
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable uniaxial, area, and volume negative thermal expansion in quartz-like and diamond-like metal–organic frameworks
Lei Wang *a,
Ying Chenb,
Hideo Miurab,
Ken Suzukib and
Cong Wang*c
*a,
Ying Chenb,
Hideo Miurab,
Ken Suzukib and
Cong Wang*c
aDepartment of Physics, University of Science and Technology Beijing, Beijing 100083, China. E-mail: leiw_phy@ustb.edu.cn; congwang@buaa.edu.cn
bFracture and Reliability Research Institute, Graduate School of Engineering, Tohoku University, Sendai 980-8579, Japan
cSchool of Integrated Circuit Science and Engineering, Beihang University, Beijing 100191, China
First published on 8th August 2022
Abstract
This paper proposes that it will be an effective way to discover and explore organic negative thermal expansion (NTE) materials based on the specific topologies in inorganic NTE materials. Various NTE behaviors from the uniaxial, area, and volume-NTE can be achieved by adjusting the topology, for instance, quartz-like and diamond-like. Zn(ISN)2 and InH(BDC) metal–organic frameworks (MOFs) with quartz-like topology have been studied by first principles calculations. The calculated area-NTE of Zn(ISN)2 and uniaxial-NTE of InH(BDC) within quasi-harmonic approximation (QHA) agree well with the experimental data. Through the calculation of Grüneisen parameters, it is shown that low-frequency optical phonons appear dominant resulting in their NTE, but the coupling to high-frequency phonons is of greater ultimate importance. The lattice vibrational modes of great contribution to area-NTE of Zn(ISN)2 and uniaxial-NTE of InH(BDC) are analyzed in detail. Also, four MOFs with diamond-like topology are predicted to exhibit volume-NTE behavior. Moreover, it is found that there is a bulk modulus anomaly in some studied MOFs with the quartz-like and diamond-like framework, where the temperature dependence of bulk modulus does not follow the inverse dependence on that of volume. These specific topologies provide key geometric frameworks for various NTE behaviors of MOFs, and meanwhile, the local structure and bond environment in MOFs can lead to abnormal interatomic force, i.e., bulk modulus anomaly. This abnormal elastic property also deserves more attention.
1. Introduction
Negative thermal expansion (NTE) materials that shrink upon heating are not only of particular interest but have important potential applications.1 By using NTE materials to compensate for the more common positive thermal expansion (PTE) behavior of other materials, zero thermal expansion behavior in NTE/PTE composites can be achieved.2–4 Crystal engineering has been concerned with the design of solids with specific, controlled properties. One such attractive property is NTE behavior. To satisfy more design requirements, all kinds of NTE from uniaxial, to area to volume are in demand. Several inorganic systems exhibiting adjustable NTE behavior are known but organic materials displaying tunable NTE behavior have rarely been reported.5,6For inorganic materials, different NTE mechanisms may be involved.7 Whereas, NTE behavior in organic materials often occurs for geometrical reasons, considering special geometrical arrangements of the atoms (molecules) and peculiarities of the bonding which restrict the thermal movement of the atoms (molecules).8,9 Such NTE materials are characterized by open, underconstrained lattices or by a network of rigid, underconstrained entities. Within the scope of inorganic materials, following the specific crystal structure, other NTE candidates can be derived from a certain NTE system. For example, the diamond-like, rutile-like, as well as zinc blende-like materials, can be traced to the NTE property.10–13 Similarly, the specific topology that dominates NTE behavior in inorganic materials can be an inspiration for looking for NTE candidates in organic materials.14,15
Quartz is an archetypal material exhibiting NTE behavior and possesses a framework structure of stiff units, namely SiO4 tetrahedra, linked through shared oxygen atoms as very flexible hinges. Both phases of low-symmetry α-quartz (low temperature) and high-symmetry β-quartz (high temperature) show the NTE property.16,17 It has been reported that Ag3[Co(CN)6] and Zn[Au(CN2]2 frameworks with the topology of β-quartz exhibit the coexistence of large uniaxial-NTE and negative linear compressibility (NLC) properties.18–23 The NTE behavior in some materials such as Cu2O, Ag2O, ZnSiP2, and so on is attributed to the tetrahedral coordination of the crystal which has similarities with diamond-like topology.10,13
Recently, metal–organic frameworks (MOFs) as a new class of materials are receiving considerable attention due to their potential applications in some fields such as catalysis, gas separation, gas storage, and so on.24 MOFs can be designed by using flexible or rigid as well as mixed organic linkers which bind to the metal atoms that could provide structural diversity with various properties. The preparation and properties of quartz-like and diamond-like MOFs have been reported.14,25–30 By the way, most MOFs comprising four-connected tetrahedral units exist in the diamond-like topology, rather than the quartz-like topology. Sun et al. first prepared two quartz-like, chiral, open MOFs, Zn(ISN)2 (ISN = isonicotinate) with the low symmetry of α-quartz, and InH(BDC) (BDC = terephthalate) with the high symmetry of β-quartz.14 Then Collings et al. experimentally found that Zn(ISN)2 exhibited area-NTE in the (a,b)-plane, and InH(BDC) displayed the uniaxial-NTE along the c-axis.25 Although there is no thermodynamic requirement that NLC and NTE must coexist, it does seem likely that a general correspondence will be observed for noncubic framework materials.20,23 Based on this clue, large NLC behavior of InH(BDC) was found experimentally along its NTE direction by Zeng et al.26 Similarly, Zn(ISN)2 would be the most likely negative area compressibility (NAC) candidate by its confirmed area-NTE. On this point, it still needs experimental verification. Here, focusing on the NTE property, the physical mechanism of the area-NTE of Zn(ISN)2 and uniaxial-NTE of InH(BDC) has not been studied theoretically. In diamond-like MOFs, the volume-NTE below room temperature of Sr(C2(COO)2) had been reported.15 Whether there will be other diamond-like MOF materials with NTE property, is still an open question. Within a specific topology, will delicate local structures and atomic linkages cause abnormal interatomic forces, that is, the elasticity anomaly? It is also worth exploring together. Based on these, in this paper, we aim to (i) uncover the underlying mechanism of area-NTE of Zn(ISN)2 and uniaxial-NTE of InH(BDC), (ii) predict other diamond-like MOFs with NTE behavior, (iii) realize tunable uniaxial, area and volume NTE by changing the topology, and (iv) investigate the temperature dependence of bulk modulus in studied MOFs. It is proposed that this will be an effective way to design elaborate organic NTE materials from the simple topology in inorganic NTE materials. Moreover, it is instructive that the switching of uniaxial-NTE to area-NTE and then to volume-NTE can be achieved with fine-tuning of the geometric frame.
2. Methods and computational details
The total energy calculations are performed using the VASP code, which is based on density-functional theory (DFT). The exchange and correlation functionals are given by the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional.31 Electron-ion interactions are represented by the projector augmented wave (PAW) method with plane waves up to an energy of 500 eV.32 For the sampling of the irreducible Brillouin zone, we use K-point grids of the only Gamma point for the geometry optimization. In all the calculations, self-consistency is achieved with a tolerance in the total energy of 0.1 meV. The total energy is minimized simultaneously concerning all the lattice parameters to determine the equilibrium lattice constant. The equilibrium volume is obtained by fitting the total energy as a function of volume to a three-order Murnaghan equation of state. At the fixed volume, all atoms are fully relaxed using the conjugate gradient (CG) algorithm.The frozen-phonon approach where phonon frequencies are calculated from Hellmann–Feynman (HF) forces is employed to yield the thermodynamic properties.33 A perturbation displacement of 0.01 Å is applied to evaluate the resulting force constants. The structures of the unit cells of studied MOFs are employed to study the lattice dynamics. Fourier interpolation is used to obtain the dynamical matrices on a 3 × 3 × 3 q-point mesh. This set is used to evaluate all quantities that involve integration over phonon wave vector q.
