
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA stretchable and healable elastomer with shape memory capability based on multiple hydrogen bonds†
Jiacheng Ma ,
Shifeng Wen* and
Zhufeng Yue
,
Shifeng Wen* and
Zhufeng Yue
School of Mechanics and Civil & Architecture, Northwestern Polytechnical University, Xi'an 710129, PR China. E-mail: wenshifeng@nwpu.edu.cn
First published on 3rd August 2022
Abstract
Although a wide range of self-healing materials have been reported by researchers, it is still a challenge to endow exceptional mechanical properties and shape memory characteristics simultaneously in a single material. Inspired by the structure of natural silk, herein, we have adopted a simple synthetic method to prepare a kind of elastomer (HM-PUs) with stiff, healable and shape memory capabilities assisted by multiple hydrogen bonds. The self-healing elastomer exhibits a maximum tensile strength of 39 MPa, toughness of 111.65 MJ m−3 and self-healing efficiency of 96%. Moreover, the recuperative efficiency of shape memory could reach 100%. The fundamental study of HM-PUs will facilitate the development of flexible electronics and medical materials.
Introduction
Synthetic polymers are a kind of material with entropic elasticity, which have been widely applied in the fields of rubber products, flexible electronics, aerospace and encapsulants. Moreover, various hydrogels constructed by polymer chains have been used in medical research.1–8 Nevertheless, polymers will presumably be damaged by external force in practical application; therefore, it is important to reinforce the robustness and retrievability of traditional polymers. Up to now, two self-healing mechanisms have been proposed in healable materials, namely extrinsic and intrinsic self-healing materials.9–17 Extrinsic self-healing materials have been limited in application on account of their limited self-healing times, and the intrinsic self-healing strategy has accordingly occupied the leading role in self-healing materials.Combining tough properties and high self-healing efficiency is a paradox. Therefore, healable materials with high self-healing efficiency and excellent mechanical properties have intrigued scientists. Unfortunately, self-healing materials based on weak dynamic bonds tend to lack good mechanical properties. To address this conundrum, scientists have devoted a good deal of attention to it. 2-Ureido-4[1H]-pyrimidinone (UPy) is a kind of quadruple hydrogen bond structure, and it is widely utilized in self-assembled polymers and molecular structure design. On account of the superiorities of the UPy structure, scientists have introduced UPy and its consequent benefits in the molecular structures of healable materials. For instance, Bao et al. reported a kind of supramolecular polymer material with good mechanical properties and highly stretchable performance by condensation polymerization assisted by UPy; with the aid of quadruple hydrogen bonds, the maximum stress could reach 3.74 MPa and the material exhibited high self-healing efficiency (∼88%).18 In Lee's work, UPy groups were used to functionalize polyurethane, and an elastomer with high tensile strength (9.44 MPa) and elongation (2340%) was prepared; furthermore, the maximum self-healing efficiency showed desirable performance (∼70%, in organic solvent).19 In both the cases above, the mechanical properties of these healable polymers functionalized by UPy groups could not meet the ideal level, and the solubility of UPy is too low in polar organic solvents to use in organic syntheses.20,21 Hence, two problems urgently need to be addressed: (1) which kind of hydrogen bond structure should be adopted to replace the UPy with quadruple hydrogen bond structures; (2) how to balance the excellent mechanical properties and high self-healing efficiency (Scheme 1).
 | ||
| Scheme 1 Schematic diagram of the silkworm fibril. | ||
In nature, biomaterials exhibit extraordinary characteristics that man-made materials cannot compare with.22–25 Silkworm silk is a kind of natural long fiber made from its secretion, which possesses excellent characters such as lightweight, good flexibility, high tenacity, admirable biodegradability, and biocompatibility, has been widely used in these areas of fabrics, composite materials, and biosensors.26–31 Most importantly, the super robustness of silkworm silk originates from the hydrogen bond arrays in β-sheet nanocrystals. Hydrogen bond arrays play two roles in silk fibril: (1) assemble nanocrystals to strength the fibril; and (2) dissipate energy effectively under external force via hydrogen bond breakage.32–34
Inspired by the structure of the silkworm fibril, multiple hydrogen bonds were introduced in polyurethane by condensation polymerization. Since the diffusion of polymer chains plays a paramount role in the self-healing process, we proposed a particular molecular design that embedded asymmetric alicyclic structure in polyurethane networks as it would affect the self-healing efficiency in cracks.35 Also, the high toughness and shape memory property of the kind of elastomer is a result of the density and configuration of the hydrogen bond arrays, and these properties are tuned by macromolecule design.
Herein, HM-PUs were synthesized from 4,4′-methylenebis(cyclohexyl isocyanate) (HMDI) as the hard segment, poly(tetramethylene ether)glycol (PTMEG) as the soft segment, and isophorone diamine (IPDA) as the chain extender and the self-healing elastomer, which not only exhibits remarkable tensile strength of 39 MPa but has a high speed of shape memory. Furthermore, this kind of elastomer even outperforms some commercial elastomers,36–38 and the fracture energy of the elastomer is higher than some alloys.42 As a kind of dynamic bond, the force-induced hydrogen bonds will dissipate energy to improve the toughness of the polymer networks. Under strain, ordered hydrogen bonds formed between polymer chains lead to the appearance of a metastable crystal, and accordingly, the toughness increases.
