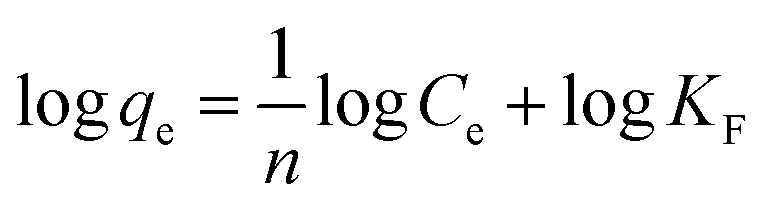DOI:
10.1039/D2RA02703H
(Paper)
RSC Adv., 2022,
12, 21780-21792
Boron doping positively enhances the catalytic activity of carbon materials for the removal of bisphenol A†
Received
28th April 2022
, Accepted 12th July 2022
First published on 8th August 2022
Abstract
Boron-doped carbon materials (BCs), low-cost and environmentally friendly carbocatalysts, were prepared for the activation of persulfate (PS) for the removal of bisphenol A (BPA). Compared with B-free carbon materials (Cs), the adsorption and catalytic activity were significantly enhanced by the boron modification. Fast and efficient removal of BPA was achieved using the BCs/PS system. The BPA removal rate constant increased linearly with the adsorption capacity of BCs. Electron paramagnetic resonance (EPR) spectroscopy and radical quenching experiments indicated that the degradation mechanisms in the BCs/PS system were different from conventional radical-based oxidation pathways. On the contrary, nonradical pathways were demonstrated to dominate the oxidation processes in the removal of BPA using the BCs/PS system. Herein, a mechanism is proposed where PS is activated by the carbon material to form a reactive electron-deficient carbocatalyst ([BCs]*) complex with a high redox potential, driving a nonradical oxidation pathway to achieve BPA removal. Through experimental investigation and the use of electrochemical techniques (cyclic voltammetry, Tafel corrosion analysis and open circuit voltages), B-doped carbon materials for the activation of PS elevate the potential of the derived nonradical [BCs]* complexes, and then accelerate the BPA removal efficiency via an electron transfer process. Utilizing adsorption and nonradical oxidation processes, the BCs/PS system possesses great potential for the removal of BPA in practical applications such as wastewater treatment.
1. Introduction
Bisphenol A (BPA) is an industrial chemical primarily used in the manufacture of polycarbonate plastics and epoxy resins, as well as in common consumer products such as bottles, binders, pipes and electronics.1,2 It is found ubiquitously in the environment and may give rise to the pollution in various fields due to its endocrine disrupting properties and from human exposure via dietary and nondietary routes3 causing organ damage.4 Conventional wastewater treatment techniques are not capable of removing BPA from untreated water and thus, researchers have been developing new technologies for the reduction of these micropollutants.5,6 Nowadays, advanced oxidation processes (AOPs), originally defined as techniques involving the generation and participation of hydroxyl radicals (˙OH, standard reduction potential of 2.8 V), are seen as effective and thorough strategies for the removal of organic contaminants due to their superior oxidation efficiencies.7,8 In particular, peroxymonosulfate (PMS, HSO5−) and persulfate (PS, S2O82−) are typical oxidizing agents that have been used recently to degrade organic pollutants by various activation methods, such as heat,9 ultrasound,10 addition of a base,11 ultraviolet light,12 and the use of transition metals13 to produce sulfate radicals (SO4˙−, standard reduction potential of 2.5–3.1 V), which are alternatives to ˙OH.
Carbon-based materials, such as reduced graphene oxide (rGO),14,15 carbon nanotubes (CNTs),16,17 carbon nanofibers (CNFs),18 and nanodiamond19,20 have been reported to be efficient for PS activation. This leads to the enhanced degradation of micropollutants without secondary pollution because this creates a material with an abundance of oxygen-containing functional groups, high electrical conductivity, large surface area and large pore volumes. Although PS, an inexpensive and efficient oxidant, can be activated via carbon-based materials, these materials still require multiple-step fabrication processes that use harsh treatments and have high costs.21
Heteroatom-doped carbon-based materials have been successfully applied for PS activation to enhance the oxygen reduction reaction (ORR) and catalytic activities toward organic micropollutant degradation via the modulation of charge electronic neutral fractures.5,8,21–24 It was reported that, compared to undoped and boron-doped carbon nanoparticles, boron/fluorine dual-doped carbon nanoparticles drove the ORR through a two-electron pathway with low generation of the unstable peroxide, HO2−.25 Balaji et al. prepared a phosphorus-doped reduced graphene oxide (P-rGO) which delivered a better ORR performance (0.75 A mg−1) when compared with a commercial HiSPEC Pt/C (0.12 A mg−1) catalyst.26 Kaipannan et al. demonstrated that an activated carbon/ZnS (ACZS) nanocomposite electrode performed well as a supercapacitor and delivered a high specific capacitance of 241 F g−1 at 1 A g−1 current density.27 Kang et al. synthesized a porous boron-bearing carbon electrocatalyst doped with Fe and nitrogen (Fe-BNC), which showed a high surface area (1300 m2 g−1) and favored a 4-electron reduction pathway for efficient ORR.28
Of the non-metallic heteroatoms available for doping, B atoms are able to break the basic sp2-hybridized structure and create new active sites for PS activation.29 Furthermore, Yang et al. demonstrated that, when used as a metal-free electrocatalyst, B-doped CNTs (B-CNTs) exhibited an enhancement of the oxygen reduction reaction compared with pristine CNTs.30 Xie et al. prepared a porous interconnected 3D B-doped graphene aerogel (BGA) which could deliver a high capacity and showed outstanding rate capability for use in lithium–sulfur batteries.31 Wu et al. synthesized a B-doped graphene material which showed good performance for in situ metal-free electrochemical advanced oxidation of BPA.5 Therefore, B-doped carbon-based materials may contribute to effective AOP methods.
In this work, we prepared a B-doped carbon-based material via a simple hydrothermal process based on a mixture of coffee grounds, sodium bicarbonate and boric acid, which were all abundant, low-cost, and eco-friendly. The as-prepared BCs were adopted to activate PS for BPA degradation. In order to investigate the efficiency of BPA removal in the BCs/PS system, we explored the following aspects of the BCs/PS system. (i) The structural properties of the BCs as characterized using scanning electron microscopy (SEM) equipped with an energy dispersive X-ray spectrometer (EDX), Brunauer–Emmett–Teller (BET) analysis, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and Fourier-transform infrared spectroscopy (FT-IR). (ii) Elucidating the BCs/PS system mechanisms through experimental investigation and electrochemical techniques (including cyclic voltammetry (CV), Tafel corrosion analysis, and open circuit voltages). (iii) Studying the effects of some crucial parameters of the BCs/PS system with regard to BPA degradation, such as the dosage of the carbon material, initial pH and water matrices.