To study the effects of temperature, one needs to focus on free energy, incorporating the effects of thermal vibrations. The free energy of a crystal can be expressed in the quasi-harmonic approximation (QHA) as
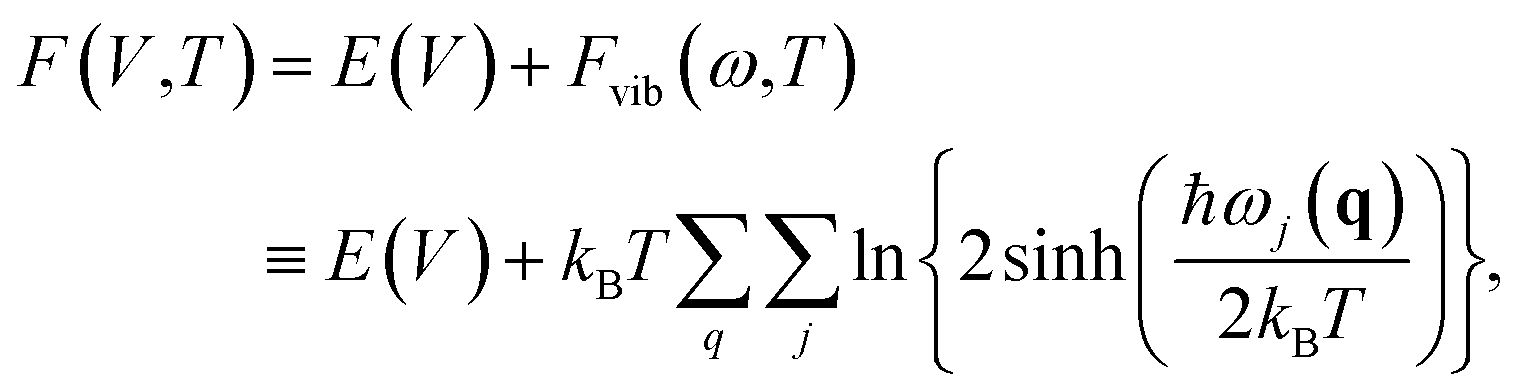
γj(q) is the mode Grüneisen parameter corresponding to the (q,j) phonon mode, defined as

The γj(q) is a primary factor for describing the contribution of a phonon mode to volumetric expansion or shrinkage. We calculate the phonon dispersion relations at different densities and approximate the derivative by the ratio of the differences to explore the contribution of the specific phonon branches. The negative values of Grüneisen parameters are unusual and mean that the frequencies ωj(q) of phonon increase (or ‘harden’) with increasing volume, are conducive to the occurrence of abnormal physical property.
ε[ωj(q)] is the mean vibrational energy of the (q,j) phonon given by
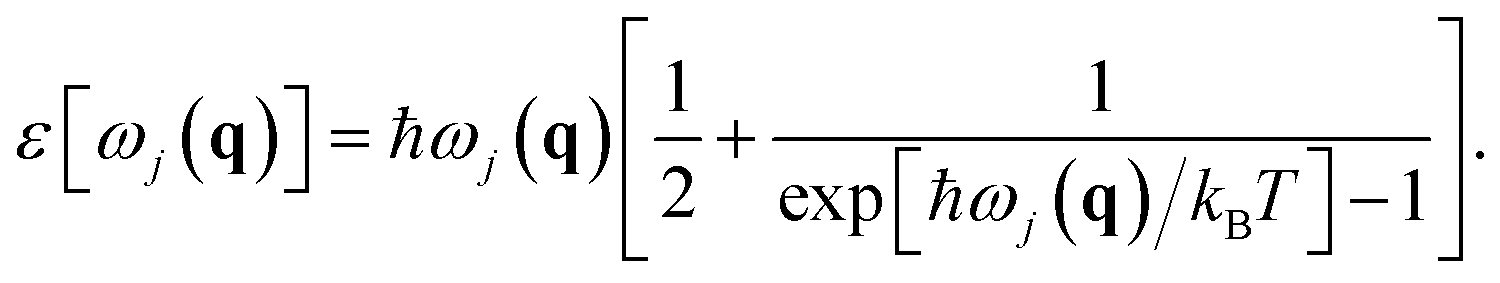
Having the equation of state (EOS), we can calculate the thermal expansion and the bulk modulus as a function of temperature. The relationship between phonons and thermal expansion is established by
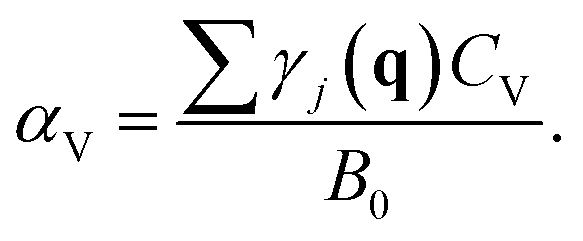
Here CV is the specific heat at constant volume and B0 is the bulk modulus. CV is given by
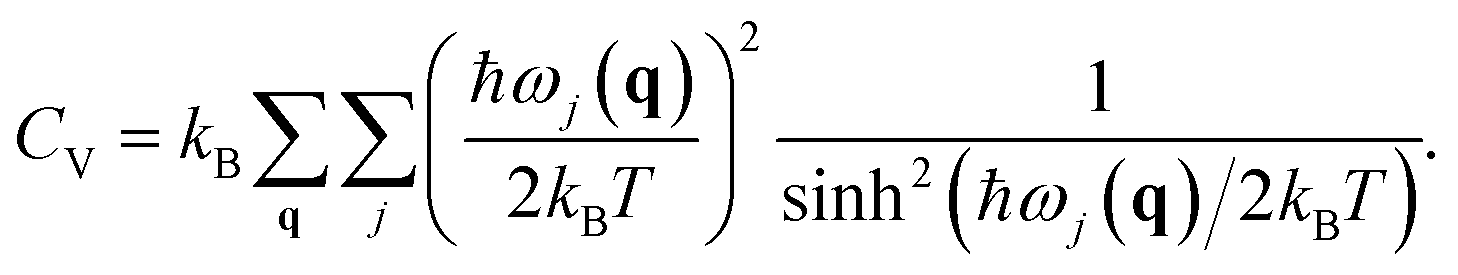
The temperature dependence of the B0 is obtained from

3. Results and discussion
3.1. The structures of quartz-like MOFs
In hexagonal Zn(ISN)2 and InH(BDC)2, their frameworks are composed of a set of tetrahedral nodes Zn or In cations, linked via molecular ISN or BDC anions to form doubly-interpenetrated quartz nets. Despite the same topology, both have contrasting geometries. Fig. 1(a) and (b) demonstrate the main peculiarity of the crystal structures of Zn(ISN)2 and InH(BDC)2: the way how bonding is formed. As seen from Fig. 1(a), Zn(ISN)2 with space group P31 adopts the similar characteristic of prototype α-quartz where Zn cations replace Si atoms in α-quartz, and long ISN anions replace O atoms. In contrast to the tetrahedral Zn-atom centers in Zn(ISN)2, InH(BDC)2 showing a highly distorted pseudo-tetrahedral geometry results in the feature structure of β-quartz (space group P6422), present in Fig. 1(b). In detail, each In-atom center in InH(BDC)2 connects to the other In-atom centers by linear terephthalate bridges. The resulting equilibrium lattice parameters of Zn(ISN)2 and InH(BDC)2 are listed in Table 1 compared with experimental data.14 There is a good agreement between the calculated results and experimental data.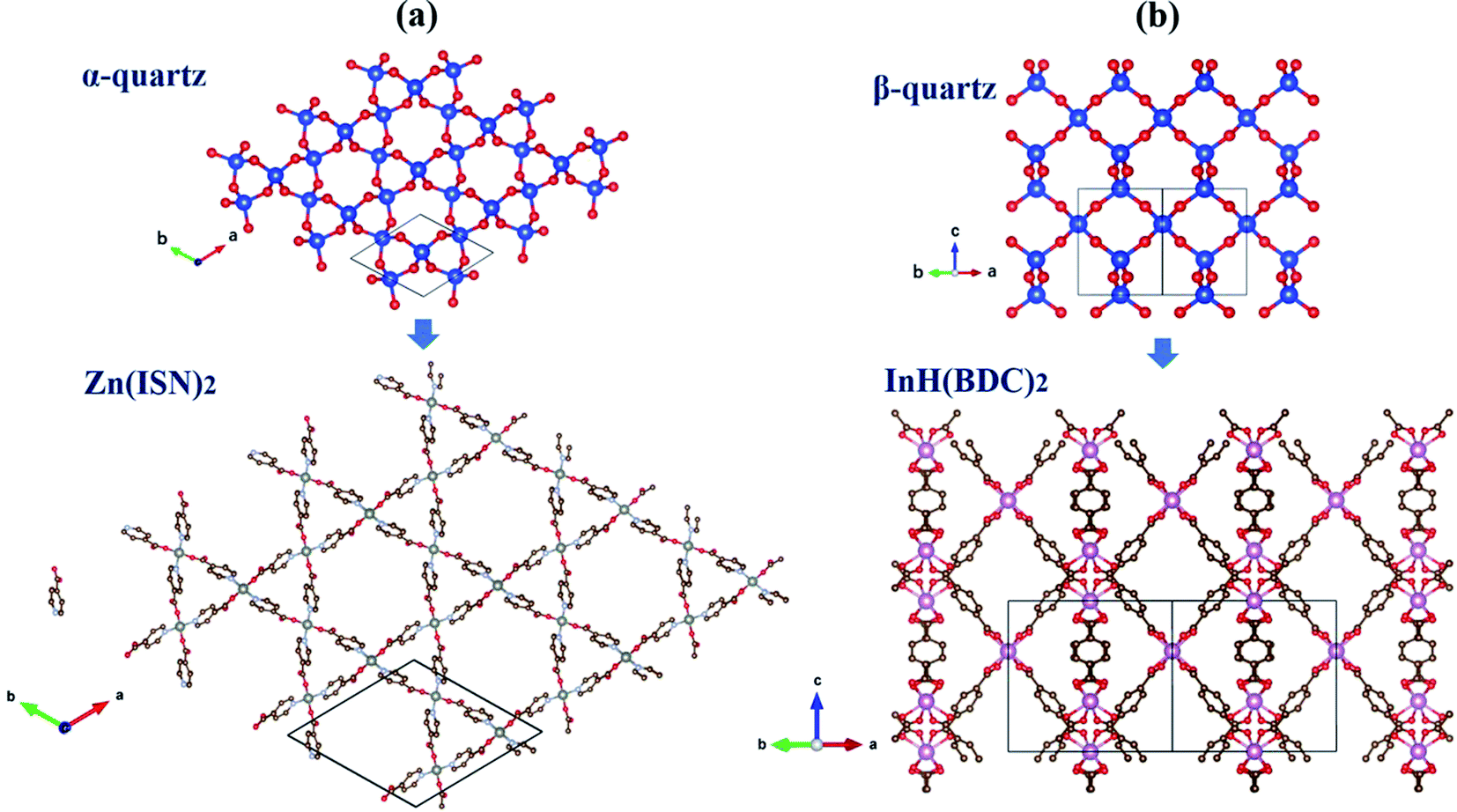 | ||
| Fig. 1 (a) Crystal frameworks (supercell based on 3 × 3 × 1 unit cells) of α-quartz, and Zn(ISN)2 with α-quartz-like topology in a view along the c-axis. (b) Crystal frameworks (supercell based on 2 × 2 × 2 unit cells) of β-quartz, and InH(BDC)2 with β-quartz-like topology in a view along [111]. H atoms are omitted for clarity. | ||
| Zn(ISN)2 | InH(BDC)2 | |||
|---|---|---|---|---|
| Cal. | Exp. | Cal. | Exp. | |
| a | 15.651 | 15.526 | 15.584 | 15.087 |
| b | 15.651 | 15.526 | 15.584 | 15.087 |
| c | 6.403 | 6.258 | 12.137 | 12.032 |
| V | 1358.21 | 1306.43 | 2553.02 | 2371.78 |
3.2. Uniaxial-NTE and area-NTE in quartz-like MOFs