More importantly, HM-PU with high transparency (∼100%), high self-healing efficiency (>90%), and high tensile strength (39 MPa) was developed; also, this material possesses remarkable shape memory characteristic (recuperative efficiency: ∼100%).
Experimental section
Materials
PTMEG (Mn = 1000 g mol−1), HMDI (>90%), IPDA (99%), and dibutyltin dilaurate (DBTDL, 95%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. N,N-Dimethylformamide (DMF) was purchased from Tianjin Dingshengxin Chemical Co., Ltd. Teflon mold was purchased from Dongguan Qihang Plastic Materials Co., Ltd. All solvents and reagents were used in organic synthesis without further purification.Synthesis of HM-PUs
As the example of HM-PU1, we adopted the two-step synthesis method. First, PTMEG (2 g, 2 mmol) was added into a three-necked flask equipped with a magnetic agitator, closely followed by heating (60 °C) and stirring (600 rpm) for 1 h to remove residual moisture. Then, DBTDL (0.02 g) and HMDI (1.09 g, 6.7 mmol) were dissolved in DMF (30 mL) and added into a three-necked flask; subsequently, the reaction system was further heated up to 70 °C in N2 atmosphere and reacting for 10 h. After that, the temperature of the reaction system was cooled to 35 °C; subsequently, IPDA (0.8 g, 4.7 mmol) was dropwise added into a flask for 3 min and stirred for 48 h. After the reaction was completed, the concentration of the solution was adjusted to 20 wt%, followed by placing it in a vacuum atmosphere and heating (80 °C) for 24 h; finally, the HM-PU1 elastomer was obtained. The reagents' molar ratio and the molecular information of HM-PU1, HM-PU2, and HM-PU3 is shown in Table S1.†Characterization
HM-PUs film was cut into a dumbbell shape by a tablet machine (CREE-6040, Dongguan CREE INST Rument Technology Co., Ltd, China); the specification of the testing area is 20 × 5× (0.52 ± 0.03) mm. The mechanical properties were tested at room temperature using a universal testing machine (WBE, Weibang instrument Co., Ltd, China), and the experiment was tested at a stretching speed of 100 mm min−1. The 1H NMR spectrum was measured on a nuclear magnetic resonance spectrometer (Bruker AVANCE III 400 MHz, Germany). Molecular weights and polymer dispersity index (PDI) were measured using gel permeation chromatography (Shimadzu, Rid-20A, Japan). The FTIR spectra were recorded on an FTIR spectrometer (Nicolet iS10, Thermo Scientific, U.S.) in the scanning range from 400 cm−1 to 4000 cm−1. The UV-vis spectra were recorded on an ultraviolet-visible spectrophotometer (TU-1810, Beijing Purkinje General Instrument Co., Ltd, China) at room temperature. DSC measurements were performed on a differential scanning calorimeter (DSC214, Netzsch, Germany) in the temperature range from −85 °C to 85 °C at a heating rate of 10 °C min−1. The TGA data was recorded on an STA 449 F3 (NETZSCH, Germany) in the temperature range from 35 °C to 800 °C at a heating rate of 10 °C min−1. XRD measurements were performed on an X-ray diffractometer (D8 DISCOVER A25, Bruker, Germany) in the diffraction angle range from 13° to 70° equipped with Co generator. The microscopic images were recorded using a VHX-5000 digital microscope (Keyence, Japan).Results and discussion
Molecular design and general characterization
We intended to synthesize a kind of molecular structure that resembled the structure of silkworm fibril as the structure of multiple hydrogen bond was a superb option to assemble linear macromolecule, and the hydrogen bonds between the carbonyl groups and hydrogen atoms can constitute the polymer matrix network with the help of hard segments as the node and soft segments as the crosslinker. Of course, this special structure we designed can adequately dissipate the energy under external force, avoiding the rupture in materials.Herein, a two-step polycondensation reaction (Scheme 2) was adopted to prepare a kind of healable elastomer that constituted the crosslinked matrix with hydrogen bonds. Also, HMDI as the hard segment and IPDA as the extender were chosen as the nodes in the matrix, and PTMEG as the soft segment is the crosslinker that joins these nodes. The asymmetric alicyclic structure of IPDA will weaken the forces of hydrogen bonds between carbonyl groups, resulting in high diffusion speed of the molecular chains and fast self-healing rate.