2. Methods and materials
2.1. Reagents and materials
Detailed information for materials and reagents are given in Text S1.†
2.2. Fabrication of carbon samples
Pre-BCs was prepared via a hydrothermal process as follows. 5 g of coffee grounds, 7 g of sodium bicarbonate and 2.1 g of boric acid were added to a beaker containing 300 mL of deionized (DI) water. The solution was magnetically stirred at 80 °C for 6 h. Then, the above mixture was dried at 80 °C for 12 h. Subsequently, the dried mixture was annealed at 700 °C for 2 h under a N2 atmosphere with a heating rate of 5 °C min−1. After naturally cooling to room temperature, the co-carbon sample was sequentially washed with DI water and ethanol and dried at 80 °C. Accordingly, the carbon samples were denoted as BCs. For comparison, a blank carbon sample was prepared by carbonization of 7 g of sodium bicarbonate and 5 g of coffee grounds at 700 °C, hereafter denoted as Cs.
2.3. Experimental procedures
Detailed information relating to the experimental procedures are shown in Text S2.†
2.4. Analytical methods
Conventional analytical methods are shown in Text S3.† Parameters for the EPR spin-trapping technique are shown in Text S4.† The concentrations of PS were determined using a spectrophotometric method (Text S5†). A CHI660E electrochemical workstation was used to conduct the electrochemical analysis (Text S6†).
3. Results and discussion
3.1. Catalytic performance of catalysts
As shown in Fig. 1, the removal of BPA using different carbon catalysts was conducted. It is notable that fullerene (C60) could barely remove BPA. BPA was rapidly removed by the BCs/PS system within 60 min, which was more efficient than the CNTs/PS system, commercial graphene (G)/PS, multi-layer graphene (MLG)/PS and Cs/PS, with these catalysts having removal efficiencies within 60 min of 33.6%, 12.6%, 48.8% and 66.6%, respectively. The apparent rate constants (kobs) for BPA removal using PS activated via various carbocatalysts follow the order: BCs (0.0710 min−1) > Cs (0.0344 min−1) > MLG (0.0342 min−1) > G (0.0103 min−1) > CNTs (0.0087 min−1) > C60 (0.0000 min−1) (Fig. 1(d)).
 |
| | Fig. 1 BPA removal efficiency via adsorption of various carbon catalysts (a), and their reaction rates within 10 min (b). ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [carbon catalysts]0 = 0.1 g L−1, initial pH = 3 ± 0.2). BPA removal efficiency via adsorption and PS oxidation of various carbon catalysts (c), and their reaction rates within 10 min (d) ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [carbon catalysts]0 = 0.1 g L−1, [PS]0 = 2 mM, initial pH = 4 ± 0.2). | |
3.2. Characterization
Under a N2 atmosphere, the thermogravimetric analysis (TGA) and differential thermogravimetry (DTG) were measured (Fig. S1†). For the coffee grounds/NaHCO3 system, a sharp weight loss between 800 and 900 °C was observed. However, the TGA and DTG curves in the coffee grounds/NaHCO3/boric acid system were different from those for the coffee grounds/NaHCO3 system. The rapid weight loss in the coffee grounds/NaHCO3/boric acid system only occurred at 25–150 °C, indicating that coffee grounds/NaHCO3/boric acid system was more stable than the coffee grounds/NaHCO3 system.
The SEM images demonstrated that the pristine Cs showed a honeycomb-like porous structure (Fig. 2(a) and (b)). Interestingly, the BCs had a coralloid-like shape after modification with boron (Fig. 2(c) and (d)). The surface pores of both the Cs and BCs were found to be mesopores. EDX elemental mapping images showed that the distribution of C, O and B elements within the carbon frameworks was uniform (Fig. 2(e)). The BET surface areas were measured using nitrogen physisorption. As illustrated in Table 1 and Fig. 3, after B doping, the specific surface area (SSA) of BCs increased from 211.712 (Cs) to 518.200 m2 g−1 and the porous structures of both Cs and BCs had pore sizes mainly in the range of mesopores (2–50 nm).
