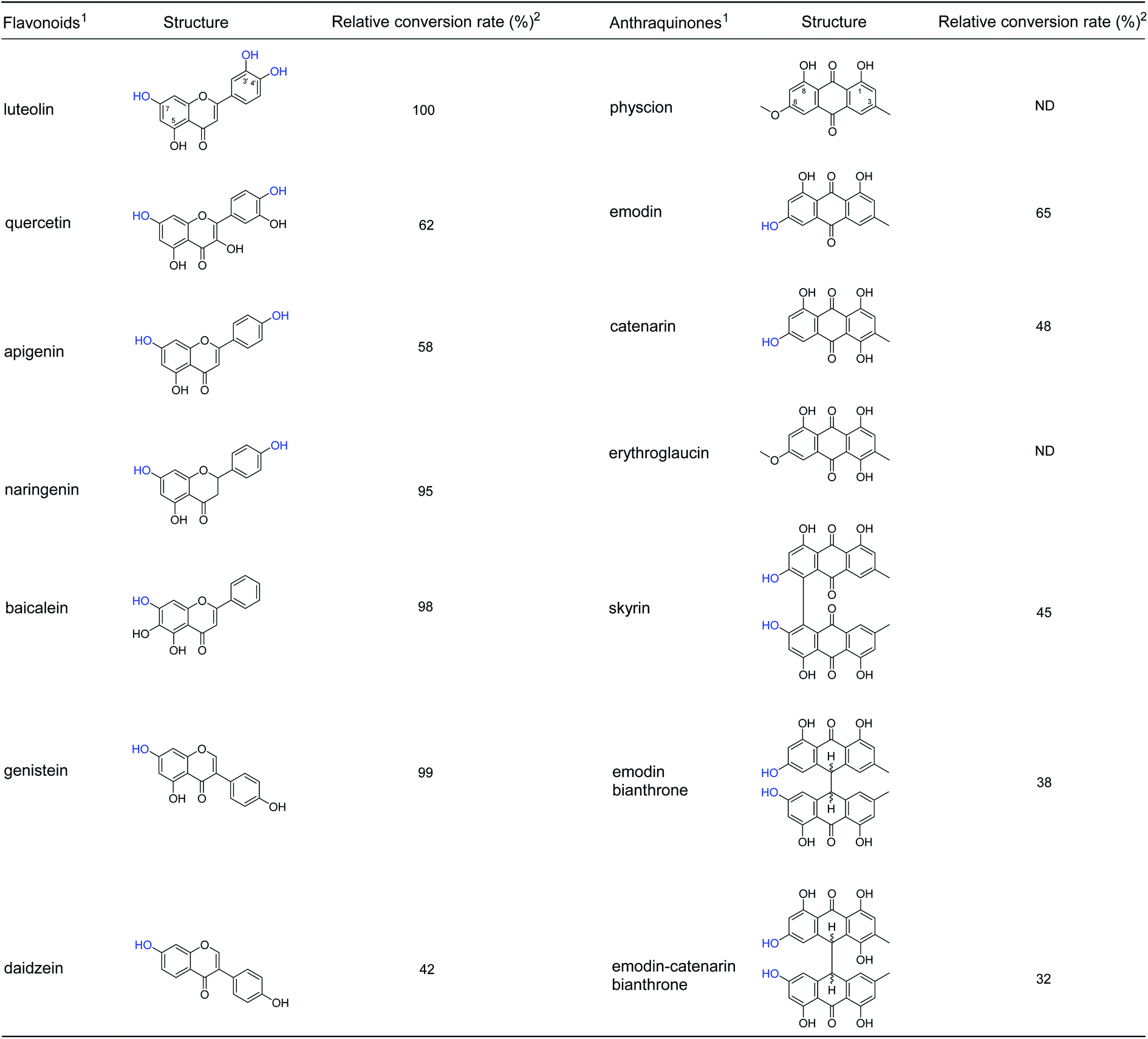 | ||
| Fig. 3 Glycosylation specificity of flavonoids and anthraquinones by A46 cells. 1The glycosylation reactions (100 mL) were conducted in 1/15 M Na2HPO4/KH2PO4 buffer (pH 6.47) containing 2% (w/v) harvested wet cells, 20% (w/v) sucrose, 100 μM substrate and 20% (v/v) DMSO. The reaction mixture was conducted at 30 °C for 24 h. 2Relative conversion rate was calculated on the bases of the remaining acceptor. | ||
4. Conclusions
In summary, Bacillus cereus A46 cells showed high stability and activity in 20% methanol, ethanol, DMSO and DMF. High conversion rate (96%) of luteolin glycosides and regulated preparation of luteolin 7-O-β-glucoside were achieved in organic solvents system. A46 cells could efficiently transfer glucose to specific hydroxyl group (the 7th position in the A ring and 3′ or 4′-position in the B ring) of the flavone structure and 5-OH group of anthraquinones. DPPH radical-scavenging activities of luteolin glucosides confirmed that the presence of the 3′,4′-dihydroxy arrangement is important for the superior antioxidant capacity of luteolin.Author contributions
X. M. Wu conceived and supervised the project. T. T. Xu designed and performed experiments, and wrote the manuscript. All authors contributed to the data analysis and manuscript preparation.Conflicts of interest
All authors declare no conflicts of interest.Acknowledgements
The work was financed by National Natural Science Foundation of China (82174089 and 81991524).References
- X. Wang, L. Wang, R. Dong, K. Huang, C. Wang, J. Gu, H. Luo, K. Liu, J. Wu, H. Sun and Q. Meng, Phytomedicine, 2021, 87, 153586 CrossRef CAS PubMed.