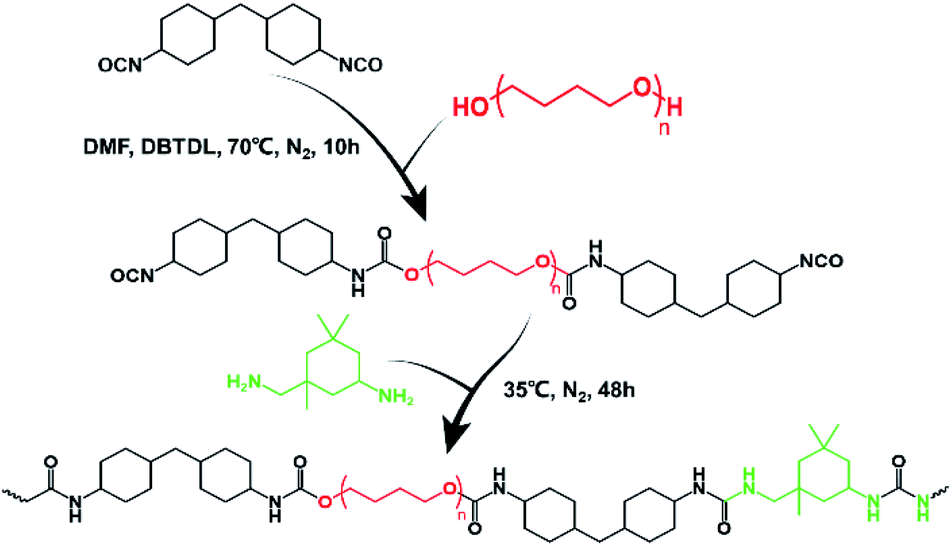 | ||
| Scheme 2 Synthetic route of HM-PUs containing multiple hydrogen bonds. | ||
In the synthetic reaction, three kinds of HM-PUs with different molar ratios of hard and soft segments were prepared, showing various properties, and the information of HM-PUs is shown in Table S1.† The GPC results show that Mn and Mw are capable of coming up to a high order of magnitude (>104), and the small PDI shows that the molecular weights of HM-PUs are evenly distributed. As the hydrogen bond structure domains are adequately small, the visible light transmittance of HM-PUs was up to 99% in the wavelength range from 300 cm−1 to 800 cm−1.39 In addition, the chemical structure of HM-PU2 was analyzed by the 1H NMR spectrum, as shown in Fig. 1c. In the FTIR spectra of HM-PUs, the –NCO peaks completely disappeared, indicating that the reaction was finished, and the –NCO groups had transformed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. It is worth noting that the characteristic peaks of CO in urethane at 1718 cm−1 and CO in urea in the wavenumber range from 1628 cm−1 to 1658 cm−1 varied with the molar ratio of hard and soft segments. The thermal properties of HM-PUs were investigated by DSC analysis (Fig. 1d); the thermograms show the variational tendency of HM-PUs due to the change in the intermolecular forces between hard and soft segments, and the glass transition temperature (Tg) of HM-PU1 reaches up to −56.4 °C than any others, indicating that hydrogen bonds and hard segments will significantly affect the diffusion of polymer chains. Also, we obtained the results of decomposition temperature to be approximately 273 °C from the TGA curves in Fig. S1.† In addition, the FTIR spectra of HM-PUs in the range from 1600 cm−1 to 1750 cm−1 are assigned to CO groups, and the absorptive region of CO is deconvoluted into four subpeaks; these subpeaks can be assigned to CO in urea and urethane groups (Table S2†).40,41 Then, the content of four kinds of CO groups are listed in Fig. 2d such as free urethane amide, urethane amide (CO), and urea amide (CO). We can come to a conclusion from the bar diagram that the CO content of HM-PU3 (19%) in the free urethane amide region is much higher than the contents of HM-PU1 (7%) and HM-PU2 (8%), and the ordered hydrogen bonds of HM-PU1 built by the CO groups in urea amide reaches the maximum value (19.5%), indicating that the intermolecular force is stronger than that of the others (HM-PU2 and HM-PU3). As these analyses mentioned above, the results of DSC and FTIR indicate that free urethane amide will accelerate the diffused rate of polymer chains, and the metastable crystal in HM-PU is formed by ordered hydrogen bonds between the urea amide groups and hydrogen atoms.
O groups. It is worth noting that the characteristic peaks of CO in urethane at 1718 cm−1 and CO in urea in the wavenumber range from 1628 cm−1 to 1658 cm−1 varied with the molar ratio of hard and soft segments. The thermal properties of HM-PUs were investigated by DSC analysis (Fig. 1d); the thermograms show the variational tendency of HM-PUs due to the change in the intermolecular forces between hard and soft segments, and the glass transition temperature (Tg) of HM-PU1 reaches up to −56.4 °C than any others, indicating that hydrogen bonds and hard segments will significantly affect the diffusion of polymer chains. Also, we obtained the results of decomposition temperature to be approximately 273 °C from the TGA curves in Fig. S1.† In addition, the FTIR spectra of HM-PUs in the range from 1600 cm−1 to 1750 cm−1 are assigned to CO groups, and the absorptive region of CO is deconvoluted into four subpeaks; these subpeaks can be assigned to CO in urea and urethane groups (Table S2†).40,41 Then, the content of four kinds of CO groups are listed in Fig. 2d such as free urethane amide, urethane amide (CO), and urea amide (CO). We can come to a conclusion from the bar diagram that the CO content of HM-PU3 (19%) in the free urethane amide region is much higher than the contents of HM-PU1 (7%) and HM-PU2 (8%), and the ordered hydrogen bonds of HM-PU1 built by the CO groups in urea amide reaches the maximum value (19.5%), indicating that the intermolecular force is stronger than that of the others (HM-PU2 and HM-PU3). As these analyses mentioned above, the results of DSC and FTIR indicate that free urethane amide will accelerate the diffused rate of polymer chains, and the metastable crystal in HM-PU is formed by ordered hydrogen bonds between the urea amide groups and hydrogen atoms.