 |
| | Fig. 2 SEM images of Cs (a) and (b), and BCs (c) and (d); (e) SEM image of BCs with EDX elemental mapping. | |
Table 1 Pore structure of Cs and BCs
| Samples |
BET (m2 g−1) |
Total pore volume (cm3 g−1) |
Average pore diameter (nm) |
| Cs |
211.712 |
0.265 |
5.003 |
| BCs |
518.200 |
0.497 |
3.837 |
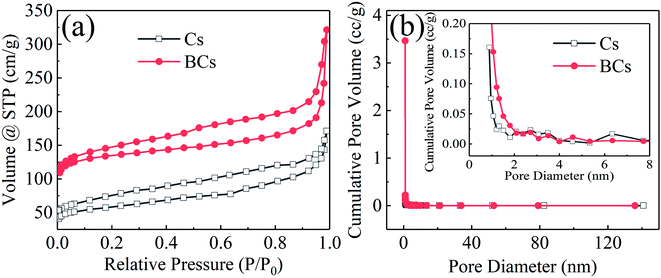 |
| | Fig. 3 (a) N2 sorption isotherms at −196 °C; (b) Barrett–Joyner–Halenda pore-size distributions of the Cs and BCs. | |
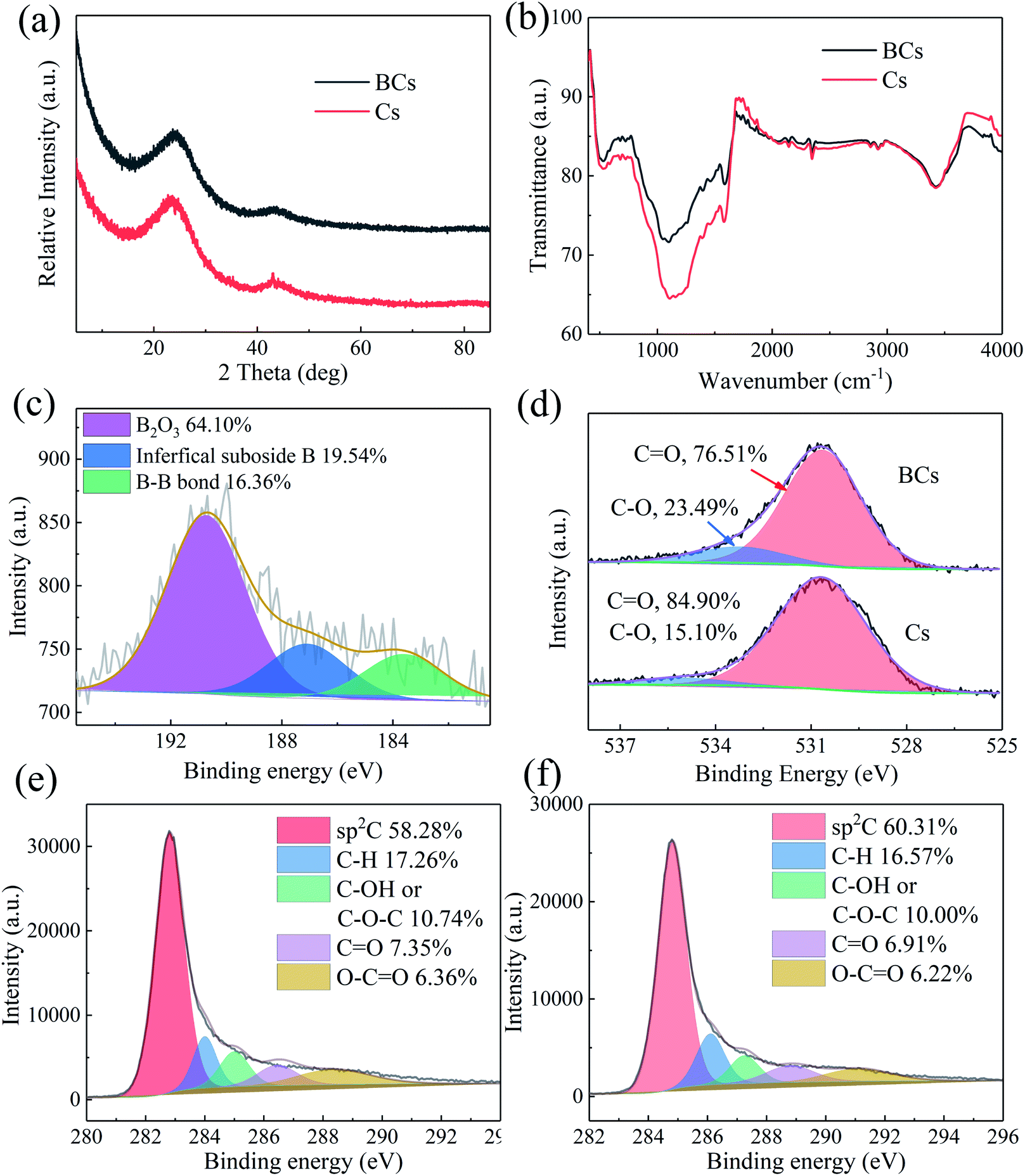
The crystalline structure of Cs and BCs was studied using XRD. As shown in Fig. 4(a), the XRD patterns of Cs and BCs have a characteristic peak at 26.5° that was attributed to the (002) plane, typical plane of graphitic carbon.32 The FT-IR spectra of BCs and Cs is shown in Fig. 4(b) and the absorption peaks at 1120, 1168, and 1397 cm−1 could be indexed to the vibrations of B–C stretching, B–O–H bending, and B–O stretching, respectively,33 confirming that a large quantity of B was doped into the carbon-based materials. Additionally, the FT-IR results showed that the frameworks of BCs and Cs obtained via hydrothermal carbonization were similar, revealing that the B modification process did not change the structure of the carbocatalysts.
 |
| | Fig. 4 XRD patterns of BCs and Cs (a); FT-IR spectra of BCs and Cs (b); B 1s high-resolution spectrum of BCs (c); high resolution O 1s spectra of BCs and Cs (d); and high resolution C 1s spectra of BCs (e) and Cs (f). | |
The full scan XPS spectra (Fig. S2†) showed the chemical states of carbocatalysts and that BCs contained three elements; Table 2 illustrates their elemental contents. As presented in Fig. 4(c), the B 1s spectrum provided evidence of boron oxide at 190.72 eV, interfacial suboxide boron at 187.09 eV and B–B bonds at 183.61 eV.34 The results confirmed that boron-containing groups existed in the BCs framework. For the O 1s spectra, shown in Fig. 4(d), peaks at 533.24 and 530.64 eV were respectively assigned to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups.35 The C 1s spectra (Fig. 4(e) for BCs and Fig. 4(f) for Cs) were deconvoluted into five peaks at around 284.8, 286.1, 287.25, 288.8 and 291.1 eV, which were assigned to the sp2-hybridized carbon bonds, C–H, C–OH or C–O–C, CO, and O–CO, respectively.36
O groups.35 The C 1s spectra (Fig. 4(e) for BCs and Fig. 4(f) for Cs) were deconvoluted into five peaks at around 284.8, 286.1, 287.25, 288.8 and 291.1 eV, which were assigned to the sp2-hybridized carbon bonds, C–H, C–OH or C–O–C, CO, and O–CO, respectively.36
Table 2 Atomic percentages of C, O and B elements on the surface of Cs and BCs obtained from XPS analysis
| Element (%) |
Cs |
BCs |
| C |
89.22 |
89.56 |
| O |
10.78 |
8.36 |
| B |
— |
2.08 |
3.3. Mechanism investigation
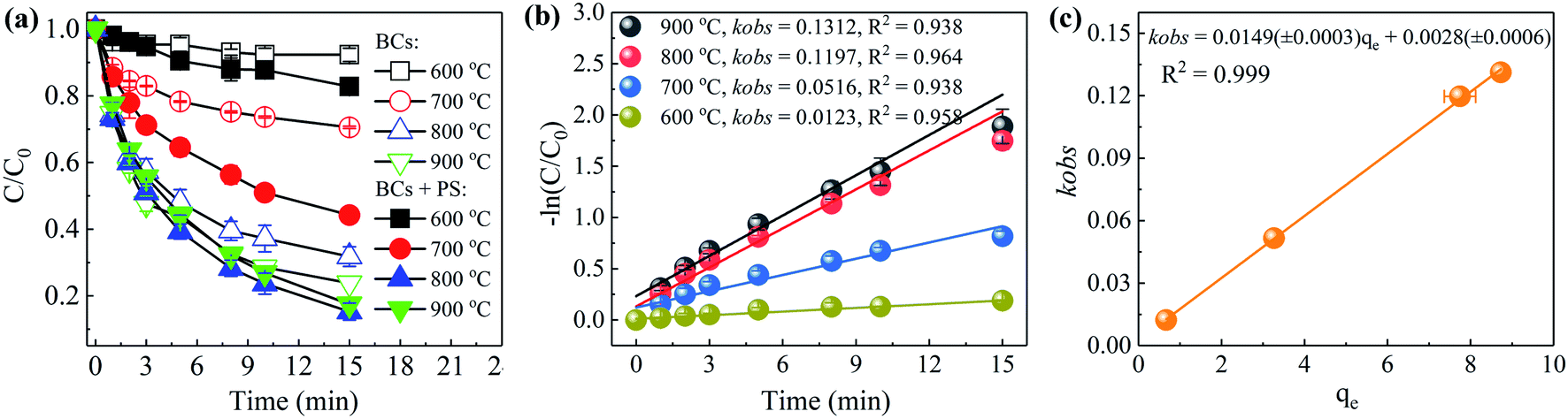
3.3.1. BPA adsorption kinetics and isotherms. It has been reported that the efficiency with which carbon/PS systems remove contaminants can be notably affected by the adsorptive capacity of the carbocatalysts.37,38 As shown in Fig. 5(a), when the calcination temperatures were 600, 700, 800 and 900 °C, the adsorption capacities for BPA removal after 15 min were 7.7%, 29.5%, 68.2% and 76.2%, respectively. The simultaneous use of PS and BCs showed that the removal rates for BPA were greatly enhanced to 17.2%, 55.8%, 84.8% and 82.6%, respectively. As shown in Fig. 5(c), the correlation between kobs and adsorption capacities of BPA (qe) was studied. The correlation coefficient was 0.999 for a series of BCs, indicating that the adsorption capacity was a key factor in the PS/BCs system. To further analyze the BPA adsorption by BCs and Cs, adsorption kinetics and adsorption isotherms were investigated.