As a whole, the Zn(ISN)2 and InH(BDC)2 show positive volume thermal expansion in the temperature range from 0 to 500 K as seen in Fig. 2(a). In our calculated results, Zn(ISN)2 has a bigger relative change in volume as about three times as InH(BDC)2. The relative changes in lattice parameters of Zn(ISN)2 and InH(BDC)2 upon heating are present in Fig. 2(b) and (c), respectively. Within the framework of QHA, our calculated values are in good agreement with the experimental data qualitatively.25 It is particularly evident in Zn(ISN)2 where the calculated area-NTE property along (a,b)-plane agrees quantitatively with the experimental data (α(a,b)-plane = −21.8 M K−1). In InH(BDC)2, uniaxial-NTE behavior along c-axis is confirmed by calculation (experimental value: αc = −35 M K−1). Relative changes along different axes of InH(BDC)2 in calculations are much smaller than experimental measurements, indicating InH(BDC)2 has so much stronger anharmonic interaction than Zn(ISN)2 that QHA can't work well. It should be noted that area-NTE is a rare phenomenon compared with uniaxial-NTE and volume-NTE. So far, only a few materials have been reported to possess this area-NTE property.36–41 It is necessary to analyze phonon anomalies from the perspective of lattice dynamics to expand the variety of this area-NTE material and explore the contrasting responses of Zn(ISN)2 and InH(BDC)2 to temperature changes.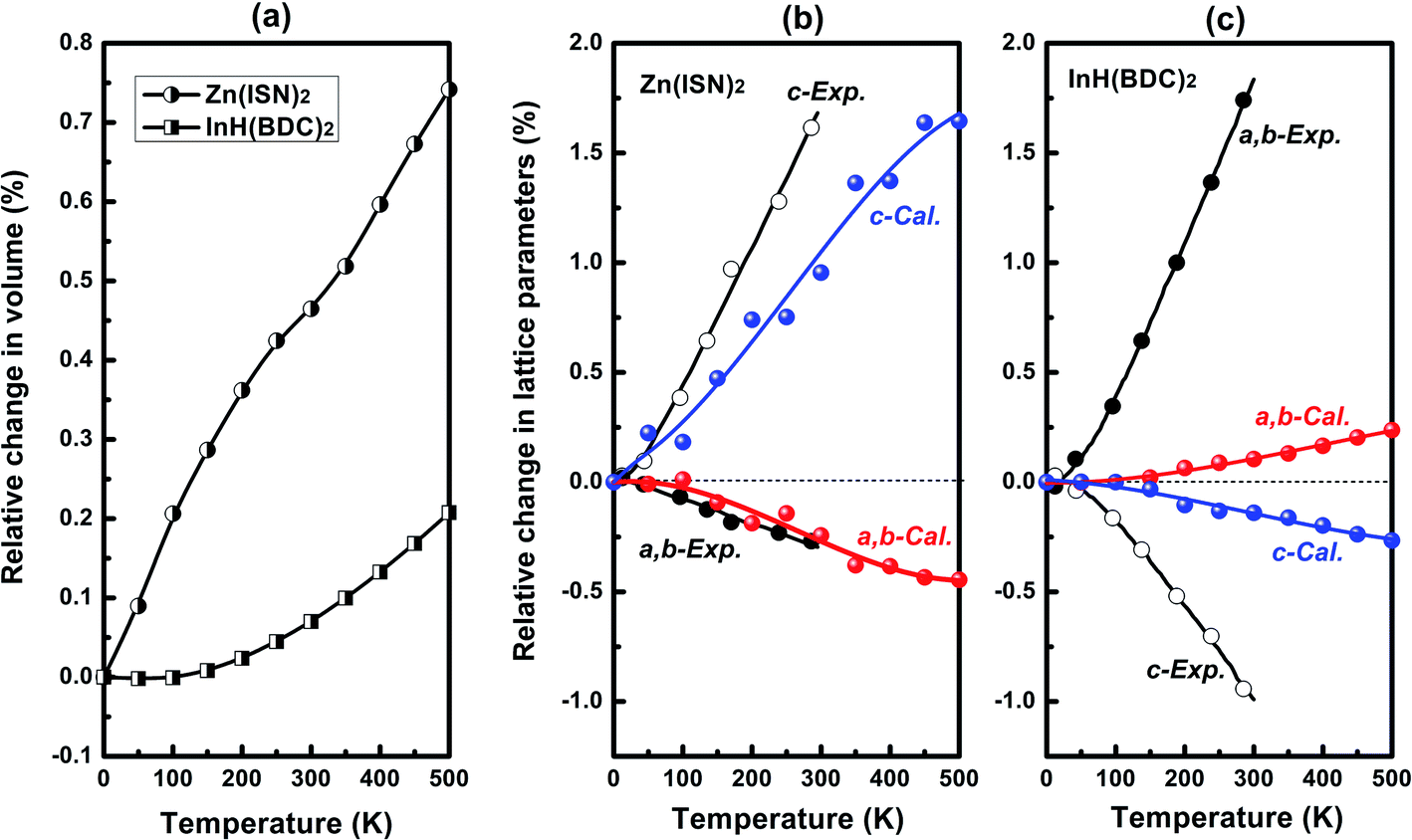 | ||
| Fig. 2 (a) Calculated relative change in volume of Zn(ISN)2 and InH(BDC)2 as a function of temperature. Calculated relative change in lattice parameters of (b) Zn(ISN)2 and (c) InH(BDC)2 with increasing temperature, compared with the experimental data.25 | ||
The Grüneisen parameters γj(q) along high-symmetry directions of Zn(ISN)2 and InH(BDC)2 are plotted in Fig. 3. The curves of Grüneisen parameters correspond to different phonons of frequencies, the magnitudes of which are represented by the depth of color. Different from other NTE materials where acoustic phonons play an important role in NTE behavior,5,42 it is found that low-frequency optical phonons contribute most to the area-NTE of Zn(ISN)2 or uniaxial-NTE of InH(BDC)2, and some high-frequency optical phonons are also associated with these abnormal properties. Fig. 3(a) and (b) have shown that compared with other ordinary phonons, low-frequency optical phonons in Zn(ISN)2 and InH(BDC)2 have strong negative Grüneisen parameters, while a few high-frequency phonons have slightly negative values. As far as we know, the case, that the joint contribution from low-frequency phonons coupled with high-frequency phonons leads to NTE, is also reflected in the A2M3O12 series.43
 | ||
| Fig. 3 (a) Calculated mode Grüneisen parameters along high-symmetry directions in Brillouin zone of (a) Zn(ISN)2 and (b) InH(BDC)2. The curves of mode Grüneisen parameters correspond to phonon modes of different frequencies which are represented by the depth of the color. | ||
To analyze the vibrational modes contributing to the area-NTE of Zn(ISN)2, the Eigen-displacement vectors of Zn(ISN)2 corresponding to optical phonons with the strongest negative Grüneisen parameter and the second-strongest negative Grüneisen parameter at G (0, 0, 0) point are explored and displayed in Fig. 4(a) and (b), respectively. Combining these two vibrational modes, we find that Zn atoms at the hinges are stationary and the amplitudes of O atoms are small, but those of C and N atoms are very strong. It is worth noting that pyridines composed of C and N atoms involve rotational vibration. Although the directions of the rotational vibrations are different in the two phonon modes, both can cause the wrinkles of the covalent chains of Zn–pyridine–Zn linkages and then lead to the shortening of the a-axis and b-axis in Zn(ISN)2 upon heated, i.e. the area-NTE of Zn(ISN)2. Moreover, the rotational vibrations of some pyridines are not along the directions of the chains of Zn–pyridine–Zn linkages but will be deflected, leading to the decreased angle θ between the a-axis and b-axis. The diagrammatic sketch of structural deformation caused by rotational vibration of pyridine is shown in Fig. 4(c).
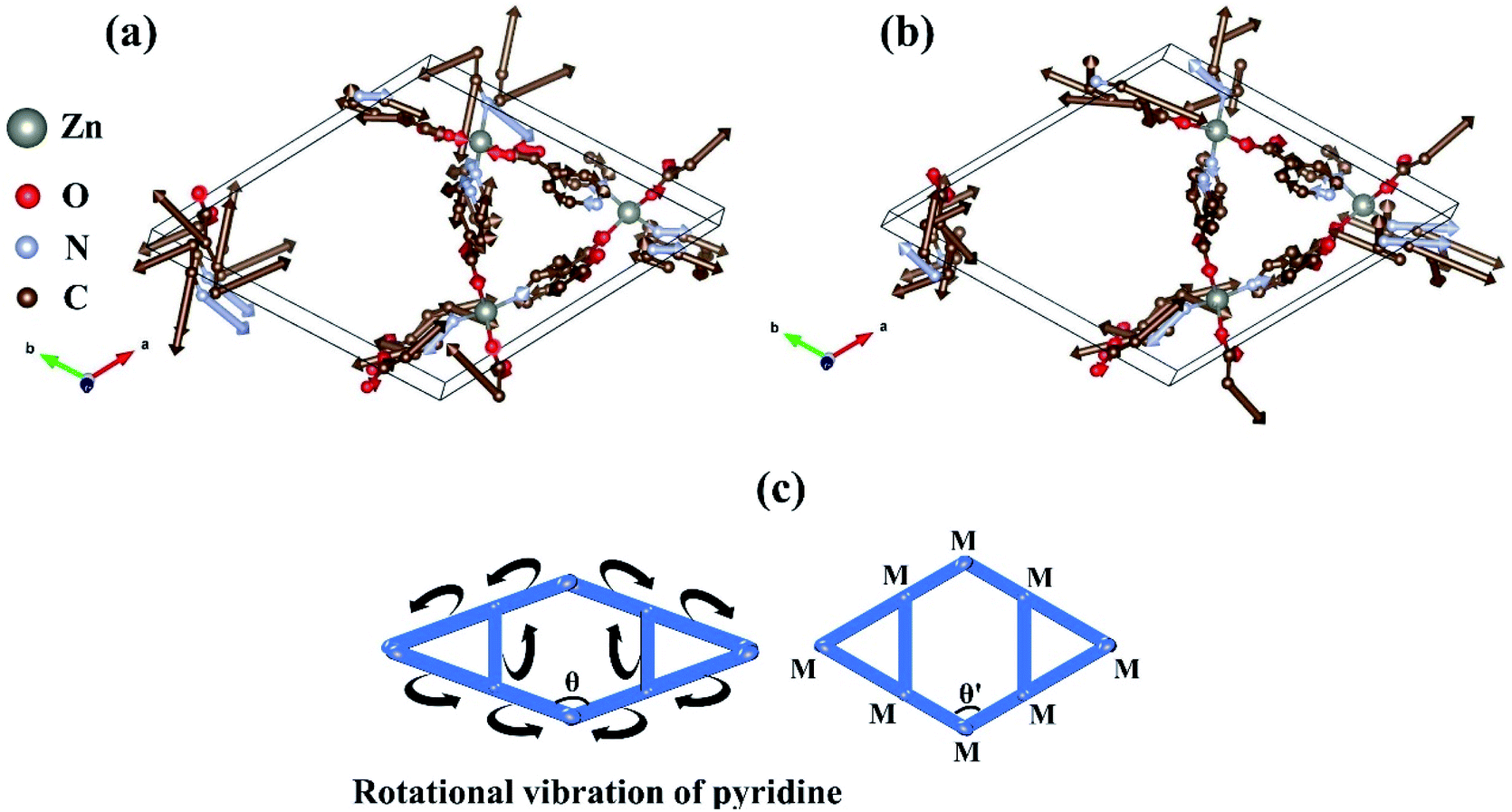 | ||