 | ||
| Fig. 1 General characterization of HM-PUs. (a) UV-vis spectra of HM-PUs in the visible light wavelength range. (b) FTIR spectra of HM-PUs in the wavenumber range from 400 cm−1 to 4000 cm−1. (c) 1H NMR spectrum of HM-PU2. (d) DSC curves of HM-PUs in the temperature range of −85 °C to 85 °C. | ||
 | ||
| Fig. 2 FTIR spectra of HM-PUs in the CO stretching region. | ||
Self-healing and mechanical properties of HM-PUs
The polymer network of HM-PU can self-heal after rupturing due to the interesting process of breakage and rebuilding of hydrogen bonds, as illustrated in Fig. 3a. Subsequently, a bloom-shaped film was cut into two pieces, followed by placing the two sections in contact in 50 °C after 5 h, and the pieces completely connected under stretching. The three samples such as HM-PU1, HM-PU2, and HM-PU3 were put into different temperature atmospheres (100 °C, 70 °C, and 50 °C), respectively, for 30 min. Thus, the optical microscopy images show that HM-PUs have a good self-healing property. | ||
| Fig. 3 Molecular network of HM-PU and optical photos. (a) Chemical structure and self-healing mechanism of HM-PU. (b) The self-healing process of the bloom-shaped film. (c) Optical microscopy photos of the scratch films showing a good appearance during the self-healing process (HM-PU1-100 °C-30 min, HM-PU2-70 °C-30 min, HM-PU3-50 °C-30 min). | ||
One attractive phenomenon we can see is that the mechanical properties are variational by adjusting the molar ratios of soft and hard segments of HM-PUs (Fig. 4 and Table S3†). In Fig. 4b, HM-PU1 shows an extreme tensile strength of 39.7 MPa, and the tensile strength, toughness, and Young's modulus obviously decreased promptly with decreasing content of IPDA as the extender. The following reasons account for this: (1) As is usual, the hydrogen bonds between urea groups are stronger than the hydrogen bonds between urethane groups, and the content of the segments of IPDA in the polymer network of HM-PU1 are more than that of the two other HM-Pus; (2) The more the number of soft segments networks, the faster the diffusion speed of the polymer chains. It is interesting to note that the HM-PUs films exhibit enormous crack tolerance due to the extraordinary energy dissipation capacity. In Fig. 4a, the HM-PU1 film with a notch of 1 mm was stretched up to 400% (approximately 4 times) of its original length, and the crack was extended as a smooth notch, indicating that the elastomer with cracks can still work at high elongation. The fracture energies of HM-PUs were calculated to be in the range from 45 kJ m−2 to 60.34 kJ m−2 in the ESI (Fig. S4†). In addition, the fracture energy is higher than the fracture energies of most elastomers and some alloys.42 In order to further evaluate the self-healing efficiency of HM-PUs, the specimens were tested by tensile tests of HM-PUs after different times at a specific temperature (Fig. 4c–e). The self-healing efficiency is defined as the ratio of the stress or elongation to that of the virgin samples, and the self-healing efficiency is characterized by the two formulas as follows.
 | (1) |
 | (2) |
 | ||
| Fig. 4 The mechanical properties of HM-PUs. (a) A notched HM-PU1 specimen at a strain of 400%. (b) Typical stress–strain curves of the virgin and notched samples. Typical stress–strain curves of virgin and self-healed samples. (c) HM-PU3 healed at 50 °C. (d) HM-PU2 healed at 70 °C. (e) HM-PU1 healed at 100 °C. | ||
The self-healing efficiency depicted in Fig. 4 and S5† shows its variation tendency in terms of stress and elongation as the variable factors of temperature and healing time, respectively. As we can see, the self-healing efficiency of HM-PUs improves with time or temperature. Also, HM-PU3 could heal in a moderate atmosphere (50 °C) for a short time, while the corresponding self-healing efficiency of the self-healed samples of HM-PU1 and HM-PU2 at 50 °C is so weak that the two samples fractured rapidly; hence, the results will not be shown in this article. When the healing temperature is set as 70 °C and 100 °C, respectively, the self-healing efficiency of HM-PU1 and HM-PU2 increases stepwise. After 10 h at 50 °C, the healing efficiency of HM-PU3 exhibits appreciable value (90% in terms of elongation, 80% in terms of stress). Compared with HM-PU1 and HM-PU2, the self-healing performance is not enough, showing a macroscopic decline. Certainly, the self-healing mechanism could be interpreted by the FTIR analysis in the section of general characterization. It should be thought to be due to the dynamic consecutive exchange and recombination of enhanced hydrogen bonds as the molecular chains diffuse to the fracture surface area, and the mechanical properties were recovered; most importantly, the key factor of diffusion of molecular chains is the number of soft segments. Thus, the healing efficiency of HM-PU3 is the highest than the two others. Also, benefiting from the dynamic property of hydrogen bonds, HM-PUs have a good recyclable character in the field of environmental protection (Fig. S6†). In summary, on account of the significant effects of hydrogen bond in the elastomer, in consequence, we can get one kind of specific material with ideal properties by adjusting the balance between the self-healing property and the mechanical properties.43,44
The diffused properties of polymer chains
In order to study the self-healing mechanism, UV-vis spectra and molecular dynamics simulation (MD) were utilized to study the properties of polymer chains.The pieces (0.4 g) of HM-PU1, HM-PU2, and HM-PU3 were added into three bottles and immersed in THF solvent (17 mL) for 8 h. The mutative process of HM-PUs fragments in THF had been recorded by a camera by taking photos at different times. In the initial state, the pieces in the bottles can be clearly observed, whereas these pieces disappeared after 8 h, exhibiting transparent liquids, indicating that a good deal of pieces had been dissolved. Subsequently, the solution in the bottles was removed, and some residues in the bottom of the bottles display three kinds of forms as the colloidal gel, colloidal gel/viscous liquid, and viscous liquid for HM-PU1, HM-PU2, and HM-PU3, respectively. Therefore, HM-PU3 is easier to dissolve in organic solvents, revealing that the diffusion coefficient of its polymer chains is higher than others on account of more soft segments and fewer hydrogen bonds. In order to represent the quantitative level of polymer chains in the procedure of diffusion, UV-vis measurements were employed. As shown in Fig. 5, obvious characteristic peaks belonging to HM-PU1, HM-PU2, and HM-PU3 at about 241 nm, 240 nm, and 238 nm, respectively, are due to carbonyl in urea and urethane groups, wherein the types of electronic transition are n–π* and n–σ. After 2 h, the absorption peak of HM-PU1 is as high as 0.59, whereas the peak of HM-PU3 as low as 0.2. The reasons we can conclude is as follows: (1) the number of CO groups in polymer chains dissolved in THF reached the highest value compared to others; (2) The hyperchromic effect of –NHR groups in urea is stronger than that of the –OCR groups in urethane. Moreover, the characteristic peak of CO shifts to the region of long wavelength due to the hyperchromic effect of –NHR groups, meaning redshift. The most important conclusion we can obtain from Fig. 5 is that the absorption peaks of HM-PU3 grow more quickly than that of HM-PU1 and HM-PU2 with time passing by, revealing the phenomenon that the polymer chains of HM-PU3 can diffuse more quickly, indicating the HM-PU3 elastomer can heal in a shorter time.