 |
| | Fig. 5 BPA removal efficiency produced by BCs at different calcination temperatures (a), the associated reaction rates (b), and the correlation of kobs to qe (c) ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [BCs] = 0.04 g L−1, [PS]0 = 2 mM, pH0 = 4.0 ± 0.1). | |
The adsorption kinetics for the removal of BPA by BCs and Cs were investigated and the models were applied as follows:
Pseudo-first-order kinetic equation:39
| |
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (1) |
where
qe and
qt are the adsorption capacities at equilibrium and at time
t (min) in mg g
−1,
k1 is the rate constant of the pseudo-first-order diffusion model, g (mg min)
−1;
Pseudo-second-order kinetic model:40
| |
 | (2) |
where
k2 is the rate constant of the pseudo-second-order model g (mg min)
−1;
Intra-particle diffusion model:40
where
kid is the rate constant of the intra-particle diffusion model mg (g min
0.5)
−1, and
C is the intercept of the intra-particle diffusion model (mg g
−1).
Table 3 shows the resultant parameters of the experimental data which were fitted to the kinetics models. As described in Table 3, Fig. 6(a)–(c), the kinetics of BPA adsorption on BCs and Cs might be simulated well using a pseudo-second-order kinetics model (correlation coefficients, R2 > 0.996 for BCs and R2 > 0.998 for Cs), indicating a chemisorption process. Additionally, according to the pseudo-second-order model (2), the maximum BPA adsorption capacities of BCs and Cs were 7.199 and 3.860 mg g−1.
Table 3 Kinetic parameters for the adsorption of BPA onto BCs and Cs
| Adsorption kinetics |
Pseudo-first-order |
Pseudo-second-order |
Intra-particle diffusion |
| k1 (g mg−1 min−1) |
R2 |
k2 (g mg−1 min−1) |
R2 |
kid (mg g−1 min−0.5) |
C (mg g−1) |
R2 |
| BCs |
0.133 |
0.735 |
0.080 |
0.996 |
0.276 |
0.399 |
0.900 |
| Cs |
0.066 |
0.744 |
0.170 |
0.998 |
0.147 |
0.204 |
0.919 |
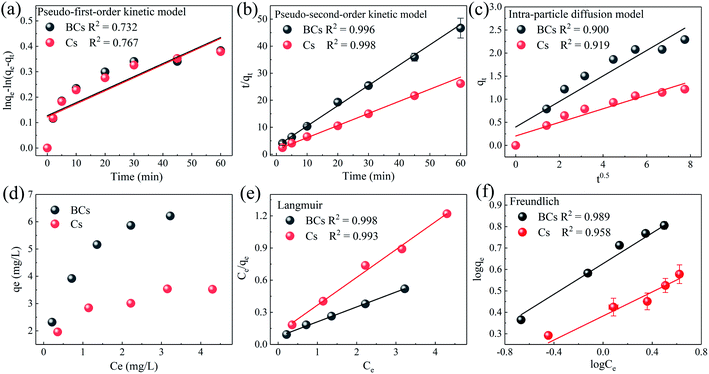 |
| | Fig. 6 Plots fitting experimental data to pseudo-first-order (a), pseudo-second-order (b), and intra-particle diffusion (c) models for BPA removal using BCs and Cs. Adsorption isotherms for the adsorption of BPA to BCs and Cs (d) Langmuir fitting (e), and Freundlich fitting (f) ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [BCs]0 = [Cs]0 = 0.1 g L−1, pH0 = 4.0 ± 0.1). | |
To further investigate BPA adsorption by the carbocatalysts, Langmuir (4) and Freundlich (5) adsorption isotherms were used to fit the adsorption equilibrium, as shown in Fig. 6(d)–(f).
Langmuir isotherm model:41
| |
 | (4) |
where
Ce is the equilibrium concentration of BPA solution mg L
−1,
qm is the maximum adsorption capacity constant for the Langmuir isotherm model in mg g
−1, and
KL is the equilibrium adsorption constant for the Langmuir isotherm model in L mg
−1.
Freundlich isotherm model:42
| |
 | (5) |
where
n is the Freundlich adsorption model exponent,
KF is the constant related to the relative sorption capacity of adsorbent in mg g
−1.
As shown in Table 4, the Langmuir isotherm equation appeared to be most suitable to describe the adsorption of BPA on BCs and Cs, yielding R2 values above 0.998 for BCs and 0.993 for Cs. This indicated that the isothermal adsorption behavior of BPA on BCs and Cs followed the Langmuir model and proved that a monolayer adsorption of BPA occurred on homogeneous BCs and Cs surfaces.43
Table 4 Adsorption isotherm parameters for BPA adsorption onto BCs and Cs
| Adsorption isotherm |
Langmuir |
Freundlich |
| qe/(mg g−1) |
KL/(L mg−1) |
R2 |
KF/(mg g−1) |
N |
R2 |
| BCs |
7.199 |
1.913 |
0.998 |
4.296 |
2.667 |
0.989 |
| Cs |
3.860 |
2.401 |
0.993 |
2.580 |
4.168 |
0.958 |
3.3.2. Kinetic analysis. The effect that the dosage of BCs and Cs used has on the removal of BPA in the BCs/PS and Cs/PS systems was investigated. The relationship was determined as shown in Fig. S3.† The adsorption rates increased as the BCs and Cs dosages increased from 0.05 to 0.25 g L−1. The BPA removal rate apparently increased due to the addition of PS. Taking BCs as an example (Fig. S3(b)†), when the BCs dosage was 0.05, 0.15, 0.20 and 0.25 g L−1, the BPA adsorption capacities after 60 min were 26.63%, 51.59%, 56.70% and 59.52%, respectively. The simultaneous use of PS and BCs resulted in BPA removal rates that were greatly enhanced to 37.97%, 87.94%, 94.77% and 98.76%, respectively. Previous studies17,44,45 proved that the heterogeneous catalytic oxidation of dissolved organic compounds can be described by a Langmuir–Hinshelwood (L–H) kinetics model (6),| |
 | (6) |
where C (mmol L−1) is the concentration of BPA, K is the equilibrium adsorption constant, and k is the rate constant.According to previous studies,17,44,45 eqn (6) can be simplified to eqn (7).
| | |
C1−n = (n − 1)kappt + C0 1−n
| (7) |
where
C0 (mmol L
−1) is the initial BPA concentration,
n is the apparent order of the reaction, and
kapp is the apparent rate constant.