| Fig. 4 Eigen-displacement vectors of phonon modes with (a) the strongest negative Grüneisen parameter and (b) the second-strongest negative Grüneisen parameter at G (0, 0, 0) point, that are involved in the area-NTE of Zn(ISN)2. Arrows are proportional to the amplitude of the atomic motion. (c) Structural deformation diagram in which rotational vibration of pyridine as extracted feature of vibration mode according to the vector analysis of (a) and (b). M represents the metal (Zn) atom. H atoms are deleted since their vibrations are not considered here. | ||
Concerning the origin of uniaxial-NTE of InH(BDC)2, some specific Eigen-displacement vectors of phonon modes are analyzed. Fig. 5(a) presents the vibrational modes of InH(BDC)2 with the strongest negative Grüneisen parameter at G (0, 0, 0) point. It is noticed that the In atoms are stationary and the amplitudes of C atoms are very small. That means that the benzene ring has little contribution to the whole. However, the O atoms connected to In atoms have a very strong amplitude, so the vibrations of the O atoms become the key to the characteristics of this phonon mode. We extract this In–O local structure as a detailed analysis object shown in Fig. 5(b). Eight O atoms connected to one In atom have symmetrical directions of vibration. Such vibration vectors result in the transverse component forces that are opposite to each other in the (a,b)-plane, thus reducing the angle θ between the chains of In–O–Benzene–In linkages and (a,b)-plane. In this case, the c-axis of InH(BDC)2 is shortened. This vibrational mode plays a decisive role in the uniaxial-NTE of InH(BDC)2, and the corresponding structural deformation diagram is shown in Fig. 5(c). Another characteristic vibrational mode of InH(BDC)2 with the second-strongest negative Grüneisen parameter at G (0, 0, 0) point is exhibited in Fig. 5(d). It is seen that In atoms at the hinges act and have strong amplitudes, while the amplitudes of O atoms are not strong and have the opposite vibrational directions to In atoms. The C atoms have various amplitudes and vibrational directions, which makes the benzene ring have distorted vibration and the chains of In–O–Benzene–In linkages will be slightly bent. The local structure of the In–O–Benzene ring is stripped from the crystal and shown in Fig. 5(e). The opposite vibration direction of In atoms and other nonmetal atoms shortens the distance between the chains of In–O–Benzene–In linkages and (a,b)-plane, which leads to the contraction of the c-axis. The corresponding deformation schematic of crystal structure in InH(BDC)2 is displayed in Fig. 5(f), where the slight deformation of the benzene ring is considered as quasi-rigid here.43
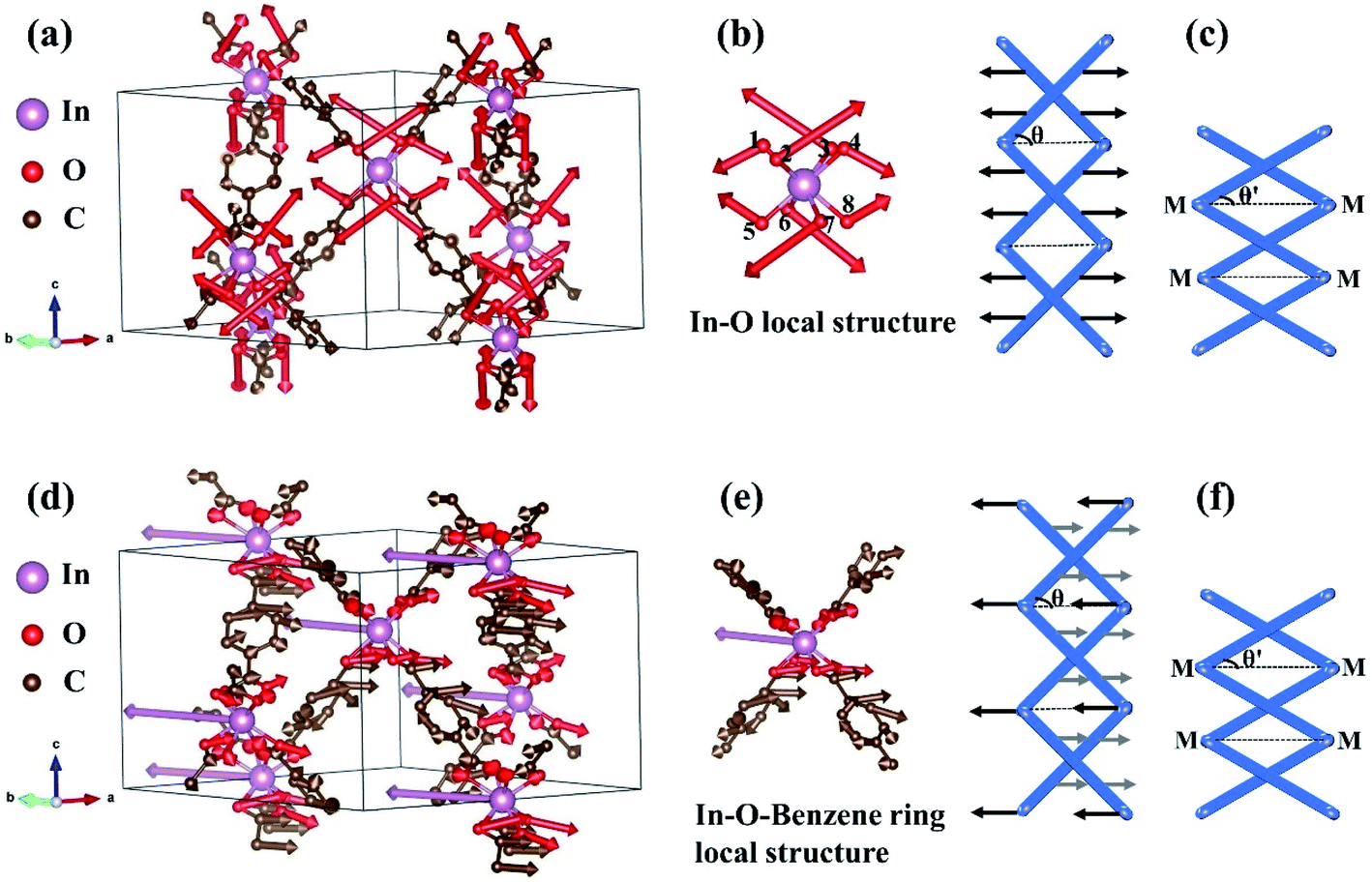 | ||
| Fig. 5 (a) Eigen-displacement vectors of phonon modes in InH(BDC)2 with the strongest negative Grüneisen parameter at G (0, 0, 0) point. The corresponding vibrational modes of In–O local structure and schematic representations of the deformations of crystal structure in InH(BDC)2 are present in (b) and (c), respectively. (d) Eigen-displacement vectors of phonon modes in InH(BDC)2 with the second-strongest negative Grüneisen parameter at G (0, 0, 0) point. (e) and (f) show the corresponding vibrational modes of In–O–Benzene ring local structure, and schematic of the deformations of crystal structure in InH(BDC)2, respectively. M represents the metal (In) atom. H atoms are deleted since their vibrations are not considered here. | ||
3.3. Bulk modulus anomaly in α-quartz-like MOFs