 | ||
| Fig. 5 (a) The mutative process of HM-PUs immersed in THF. UV-vis spectra of the solution of HM-PUs in THF after certain times. (b) The UV-vis spectra of HM-PUs after 2 h. (c) The UV-vis spectra of HM-PU1 in the time range of 2 h to 8 h. (d) The UV-vis spectra of HM-PU2 in the time range of 2 h to 8 h. (e) The UV-vis spectra of HM-PU3 in the time range of 2 h to 8 h. | ||
In order to describe the diffused behavior of the molecular chains accurately, molecular dynamics simulation (MD) was used to simulate their dynamic behaviors. In these cases, all MD simulations for the two cell systems were carried out with the commercial software Materials Studio (MS) (Accelrys Inc., San Diego, Version 2019) with COMPASS force field. The amorphous cells were built by the amorphous cell module containing ten molecular chains, which include the single molecular chain with one PTMEG segment, one IPDA extender, and that with two PTMEG segments, one IPDA extender for PU1:1 and PU2:1, respectively (Fig. 6a and b). The density of all the cells is 0.5 g cm−3. Then, the two cells were constructed utilizing COMPASS force field to assign the charges, followed by minimizing the structures via Forcite module until convergence. They were carried out for MD using the NPT ensemble for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ps with 1 fs time step, and one frame was recorded every 10 steps; eventually, full trajectories were saved. The MSD can be calculated using the following formula45
000 ps with 1 fs time step, and one frame was recorded every 10 steps; eventually, full trajectories were saved. The MSD can be calculated using the following formula45
 | (3) |
 | ||
| Fig. 6 Snapshots of the MD simulation of the cell structures of HM-PU with ten representative polymer chains. (a) One PTMEG segment and one IPDA extender. (b) Two PTMEG segments and one IPDA extender. (c) The energies of simulated cell systems. (d) The results of the mean-squared displacements (MSD) of HM-PU. | ||
Whereafter, the two structures were annealed via one annealing cycle of a linear heating and cooling process from 300 K to 600 K and from 600 K to 300 K, respectively. The simulation was implemented under the NPT ensemble for 25000 steps and the time step was 1 fs. Subsequently, the cohesive energy per chain, defined as the average energy of every molecular chain diffusing from the condensed state to an infinite distance from one another, was calculated. In the amorphous cells we constructed above, the cohesive energy per molecular chain can be calculated by the following formula46
 | (4) |
To sum up, the MSD and cohesive energy were calculated using MD simulation; the cohesive energy of PU1:1 (73.53 kcal mol−1) is much higher than that of PU2:1 (67.97 kcal mol−1), indicating that PU2:1 per chain needs more energy to be at an infinite distance, and the tendencies of PU can also be observed in the MSD curves.
It can be said that the UV-vis spectra and MD simulation were used to verify the inner mechanism of the healing process, revealing a phenomenon that the more the number of hard segment polymer chains, the lower the diffused rate.