In this work, the BPA degradation kinetics when using the Cs/PS and BCs/PS systems are as shown in Fig. 7. When the carbon material dosages were 0.05, 0.15, 0.20 and 0.25 g L−1, the resulting R2 values were 0.9785, 0.9963, 0.9962, 0.9938 for Cs and 0.9973, 0.9973, 0.9978, 0.9993 for BCs, respectively. These results demonstrate that the removal of BPA using the Cs/PS and BCs/PS systems fitted eqn (7) well. This indicates that adsorption and oxidation of BPA on Cs and BCs could be theoretically evaluated using the L–H kinetics model and that the boron-modified carbon materials could promote the activation of PS activation for BPA degradation.
 |
| | Fig. 7 Kinetic analysis for BPA removal by Cs/PS (a) and BCs/PS (b) ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [BCs]0 = [Cs]0 = 0.05–0.25 g L−1, [PS]0 = 2 mM, pH0 = 4.0 ± 0.1). | |
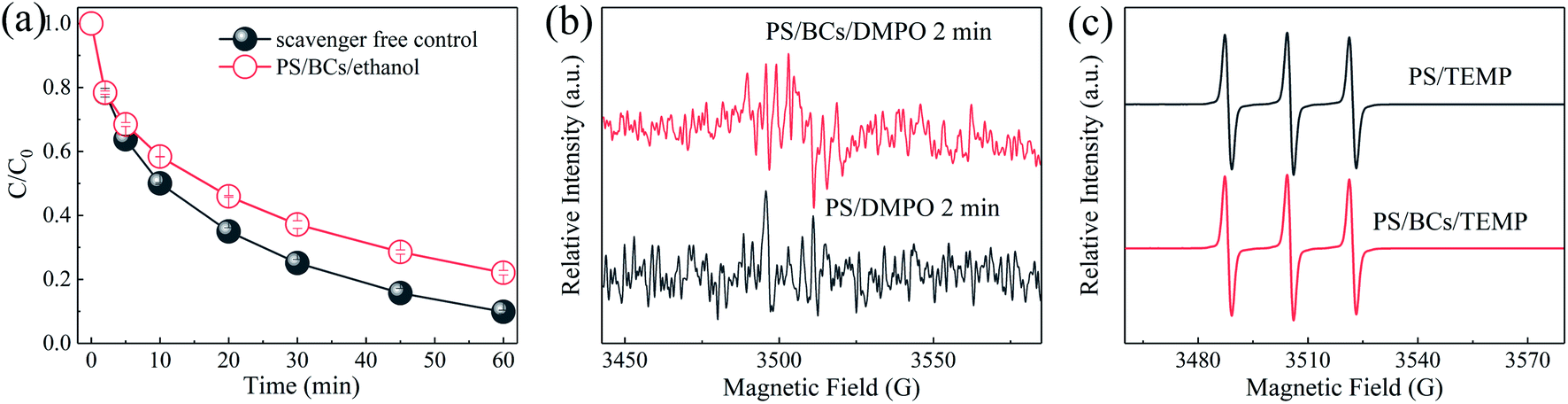
3.3.3. Identification of reactive species. Organic contaminant degradation via the activation of PS activation using a carbocatalyst has been reported previously and the reactive species generally include singlet oxygen,8,35 ˙OH, SO4˙−,22 and the reactive electron-deficient carbocatalysts ([carbon]*).46,47 Ethanol was employed as a typical scavenger for both SO4˙− (k = 1.6–7.7 × 107 M−1 s−1) and ˙OH (k = 1.2–2.8 × 109 M−1 s−1) and in this study, as shown in Fig. 8(a), a negligible effect on the BPA removal was observed when ethanol was added into the PS/BCs system. Fig. 8(b) shows that the EPR signals for DMPO-OH48 and DMPO-SO449,50 were so weak that they couldn’t be detected, indicating that the BPA degradation might proceed via a non-radical pathway. Furthermore, the EPR results presented in Fig. 8(c) showed that the PS/BCs/TEMP system resulted in a TEMPN signal intensity that was comparable with that of the PS/TEMP system, demonstrating that singlet oxygen was not the primary reactive species in the non-radical pathway for BPA removal.
 |
| | Fig. 8 (a) Quenching test in the PS/BCs system ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [BCs]0 = 0.1 g L−1, [PS]0 = 1 mM, [ethanol]0 = 100 mM, pH0 = 4.0 ± 0.1); (b) EPR spectra obtained from the PS/BCs systems in aqueous solution ([BCs]0 = 1.0 g L−1, [PS]0 = 10 mM, [DMPO] = 20 mM); and (c) EPR spectra of TEMP-1O2 adduct (TEMPN) formed in aqueous solutions containing PS, BCs, and TEMP ([BCs]0 = 1 g L−1, [PS]0 = 10 mM, [TEMP] = 20 mM). | |
3.3.4. Electrochemical analysis. The propensity of a material toward electron donation is closely related to the extent of BPA removal. In our study, in order to investigate the existence of [carbon]*, an electrochemical analysis was applied. We proposed that the BPA removal capacity was influenced by the electron transfer capacity of our material and an electrochemical analysis proved the hypothesis.CV analysis is widely used to identify the redox potentials of compounds. The CV spectra of BCs and Cs are shown in Fig. 9(a). Ordinarily, there are two pairs of typical oxidation-reduction peaks in CV profiles. The potential of the redox peak for BCs was higher than that for Cs, implying that BCs has a higher current density and more remarkable catalytic activity than Cs.46 The occurrence of direct electron transfer was also suggested by the Tafel corrosion analysis. The results showed that BCs had a remarkable electron transfer rate for BPA removal because its corrosive potential was lower than that of Cs (Fig. 9(b)).51
 |
| | Fig. 9 CV curves of Cs and BCs obtained at the scan rate of 10 mV s−1 (a); Tafel corrosion scan curves for Cs and BCs (b); and open circuit voltages of Cs (c) and BCs (d) (glassy carbon electrodes, [BPA]0 = 0.10 mM, T = 25 ± 1 °C, [PS]0 = 1.00 mM). | |
The open circuit voltages of Cs (Fig. 9(c)) and BCs (Fig. 9(d)) indicate that BCs had an open circuit voltage of approximately 0.2 V (vs. NHE), double that of Cs (approximately 0.1 V). Furthermore, the potential of [carbon]* can also be identified from the equilibrium values of the open circuit voltage. Thus, the open circuit potentials of [BCs]* and [Cs]* were measured to evaluate the electron transfer process. If PS was added into the reaction mixture, the open circuit potential raised sharply and the potential of [BCs]* was raised to around 0.75 V (0.55 V for Cs). In addition, when the BPA was added into the reaction mixture the equilibrium potential rose further. As mentioned previously, in the system where PS is activated by BCs, the potential of the [BCs]* complexes was profoundly elevated, therefore increasing the BPA removal efficiency via an electron transfer pathway.