Adequate knowledge of the temperature dependence of the bulk modulus is necessary for understanding the thermoelastic and anharmonic properties of crystals. Elasticity provides more information than the volume in understanding temperature dependence of the equation of state (EOS) because the compressibility or bulk modulus is defined by the derivative of the volume. Thus, the bulk modulus is more sensitive to the variation in EOS than volume. The temperature dependence of bulk modulus of Zn(ISN)2 and InH(BDC)2 are calculated and shown in Fig. 6(a) and (b), respectively. At 0 K, the bulk modulus of Zn(ISN)2 is about 60 GPa as much as five times that of InH(BDC)2, showing Zn(ISN)2 has a natural stronger interatomic force than InH(BDC)2. Normally, the changing trend of bulk modulus in the external field (temperature or pressure) is inversely dependent on that of volume. It follows that the bulk modulus decreases (increases) with the increase of temperature in PTE materials (volume-NTE materials). For instance, a typical isotropic MOF-5 compound with volume-NTE exhibits that the bulk modulus increases upon heating.44 Concerning Zn(ISN)2 and InH(BDC)2, they show a completely different trend of bulk modulus with the increase of temperature. It is noted that the bulk modulus anomaly occurs in Zn(ISN)2 where the changing trend of the bulk modulus is consistent abnormally with that of volume when heated.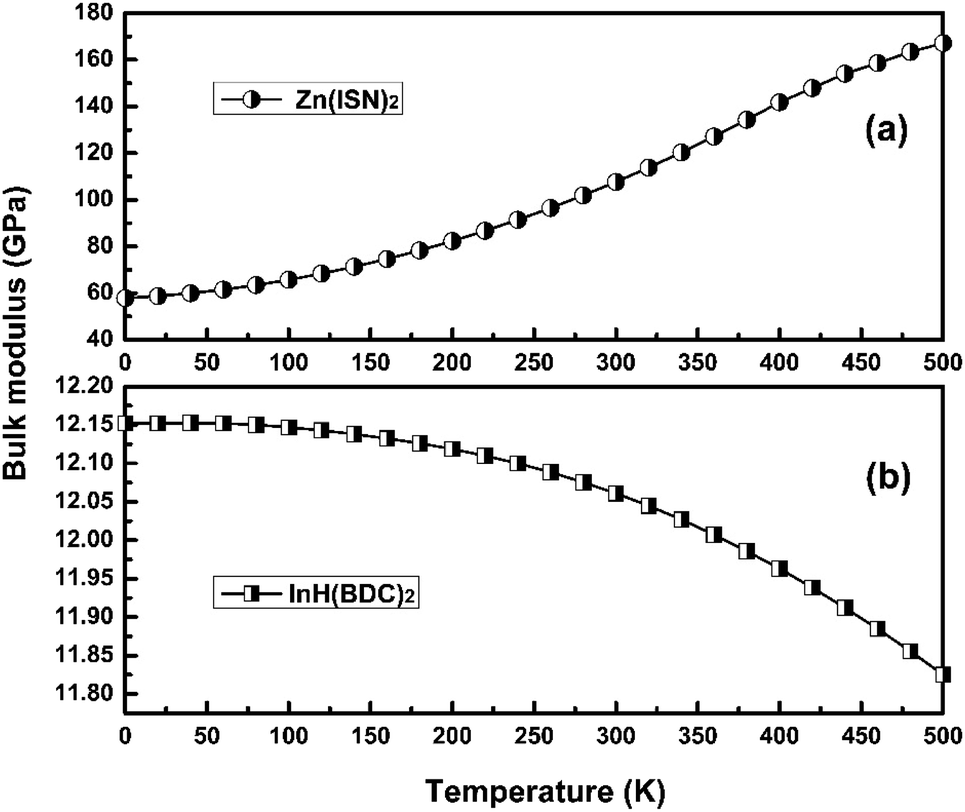 | ||
| Fig. 6 The temperature dependence of bulk modulus of (a) Zn(ISN)2 and (b) InH(BDC)2. | ||
From theoretical considerations, an equation is derived by Anderson in the fundamental constant of a solid:45

The isothermal Anderson–Grüneisen parameter δT, a parameter to estimate the abnormal temperature dependence of bulk modulus, is defined as:
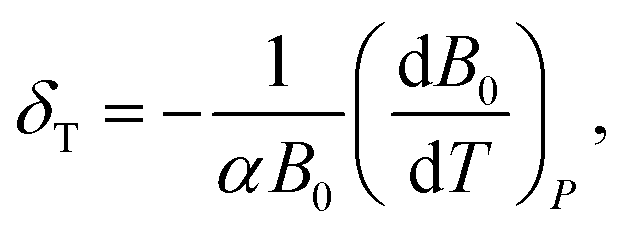
The Anderson–Grüneisen parameter δT, similar in many respects to the Grüneisen parameter γ, yields important information about the interatomic forces and enables us to predict the macroscopic behavior of different thermal properties for which, at present, experimental data or theory alone is inadequate. Commonly, δT is positive, indicating a normal temperature dependence of bulk modulus. If δT is negative, it will show the abnormal relationship between bulk modulus and temperature. The negative δT of Zn(ISN)2 and the positive δT of InH(BDC)2 can be obtained by the simple symbolic estimation of the formula. The interatomic forces in Zn(ISN)2 with α-quartz-like topology are gradually increasing, indicating Zn(ISN)2 is not easily compressional when heated. Here, it can be predicted that the bulk modulus anomaly is more likely to occur in some other MOFs with α-quartz-like topology.
3.4. Volume-NTE in diamond-like MOFs
Another typical structural motif of MOFs is the diamond-like topology, by which MOFs access a variety of geometries. A series of MOFs with diamond-like topology are selected and calculated to explore possible anomalous NTE properties. Finally, we lock in four MOFs with volume-NTE behavior, which are Ag(4-cnpy)2 (space group: Pbca), Co(5-X-2-pymo)2 (space group: I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d), Cu(2,5-Me2pyz)2 (space group: P21/n), and Zn(PT)2 (space group: P21/n).27–30 The crystal frameworks of these four MOFs are shown in Fig. 7(a)–(d), respectively. Although these MOFs with diamond-like topology have volume-NTE behavior, their temperature ranges of volume-NTE are quite different. It is exhibited in Fig. 8 that the volume-NTE of Ag(4-cnpy)2 and Co(5-X-2-pymo)2 can be extended to 1000 K, while that of Cu(2,5-Me2pyz)2 and Zn(PT)2 only exist in the region of 0–500 K. Moreover, the volume-NTE of Ag(4-cnpy)2 and Co(5-X-2-pymo)2 is much stronger than that of Cu(2,5-Me2pyz)2 and Zn(PT)2. Previous literature has speculated that these materials are potential area-NTE/NAC or uniaxial-NTE/NLC materials,25 but through our study of their axial behavior, we find that these materials only show volume anomalies, not axial anomalies. This still needs further experimental verification.