- H. Xu, B. S. Linn, Y. Zhang and J. Ren, React. Oxygen Species, 2019, 7, 136–147 CAS.
- A. M. O. Hasym, N. M. Nor, L. H. M. Adnan, N. Z. B. Ahmad, A. W. Septama, N. N. Najihah, O. M. Lwin and N. Simbak, Sains Malays., 2021, 50, 821–828 CrossRef.
- D. K. Singh, S. Tousif, A. Bhaskar, A. Devi, K. Negi, B. Moitra, A. Ranganathan, V. P. Dwivedi and G. Das, PLoS Pathog., 2021, 17, e1009805 CrossRef CAS PubMed.
- L. Li, R. Zhou, H. Lv, L. Song, X. Xue and L. Wu, ACS Chem. Neurosci., 2021, 12, 3314–3322 CrossRef CAS PubMed.
- Y. Wang, Q. Wang, W. Feng, Q. Yuan, X. Qi, S. Chen, P. Yao, Q. Dai, P. Xia, D. Zhang and F. Sun, Drug Delivery, 2021, 28, 1695–1708 CrossRef CAS PubMed.
- P. Zhu, L. Chen, Y. Zhao, C. Gao, J. Yang, X. Liao, D. Liu and B. Yang, J. Mol. Struct., 2021, 1237, 130339 CrossRef CAS.
- T. Vogt and P. Jones, Trends Plant Sci., 2001, 5, 380–386 CrossRef.
- T. Mizuno, K. Sugahara, C. Tsutsumi, M. Iino, S. Koi, N. Noda and T. Iwashina, Phytochemistry, 2021, 181, 112581 CrossRef CAS PubMed.
- S. A. Youssef, Z. Z. Ibraheim and A. A. Attia, Bull. Pharm. Sci., Assiut Univ., 1995, 18, 33–38 CrossRef CAS.
- D. N. Olennikov and N. K. Chirikova, Chem. Nat. Compd., 2015, 51, 1067–1069 CrossRef CAS.
- A. P. Rauter, A. Martins, C. Borges, J. Ferreira, J. Justino, M.-R. Bronze, A. V. Coelho, Y. H. Choi and R. Verpoorte, J. Chromatogr. A, 2005, 1089, 59–64 CrossRef CAS PubMed.
- S. S. Cicek, J. Willer, F. Preziuso, F. Sonnichsen, R. Greil, U. Girreser, C. Zidorn and K. Johrer, RSC Adv., 2021, 11, 10489–10496 RSC.
- M. Yang, G. J Davies and B. G. Davis, Angew. Chem., Int. Ed., 2007, 46, 3885–3888 CrossRef CAS PubMed.
- K. J. Ho, B. G. Kim, J. A. Kim, Y. Park, Y. J. Lee, Y. Lim and J.-H. Ahn, J Microbiol. Biotechnol., 2007, 17, 539–542 Search PubMed.
- J. H. Ko, B. G. Kim, H.-G. Hur, Y. Lim and J.-H. Ahn, Plant Cell Rep., 2006, 25, 741–746 CrossRef CAS PubMed.
- J. H. Ko, B. G. Kim and J.-H. Ahn, FEMS Microbiol. Lett., 2006, 258, 263–268 CrossRef CAS PubMed.
- X. M. Wu, J. L. Chu, B. Wu, S. Zhang and B. F. He, Bioresour. Technol., 2013, 129, 659–662 CrossRef CAS PubMed.
- X. M. Wu, J. L. Chu, J. Y. Liang and B. F. He, RSC Adv., 2013, 3, 19027–19032 RSC.
- Y. B. Han, W. Bai, C. X. Ding, J. Liang, S.-H. Wu and R. X. Tan, J. Am. Chem. Soc., 2021, 143, 14218–14226 CrossRef CAS PubMed.
- X. M. Wu, Q. Y. Guan, Y. B. Han, X. C. Wang, W. Y. Zhuang and R. X. Tan*, Angew. Chem., Int. Ed., 2022, 61, e202114919 CAS.
- Y. R. Lu and Y. Foo, Phytochemistry, 2000, 55, 263–267 CrossRef.
- K. A. Mitchell, K. R. Markham and M. J. Bayly, Phytochemistry, 2001, 56, 453–461 CrossRef CAS PubMed.
- K. R. Markham, B. Ternai, R. Stanley, H. Geiger and T. J. Mabry, Tetrahedron, 1978, 34, 1389–1397 CrossRef CAS.
- C. Tournaire, S. Croux, M.-T. Maurette, I. Beck, M. Hocquaux, A. M. Braun and E. Oliveros, J. Photochem. Photobiol., B, 1993, 19, 205–215 CrossRef CAS.
- S. Nagai, K. Ohara and K. Mukai, J. Phys. Chem. B, 2005, 109, 4234–4240 CrossRef CAS PubMed.
- W. M. Wu, L. Lu, Y. Long, T. Wang, L. Liu, Q. Chen and R. Wang, Food Chem., 2007, 105, 107–115 CrossRef CAS.
- M. Lucarini, V. Mugnaini and G. F. Pedulli, J. Org. Chem., 2002, 67, 928–931 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03300c |
| This journal is © The Royal Society of Chemistry 2022 |