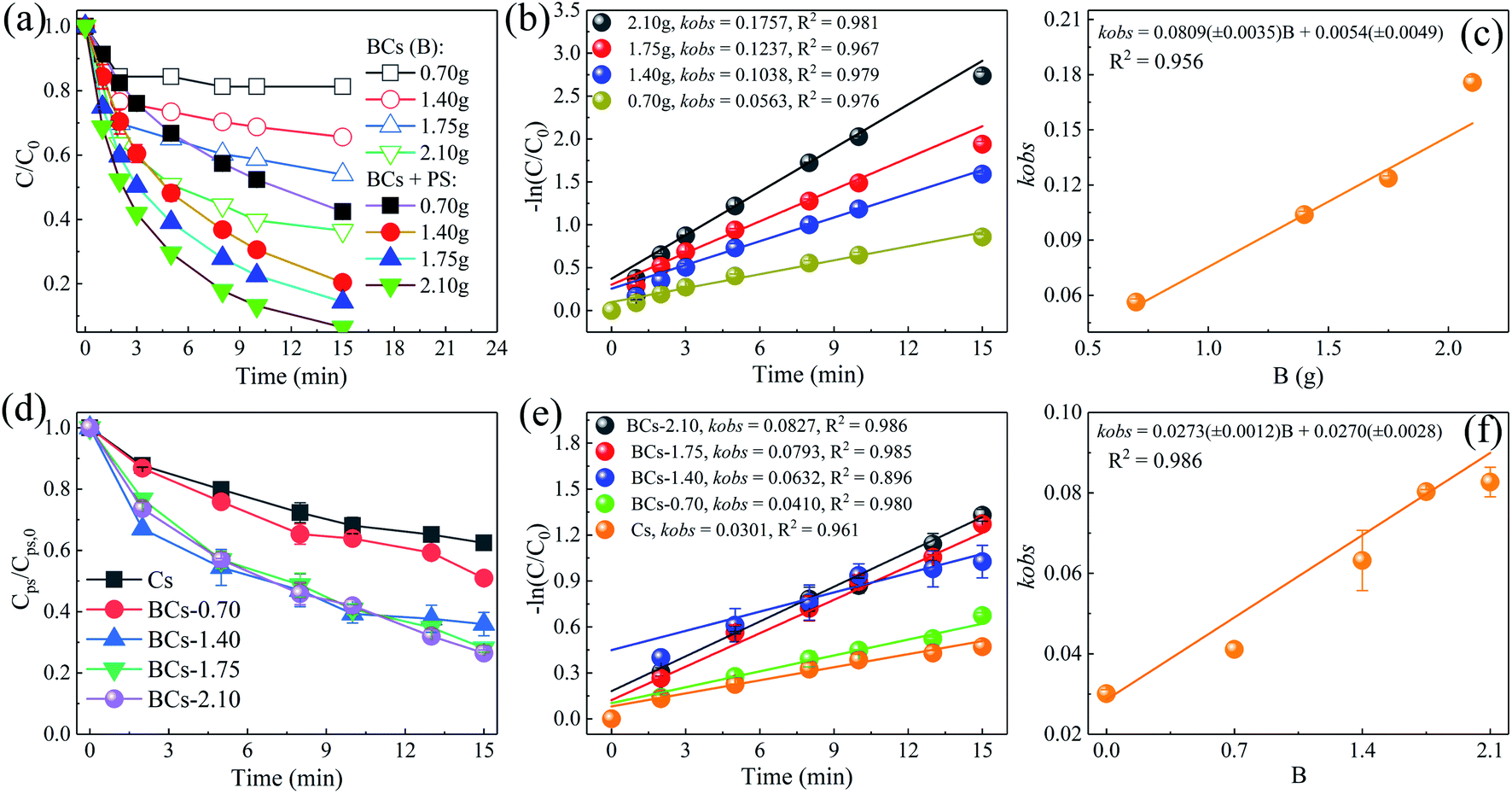
3.3.5. Exploring the role of B in the PS/BCs system. As displayed in Fig. 10(a), the adsorption capacity increased obviously when the amount of B doping increased. In the presence of PS, the BPA removal efficiency was enhanced significantly. When the amount of B doping was 0.7, 1.4, 1.75, and 2.1 g, the BPA removal efficiencies were 57.6%, 79.6%, 85.6%, and 93.5% after 15 min, respectively. Fig. 10(d) indicates that the adsorption efficiency of PS was enhanced more by BCs compared with by Cs. The pseudo-first order kinetics with respect to the level of B doping were found to give relatively good linearity (Fig. 10(c), R2 = 0.956 and Fig. 10(f), R2 = 0.986). These results confirmed the important role that the level of B doping plays as it not only increased the BPA adsorption rate, but also the PS adsorption quantity. This result indicates that the introduction of B into carbon material clearly enhanced the catalytic activities of the carbon catalysts.
 |
| | Fig. 10 Effect of different B doping levels on BPA removal in the PS/BCs systems (a); the associated reaction rates (b); and the correlation of kobs to the level of B doping (c) ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [BCs] = 0.04 g L−1, [PS]0 = 2 mM, pH0 = 4.0 ± 0.1). Effect of different B doping levels on PS adsorption without BPA in the PS/BCs systems (d); the associated reaction rates (e); and the correlation of kobs to the level of B doping (f) (T = 25 ± 1 °C, [BCs] = 0.04 g L−1, [PS]0 = 2 mM, pH0 = 4.0 ± 0.1). | |
Based on the above discussions, we proposed a nonradical mechanism, as depicted in Scheme 1. (i) The unpaired electrons on BCs are removed by PS and subsequently form a metastable intermediate, [BCs]*, which could increase the redox potential of BCs; (ii) [BCs]* fills the newly created vacancies by capturing electrons from BPA, thus oxidizing BPA; and (iii) in certain circumstances, when the quantity of B doping is increased, the potential of the [BCs]* complex becomes more positive, giving it a higher oxidation capacity and accelerating the electron transfer from BPA, thus boosting the BPA removal rate.
 |
| | Scheme 1 Proposed mechanism for the degradation of BPA. | |
3.4. Possible BPA degradation pathways
The intermediate products generated after 30 min of BPA degradation were identified using liquid chromatography with mass spectrometry (LC-MS). Various intermediates were detected, and the possible intermediate products are proposed in Table S1.†2,52–57 Three possible oxidative pathways for the degradation of BPA are proposed. As displayed in Fig. 11, in pathway I, BPA is directly changed into two intermediates m/z 96.00 (B2, phenol) and m/z 131.81 (B3, 4-vinylphenol) via a C–C cleavage process. These intermediates are further oxidized into B5 (hydroquinone, m/z = 109.00) and B7 (4-hydroxyacetophenone, m/z = 134.98), respectively. For pathway II, the BCs-PS system could involve demethylation to generate B1 (4-[4-(hydroxymethyl)phenyl] phenol, m/z = 202.95). In subsequent steps, B1 is further decomposed to B5, which could be gradually oxidized to B6 (p-benzoquinone, m/z = 107.93). The third pathway is led by a dehydroxylation reaction, where the intermediate B4 (2,2-bis(phenyl)propane, m/z = 197.00) is formed from BPA. This is then converted to B8 (1-methylethenylbenzene, m/z = 117.87) via a further oxidation process. In addition, these intermediates would then be degraded by further oxidation and ring opening process. Five peaks with m/z values 117.87 (succinic acid), 113.98 (divinyloxyethane), 128.93 (5-oxohex-2-enoic acid (OEA)), 90.81 (ethanedioic acid) and 62.38 (ethylene glycol) are derived from intermediates B5, B6, B7 and B8. The five smaller intermediates can be finally mineralized into CO2 and H2O.