2d), Cu(2,5-Me2pyz)2 (space group: P21/n), and Zn(PT)2 (space group: P21/n).27–30 The crystal frameworks of these four MOFs are shown in Fig. 7(a)–(d), respectively. Although these MOFs with diamond-like topology have volume-NTE behavior, their temperature ranges of volume-NTE are quite different. It is exhibited in Fig. 8 that the volume-NTE of Ag(4-cnpy)2 and Co(5-X-2-pymo)2 can be extended to 1000 K, while that of Cu(2,5-Me2pyz)2 and Zn(PT)2 only exist in the region of 0–500 K. Moreover, the volume-NTE of Ag(4-cnpy)2 and Co(5-X-2-pymo)2 is much stronger than that of Cu(2,5-Me2pyz)2 and Zn(PT)2. Previous literature has speculated that these materials are potential area-NTE/NAC or uniaxial-NTE/NLC materials,25 but through our study of their axial behavior, we find that these materials only show volume anomalies, not axial anomalies. This still needs further experimental verification.
 | ||
| Fig. 7 The crystal framework structures (supercell based on 2 × 2 × 2 unit cells) of diamond-like MOFs of (a) Ag(4-cnpy)2, (b) Co(5-X-2-pymo)2, (c) Cu(2,5-Me2pyz)2, and (d) Zn(PT)2. H atoms are omitted for clarity. Abbreviations used: 4-cnpy = 4-cyanopyridine, 5-X-2-pymo = 5-halo-pyrimidin-2-ol (X = Be/I), 2,5-Me2pyz = 2,5-dimethylpyrazine, PT = 5-propyltetrazole. | ||
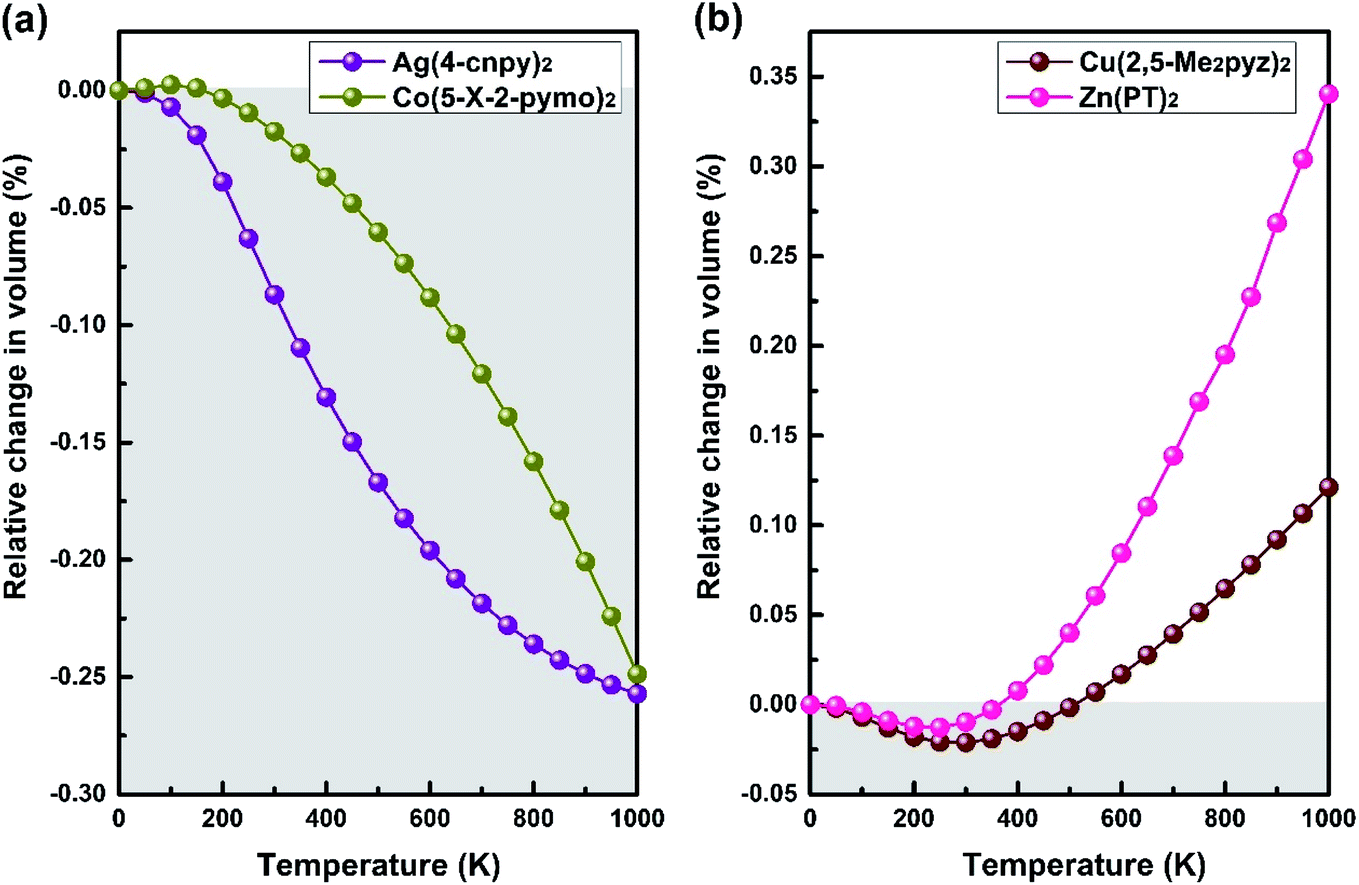 | ||
| Fig. 8 (a) Calculated relative change in volume of Ag(4-cnpy)2 and Co(5-X-2-pymo)2, which exhibit the volume-NTE up to 1000 K. (b) Calculated relative change in volume of Cu(2,5-Me2pyz)2 and Zn(PT)2, whose volume-NTE behaviors exist in the region of 0–500 K. | ||
So far, various abnormal behaviors from uniaxial-NTE, area-NTE to volume-NTE in MOFs have been achieved based on quartz-like and diamond-like topologies. Moreover, in the preparation of MOFs with these specific topologies, the flexibility of the framework can be further adjusted by changing the length of the internal organic chain, thus realizing the regulation of the size of various NTE behaviors.5
3.5. Bulk-modulus anomaly in diamond-like MOFs
The temperature dependence of bulk modulus in these MOFs with the diamond-like framework is also studied. It is found that the bulk modulus anomaly will still appear. In Fig. 9(a), the bulk modulus of Ag(4-cnpy)2 will increase with increased temperature. This is normal elastic behavior, that is, the changing trend of the bulk modulus is inversely dependent on that of volume. The volume of Ag(4-cnpy)2 decreases with the increase of temperature, and then the interatomic force increases, and the bulk modulus finally shows an increasing trend. Above 500 K, the PTE behavior of Cu(2,5-Me2pyz)2 begins to appear as seen in Fig. 8(b), but its bulk modulus has no inflection point and continues to increase with the increase of temperature in Fig. 9(b), showing the abnormal bulk modulus in the high-temperature region. About Zn(PT)2 and Co(5-X-2-pymo)2, both of them exhibit bulk-modulus anomaly within the temperature range studied in Fig. 9(c) and (d), where the bulk modulus – temperature relationship is consistent anomalously with the volume – temperature relationship. In other NTE materials, abnormal elastic properties are rare, but in MOFs, it seems that elastic properties anomaly and abnormal NTE behaviors are more likely to be accompanied. These specific topologies (quartz-like, diamond-like, and so on) provide an important geometric framework for various NTE behaviors of MOFs, but at the same time, the local structure and bond environment in MOFs may lead to other abnormal properties, such as bulk-modulus anomaly. If the bond linkage is close to linear, it will cause the normal changing trend of interatomic force in the external field. If there is an angle between the links of these bonds, the changing trend of abnormal interatomic forces will probably appear, namely, a variety of abnormal elastic properties are possible. Here, we also hope that this elastic property of MOFs can attract more attention and exploration. | ||