The studies of residual strain and energy dissipation of HM-PUs
In order to investigate the energy dissipations and residual strains of HM-PUs, they were further subjected to the mechanical tests of cyclic loading/unloading with various maximum strains; the cyclic speed was 100 mm min−1 (Fig. 7), and all the samples could recover quickly. In Fig. 7b, the residual strain and energy dissipation increase with the maximum due to a great deal of hydrogen bonds, and some hydrogen bonds could fracture as the strain increases.47 To explore the elastic properties of HM-PU, the residual strain (εrs) was executed to be normalized by the maximum strain (εms), and the computational formula is εrs/εms. The data of normalized strain (εrs/εms) can be calculated from Fig. 7a; the εrs/εms of HM-PU1 increased with the maximum rapidly. Also, the slope for εrs/εms of HM-PU2 and HM-PU3 is almost zero, indicating that their rupture degree and microphase separation is not enhanced at high elongation; nevertheless, HM-PU1 exhibits the reverse phenomenon. In addition, the variational tendency of energy dissipation is analogous to that of residual strain (Fig. 7d and S2†). In summary, the polymer chains with more hard segments will dissipate more energy in the mechanical deformation process.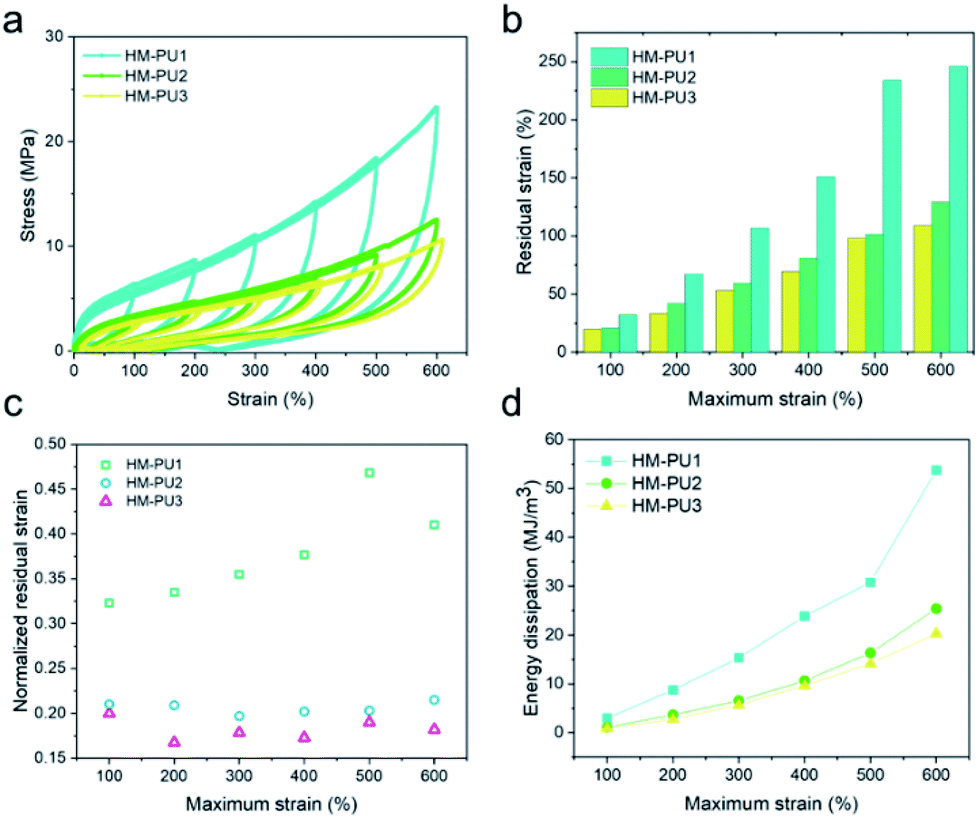 | ||
| Fig. 7 (a) Typical cyclic curves of HM-PUs. (b) Residual strain of HM-PUs at different maximum strains. (c) Normalized residual strain obtained from (a). (d) Energy dissipations of HM-PUs at different maximum strains. | ||
The shape memory properties of HM-PUs
On account of dense multiple hydrogen bonds in polymer chains of HM-PU, they could reversibly alternate between one state of the transformed shape and another state of the original untransformed shape through the stimulus of heat.48In Fig. 8, we designed three models such as “OK”-shaped film, spiral film, and box-shaped film. All the films were placed under 70 °C to deform, followed by placing them at room temperature, and the shapes were fixed; subsequently, these distorted films recovered to their initial shapes under the stimulus of 70 °C temperature. For the purpose of quantifying the rate of the shape memory, the film of HM-PU1 was deformed at 70 °C, whereafter it was fixed in room temperature atmosphere, followed by heating it at 70 °C once again, and the process of changing the angle was monitored via a camera. The angle of this film recovered from 0° to 180° in 25 s, and the interval times between the two angles we recorded to show a linear increase, indicating that the efficiency of shape recovery decreases with time.
 | ||
| Fig. 8 The shape memory behavior of HM-PU1. (a) Reversible shape transition of different shapes (“OK”, “spiral”, and “box”) stimulated at 70 °C. (b) The changing process of the angle. | ||
Furthermore, the HM-PU1 film stretched to 300% was employed in the process of shape fixing and recovery, and underwent six shape memory cycles to test the durability of shape memory (Fig. 9). As shown in Fig. 9c, the recovery property of HM-PU1 is excellent; even after undergoing six cycles, the wastage of shape memory performance is negligible. But the fixed shapes of HM-PU2 and HM-PU3 cannot be maintained in the elongation of 300% and rebound to a value lower than 300% because the energies of hydrogen bonds in the stretched polymer chains cannot completely counteract the energy of resilience of the stretched polymer network. In order to reveal the mechanism of the shape memory behavior, the XRD was used to test HM-PU1 at different elongations. In the XRD curves (Fig. 9f), the characteristic peaks changed with elongation, and some weak peaks appear at 40°; these peaks disappeared until the film was heated to the initial elongation, revealing that the microstructure of HM-PU1 undergoes a conversion from the uncrystallized form to the crystalline form and as well from the crystalline form to the uncrystallized form in the shape memory process (Fig. S7†).49 Based on the mechanism, four strings of HM-PU1 were used to suture two pieces of papers, followed by heating it by a heat gun, and the gap between the papers was disappeared quickly; thus, it can be seen that this kind of material can be applied in the medical field such as in sutures.
 | ||
| Fig. 9 The shape memory properties of HM-PUs. (a) Photos of HM-PU1 in the original state, the state stretched to 300% and the recovered state at 70 °C. (b) The two pieces of papers were sutured by four bands of HM-PU1 films. The HM-PU films were stretched to 300% and fixed at room temperature, then recovered to their original state for six cycles at 70 °C. (c) HM-PU1. (d) HM-PU2. (e) HM-PU3. (f) XRD of HM-PU1 in the original state and the states at 50%, 100%, 200% and 300%. | ||
Conclusions
In summary, inspired by silkworm fibril, we have developed one kind of elastomer with a number of excellent properties such as high transmittance (99%), high tensile strength at break (39 MPa), splendid toughness (111.65 MJ m−3), high self-healing efficiency, notch insensitive character, and shape memory property. The great mechanical properties are originated from the hydrogen bond arrays, and the network that soft segments contribute to the high elasticity. Due to the special structure comprising several dynamic multiple hydrogen bonds, it endows this elastomer with sustainability and durability. The elastomer we designed by a simple synthetic method is a kind of material with self-healing and shape memory properties, not only paving the way for designing and manufacturing the materials with extraordinary mechanical properties, but also for application in the fields of flexible electronics, medical apparatus, and shape memory robots.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support for this work from the National Natural Science Foundation of China (No. 51872237 and 21301161).Notes and references
- S. B. Patil, S. Z. Inamdar, K. R. Reddy, A. V. Raghu, K. G. Akamanchi, A. C. Inamadar, K. K. Das and R. V. Kulkarni, In-vitro and Ex-vivo Evaluation, Drug Delivery Lett., 2020, 10(3), 185–196 CAS.
- D. Kasai, R. Chougale, S. Masti, G. Gouripur, R. Malabadi, R. Chalannavar, A. V. Raghu, D. Radhika and H. Shanavaz, Green Mater., 2021, 9(2), 49–68 CrossRef.
- R. Chougale, D. Kasai, S. Nayak, S. Masti, A. Nasalapure and A. V. Raghu, Green Mater., 2020, 8(1), 40–48 CrossRef.
- A. V. Nasalapure, R. K. Chalannavar, D. R. Kasai, K. R. Reddy and A. V. Raghu, Nano Express, 2021, 2(3), 030003 CrossRef.
- G. Divyashri, R. V. Badhe, B. Sadanandan, V. Vijayalakshmi, M. Kumari, P. Ashrit, D. Bijukumar, M. T. Mathew, K. Shetty and A. V. Raghu, Polym. Adv. Technol., 2022, 33(7), 2025–2043 CrossRef CAS.
- A. Nagaraja, M. D. Jalageri, Y. M. Puttaiahgowda, K. R. Reddy and A. V. Raghu, J. Microbiol. Methods, 2019, 163, 105650 CrossRef CAS PubMed.
- S. B. Patil, S. Z. Inamdar, K. K. Das, K. G. Akamanchi, A. V. Patil, A. C. Inamadar, K. R. Reddy, A. V. Raghu and R. V. Kulkarni, J. Drug Delivery Sci. Technol., 2020, 56, 101525 CrossRef CAS.
- K. Wang, J. H. Wang, L. Li, L. Q. Xu, N. Feng, Y. Wang, X. Fei, J. Tian and Y. Li, ACS Biomater. Sci. Eng., 2020, 6(2), 1259–1268 CrossRef CAS PubMed.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18(8), 997–1000 CrossRef CAS.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6(8), 581–585 CrossRef CAS PubMed.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2(8), 694–699 CrossRef CAS PubMed.
- S. B. Ji, W. Cao, Y. Yu and H. P. Xu, Adv. Mater., 2015, 27(47), 7740–7745 CrossRef CAS PubMed.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. A. Bao, Nat. Chem., 2016, 8(6), 619–625 CrossRef PubMed.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48(11), 3539–3546 CrossRef CAS.
- H. M. Chen, J. J. Koh, M. M. Liu, P. J. Li, X. T. Fan, S. Q. Liu, J. C. C. Yeo, Y. J. Tan, B. C. K. Tee and C. B. He, ACS Appl. Mater. Interfaces, 2020, 12(28), 31975–31983 CrossRef CAS PubMed.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25(20), 2849–2853 CrossRef CAS PubMed.
- L. L. Chen, W. W. Feng, M. X. Li, Z. K. Jin, Y. C. Zhang, Z. Cheng, Y. C. Liu and C. Wang, Adv. Funct. Mater., 2022, 32(1), 2107538 CrossRef CAS.
- X. Z. Yan, Z. Y. Liu, Q. H. Zhang, J. Lopez, H. Wang, H. C. Wu, S. M. Niu, H. P. Yan, S. H. Wang, T. Lei, J. H. Li, D. P. Qi, P. G. Huang, J. P. Huang, Y. Zhang, Y. Y. Wang, G. L. Li, J. B. H. Tok, X. D. Chen and Z. A. Bao, J. Am. Chem. Soc., 2018, 140(15), 5280–5289 CrossRef CAS PubMed.
- M. W. M. Tan, G. Thangavel and P. S. Lee, Adv. Funct. Mater., 2021, 31(34), 2103097 CrossRef CAS.
- X. P. Chen, P. Z. Fei, K. A. Cavicchi, W. W. Yang and N. Ayres, Colloid Polym. Sci., 2014, 292(2), 477–484 CrossRef CAS.
- J. X. Cui and A. Del Campo, Chem. Commun., 2012, 48(74), 9302–9304 RSC.
- B. H. Ji and H. J. Gao, Annu. Rev. Mater. Res., 2010, 40, 77–100 CrossRef CAS.
- M. E. Launey, M. J. Buehler and R. O. Ritchie, Annu. Rev. Mater. Res., 2010, 40, 25–53 CrossRef CAS.
- M. A. Meyers, P. Y. Chen, A. Y. M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53(1), 1–206 CrossRef CAS.
- A. M. Kushner and Z. B. Guan, Angew. Chem., Int. Ed., 2011, 50(39), 9026–9057 CrossRef CAS PubMed.
- F. G. Omenetto and D. L. Kaplan, Science, 2010, 329(5991), 528–531 CrossRef CAS PubMed.
- H. Tao, D. L. Kaplan and F. G. Omenetto, Adv. Mater., 2012, 24(21), 2824–2837 CrossRef CAS PubMed.
- A. Bucciarelli, V. Mulloni, D. Maniglio, R. K. Pal, V. K. Yadavalli, A. Motta and A. Quaranta, Opt. Mater., 2018, 78, 407–414 CrossRef CAS.
- G. Guidetti and F. G. Omenetto, MRS Commun. Commun., 2020, 10(2), 201–214 CrossRef CAS.
- B. D. Lawrence, M. Cronin-Golomb, I. Georgakoudi, D. L. Kaplan and F. G. Omenetto, Biomacromolecules, 2008, 9(4), 1214–1220 CrossRef CAS PubMed.
- G. Matzeu, L. Mogas-Soldevila, W. Y. Li, A. Naidu, T. H. Turner, R. Gu, P. R. Blumeris, P. Song, D. G. Pascal, G. Guidetti, M. Li and F. G. Omenetto, Adv. Mater., 2020, 32(28), 2001258 CrossRef CAS PubMed.
- F. Paquet-Mercier, T. Lefevre, M. Auger and M. Pezolet, Soft Matter, 2013, 9(1), 208–215 RSC.
- T. Yoshioka, Y. Takasu, H. Sezutsu and T. Kameda, ACS Biomater. Sci. Eng., 2018, 4(3), 832–835 CrossRef CAS PubMed.
- S. Keten and M. J. Buehler, Nano Lett., 2008, 8(2), 743–748 CrossRef CAS PubMed.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30(1), 1705145 CrossRef PubMed.
- W. H. Fan, Y. Jin, L. J. Shi, W. N. Du, R. Zhou, S. Q. Lai, Y. C. Shen and Y. P. Li, ACS Appl. Mater. Interfaces, 2020, 12(5), 6383–6395 CrossRef CAS PubMed.
- L. Z. Zhang, Z. H. Liu, X. L. Wu, Q. B. Guan, S. Chen, L. J. Sun, Y. F. Guo, S. L. Wang, J. C. Song, E. M. Jeffries, C. L. He, F. L. Qing, X. G. Bao and Z. W. You, Adv. Mater., 2019, 31(23), 1901402 CrossRef PubMed.
- F. Li, Q. Q. Ye, Q. Gao, H. Chen, S. Q. Shi, W. R. Zhou, X. N. Li, C. L. Xia and J. Z. Li, ACS Appl. Mater. Interfaces, 2019, 11(17), 16107–16116 CrossRef CAS PubMed.
- T. Naganuma and Y. Kagawa, Compos. Sci. Technol., 2002, 62(9), 1187–1189 CrossRef CAS.
- M. M. Coleman, D. J. Skrovanek, J. B. Hu and P. C. Painter, Macromolecules, 1988, 21(1), 59–65 CrossRef CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19(8), 2149–2157 CrossRef CAS.
- W. Cui, D. R. King, Y. W. Huang, L. Chen, T. L. Sun, Y. Z. Guo, Y. Saruwatari, C. Y. Hui, T. Kurokawa and J. P. Gong, Adv. Mater., 2020, 32(31), 1907180 CrossRef CAS PubMed.
- A. V. Raghu, G. S. Gadaginamath, N. T. Mathew, S. B. Halligudi and T. M. Aminabhavi, React. Funct. Polym., 2007, 67(6), 503–514 CrossRef CAS.
- A. V. Raghu, G. S. Gadaginamath, S. S. Jawalkar, S. B. Halligudi and T. M. Aminabhavi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(20), 6032–6046 CrossRef CAS.
- M. A. Ratner, Phys. Today, 1997, 50(7), 66 Search PubMed.
- Z. W. Li, Z. Y. Lu, Z. Y. Sun, Z. S. Li and L. J. An, J. Phys. Chem. B, 2007, 111(21), 5934–5940 CrossRef CAS PubMed.
- K. P. Cui, T. L. Sun, X. B. Liang, K. Nakajima, Y. N. Ye, N. Chen, T. Kurokawa and J. P. Gong, Phys. Rev. Lett., 2018, 121(18), 185501 CrossRef PubMed.
- C. B. Cooper, S. Nikzad, H. P. Yan, Y. Ochiai, J. C. Lai, Z. A. Yu, G. Chen, J. Kang and Z. N. Bao, ACS Cent. Sci., 2021, 7(10), 1657–1667 CrossRef CAS PubMed.
- Y. Eom, S. M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12(1), 621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03250c |
| This journal is © The Royal Society of Chemistry 2022 |