 |
| | Fig. 11 Potential BPA degradation pathways. | |
3.5. Impacts of water matrices
The composition of waste water could limit the effectiveness of the BCs/PS system, especially because of the dissolved inorganic ions or particulates.58,59 Therefore, the effects of different water matrices on BPA removal were also investigated to estimate the interactions between the BCs/PS system and different water matrices, including deionized water (DW), running water (RW), landscape water (LW), Mingyuan lake water (MYW), and Jiang’an river water (JAW). Table S2† displays the characteristics of four actual water samples. The BPA removal efficiency in the DW, RW, JAW, LW and MYW samples were 49.50%, 43.20%, 42.76%, 38.65% and 42.53%, respectively. As shown in Fig. 12, in the presence of PS, the BPA removal efficiencies were respectively 83.29%, 69.59%, 68.78% and 71.60% in RW, JAW, LW and MYW, profoundly lower than in DW (94.15%). This is because the presence of dissolved organic matter and inorganic species could compete with the PS for the reaction sites on the surface of BCs where [BCs]* is produced. This process could also limit the effective adsorption of BPA on the BCs surface, consequently causing a decrease in the removal rate.
 |
| | Fig. 12 BPA removal in simulated wastewaters ([BPA]0 = 0.02 mM, T = 25 ± 1 °C, [BCs]0 = [Cs]0 = 0.1 g L−1, [PS]0 = 2 mM, pH0 = 4.0 ± 0.1). | |
4. Conclusions
In this work, B-doped carbocatalysts were prepared for the activation of persulfate (PS) for micropollutant degradation. The results demonstrate that B-doped carbon materials have, when compared with B-free carbon materials, enhanced adsorption and catalytic activity for bisphenol A (BPA) removal. The adsorption process plays a critical role in the subsequent degradation process and the quantity of B doping was identified as the primary factor to attain a greater adsorption quantity of PS and a faster BPA adsorption rate. An electrochemical analysis (cyclic voltammetry, Tafel corrosion analysis and open circuit voltages) proved that boron-doped carbon materials (BCs) could elevate the potential of the derived nonradical [BCs]* complexes, and that an electron transfer pathway for BPA removal was enhanced via the higher potential [BCs]* complexes. This discovery is critical as it reveals the importance of B-doping in carbon materials/PS systems for the removal of BPA via nonradical pathways. Moreover, the outstanding performance of B-doped carbon materials means that metal-free catalysis for PS activation can be utilized as an environmentally friendly process for micropollutant reduction.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgements
The authors thank Mr Fanghao Liao and Miss Jiayi Yang from Southwest Branch of China Construction Third Engineering Bureau Group Co., Ltd. for help in data analysis.
References
- G. Ozyildiz, T. Olmez-Hanci and I. Arslan-Alaton, Appl. Catal., B, 2019, 254, 135–144 CrossRef CAS.
- F. Wang, Y. Lai, Q. Fang, Z. Li, P. Ou, P. Wu, Y. Duan, Z. Chen, S. Li and Y. Zhang, Appl. Catal., B, 2020, 262, 118099 CrossRef CAS.
- Y. Zhang, D. Zhang, L. Zhou, Y. Zhao, J. Chen, Z. Chen and F. Wang, Chem. Eng. J., 2018, 336, 690–700 CrossRef CAS.
- N. Bolong, A. F. Ismail, M. R. Salim and T. Matsuura, Desalination, 2009, 239, 229–246 CrossRef CAS.
- P. Wu, Y. Zhang, Z. Chen, Y. Duan and S. Li, Appl. Catal., B, 2019, 255, 117784 CrossRef CAS.
- Y. Shao, L. Zhou, Q. Wu, C. Bao and M. Liu, J. Hazard. Mater., 2017, 339, 418–426 CrossRef CAS PubMed.
- Z. Peng, Z. Jing, Y. Zhang, G. Zhang, W. Li, C. Wei, J. Liang, Y. Liu and S. Shu, J. Hazard. Mater., 2017, 344, 1209–1219 Search PubMed.
- X. Huo, P. Zhou, J. Zhang, Y. Liu, X. Cheng, Y. Liu, W. Li and Y. Zhang, J. Hazard. Mater., 2020, 391, 122055 CrossRef CAS PubMed.
- Y. Fan, Y. Ji, D. Kong, J. Lu and Q. Zhou, J. Hazard. Mater., 2015, 300, 39–47 CrossRef CAS.
- R. Yin, W. Guo, H. Wang, J. Du, X. Zhou, Q. Wu, H. Zheng, J. Chang and N. Ren, Chem. Eng. J., 2018, 335, 145–153 CrossRef CAS.
- Q. Chengdu, L. Xitao, M. Jun, L. Chunye and X. Li, Chemosphere, 2016, 151, 280–288 CrossRef PubMed.
- F. Rehman, M. Sayed, J. A. Khan, N. S. Shah, H. M. Khan and D. D. Dionysiou, J. Hazard. Mater., 2018, 357, 506–514 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- H. Sun, S. Liu, G. Zhou, H. M. Ang, M. Tadé and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466 CrossRef CAS PubMed.
- H. Sun, Y. Wang, S. Liu, L. Ge, L. Wang, Z. Zhu and S. Wang, Chem. Commun., 2013, 49, 9914–9916 RSC.
- X. Cheng, H. Guo, Y. Zhang, G. V. Korshin and B. Yang, Water Res., 2019, 157, 406–414 CrossRef CAS PubMed.
- X. Cheng, H. Guo, Y. Zhang, X. Wu and Y. Liu, Water Res., 2017, 113, 80–88 CrossRef CAS.
- K. A. Lin, J. T. Lin, X. Y. Lu, C. Hung and Y. F. Lin, J. Colloid Interface Sci., 2017, 505, 728–735 CrossRef CAS PubMed.
- X. Duan, Z. Ao, D. Li, H. Sun, L. Zhou, A. Suvorova, M. Saunders, G. Wang and S. Wang, Carbon, 2016, 103, 404–411 CrossRef CAS.
- H. Lee, H. I. Kim, S. Weon, W. Choi, Y. S. Hwang, J. Seo, C. Lee and J. H. Kim, Environ. Sci. Technol., 2016, 50, 10134 CrossRef CAS PubMed.
- W. Tian, H. Zhang, X. Duan, H. Sun, M. O. Tade, H. M. Ang and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7184–7193 CrossRef CAS PubMed.
- X. Duan, H. Sun and S. Wang, Acc. Chem. Res., 2018, 51, 678–687 CrossRef CAS.
- W. Tian, H. Zhang, H. Sun, M. O. Tadé and S. Wang, Chem. Eng. J., 2018, 347, 432–439 CrossRef CAS.
- W. Yu, Y. Feng, X. Liu, G. Tan and X. Dan, Appl. Surf. Sci., 2018, 435, 281–289 CrossRef.