| Fig. 9 The temperature dependence of bulk modulus of (a) Ag(4-cnpy)2, (b) Cu(2,5-Me2pyz)2, (c) Zn(PT)2, and (d) Co(5-X-2-pymo)2. | ||
4. Conclusion
In conclusion, various NTE behaviors from the uniaxial, area, and volume-NTE can be realized in MOFs with quartz-like and diamond-like frameworks. The area-NTE of Zn(ISN)2 and uniaxial-NTE of InH(BDC) have been studied. Their thermal behaviors in calculations agree well with the experimental data. According to the calculation of Grüneisen parameters, low-frequency optical phonons contribute most to area-NTE of Zn(ISN)2 and uniaxial-NTE of InH(BDC). It is worth noting that some high-frequency phonons also play a role. The vibrational eigenvectors of some key phonons are analyzed in detail. The rotational vibration of pyridine in Zn(ISN)2, and transverse vibration of In–O local structure and In–O–Benzene ring local structure in InH(BDC) are identified. Furthermore, four MOFs with the diamond-like framework are predicted to possess volume-NTE behavior. It still needs to be verified by experiments. In some studied MOFs, the abnormal bulk modulus with increasing temperature is found. Although these specific topologies provide an important geometric framework for various NTE behaviors of MOFs, the local structure and bond environment may be associated with the abnormal interatomic force, leading to the bulk modulus anomaly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC) (No. 51732001), Fundamental Research Funds for the Central Universities (No. FRF-TP-20-041A2). The authors would like to express their sincere thanks to the Center for Computational Materials Science of the Institute for Materials Research, Tohoku University for their support of the supercomputing facilities.References
- M. T. Dove and H. Fang, Negative thermal expansion and associated anomalous physical properties: review of the lattice dynamics theoretical foundation, Rep. Prog. Phys., 2016, 79, 066503 CrossRef PubMed.
- C. Wang, L. Chu, Q. Yao, Y. Sun, M. Wu, L. Ding, J. Yan, Y. Na, W. Tang and G. Li, Tuning the range, magnitude, and sign of the thermal expansion in intermetallic Mn3(Zn, M)xN (M=Ag, Ge), Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 220103 CrossRef.
- J. Chen, X. Xing, C. Sun, P. Hu, R. Yu, X. Wang and L. Li, Zero thermal expansion in PbTiO3-based perovskites, J. Am. Chem. Soc., 2008, 130, 1144–1145 CrossRef CAS PubMed.
- K. Takenaka, Negative thermal expansion materials: technological key for control of thermal expansion, Sci. Technol. Adv. Mater., 2012, 13, 013001 CrossRef PubMed.
- L. Wang, M. T. Dove, J. Shi, B. Sun, D. Hu and C. Wang, Adjustable uniaxial zero thermal expansion and zero linear compressibility in unique hybrid semiconductors: the role of the organic chain, Dalton Trans., 2020, 49, 719–728 RSC.
- L. Wang and C. Wang, Negative/zero thermal expansion in black phosphorus nanotubes, Phys. Chem. Chem. Phys., 2018, 20, 28726–28731 RSC.
- G. Barrera, J. Bruno, T. Barron and N. Allan, Negative thermal expansion, J. Phys.: Condens. Matter, 2005, 17, R217–R252 CrossRef CAS.
- K. Wang, M. Feng, L. Zhou, J. Li, X. Qi and X. Huang, A hybrid with uniaxial negative thermal expansion behaviour: the synergistic role of organic and inorganic components, Chem. Commun., 2014, 50, 14960–14963 RSC.
- J. Li, W. Bi, W. Ki, X. Huang and S. Reddy, Nanostructured crystals: unique hybrid semiconductors exhibiting nearly zero and tunable uniaxial thermal expansion behavior, J. Am. Chem. Soc., 2007, 129, 14140–14141 CrossRef CAS PubMed.