- C. Chokradjaroen, S. Kato, K. Fujiwara, H. Watanabe, T. Ishii and T. Ishizaki, Sustainable Energy Fuels, 2020, 4, 4570–4580 RSC.
- S. S. Balaji, P. A. Ganesh, M. Moorthy and M. Sathish, Bull. Mater. Sci., 2020, 43, 151 CrossRef CAS.
- S. Kaipannan, P. A. Ganesh, K. Manickavasakam, S. Sundaramoorthy and S. Marappan, J. Energy Storage, 2020, 32, 101774 CrossRef.
- A. Xk, A. Gf, A. Fs, A. Xd, W. A. Lei, A. Xzf and B. Jlila, Int. J. Hydrogen Energy, 2021, 4, 36221–36231 Search PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed. Engl., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- Y. Xie, Z. Meng, T. Cai and W. Q. Han, ACS Appl. Mater. Interfaces, 2015, 7, 25202–25210 CrossRef CAS PubMed.
- J. Kang, X. Duan, C. Wang, H. Sun, X. Tan, M. O. Tade and S. Wang, Chem. Eng. J., 2018, 332, 398–408 CrossRef CAS.
- Y. Liu, W. Li, P. Wu, C. Ma, X. Wu, M. Xu, S. Luo, Z. Xu and S. Liu, Sens. Actuators, B, 2019, 281, 34–43 CrossRef CAS.
- P. Zhou, W. Ren, G. Nie, X. Li, X. Duan, Y. Zhang and S. Wang, Angew. Chem., Int. Ed. Engl., 2020, 59, 16517–16526 CrossRef CAS PubMed.
- X. Cheng, H. Guo, Y. Zhang, Y. Liu, H. Liu and Y. Yang, J. Colloid Interface Sci., 2016, 469, 277–286 CrossRef CAS PubMed.
- W. Tian, H. Zhang, X. Duan, H. Sun, M. O. Tade, H. M. Ang and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7184 CrossRef CAS PubMed.
- S. Zhu, X. Huang, F. Ma, L. Wang and X. Duan, Environ. Sci. Technol., 2018, 52, 8649–8658 CrossRef CAS PubMed.
- X. Wang, Y. Qin, L. Zhu and H. Tang, Environ. Sci. Technol., 2015, 49, 6855–6864 CrossRef CAS PubMed.
- Y. Lee, J. Kim and H. Shin, Sep. Sci. Technol., 2013, 48, 1093–1101 CrossRef CAS.
- F. Zhang, J. Lan, Y. Yang, T. Wei, R. Tan and W. Song, J. Nanopart. Res., 2013, 15, 1–10 Search PubMed.
- Y. Shao, X. Wang, Y. Kang, Y. Shu, Q. Sun and L. Li, Acta Sci. Circumstantiae, 2014, 429, 25–33 CAS.
- S. H. Huo and X. P. Yan, J. Mater. Chem., 2012, 22, 7449–7455 RSC.
- D. P. Li, Y. R. Zhang, X. X. Zhao and B. X. Zhao, Chem. Eng. J., 2013, 232, 425–433 CrossRef CAS.
- N. G. Asenjo, R. Santamaría, C. Blanco, M. Granda, P. álvarez and R. Menéndez, Carbon, 2013, 55, 62–69 CrossRef CAS.
- L. Li, L. Gong, Y. X. Wang, Q. Liu, J. Zhang, Y. Mu and H. Q. Yu, Water Res., 2016, 98, 235–241 CrossRef CAS PubMed.
- W. Ren, L. Xiong, X. Yuan, Z. Yu and S. Wang, Environ. Sci. Technol., 2019, 53, 14595–14603 CrossRef CAS PubMed.
- W. Ren, L. Xiong, G. Nie, H. Zhang and S. Wang, Environ. Sci. Technol., 2019, 54, 1267–1275 CrossRef.
- P. Guilong, M. Zhang, S. Deng, D. Shan and H. Qiang, Chem. Eng. J., 2018, 341, 361–370 CrossRef.
- P. Zhou, W. Li, J. Zhang, G. Zhang, X. Cheng, Y. Liu, X. Huo and Y. Zhang, J. Taiwan Inst. Chem. Eng., 2019, 100, 202–209 CrossRef CAS.
- P. Zhou, J. Zhang, Z. Xiong, Y. Liu and Y. Zhang, Appl. Catal., B, 2019, 265, 118264 CrossRef.
- M. Zhao, C. Zhang, X. Yang, L. Liu, X. Wang, W. Yin, Y. Li, S. Wang and W. Fu, J. Hazard. Mater., 2020, 396, 122712 CrossRef CAS PubMed.
- A. Bacha, I. Nabi, H. Cheng, K. Li, S. Ajmal, T. Wang and L. Zhang, Chem. Eng. J., 2020, 389, 124482 CrossRef CAS.
- X. Dong, B. Ren, Z. Sun, C. Li, X. Zhang, M. Kong, S. Zheng and D. D. Dionysiou, Appl. Catal., B, 2019, 253, 206–217 CrossRef CAS.
- S. Wang, J. Tian, Q. Wang, F. Xiao and F. Cui, Appl. Catal., B, 2019, 256, 117783 CrossRef CAS.
- L. Yang, X. Bai, J. Shi, X. Du and P. Jin, Appl. Catal., B, 2019, 256, 117759 CrossRef CAS.
- X. Zhang, R. Zhao, N. Zhang, Y. Su and C. Du, Appl. Catal., B, 2019, 263, 118316 CrossRef.
- J. Lin, Y. Hu, L. Wang, D. Liang and S. Shao, Chem. Eng. J., 2019, 382, 122931 CrossRef.
- E. Díez-Mato, F. C. Cortezón-Tamarit, S. Bogialli, D. García-Fresnadillo and M. D. Marazuela, Appl. Catal., B, 2014, 160–161, 445–455 CrossRef.
- L. Hu, G. Zhang, M. Liu, Q. Wang and P. Wang, Chem. Eng. J., 2018, 338, 300–310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Texts describe the materials and reagents, experimental procedures, conventional analytical methods, EPR measurements, the methods for detection of PS concentrations, and electron transfer experiments. Tables show the intermediates detected using LC-MS and their associated m/z values, characteristics of various water matrices, and pore structure of the used BCs. Figures show TGA and DTG curves for the precursors heated under an N2 atmosphere, XPS survey spectra, BPA removal efficiency at varying doses of BCs and Cs, BPA removal efficiency using Cs and BCs at varying initial pHs, and recycling test for the removal of BPA only with adsorption by BCs. See https://doi.org/10.1039/d2ra02703h |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Jinhong Gua,
Lei Weia,
Zhenping Suna,
Fuxiang Du*a,
Chao Daia,
Xiongfei Wua,
Zhiguang Liua and
Jian Rena
*ab,
Jinhong Gua,
Lei Weia,
Zhenping Suna,
Fuxiang Du*a,
Chao Daia,
Xiongfei Wua,
Zhiguang Liua and
Jian Rena