- G. Artioli, M. Dapiaggi, P. Fornasini, A. Sanson, F. Rocca and M. Merli, Negative thermal expansion in cuprite-type compounds: a combined synchrotron XRPD, EXAFS, and computational study of Cu2O and Ag2O, J. Phys. Chem. Solids, 2006, 67, 1918–1922 CrossRef CAS.
- T. Chatterji, M. Zbiri and T. C. Hansen, Negative thermal expansion in ZnF2, Appl. Phys. Lett., 2011, 98, 181911 CrossRef.
- L. Wang, P. Yuan, F. Wang, Q. Sun, Z. Guo, E. Liang and Y. Jia, First-principles investigation of negative thermal expansion in II-VI semiconductors, Mater. Chem. Phys., 2014, 148, 214–222 CrossRef CAS.
- P. Deus and H. Schneider, Thermal expansion of the diamond-like semiconducting compound ZnSiP2 within the temperature range 20 to 300 K, Phys. Status Solidi A, 1983, 79, 411–415 CrossRef CAS.
- J. Sun, L. Weng, Y. Zhou, J. Chen, Z. Chen, Z. Liu and D. Zhao, QMOF-1 and QMOF-2: three-dimensional metal–organic open frameworks with a quartzlike topology, Angew. Chem., Int. Ed., 2002, 41, 4471–4473 CrossRef CAS PubMed.
- F. Hohn, I. Pantenburg and U. Ruschewitz, Sr[C2(COO)2]: the first anhydrous salt of acetylenedicarboxylic acid, Chem.–Eur. J., 2002, 8, 4536–4541 CrossRef CAS PubMed.
- G. A. Lager, J. Jorgensen and F. Rotella, Crystal structure and thermal expansion of α-quartz SiO2 at low temperatures, J. Appl. Phys., 1982, 53, 6751–6756 CrossRef CAS.
- P. Welche, V. Heine and M. Dove, Negative thermal expansion in beta-quartz, Phys. Chem. Miner., 1998, 26, 63–77 CrossRef CAS.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Colossal positive and negative thermal expansion in the framework material Ag3[Co(CN)6], Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- A. L. Goodwin, D. A. Keen and M. G. Tucker, Large negative linear compressibility of Ag3[Co(CN)6], Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18708–18713 CrossRef CAS PubMed.
- L. Wang, C. Wang, H. Luo and Y. Sun, The correlation between uniaxial negative thermal expansion and negative linear compressibility in Ag3[Co(CN)6], J. Phys. Chem. C, 2017, 121, 333–341 CrossRef CAS.
- A. L. Goodwin, B. J. Kennedy and C. J. Kepert, Thermal expansion matching via framework flexibility in zinc dicyanometallates, J. Am. Chem. Soc., 2009, 131, 6334–6335 CrossRef CAS PubMed.
- A. B. Cairns, J. Catafesta, C. Levelut, J. Rouquette, A. Van Der Lee, L. Peters, A. L. Thompson, V. Dmitriev, J. Haines and A. L. Goodwin, Giant negative linear compressibility in zinc dicyanoaurate, Nat. Mater., 2013, 12, 212–216 CrossRef CAS PubMed.
- L. Wang, H. Luo, S. Deng, Y. Sun and C. Wang, Uniaxial negative thermal expansion, negative linear compressibility, and negative Poisson's ratio induced by specific topology in Zn[Au(CN)2]2, Inorg. Chem., 2017, 56, 15101–15109 CrossRef CAS PubMed.
- S. L. James, Metal-organic frameworks, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- I. E. Collings, M. G. Tucker, D. A. Keen and A. L. Goodwin, Geometric switching of linear to area negative thermal expansion in uniaxial metal–organic frameworks, CrystEngComm, 2014, 16, 3498–3506 RSC.
- Q. Zeng, K. Wang and B. Zou, Large negative linear compressibility in InH(BDC)2 from framework hinging, J. Am. Chem. Soc., 2017, 139, 15648–15651 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani, D. M. Proserpio and A. Sironi, Interpenetrating diamondoid frameworks of silver (I) cations linked by N,N′-bidentate molecular rods, J. Chem. Soc., Chem. Commun., 1994, 24, 2755–2756 RSC.
- T. Otieno, S. J. Rettig, R. C. Thompson and J. Trotter, Complex polymeric cations of copper (I) with graphite-and diamond-related lattices; crystal structures of poly-tris (. mu.-2, 5-dimethylpyrazine) dicopper (I) hexafluorophosphate and poly-bis (. mu.-2, 5-dimethylpyrazine) copper (I) hexafluorophosphate, Inorg. Chem., 1993, 32, 1607–1611 CrossRef CAS.
- L. Ma, Y. C. Qiu, G. Peng, J. B. Cai and H. Deng, In situ tetrazole ligand synthesis of two-fold interpenetrating zinc coordination frameworks, Eur. J. Inorg. Chem., 2011, 23, 3446–3453 CrossRef.
- N. Masciocchi, S. Galli, A. Sironi, E. Cariati, M. A. Galindo, E. Barea, M. A. Romero, J. M. Salas, J. A. Navarro and F. Santoyo-González, Tuning the structural and magnetic properties of thermally robust coordination polymers, Inorg. Chem., 2006, 45, 7612–7620 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- A. Togo, F. Oba and I. Tanaka, First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- L. Wang, C. Wang, Y. Sun, S. Deng, K. Shi, H. Lu, P. Hu and X. Zhang, First-principles study of Sc1−xTixF3 (x ≤ 0.375): negative thermal expansion, phase transition, and compressibility, J. Am. Ceram. Soc., 2015, 98, 2852–2857 CrossRef CAS.
- L. Wang, P. Yuan, F. Wang, Q. Sun, E. Liang, Y. Jia and Z. Guo, Negative thermal expansion in TiF3 from the first-principles prediction, Phys. Lett. A, 2014, 378, 2906–2909 CrossRef CAS.
- I. Grobler, V. J. Smith, P. M. Bhatt, S. A. Herbert and L. J. Barbour, Tunable anisotropic thermal expansion of a porous zinc (II) metal–organic framework, J. Am. Chem. Soc., 2013, 135, 6411–6414 CrossRef CAS PubMed.
- D. Das, T. Jacobs and L. J. Barbour, Exceptionally large positive and negative anisotropic thermal expansion of an organic crystalline material, Nat. Mater., 2010, 9, 36–39 CrossRef CAS PubMed.
- M. J. Cliffe and A. L. Goodwin, PASCal: a principal axis strain calculator for thermal expansion and compressibility determination, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- S. J. Hibble, A. M. Chippindale, A. H. Pohl and A. C. Hannon, Surprises from a simple material-the structure and properties of nickel cyanide, Angew. Chem., Int. Ed., 2007, 119, 7246–7248 CrossRef.
- S. A. Hodgson, J. Adamson, S. J. Hunt, M. J. Cliffe, A. B. Cairns, A. L. Thompson, M. G. Tucker, N. P. Funnell and A. L. Goodwin, Negative area compressibility in silver (I) tricyanomethanide, Chem. Commun., 2014, 50, 5264–5266 RSC.
- M. Y. Seyidov and R. A. Suleymanov, Negative thermal expansion due to negative area compressibility in TlGaSe2 semiconductor with layered crystalline structure, J. Appl. Phys., 2010, 108, 063540 CrossRef.
- L. Wang, C. Wang and Y. Chen, Black phosphorene exhibiting negative thermal expansion and negative linear compressibility, J. Phys.: Condens. Matter, 2019, 31, 465003 CrossRef CAS PubMed.
- L. Wang, F. Wang, P. Yuan, Q. Sun, E. Liang, Y. Jia and Z. Guo, Negative thermal expansion correlated with polyhedral movements and distortions in orthorhombic Y2Mo3O12, Mater. Res. Bull., 2013, 48, 2724–2729 CrossRef CAS.
- L. Wang, C. Wang, Y. Sun, K. Shi, S. Deng and H. Lu, Large negative thermal expansion provided by metal-organic framework MOF-5: a first-principles study, Mater. Chem. Phys., 2016, 175, 138–145 CrossRef CAS.
- M. Madan, Temperature dependence of the bulk modulus of alkali halides, J. Appl. Phys., 1971, 42, 3888–3893 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |