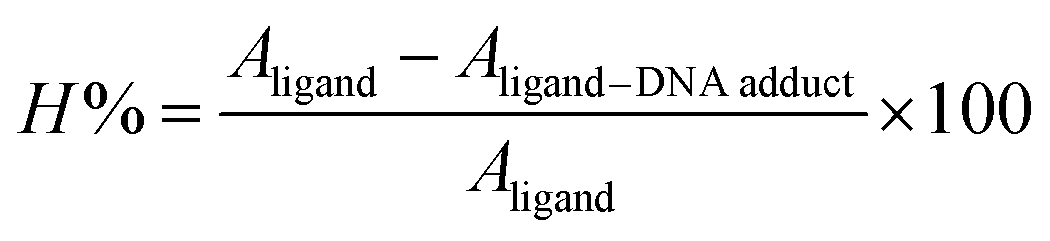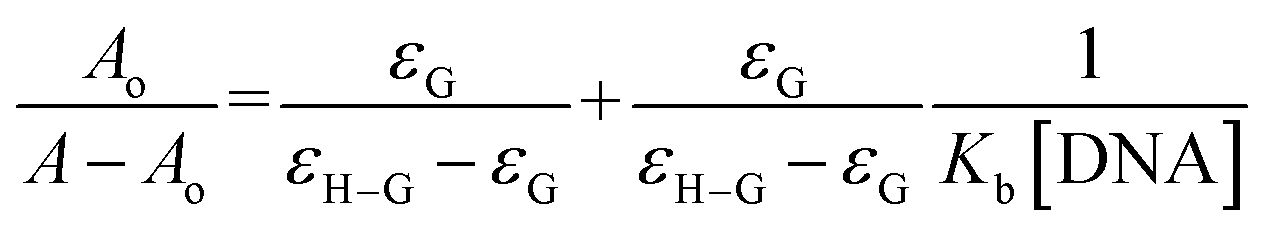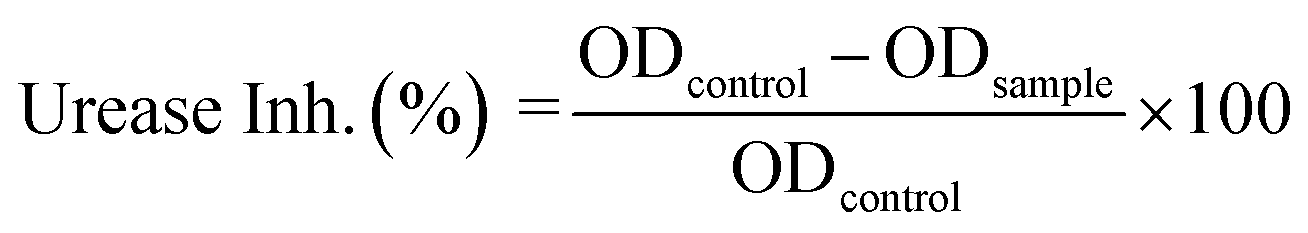DOI:
10.1039/D2RA03160D
(Paper)
RSC Adv., 2022,
12, 17194-17207
Structure and surface analyses of a newly synthesized acyl thiourea derivative along with its in silico and in vitro investigations for RNR, DNA binding, urease inhibition and radical scavenging activities†
Received
19th May 2022
, Accepted 6th June 2022
First published on 10th June 2022
Abstract
N-((4-Acetylphenyl)carbamothioyl)-2,4-dichlorobenzamide (4) was synthesized by the treatment of 2,4-dichlorobenzoyl chloride with potassium thiocyanate in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in dry acetone to afford the 2,4-dichlorobenzoyl isothiocyanate in situ which on reaction with acetyl aniline furnished (4) in good yield and high purity. The compound was confirmed by FTIR, 1H-NMR, and 13C-NMR and single crystal X-ray diffraction studies. The planar rings were situated at a dihedral angle of 33.32(6)°. The molecules, forming S(6) ring motifs with the intramolecular N–H⋯O hydrogen bonds, were linked through intermolecular C–H⋯O and N–H⋯S hydrogen bonds, enclosing R22(8) ring motifs, into infinite double chains along [101]. C–H⋯π and π⋯π interactions with an inter-centroid distance of 3.694 (1) Å helped to consolidate a three-dimensional architecture. Hirshfeld surface (HS) analysis further indicated that the most important contributions for the crystal packing were from H⋯C/C⋯H (20.9%), H⋯H (20.5%), H⋯Cl/Cl⋯H (19.4%), H⋯O/O⋯H (13.8%) and H⋯S/S⋯H (8.9%) interactions. Thus C–H⋯π (ring), π⋯π, van der Waals interactions and hydrogen bonding played the major roles in the crystal packing. The electronic structure and computed DFT (density functional theory) parameters identified the reactivity profile of compound (4). In silico binding of (4) with RNA indicated the formation of a stable protein–ligand complex via hydrogen bonding, while DNA docking studies inferred (4) as a potent groove binder. The experimentally observed hypochromic change (57.2%) in the UV-visible spectrum of (4) in the presence of varying DNA concentrations together with the evaluated binding parameters (Kb; 7.9 × 104 M−1, ΔG; −28.42 kJ mol−1) indicated spontaneous interaction of (4) with DNA via groove binding and hence supported the findings obtained through docking analysis. This compound also showed excellent urease inhibition activity in both in silico and vitro studies with an IC50 value of 0.0389 ± 0.0017 μM. However, the radical scavenging efficiency of (4) was found to be modest in comparison to vitamin C.
:1 molar ratio in dry acetone to afford the 2,4-dichlorobenzoyl isothiocyanate in situ which on reaction with acetyl aniline furnished (4) in good yield and high purity. The compound was confirmed by FTIR, 1H-NMR, and 13C-NMR and single crystal X-ray diffraction studies. The planar rings were situated at a dihedral angle of 33.32(6)°. The molecules, forming S(6) ring motifs with the intramolecular N–H⋯O hydrogen bonds, were linked through intermolecular C–H⋯O and N–H⋯S hydrogen bonds, enclosing R22(8) ring motifs, into infinite double chains along [101]. C–H⋯π and π⋯π interactions with an inter-centroid distance of 3.694 (1) Å helped to consolidate a three-dimensional architecture. Hirshfeld surface (HS) analysis further indicated that the most important contributions for the crystal packing were from H⋯C/C⋯H (20.9%), H⋯H (20.5%), H⋯Cl/Cl⋯H (19.4%), H⋯O/O⋯H (13.8%) and H⋯S/S⋯H (8.9%) interactions. Thus C–H⋯π (ring), π⋯π, van der Waals interactions and hydrogen bonding played the major roles in the crystal packing. The electronic structure and computed DFT (density functional theory) parameters identified the reactivity profile of compound (4). In silico binding of (4) with RNA indicated the formation of a stable protein–ligand complex via hydrogen bonding, while DNA docking studies inferred (4) as a potent groove binder. The experimentally observed hypochromic change (57.2%) in the UV-visible spectrum of (4) in the presence of varying DNA concentrations together with the evaluated binding parameters (Kb; 7.9 × 104 M−1, ΔG; −28.42 kJ mol−1) indicated spontaneous interaction of (4) with DNA via groove binding and hence supported the findings obtained through docking analysis. This compound also showed excellent urease inhibition activity in both in silico and vitro studies with an IC50 value of 0.0389 ± 0.0017 μM. However, the radical scavenging efficiency of (4) was found to be modest in comparison to vitamin C.
1 Introduction
A thiourea moiety has a certain binding site which includes a hydrogen bonding site (NH), corelative binding areas (S) and secondary binding areas {1,3 substituents}.1 A ring system becomes flexible when a thiourea moiety is introduced which enhances the anticancer potential towards several cancers including colon, breast, liver, lungs, renal and ovarian cancer.2–5 Since sulphur is a weak acceptor towards a hydrogen bond, it can enhance blocking. It can also increase the bonding at the same time due to the bidentate binding mode of thiourea protons.6 Thiourea moieties bearing nitrogen and sulphur atoms are aromatase enzyme inhibitors. Aromatase enzyme is the key element to produce increased amounts of estrogen. Consequently, by inhibiting aromatase enzyme, the quantity of estrogen can selectively be decreased.7 Due to this inhibitory property, it plays a vital role in healing breast cancer. N-substituted phenyl thioureas help against melanoma (skin cancer) by inhibiting the production of melanin.8,9 These recent anticancer activities of thioureas encourage chemists and pharmacologists to design and synthesize new thiourea derivatives.
Acyl thiourea derivatives can coordinate metals as a bidentate ligand using both the carbonyl oxygen and the thiocarbonyl sulfur or a monodentate ligand via the thiocarbonyl sulfur only.10 A wide range of 1-(acyl/aroyl)-3-(substituted) thioureas are extremely flexible starting materials for the synthesis of a many of heterocyclic compounds due to the presence of two free hydrogen atoms, one at each nitrogen atom.11 Acyl thiourea derivatives have received considerable attention over the years and a vast spectrum of biological activities including antitumor,12 herbicidal,13 antiviral,14 antiparasitic,15 antimicrobial,16 insecticidal,17 and fungicidal18 properties are shown by 1-(acyl/aroyl)-3-(substituted) thioureas. In addition, the anticancer properties of acyl thiourea derivatives have also been reported in ref. 19–21.
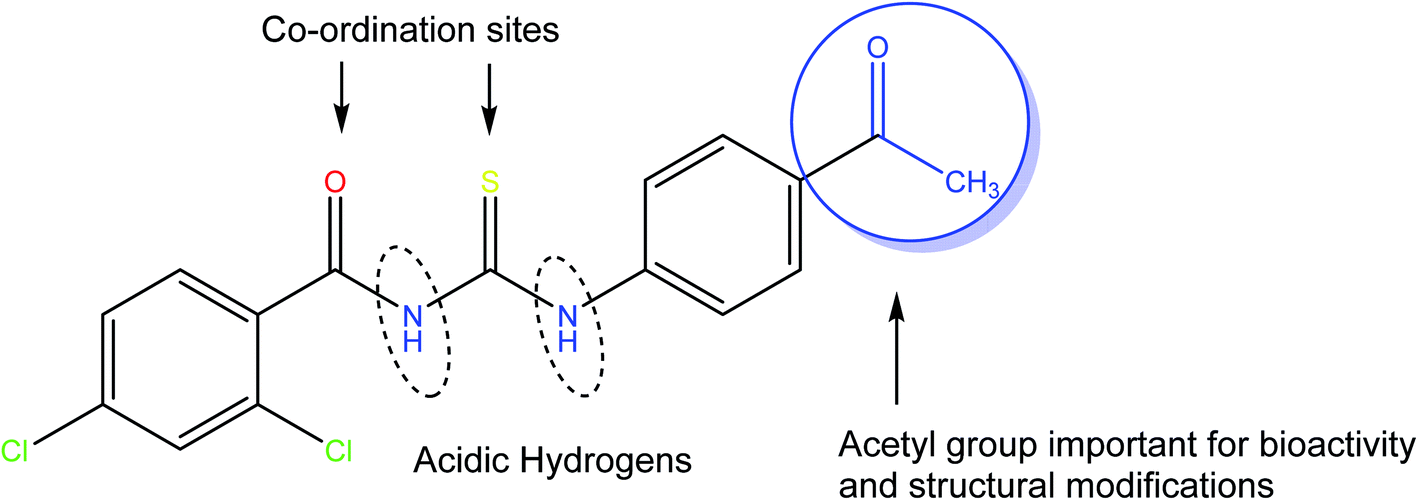
Herein, we are reporting the synthesis, crystal structure, electronic structure, and surface analysis for a new acyl thiourea derivative namely N-((4-acetylphenyl) carbamothioyl)-2,4-dichlorobenzamide (4). The availability of free acetyl group is not only important for biological activities but also broadens its synthetic scope for further structural modifications. The presence of carbonyl and thiocarbonyl are sites for coordination with metals and thiouiredic NH proton flanked by carbonyl and thiocarbonyl is more acidic compared to thioamide H and both are removable by base and are involved in inter and intra molecular H-bonding (Fig. 1). It was further investigated for in silico and in vitro RNR and DNA binding, urease inhibition and free radical scavenging activities.
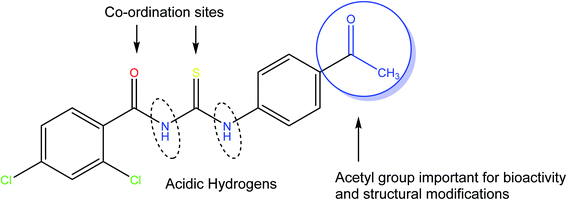 |
| | Fig. 1 The molecular architecture of the title novel thiourea molecule (4). | |
2 Experimental
2.1 Materials and methods
Analytical grade chemicals and reagents were used throughout the experimental processes. Standard methods were used for the purification and drying of the solvents. Melting point was determined using MP-D – a Gallen Kamp melting point apparatus. Infrared spectrum was obtained on FTIR spectrophotometer (460-Shimadzu) using KBr disc. 1H-NMR spectrum was recorded on 300 MHz Bruker NMR spectrometer in acetone-d6 whilst 13C-NMR spectrum was obtained at 75 MHz using tetramethyl silane (TMS) as an internal reference. Mass spectrum was recorded in the electron impact mode (EI, 70 eV) on a GC/MS Agilent Technologies 6890N instrument. Thin-layer chromatography (TLC) was performed on precoated silica gel aluminum plate (layer thickness 0.2 mm, HF 254, Reidalde-Haen from Merck). The chromatograms were visualized using ultraviolet light (254 and 260 nm). For DNA binding and urease inhibition studies, the Salmon fish sperm DNA and Jack bean urease enzyme, respectively, were obtained from Sigma-Aldrich. UV-visible spectrophotometer (Shimadzu-1800) was used in DNA binding studies, while a microplate reader (OPTI Max, Tunable) was used for urease and free radical inhibition studies.
2.2 Synthesis of N-((4-acetylphenyl) carbamothioyl)-2,4-dichlorobenzamide (4)
The novel acyl thiourea derivative (4) was synthesized from corresponding aromatic acid chloride. Under the inert environment, the acid chloride (1) (0.5 mmol in 20 mL anhydrous acetone) was drop wisely added to the potassium thiocyanate (10 mmol in 20 mL anhydrous acetone) and the reaction mixture refluxed for 3 hours. Then the reaction mixture was cooled to room temperature, and a solution of 4-aminoacetophenone (3) (0.5 mmol in acetone) was added. This reaction mixture was further stirred for 24 hours, and the reaction's progress was thoroughly monitored by TLC. On completion of reaction, the mixture was poured into the chilled water which precipitated the product that was filtered out. Finally, the crude product was recrystallized with EtOH to afford (4) as colorless crystals.
2.3 Characterization data
White solid; m. p.: 207–212 °C; yield: 60% Rf: 0.48, ethyl acetate: n-hexane (4:6); IR (cm−1): 3237 (NH stretch), 2958 (sp2 CH-Ar), 2939 (sp3 CH), 1657 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1529 (CO), 1179 (CS); 753 (C–Cl bend); 1H-NMR (300 MHz, acetone): δ 12.96 (s, 1H, NH), 10.18 (s, 1H, NH), 8.80–7.78 (m, 7H, Ar-H),2.52 (s, 3H, CH3); 13C-NMR (75 MHz, acetone): δ 197.89 (CO); 177.75 (CS); 175.79 (CO); 142.96, 137.34, 135.38, 130.46, 129.41, 129.29, 129.07, 127.03, 126.48 (Ar-C); 26.61 (CH3).
O), 1529 (CO), 1179 (CS); 753 (C–Cl bend); 1H-NMR (300 MHz, acetone): δ 12.96 (s, 1H, NH), 10.18 (s, 1H, NH), 8.80–7.78 (m, 7H, Ar-H),2.52 (s, 3H, CH3); 13C-NMR (75 MHz, acetone): δ 197.89 (CO); 177.75 (CS); 175.79 (CO); 142.96, 137.34, 135.38, 130.46, 129.41, 129.29, 129.07, 127.03, 126.48 (Ar-C); 26.61 (CH3).
2.4 X-ray crystal structure and refinement
A diffractometer (Rigaku Oxford Diffraction Xcalibur, Eos, Gemini) equipped with Cu Kα radiation (λ = 1.54184 Å) was used to collect the crystallographic data of (4). For solving and refining the structure, the multi-scan absorption correction (CrysAlis PRO 1.171.38.46)22 applied data were processed by two SHELX program packages, SHELXT and SHELXL,23,24 while for drawing, the ORTEP-3 (ref. 25) and PLATON26 programs were used. The positions of hydrogen atoms were geometrically evaluated as 0.88 Å for NH, 0.95 Å for CH and 0.98 Å for CH3. A riding model with the limitations of Uiso (H) = k × Ueq (C, N) was used for refining, where k = 1.2 for NH and CH hydrogens and k = 1.5 for CH3 hydrogens.
2.5 Hirshfeld surface (HS) analysis
HS analysis is the most satisfactory method for studying interactions in a crystal structure.27,28 The visualization of the inter-molecular interactions in the crystal structure of (4) was carried out by using Crystal Explorer-17.5.29 The two HS distances di and de were measured which are taken from the nearest nucleus inside and outside the surface, respectively. The red, blue, and white colors have been chosen to visualize the normalized contact distance (dnorm). The input file was obtained in CIF format and is provided as ESI.†
2.6 Density function theory (DFT)analysis
The ground state geometry of compound (4) was optimized by DFT calculations using B3LYP functional and 6-31G (d, p) basis set.30 Gaussian 09W software31 was used for DFT calculations. Output check files were analyzed in Gauss View 6.0.32 The energies of highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were used to determine HOMO/LUMO energy gap which is important in predicting the nature and reactivity of a compound. Polarizability and dipole moment were also evaluated along with the optimization energy.
2.7 Molecular docking methodology
Structure based molecular docking studies were carried out using Molecular Operating Environment 2015.10 (MOE).33 The targeted protein ribonucleotide reductase RNR (PDB ID: 2BQ1), crystallographic structure of DNA (PDB ID: 127D) and the structure of jack bean urease (PDB ID: 4h9m) were downloaded from protein data bank (https://www.rcsb.org/). All the respective proteins were prepared using protein preparation wizard of MOE. Initially, water molecules were removed, and protein was 3D protonated and energy was minimized to remove any steric clashes present inside the protein. The structure of optimized protein was superimposed to reference protein to determine any structural deviation after optimization process. It was observed that optimization removed steric clashes present inside protein and root mean square deviation (RMSD) value was observed to be less than 2 angstrom which demonstrate the amino acid residues remain unchanged which is suitable for ligand binding. Following the protein preparation, amino acid residues of active pockets of the respective proteins were isolated using site finder utility of MOE. In case of DNA dodecamer d (CGCGAATTCGCG), co-crystal ligand Hoechst 33258 lies in the narrow minor grove of B-DNA in the AATT region. We have utilized the dimensions of Hoechst 33258 and searched for base pairs of active sites using site finder utility of MOE software. The structure of synthesized compound (4) was generated using Chemdraw ultra-12.0. The optimization and energy minimization of generated structure was carried out using Chemdraw 3D software. The output structure was docked into active pocket of protein using default parameters of MOE. Triangular matcher was used as placement and to reduce the search space and to improve the docking accuracy,33 the refinement was set to rigid receptor.34 London dg was used as scoring function and 100 poses were generated for compound.35
2.8 Spectrophotometric DNA binding analysis
UV-visible spectrophotometric titration experiments were performed to see the binding interaction of the synthesized compound (4) with DNA at physiological pH (7.0) and temperature (37 °C). The compound (4) solution was prepared in 10% DMSO. Firstly, the DNA concentration was determined, and its purity was assured by taking the spectrum of DNA solution on UV-visible spectrophotometer. The absorbance at λmax of 260 nm was used in Beer equation (C = A260/ε × ι) at molar absorptivity of 6600 L mol−1 cm−1 to calculate the concentration of DNA, which was found to be 5 × 10−5 M, while the absorbance ratio A260/A280, which was turned out to be 1.6, confirmed that DNA is free of all other cell organelles.36 UV-visible spectrum of (4) was recorded, individually at its optimized concentration of 5 × 10−6 M, and then DNA was titrated by gradually increasing its concentration within the reaction mixture from 10–80 μM. In the titration experiments, aliquots of DNA solution in microliter (μL) quantities were added to get the desired micromolar (μM) quantities of DNA in compound's solution. The change in the spectral response of (4) was recorded after each 10 μM addition of DNA. Prior to the spectral run, the establishment of an equilibrium for compound–DNA adduct was assured by keeping the reaction mixture at rest within the cuvette for at least 4–5 minutes under physiological temperature of 37 °C.
2.9 Urease inhibition assay
In 50 μL buffer (100 mM urea, 0.01 M K2HPO4, 1 mM EDTA and 0.01 M LiCl, pH 8.2), added 20 μL each of Jack bean urease enzyme (05 U mL−1) and compound (4). The reaction mixture was incubated for 30 min at 37 °C in 96-well plates. Then 50 μL each of phenol (1%, w/v phenol and 0.005%, w/v sodium nitroprusside) and alkali (0.5%, w/v NaOH and 0.1% sodium hypochlorite NaOCl) reagents were added into each well. The absorbance was measured on a microplate reader after 10 min at 625 nm. Thiourea was used as the reference standard for urease inhibition. All reactions were performed in triplicate.
2.10 Free radical scavenging assay
To prepare the assay solution, 20 μL of increasing concentrations of (4) was added into 100 μL of 150 μM 2,2-diphenyl-1 picrylhydrazyl (DPPH) and the volume in each well was adjusted to 200 μL with DMSO. This mixture was incubated at room temperature for 30 minutes. Vitamin C (ascorbic acid) was used as reference standard in this assay. Using same micro plate reader as in urease inhibition assay, the absorbance measurements were carried out at 517 nm. Experiments were run in triplicate for each concentration and the percent DPPH radical scavenging of each concentration of (4) was evaluated.
3 Results and discussion
3.1 Synthesis
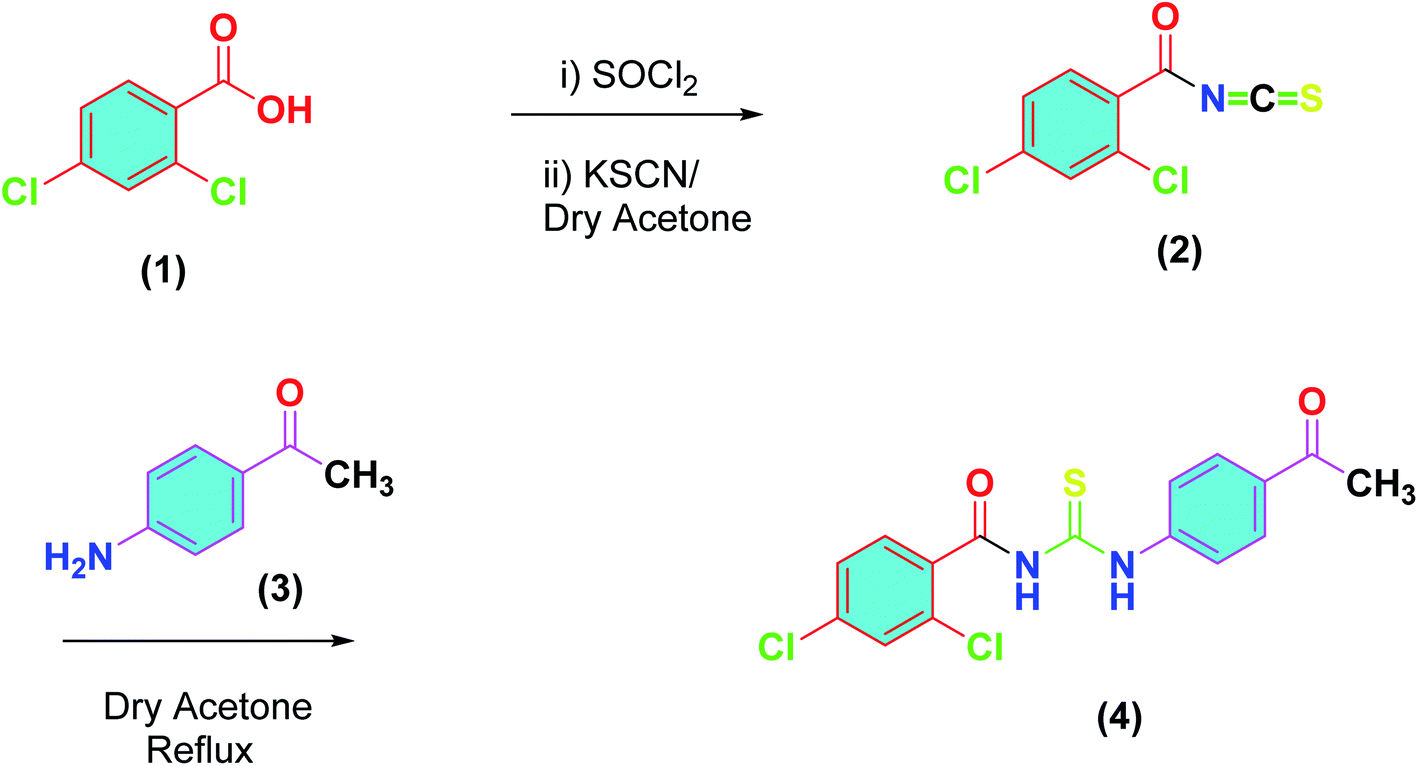
Following the procedure reported by us earlier, outlined in Scheme 1,37 the synthesis of title compound (4) was achieved. Thus, 2,4-dichlorobenzoic acid (1) was converted into acid chloride and the latter was reacted with potassium thiocyanate in dry acetone to furnish the 2,4-dichlorobenzoyl isothiocyanate (2). A solution of 4-acetyl aniline (3) in dry in acetone was added to (2) and the reaction mixture was refluxed for 3 hours, monitored by TLC. On completion, cooled to room temperature, and precipitated in the chilled water. The precipitates were filtered and recrystallized from ethanol to afford the purified thiourea derivative (4) in excellent yield.
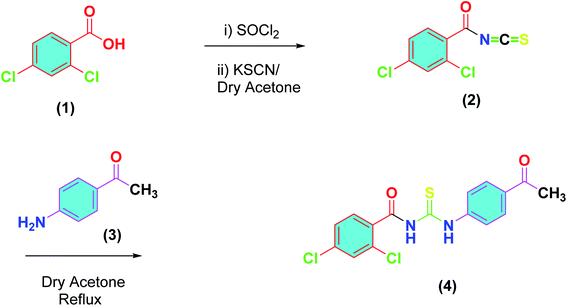 |
| | Scheme 1 Synthetic scheme for acyl thiourea derivative (4). | |
The FTIR analysis revealed characteristic absorption band for N–H at 3191–3195 cm−1, C–H aromatic at 3033–3037 cm−1, CO at 1692–1698 cm−1 and CS at 1174–1178 cm−1. 1H-NMR spectrum showed two characteristic signals of N–H protons. Thus, a broad singlet of 3-NH proton was observed at δ 13.21 ppm and 1-NH at 10.42 ppm. Four multiplets for four protons of aromatic appeared in the range δ 7.22–8.92 ppm, triplet for two protons of 1′-C appeared at δ 2.60, quintet for two protons of 2′-C observed at δ1.70, sextet for two protons of 3′-C observed at δ 1.41 ppm and triplet of three protons of 4′-CH3 appeared at 0.93 ppm. In 13C-NMR, three characteristic signals, one for thiocarbonyl and two for carbonyl carbons were observed. These signals appeared at δ 177.7 ppm, δ 175.1 ppm respectively. Five carbons of aromatic system were observed in the range of δ 151.7–115.3, and the acetyl appeared at δ 36.18.
3.2 X-ray structure
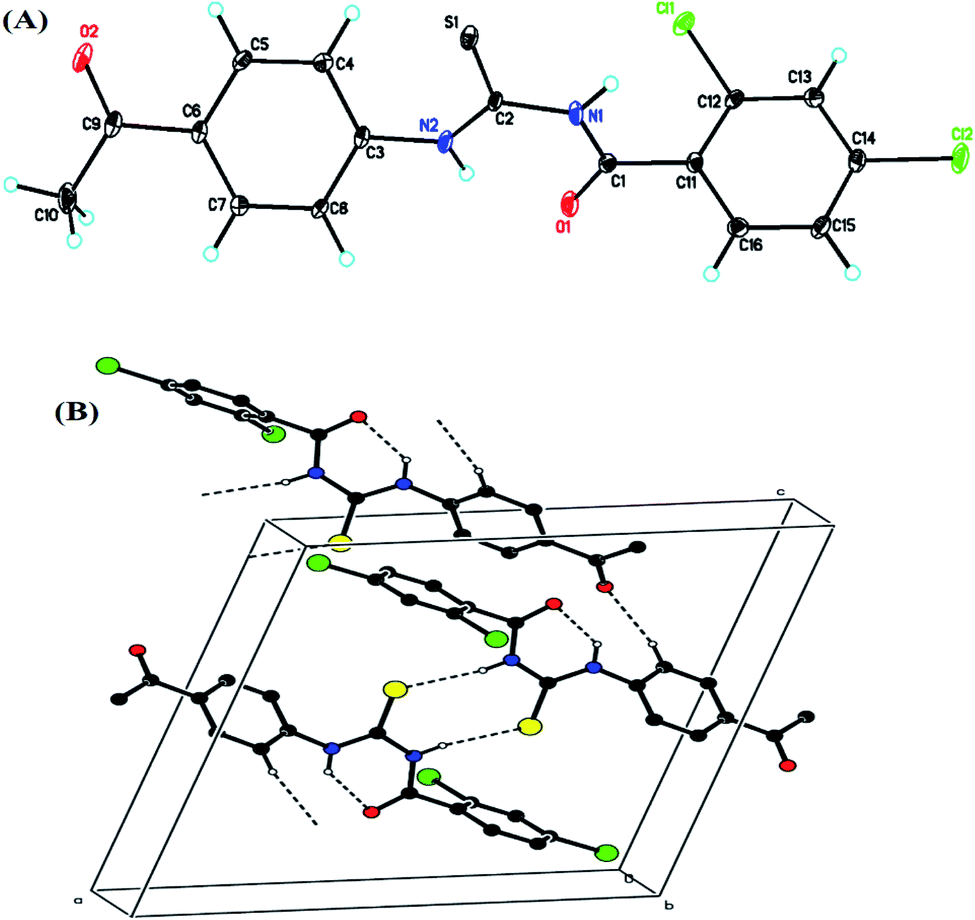
All the crystal details of (4) are provided in Table 1, while hydrogen bond geometry (Å, °) and selected interatomic distances (Å) are given in Table 2. In the molecule, (Fig. 2A), the rings, A (C3–C8) and B (C11–C16), were situated at a dihedral angle of A/B = 33.32(6)°. Atoms N2, O2, C9 and C1, Cl1, Cl2 were 0.0678(19) Å, 0.0739(19) Å, 0.0589(23) Å and −0.0544(21) Å, 0.0084(5) Å, −0.0213(6) Å away from the adjacent rings, A and B, respectively. In the crystal, the molecules, forming S(6) ring motifs38 with the intramolecular N–H⋯O hydrogen bonds, Table 2, were connected through intermolecular C–H⋯O and N–H⋯S hydrogen bonds, Table 2, enclosing R22(8) ring motifs38 into infinite double chains along [101], Fig. 2B. A three-dimensional architecture was consolidated with the help of π⋯π interactions {between A rings of adjacent molecules with an inter-centroid distance of 3.694 (1) Å} and C–H⋯π, Table 2.
Table 1 Crystal details of compound (4)a
| Crystal data |
| Computer programs: CrysAlis PRO 1.171.38.46 (Rigaku OD, 2015), SHELXT (Sheldrick, 2015), SHELXL (Sheldrick, 2015), Olex2 (Dolomanov et al., 2009). |
| Chemical formula |
C16H12Cl2N2O2S |
| Mr |
367.24 |
| Crystal system, space group |
Monoclinic, P21/n |
| Temperature (K) |
173 |
| a, b, c (Å) |
13.6684 (8), 9.1256 (4), 14.0839 (8) |
| β (°) |
111.168 (7) |
| V (Å3) |
1638.18 (17) |
| Z |
4 |
| Radiation type |
CuKα |
| μ (mm−1) |
4.85 |
| Crystal size (mm) |
0.24 × 0.14 × 0.12 |
|
| Data collection |
| Diffractometer |
Rigaku Oxford Diffraction |
| Absorption correction |
Multi-scan, CrysAlis PRO 1.171.38.46 (Rigaku Oxford Diffraction, 2015) empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm |
| Tmin, Tmax |
0.489, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections |
6234, 3112, 2743 |
| Rint |
0.039 |
| (sinθ/λ)max (Å−1) |
0.615 |
|
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S |
0.043, 0.122, 1.06 |
| No. of reflections |
3112 |
| No. of parameters |
209 |
| H-atom treatment |
H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) |
0.56, −0.36 |
Table 2 Hydrogen-bond geometry (Å, °) and selected interatomic distances (Å) in compound (4)
| Hydrogen-bond |
| Cg2 is the centroid of the B (C11–C16) ring |
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
D–H⋯A |
| N1–H1⋯S1ii |
0.88 |
2.53 |
3.3945 (19) |
168 |
| N2–H2⋯O1 |
0.88 |
1.98 |
2.687 (2) |
137 |
| C8–H8⋯O2iv |
0.95 |
2.39 |
3.290 (3) |
159 |
| C10–H10C⋯Cg2vi |
0.98 |
2.88 |
3.659 (3) |
139 |
| Symmetry codes: (ii) −x + 1, −y + 1, −z + 1; (iv) x − 1/2, −y + 3/2, z − 1/2; (vi) −x + 3/2, y + 1/2, −z + 1/2 |
|
| Interatomic distances |
| Cl1⋯N1 |
3.3576 (19) |
O1⋯C10iv |
3.222 (3) |
|
| H5⋯Cl1i |
2.98 |
O1⋯H2 |
1.99 |
|
| S1⋯C4 |
3.206 (3) |
O2⋯H5 |
2.44 |
|
| S1⋯C11ii |
3.501 (2) |
H8⋯O2iv |
2.40 |
|
| S1⋯C12ii |
3.295 (2) |
C6⋯C4v |
3.405 (3) |
|
| S1⋯H4 |
2.81 |
C1⋯H2 |
2.46 |
|
| S1⋯H1ii |
2.53 |
C2⋯H4 |
2.89 |
|
| H13⋯S1iii |
2.94 |
C7⋯H10B |
2.83 |
|
| H15⋯S1iv |
2.95 |
C10⋯H7 |
2.66 |
|
| O1⋯N2 |
2.688 (2) |
H10C⋯C14vi |
2.85 |
|
| O1⋯C6iv |
3.221 (3) |
H7⋯H10B |
2.32 |
|
| Symmetry codes: (i) x + 1/2, −y + 1/2, z + 1/2; (ii) −x + 1, −y + 1, −z + 1; (iii) x − 1/2, −y + 1/2, z − 1/2; (iv) x − 1/2, −y + 3/2, z − 1/2; (v) −x + 2, −y + 1, −z + 1; (vi) −x + 3/2, y + 1/2, −z + 1/2 |
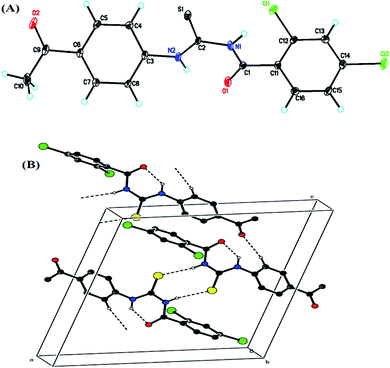 |
| | Fig. 2 (A) Crystal structure of (4) with the atoms numbering scheme; the thermal ellipsoids are drawn at the 50% probability level, (B) crystal's partial packing viewed down the b-axis. The dashed lines (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) showed N–H⋯O (intramolecular) and C–H⋯O, N–H⋯S (intermolecular) hydrogen bonds. For clarity, the nonbonding H-atoms were omitted. ) showed N–H⋯O (intramolecular) and C–H⋯O, N–H⋯S (intermolecular) hydrogen bonds. For clarity, the nonbonding H-atoms were omitted. | |
3.3 HS studies
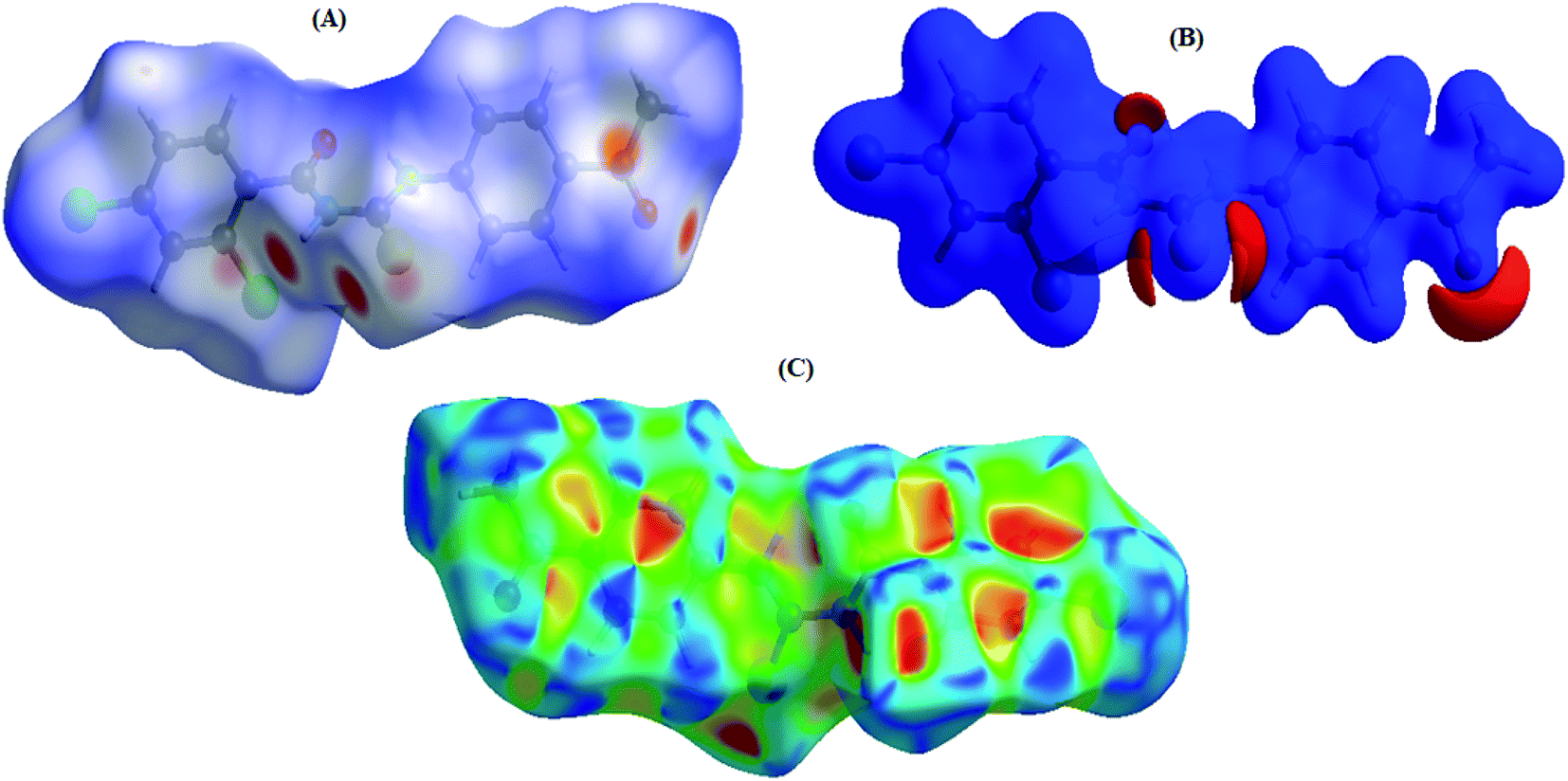
Fig. 3A represents the Hirshfeld surface, which was mapped over dnorm, where the white, red, and blue surfaces, respectively, indicated contacts with distances, equal to, shorter (in close contact), and longer (distinct contact) than the van der Waals radii.39 The appearance of shiny-red spots designated them to be respective donor and/or acceptor. These spots also appeared as blue and red regions and corresponded to positive and negative electrostatic potentials, respectively, on the HS plotted over electrostatic potential,40 Fig. 3B. The blue and red regions specified the regions for hydrogen-bond donors and hydrogen-bond acceptors, respectively. The presence red and blue triangles on the shape index of Hirshfeld surface is used to visualize the π⋯π stacking. If both triangles are adjacent to each other, then π⋯π interactions are obvious in the structure but if they are not adjacent to each other, no such interactions exist. The red and blue triangles laying near to each other in the shape index of HS, as shown in Fig. 3C, suggested, very clearly, that there are π⋯π interactions in (4).
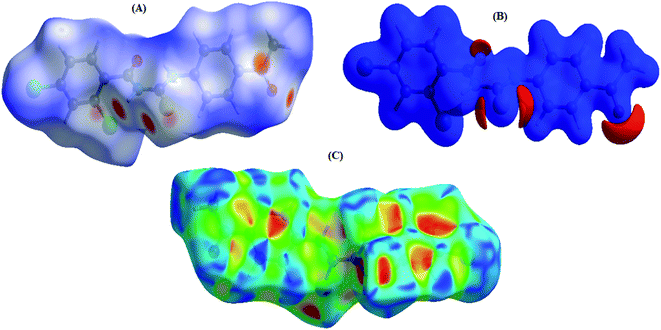 |
| | Fig. 3 (A) 3-D view of HS of (4) mapped over dnorm in the range of −0.3442 to 1.6171 a.u., (B) 3-D view of HS of (4) mapped over electrostatic potential energy ranging from −0.0500—0.0500 a.u. {at the Hartree–Fock (HF) level of theory (basis set; STO-3 G)}.The blue and red regions represent positive (hydrogen-bond donors) and negative (hydrogen-bond acceptors) potentials, respectively, (C) HS of (4) plotted over shape-index. | |
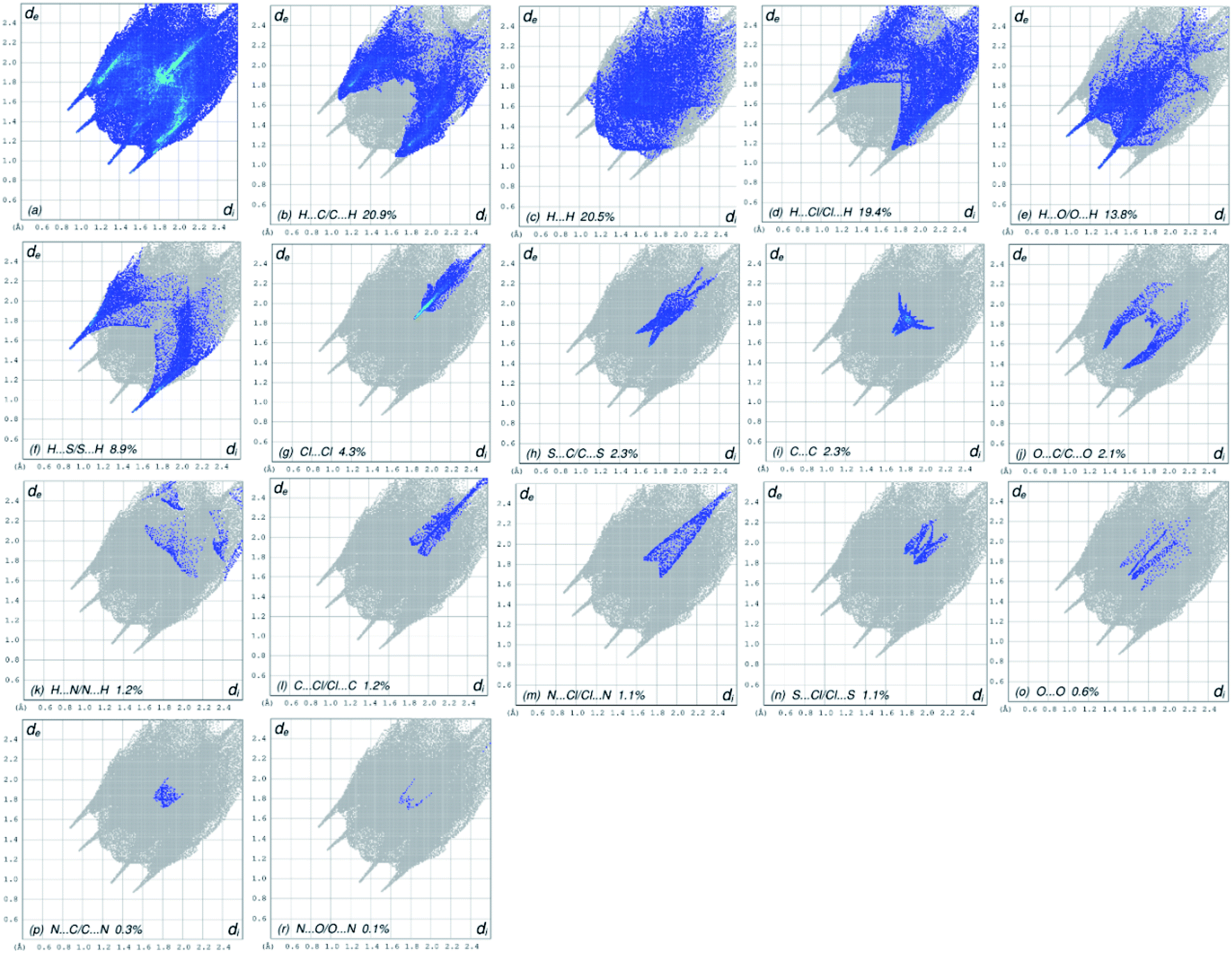
The overall two-dimensional fingerprint plot, Fig. 4a, and those delineated into H⋯C/C⋯H, H⋯H, H⋯Cl/Cl⋯H, H⋯O/O⋯ H, H⋯S/S⋯H, Cl⋯Cl, S⋯C/C⋯S, C⋯C, O⋯C/C⋯O, H⋯N/N⋯H, C⋯Cl/Cl⋯C, N⋯Cl/Cl⋯N, S⋯Cl/Cl⋯S, O⋯O, N⋯C/C⋯N and N⋯O/O⋯N,41 are demonstrated in Fig. 4b–r, respectively, together with their relative contributions to the HS. In the absence of C–H⋯π interactions, H⋯C/C⋯H interactions contributed 20.9% to the overall crystal packing, arising from the H⋯C/C⋯H contacts (Table 2), which is reflected in Fig. 4b with the tips at de + di = 1.65 Å. The H⋯H interactions contributed 20.5% to the overall crystal packing, arising from the H⋯H contacts (Table 2), which is reflected in Fig. 4c with the tip at de = di = 1.28 Å. The pair of characteristic wings in the fingerprint plot delineated into H⋯Cl/Cl⋯H contacts (Table 2 and Fig. 4c, 19.4% contribution to the HS) have symmetrical distribution of points with the tips at de + di = 2.86 Å. The pair of spikes in the fingerprint plot delineated into H⋯O/O⋯H contacts (Table 2) have symmetrical distribution of points (13.8% contribution, Fig. 4d) with the tips at de + di = 2.25 Å. The H⋯S/S⋯H contacts (Table 2 and Fig. 4e, 8.9% contribution to the HS) have symmetrical distribution of points with the tips at de + di = 2.39 Å. The Cl⋯Cl contacts (Table 2 and Fig. 4g, 4.3% contribution to the HS) have needle-shaped distribution of points with the tip at de = di = 1.83 Å. The S⋯C/C⋯S contacts (Table 2 and Fig. 4h, 2.3% contribution to the HS) have scissor-shaped distribution of points with the tips at de + di = 3.07 Å. The C⋯C contacts (Table 2 and Fig. 4i, 2.3% contribution to the HS) have nearly badman-shaped distribution of points with the tips at de + di = 2.90 Å. The O⋯C/C⋯O contacts (Table 2 and Fig. 4j, 2.1% contribution to the HS) have symmetrical distribution of points with the tips at de + di = 2.90 Å. Finally, the H⋯N/N⋯H (1.2% contribution to the HS, Fig. 4k), C⋯Cl/Cl⋯C (1.2% contribution to the HS, Fig. 4l), N⋯Cl/Cl⋯N (1.1% contribution to the HS, Fig. 4m), S⋯Cl/Cl⋯S (1.1% contribution to the HS, Fig. 4n), O⋯O (0.6% contribution to the HS, Fig. 4o), N⋯C/C⋯N (0.3% contribution to the HS, Fig. 4p) and N⋯O/O⋯N (0.1% contribution to the HS, Fig. 4r) contacts have scattered points of very low densities.
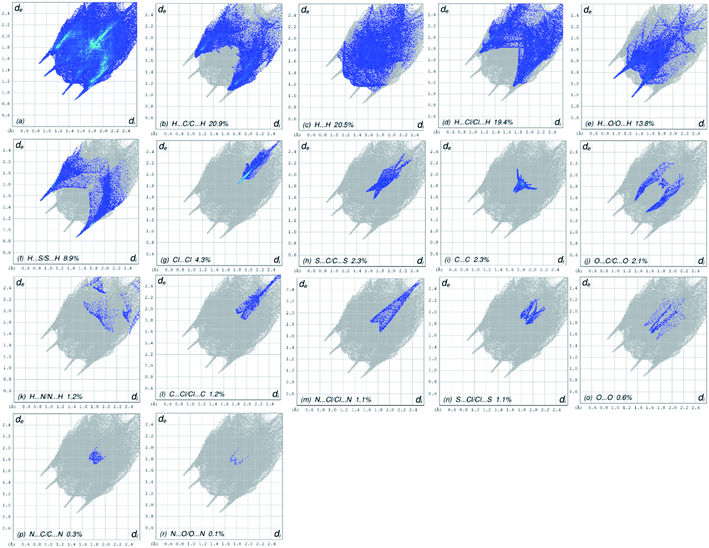 |
| | Fig. 4 Two-dimensional fingerprint plots for (4), showing (a) all interactions, and described into (b) H⋯C/C⋯H, (c) H⋯H, (d) H⋯Cl/Cl⋯H, (e) H⋯O/O⋯ H, (f) H⋯S/S⋯H, (g) Cl⋯Cl, (h) S⋯C/C⋯S, (i) C⋯C, (j) O⋯C/C⋯O, (k) H⋯N/N⋯H, (l) C⋯Cl/Cl⋯C, (m) N⋯Cl/Cl⋯N, (n) S⋯Cl/Cl⋯S, (o) O⋯O, (p) N⋯C/C⋯N and (r) N⋯O/O⋯N interactions. The di and de values are the closest internal and external distances (in Å) from given points on the HS contacts. | |
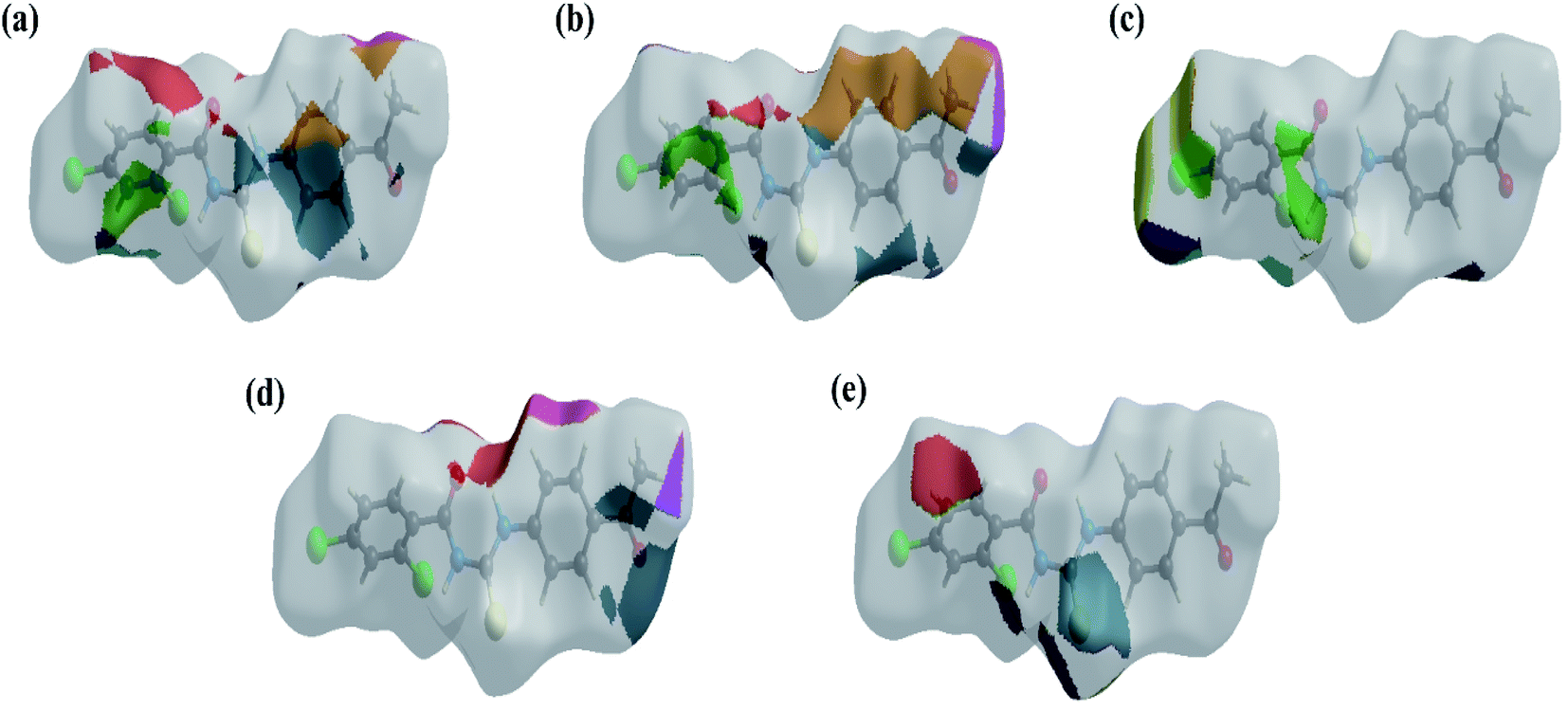
The HS images with the dnorm function plotted on the surface are shown for the H⋯C/C⋯H, H⋯H, H⋯Cl/Cl⋯H, H⋯O/O⋯H and H⋯S/S⋯H interactions, respectively, in Fig. 5a–e. The HS studies confirmed the importance of hydrogen atom contacts in the establishment of crystal packing and revealed that hydrogen bonding and van der Waals interactions played the key roles in the crystal packing due to the presence of large number of H⋯C/C⋯H, H⋯H, H⋯Cl/Cl⋯ H and H⋯O/O⋯H interactions.
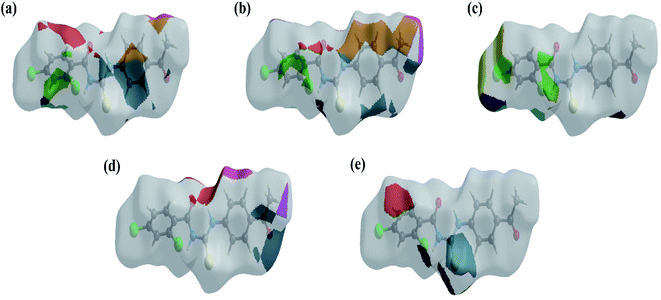 |
| | Fig. 5 The HS images with the dnorm function plotted on the surface for (a) H⋯C/C⋯H, (b) H⋯H, (c) H⋯Cl/Cl⋯H, (d) H⋯O/O⋯H and (e) H⋯S/S⋯H interactions. | |
3.4 DFT studies
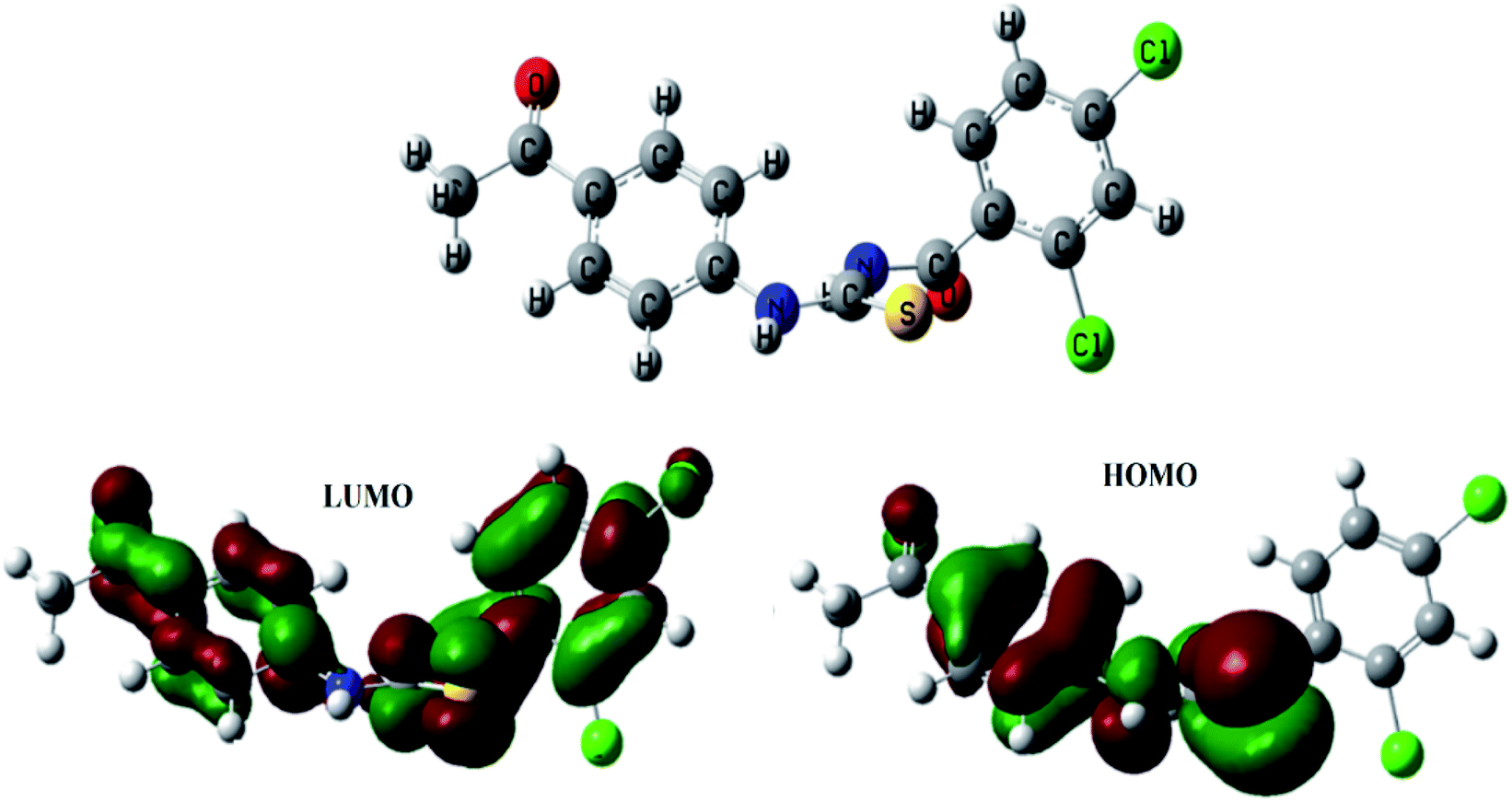
After performing DFT calculations with the basis set of 6-31G (d, p), the optimized structure and HOMO–LUMO orbitals of (4) were obtained as shown in Fig. 6. Atoms were arranged to such geometrical configuration where energy was minimized. The computed parameters are given in Table 3. The energy gap between LUMO and HOMO determine the electronic structure that could further lead to predict the kinetic stability and reactivity of a compound.42 The compound (4) was detected to be unstable one because of the small energy gap (ΔE) value; hence could be inferred as reactive in nature, Table 3. The small value of hardness (η), while a greater value of softness (S) further indicated the polarizable nature of the compound (4), as also obvious from computed polarizability and dipole moment values and validated its instability that is necessary for its binding interactions with protein/or DNA molecule, Table 3,.42 The HOMO structure is showing delocalization on benzamide moiety, while in LUMO the delocalization is more prominent on propionamide moiety as it is more electronegative part due to presence of two oxygen atoms.
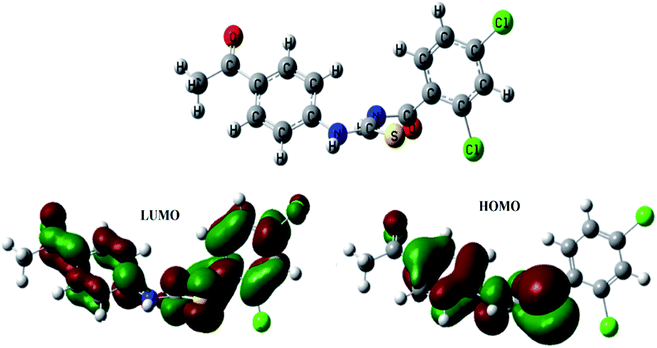 |
| | Fig. 6 Optimized structure of (4) along with bonding molecular orbital (HOMO), anti-bonding molecular orbital (LUMO). | |
Table 3 The computed DFT parameters of (4) by using basis set of 6-31G (d, p)
| EHOMO (eV) |
−0.222 |
Hardness (η) |
0.071 |
| ELUMO (eV) |
−0.080 |
Softness (S) |
7.040 |
| ΔEgap (eV) |
0.1419 |
Polarizability (α) (a.u.) |
240.156 |
| Optimization energy (eV) |
−2195.498 |
Dipole moment (D) |
4.176 |
3.5 Molecular docking studies
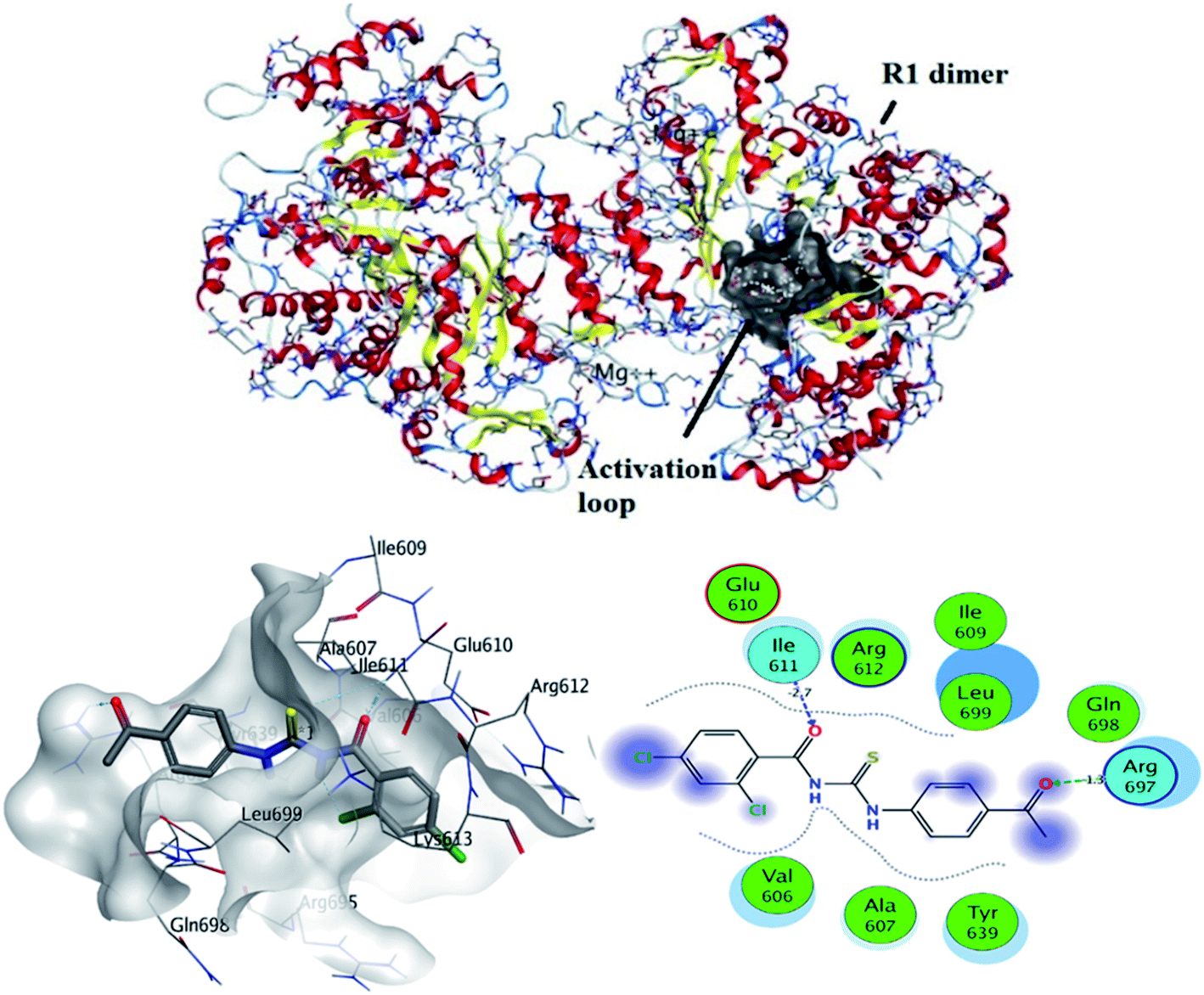
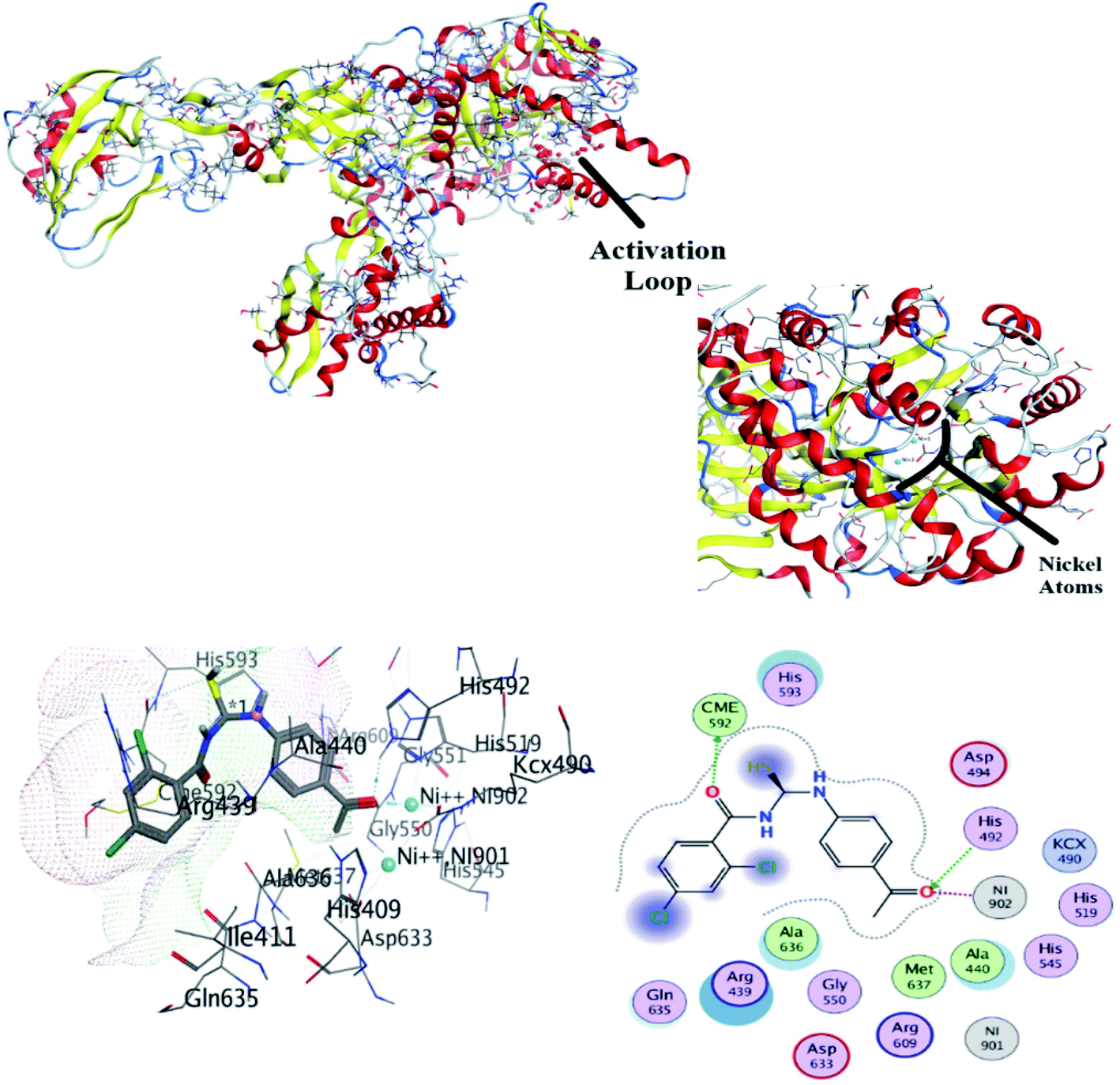
3.5.1 In silico binding of (4) with RNR. Molecular docking is considered the most power theoretical method for structure-based drug designing.43 Computational approach not only helpful for the validation of experimental findings for compound – protein/or DNA binding interactions but may support to divulge the candidacy of a compound as potent anticancer drug.44–47 Ribonucleotide reductase (RNR) plays important role in de novo synthesis of DNA precursors by catalyzing reduction of ribonucleotide in their respective deoxy ribonucleotide. RNR from higher organisms consists of two dimers i.e., R1 and R2. R1 is a larger dimer which contain binding and allosteric site for binding of substrate. Moreover, it is consisted of important cysteine residue which mediate radical transfer between R1 and R2 subunits. Radical transfer is mediated by binding of tyrosyl.48 R1 subunit of RNR protein contains important amino acid residues, which were involved in activation of protein. These amino acid residues were selected and docked with compound (4).The most stable conformation based on docking score was selected for further analysis. It can be seen from Fig. 7 that docked conformation of (4) showed important bonded and non-bonded interactions with amino acid residues of active site. The amino acid residues involved in non-bonding interactions were as follows; VAL606, TYR639, ALA607, GLU610, GLN698, ARG612, LEU699, ILE609 and GLN698. Whereas ILE611 and ARG697 amino acid residues were involved in formation of hydrogen bonding with electronegative oxygen atom of the compound (4). Hydrogen bonding was the major electrostatic attraction which was responsible for the formation of stable protein–ligand complex. Moreover, compound (4) produced docking score of −22.23 kJ mol−1.
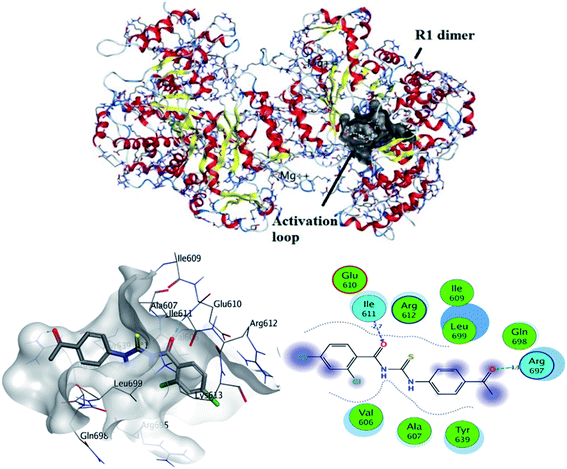 |
| | Fig. 7 Top: crystallographic structure of ribonucleotide reductase (RNR), black clouds indicating activation loop. Bottom: most probable 2D and 3D interactions of (4) within activation loop of RNR protein. | |
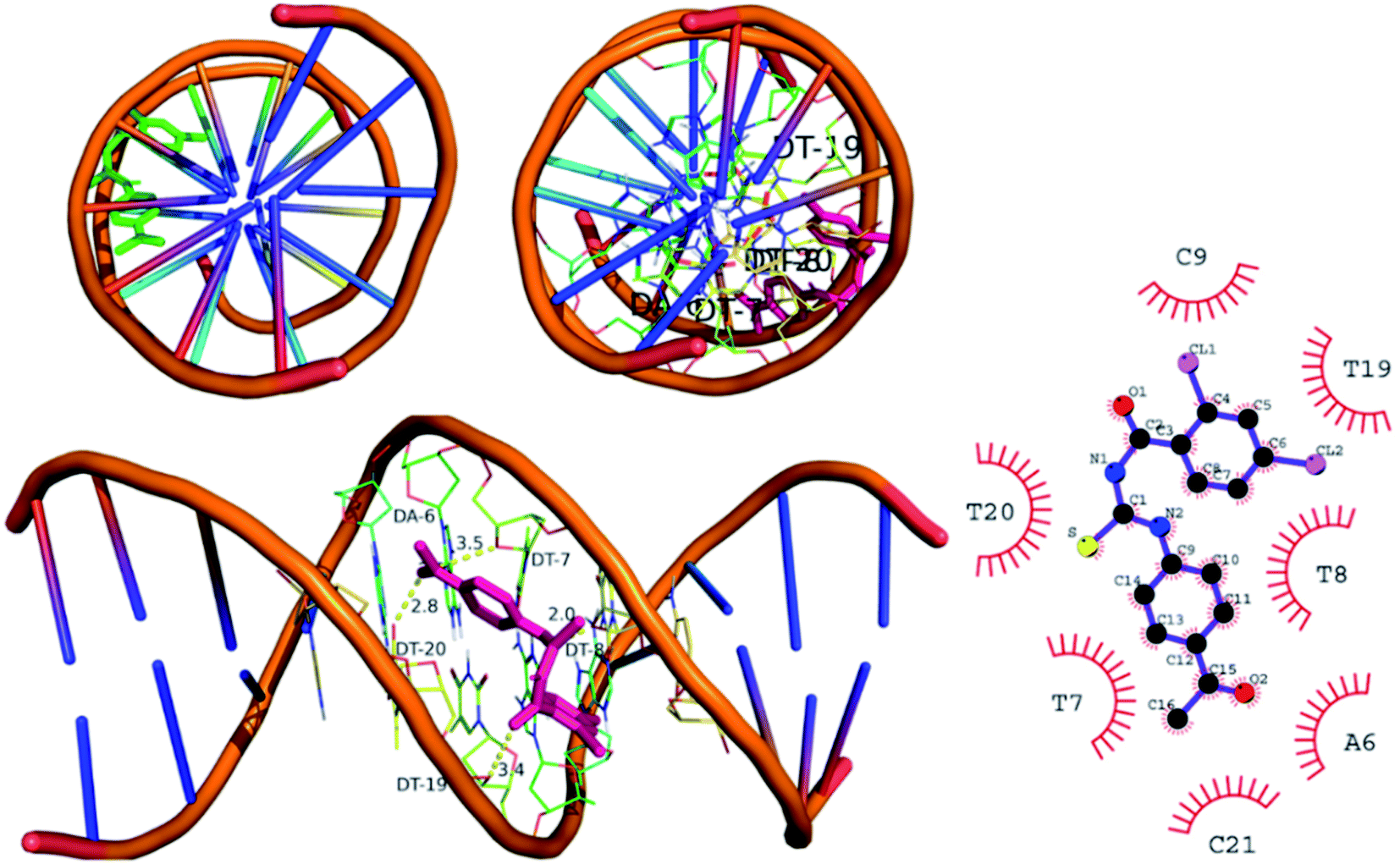
3.5.2 In silico binding of (4) with DNA. The DNA was in complex with minor groove binding drug Hoechst 33258. The DNA have specific nucleophilic bases sites (N7, N3 of purines) and O6 position of guanine base pairs present in minor groove of DNA. These sites are majorly responsible for DNA functioning and carcinogenic conversion of the cell. The molecular docking was further performed to investigate the binding interaction of (4) with DNA minor groove, Fig. 8. The DNA structure was co-crystallized with minor groove binder Hoechst. The docking protocol was validated by re-docking a co-crystal ligand i.e., Hoechst (minor groove binder) with DNA structure. The re-docking protocol showed RMSD value less than 2 Å which is an acceptable value to authenticate the comparable docked and re-docked confirmation.49 The compound 4 was docked into DNA groove which exhibited strong interactions with important bases (mainly purines) of DNA. It was observed that compound (4) showed interactions with AAT sequence of DNA groove. These studies revealed compound (4) as a potent groove binder. Moreover, it produced sable conformation with docking score of −35.8 kJ mol−1.
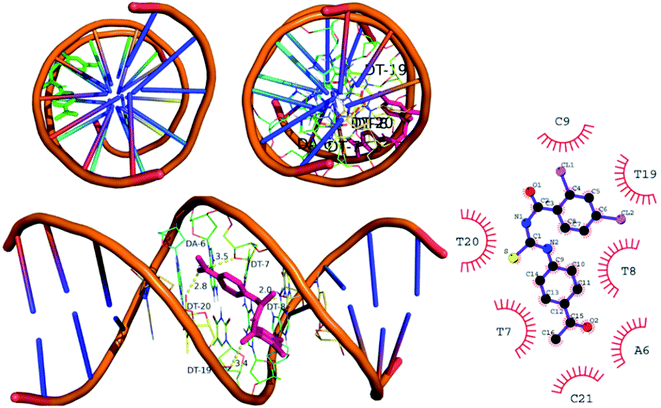 |
| | Fig. 8 Left upper and left bottom: docked conformation of (4) indicating DNA groove binding mode. Right: 2D conformation. | |
3.5.3 In silico binding of (4) with urease. The molecular docking was also done to explore the binding interaction of (4) with urease enzyme, Fig. 9. Compound 4 was observed to be the potent inhibitor of urease with two hydrogen bond interactions. His492 was bounded with ethanone moiety of (4) by hydrogen bond interaction. The other hydrogen bond interaction was observed by benzamide of (4) and CME592, Fig. 9. Stable conformation found with docking score of −35.12 kJ mol−1. This docking protocol was validated by re-docking the urease protein with co-crystal ligand acetohydroxamic acid (HAE) with finding the docking score of −11.24 kJ mol−1.
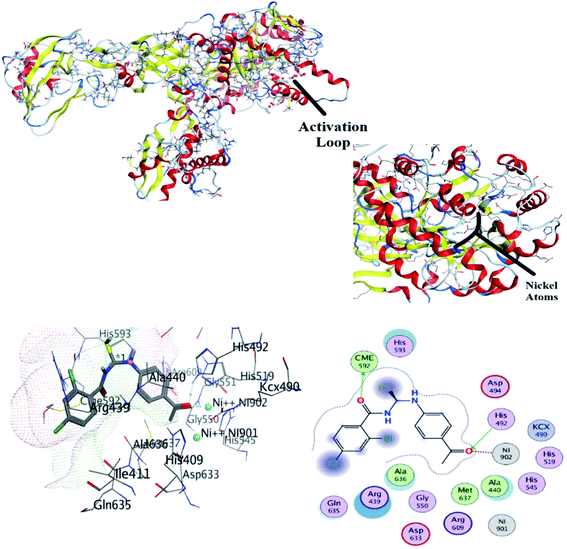 |
| | Fig. 9 Top: crystallographic structure of jack bean urease. Bottom: 3D and 2D conformation of (4) docked with urease. | |
3.6 Spectroscopic studies for (4)–DNA binding
DNA binding interaction by molecular docking was further identified experimentally by UV-visible spectroscopy, where the changes in the UV-visible spectrum of (4) was examined in the presence of ds DNA. Spectral changes in a ligand molecule in the presence of DNA could result in hyper-/hypo-chromic effect along with no peak shift or peak shift either towards longer or shorter wavelength. Fall/or rise in peak intensity along with red and blue shift in the wavelength indicated the formation of compound–DNA adduct via intercalative interaction, the rise in the peak intensity could infer the possibility of electrostatic binding of the cationic part of ligand with anionic part (PO43−) of DNA backbone, while spectral variation in terms of hypo/or hyper-chromic effect with no or insignificant change in the peak position (6–8 nm) referred to weaker interactions like van der Waals interaction between ligand and DNA groove walls or hydrogen bonding interaction with the DNA base pairs via DNA groove.50–52
The individual spectral profiles of (4) and ds DNA indicated a single peak at 328 nm and 260 nm, respectively. The compound (4) and ds DNA absorption bands are about 68 nm away and this difference in the λmax values showed clearly distinguishable regions of absorption; hence compound (4) interaction with DNA could easily be monitored. The change in (4) spectrum was observed as a gradual drop in the absorbance after titrating varying DNA concentrations, Fig. 10. By using the values of absorbance of (4) before and after the formation of compound–DNA adduct, the percent decline in the peak intensity was evaluated by the following equation.51
| |
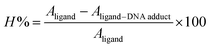 | (1) |
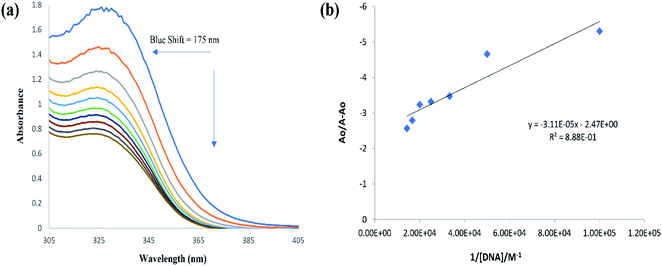 |
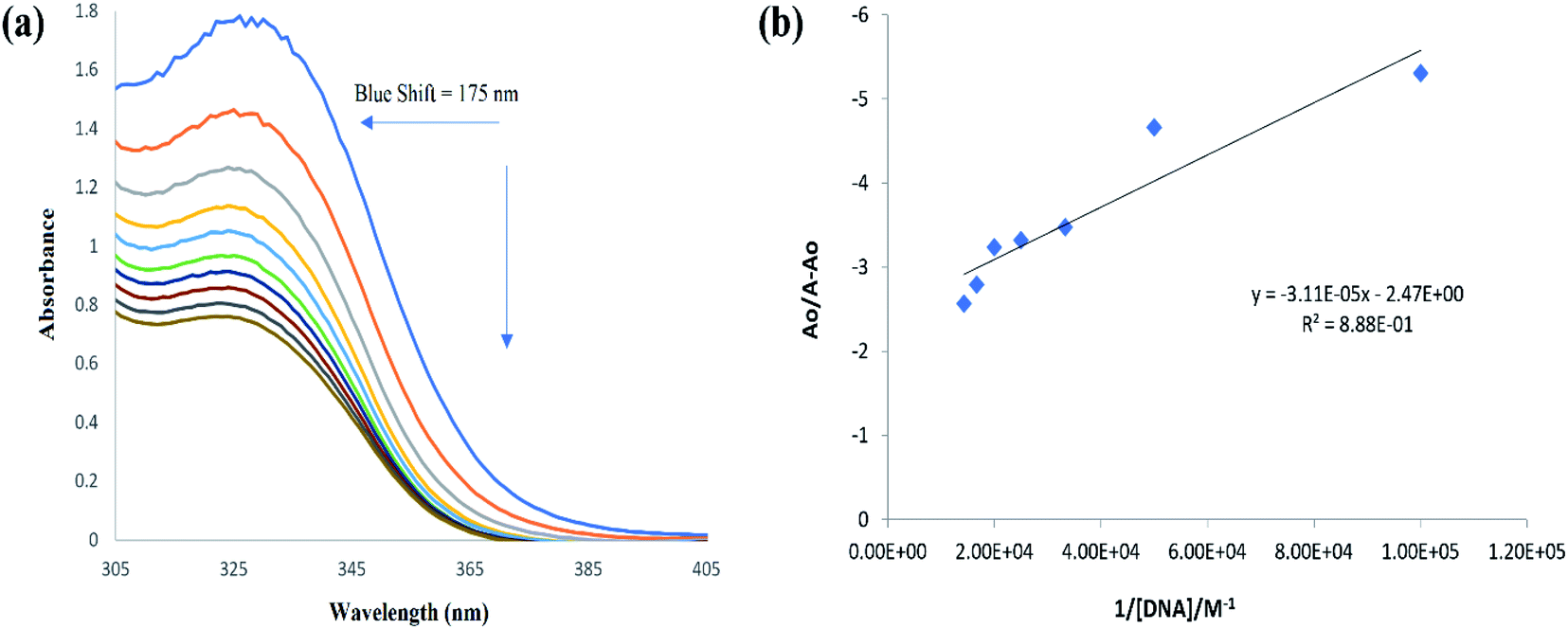
| | Fig. 10 (a) UV-visible spectral responses of (4) and (4)–DNA adduct. Changes in the peak intensity and peak shift are represented by the direction of arrows. (b) Ao − A/Ao vs. 1/[DNA] graph for binding constant calculation. | |
The resultant hypochromic effect was found to be 57.2% followed by an insignificant blue shift of magnitude 1.75 nm. The strength of compound–DNA interaction has been presumed to be associated with the magnitude of peak shifting.50
Using the absorbance of (4), before (Ao) and after DNA additions (A), in the Hildebrand eqn (2), Kb (binding constant) was evaluated and further used to calculate ΔG (Gibbs free energy change) from Van't hoff eqn (3).37,39,40,42,44–47,50,51
| |
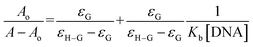 | (2) |
| | |
ΔG = −RTlnKb
| (3) |
From the plot Ao/A − Ao vs. 1/[DNA], Fig. 10, the intercept to slope ratio was used to determine the value of binding constant. The Kb (M−1) and ΔG (kJ mol−1) values were evaluated to be 7.9 × 104 and −28.42, respectively. The negative value of ΔG indicated the binding spontaneity while the evaluated magnitude of binding constant in current studies for (4)–DNA binding has been reported for both intercalation and groove binding interaction.44–47,50–54 The spectral variations and related binding parameters exhibited that the compound (4) interacted with DNA via weak interaction that was most probably occur via intercalation/or groove binding. However, insignificant peak shifting in (4)–DNA spectra and docking analysis identified compound (4) as groove binder.
3.7 Urease inhibitory studies
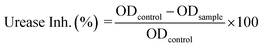
In vitro urease inhibition efficiency of (4) was determined by a method described earlier, where the amount of ammonia produced with indophenols was measured.55 The urease inhibition efficiency of (4) was calculated by using optical densities in the absence (ODcontrol) and presence of sample (ODsample) in the following formula:| |
 | (4) |
The compound (4) has shown highly significant urease inhibitory efficiency with IC50 ± SEM value of 0.0389 ± 0.0017 μM as compared to thiourea used as the standard inhibitor (18.2 ± 0.297 μM). With this least IC50 value, compound (4) not only showed its excellent inhibitory activity towards urease enzyme in comparison to standard inhibitor but also this value was found less than that of some other thiourea derivatives reported for their potent urease inhibitory activity which include transition metal complexes of thioureas (the values for 12 complexes ranges from 14.6 ± 3.3 to 68.9 ± 4.92 μM),56 thiourea and urea derivatives (the values for 33 derivatives ranges from 10.11 ± 0.11 to 72.99 ± 3.71 μM),57 atenolol thiourea hybrid (the values for 23 derivatives ranges from 11.73 ± 0.28 to 212.24 ± 0.42 μM),58 N-monoarylacetothioureas (among b1–b29, b19 was claimed excellent against both extracted urease and urease in intact cell with IC50 values of 0.16 ± 0.05 and 3.86 ± 0.10 μM, respectively).59 In vitro and in silico results along with comparison of the IC50 with the reported literature, indicated compound (4) a potential candidate that can be used as template for formulating drug for the treatment of urinary tract diseases.
3.8 Free radical scavenging activity
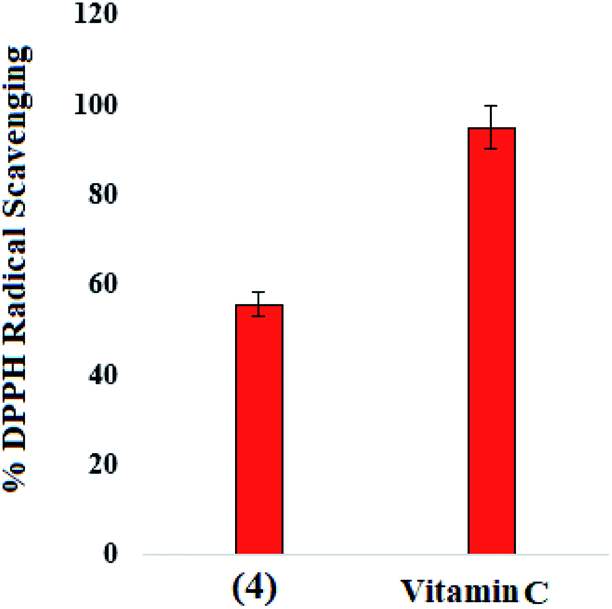
Compound (4) and reference drug vitamin C were assessed for DPPH free radical scavenging activity with some modification in the already reported method.60 Compound (4) have shown moderate free radical scavenging (55.62%) as compared to the vitamin-C scavenging potential (94.90%) at 100 μg mL−1 concentration, Fig. 11.
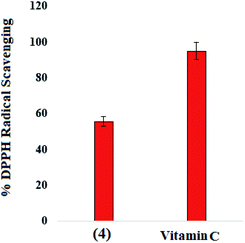 |
| | Fig. 11 Percent free radical scavenging activity of (4) and reference scavenger for DPPH radical. | |
4 Conclusions
A new acyl thiourea derivative N-((4-acetylphenyl)carbamothioyl)-2,4-dichlorobenzamide (4) was synthesized, characterized and the structural and conformational properties were determined by X-ray single crystal diffraction studies. The X-ray studies designated it to belong to monoclinic system P21/n space group. The planar rings were positioned at a dihedral angle of 33.32(6)°, while the molecules having S(6) ring motifs within the crystal were further associated with infinite double chains via intermolecular C–H⋯O and N–H⋯S hydrogen bonds enveloping the R22(8) ring motifs. For the crystal packing, the interactions H⋯C/C⋯H (20.9%), H⋯H (20.5%), H⋯Cl/Cl⋯H (19.4%), H⋯O/O⋯H (13.8%) and H⋯S/S⋯H (8.9%), as observed from HS analysis, were found to be the most important contributions. Based on DFT findings, compound (4) could be evaluated as polar and reactive in nature. In silico studies revealed binding of (4) with RNR, DNA, and urease via hydrogen bonding with electronegative oxygen atom of the (4), via groove binding, and via two hydrogen bond interactions (His492 and CME592), respectively. In vitro DNA binding and urease inhibition studies were carried out to further corroborate the in silico analysis. The experimental findings for the binding interaction of (4) with DNA by UV-visible spectral analysis also designated the compound (4) as a potential groove binder. The urease enzyme inhibition efficiency of (4) was found to be marvellous in comparison to reported urea and thiourea derivatives, but DPPH radical scavenging by (4) was found comparatively less than that with vitamin C. The urease inhibition and binding studies showed that this new compound has valuable anti-urease and anticancer activities which can suggest in vivo experiments for the further validation of current results. However, in silico justification via RNA docking studies confirmed that this compound can be used as a lead molecule for the treatment of ulcer and other associated complications.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
All contributions for this collaborative research are highly acknowledged.
References
- A. Mishra and S. Batra, Thiourea and guanidine derivatives as antimalarial and antimicrobial agents, Curr. Top. Med. Chem., 2013, 13, 2011–2025 CrossRef CAS PubMed.
- M. Ghorab, M. El-Gaby, M. S Alsaid, Y. Elshaier, A. M Soliman, F. F. El-Senduny, F. A Badria and A. Y. A. Sherif, Novel thiourea derivatives bearing sulfonamide moiety as anticancer agents through COX-2 inhibition, Adv. Anticancer Agents Med. Chem., 2017, 17, 1411–1425 CAS.
- S. Liu, M. C. Louie, V. Rajagopalan, G. Zhou, E. Ponce, T. Nguyen and L. Green, Synthesis and evaluation of the diarylthiourea analogs as novel anti-cancer agents, Bioorg. Med. Chem. Lett., 2015, 25, 1301–1305 CrossRef CAS PubMed.
- U. Zahra, A. Saeed, T. A. Fattah, U. Flörke and M. F. Erben, Recent trends in chemistry, structure, and various applications of 1-acyl-3-substituted thioureas: a detailed review, RSC Adv., 2022, 12(20), 12710–12745 RSC.
- M. M. Fallatah, S. Liu, M. B. Sevigny, H. Zou and M. C. Louie, Novel flexible heteroarotinoid, SL-1-18, promotes ERα degradation to inhibit breast cancer cell growth, Cancer Lett., 2017, 408, 82–91 CrossRef CAS PubMed.
- R.-Z. Huang, B. Zhang, X.-C. Huang, G.-B. Liang, J.-M. Qin, Y.-M. Pan, Z.-X. Liao and H.-S. Wang, Synthesis and biological evaluation of terminal functionalized thiourea-containing dipeptides as antitumor agents, RSC Adv., 2017, 7, 8866–8878 RSC.
- R. Pingaew, V. Prachayasittikul, N. Anuwongcharoen, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Synthesis and molecular docking of N,N′-disubstituted thiourea derivatives as novel aromatase inhibitors, Bioorg. Chem., 2018, 79, 171–178 CrossRef CAS PubMed.
- S. Hwang, S. Y. Choi, J. H. Lee, S. Kim, J. In, S. K. Ha, E. Lee, T.-Y. Kim, S. Y. Kim and S. Choi, Identification of a potent and noncytotoxic inhibitor of melanin production, Bioorg. Med. Chem., 2010, 18, 5602–5609 CrossRef CAS PubMed.
- P. Thanigaimalai, K.-C. Lee, V. K. Sharma, C. Joo, W.-J. Cho, E. Roh, Y. Kim and S.-H. Jung, Structural requirement of phenylthiourea analogs for their inhibitory activity of melanogenesis and tyrosinase, Bioorg. Med. Chem. Lett., 2011, 21, 6824–6828 CrossRef CAS PubMed.
- R. Gandhaveeti, R. Konakanchi, P. Jyothi, N. S. P. Bhuvanesh and S. Anandaram, Unusual coordination mode of aroyl/acyl thiourea ligands and their π-arene ruthenium(II) piano-stool complexes: synthesis, molecular geometry, theoretical studies and biological applications, Appl. Organomet. Chem., 2019, 33, e4899 CrossRef.
- A. Saeed, U. Flörke and M. F. Erben, A review on the chemistry, coordination, structure and biological properties of 1-(acyl/aroyl)-3-(substituted) thioureas, J. Sulfur Chem., 2014, 35, 318–355 CrossRef CAS.
- X.-P. Rao, Y. Wu, Z.-Q. Song, S.-B. Shang and Z.-D. Wang, Synthesis and antitumor activities of unsymmetrically disubstituted acylthioureas fused with hydrophenanthrene structure, Med. Chem. Res., 2011, 20, 333–338 CrossRef CAS.
- S.-Y. Ke and S.-J. Xue, Synthesis and herbicidal activity of N-(o-fluorophenoxyacetyl) thioureas derivatives and related fused heterocyclic compounds, Arkivoc, 2006, 10, 63–68 Search PubMed.
- J. R. Burgeson, A. L. Moore, J. K. Boutilier, N. R. Cerruti, D. N. Gharaibeh, C. E. Lovejoy, S. M. Amberg, D. E. Hruby, S. R. Tyavanagimatt, R. D. Allen and D. Dai, SAR analysis of a series of acylthiourea derivatives possessing broad-spectrum antiviral activity, Bioorg. Med. Chem. Lett., 2012, 22, 4263–4272 CrossRef CAS PubMed.
- C. Limban, A. Vasile, I. C. Chirita and M. Caproiu, Preparation of new thiourea derivatives with potential anti-parasitic and antimicrobial activity, Rev. Chim., 2010, 61, 946 CAS.
- Z. Zhong, R. Xing, S. Liu, L. Wang, S. Cai and P. Li, Synthesis of acyl thiourea derivatives of chitosan and their antimicrobial activities in vitro, Carbohydr. Res., 2008, 343, 566–570 CrossRef CAS PubMed.
- Q. Feng, Z.-L. Liu, L.-X. Xiong, M.-Z. Wang, Y.-Q. Li and Z.-M. Li, Synthesis and insecticidal activities of novel anthranilic diamides containing modified N-pyridylpyrazoles, J. Agric. Food Chem., 2010, 58, 12327–12336 CrossRef CAS PubMed.
- S. Y. Abbas, M. A. S. El-Sharief, W. M. Basyouni, I. M. Fakhr and E. W. El-Gammal, Thiourea derivatives incorporating a hippuric acid moiety: synthesis and evaluation of antibacterial and antifungal activities, Eur. J. Med. Chem., 2013, 64, 111–120 CrossRef CAS PubMed.
- İ. Koca, A. Özgür, K. A. Coşkun and Y. Tutar, Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety, Bioorg. Med. Chem., 2013, 21, 3859–3865 CrossRef PubMed.
- J. N. Asegbeloyin, E. E. Oyeka, O. Okpareke and A. Ibezim, Synthesis, structure, computational and in silico anticancer studies of N,N-diethyl-N′-palmitoylthiourea, J. Mol. Struct., 2018, 1153, 69–77 CrossRef CAS.
- B. M. Mistry, H.-S. Shin, M. Pandurangan and R. V. Patel, Synthesis of Acyl Thiourea Derivatives of 7-Trifluoromethyl-2-Pyridylquinazolin-4 (3H)-one as Anticancer Agents, Chem. Res., 2017, 41, 598–602 CAS.
- C. Pro, Version 1.171. 38.46, Rigaku Oxford Diffraction, 2015 Search PubMed.
- G. M. Sheldrick, SHELXT–Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, WinGX and ORTEP for Windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- R. Ujan, N. Arshad, F. Perveen, Q. Abbas, P. A. Channar, A. Saeed, S. I. Farooqi, K. A. Channar, T. Hökelek and U. Flörke, Single crystal, Hirshfeld surface, DFT analyses of (E)-2-(2-chloro-6-fluorobenzylidene) hydrazinecarbothioamide: elastase inhibition and DNA binding studies, J. Phys. Org. Chem., 2022, 35, e4296 CrossRef CAS.
- A. Sagaama and N. Issaoui, Design, molecular docking analysis of an anti-inflammatory drug, computational analysis and intermolecular interactions energy studies of 1-benzothiophene-2-carboxylic acid, Comput. Biol. Chem., 2020, 88, 107348 CrossRef CAS PubMed.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, 2017 Search PubMed.
- U. Vanitha, R. Elancheran, V. Manikandan, S. Kabilan and K. Krishnasamy, Design, synthesis, characterization, molecular docking and computational studies of 3-phenyl-2-thioxoimidazolidin-4-one derivatives, J. Mol. Struct., 2021, 1246, 131212 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, et al., Gaussian 09 (D01), Inc., Wallingford CT, 2009. vol. 121, pp. 150–166 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.0.16, Semichem Inc. Shawnee Mission KS, 2016 Search PubMed.
- Molecular Operating Environment (MOE), 2015.10, Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015 Search PubMed.
- Y. Zhang, T. Zhang, S. Tu, Z. Zhang and F. Meng, Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations, Molecules, 2020, 25, 4094 CrossRef CAS PubMed.
- C. R. Corbeil, C. I. Williams and P. Labute, Variability in docking success rates due to dataset preparation, J. Comput.-Aided Mol. Des., 2012, 26, 775–786 CrossRef CAS PubMed.
- M. Raja, M. Muralidharan and M. Arunachalam, Comparative analysis of DNA extracted from fish species preserved in formalin in two different periods, Turk. J. Biol., 2011, 35, 331–336 CAS.
- S. I. Farooqi, N. Arshad, F. Perveen, P. A. Channar, A. Saeed and A. Javed, Aroylthiourea derivatives of ciprofloxacin drug as DNA binder: theoretical, spectroscopic and electrochemical studies along with cytotoxicity assessment, Arch. Biochem. Biophys., 2019, 666, 83–98 CrossRef CAS PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. Chang, Patterns in hydrogen bonding: functionality and graph set analysis in crystals, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- A. Ahmad, N. Arshad, F. Perveen, R. Ujan, A. Saeed, P. A. Channar, S. I. Farooqi, G. Shabir, T. Hökelek and M. Bolte, Isomeric nitro substituted symmetrical benzamides: crystal structures, Hirshfeld surface analysis, 3D energy frameworks, DNA binding and cell line studies, J. Mol. Struct., 2022, 1247, 131396 CrossRef CAS.
- N. Arshad, A. Saeed, F. Perveen, R. Ujan, S. I. Farooqi, P. A. Channar, G. Shabir, H. R. El-Seedi, A. Javed, M. Yamin and M. Bolte, Synthesis, X-ray, Hirshfeld surface analysis, exploration of DNA binding, urease enzyme inhibition and anticancer activities of novel adamantane-naphthyl thiourea conjugate, Bioorg. Chem., 2021, 109, 104707 CrossRef CAS PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces, Chem. Commun., 2007, 37, 3814–3816 RSC.
- P. A. Channar, N. Arshad, F. A. Larik, S. I. Farooqi, A. Saeed, T. Hökelek, B. Batool, R. Ujan, H. S. Ali and U. Flörke, 4-(4-Bromophenyl) thiazol-2-amine: crystal structure determination, DFT calculations, visualizing intermolecular interactions using Hirshfeld surface analysis, and DNA binding studies, J. Phys. Org. Chem., 2019, 32, e3968 CrossRef.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Molecular docking: a powerful approach for structure-based drug discovery, Curr. Comput.-Aided Drug Des., 2011, 7, 146–157 CrossRef CAS PubMed.
- S. I. Farooqi, N. Arshad, P. A. Channar, F. Perveen, A. Saeed, F. A. Larik, A. Javed and M. Yamin, New aryl Schiff bases of thiadiazole derivative of ibuprofen as DNA binders and potential anticancer drug candidates, J. Biomol. Struct. Dyn., 2021, 39, 3548–3564 CrossRef CAS PubMed.
- N. Arshad, P. A. Channar, A. Saeed, S. I. Farooqi, A. Javeed, F. A. Larik, W. A. Abbasi and U. Flörke, Structure elucidation, DNA binding, DFT, molecular docking and cytotoxic activity studies on novel single crystal (E)-1-(2-fluorobenzylidene) thiosemicarbazide, J. Saudi Chem. Soc., 2018, 22, 1003–1013 CrossRef CAS.
- N. Arshad, M. Rafiq, R. Ujan, A. Saeed, S. I. Farooqi, F. Perveen, P. A. Channar, S. Ashraf, Q. Abbas and A. Ahmed, Synthesis, X-ray crystal structure elucidation and Hirshfeld surface analysis of N-((4-(1H-benzo [d] imidazole-2-yl) phenyl) carbamothioyl) benzamide: investigations for elastase inhibition, antioxidant and DNA binding potentials for biological applications, RSC Adv., 2020, 10, 20837–20851 RSC.
- N. Arshad, M. I. Mir, F. Perveen, A. Javed, M. Javaid, A. Saeed, P. A. Channar, S. I. Farooqi, S. Alkahtani and J. Anwar, Investigations on Anticancer Potentials by DNA Binding and Cytotoxicity Studies for Newly Synthesized and Characterized Imidazolidine and Thiazolidine-Based Isatin Derivatives, Molecules, 2022, 27, 354 CrossRef CAS PubMed.
- A. R. Offenbacher and B. A. Barry, A proton wire mediates proton coupled electron transfer from hydroxyurea and other hydroxamic acids to tyrosyl radical in class Ia ribonucleotide reductase, J. Phys. Chem. B, 2020, 124, 345–354 CrossRef CAS PubMed.
- E. D. Boittier, Y. Y. Tang, M. E. Buckley, Z. P. Schuurs, D. J. Richard and N. S. Gandhi, Assessing molecular docking tools to guide targeted drug discovery of CD38 inhibitors, Int. J. Mol. Sci., 2020, 21, 5183 CrossRef CAS PubMed.
- M. Sirajuddin, S. Ali and A. Badshah, Drug–DNA interactions and their study by UV-visible, fluorescence spectroscopies and cyclic voltametry, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- N. Arshad, N. Abbas, F. Perveen, B. Mirza, A. M. Almuhaini and S. Alkahtani, Molecular docking analysis and spectroscopic investigations of zinc (II), nickel (II) N-phthaloyl-β-alanine complexes for DNA binding: evaluation of antibacterial and antitumor activities, J. Saudi Chem. Soc., 2021, 25, 101323 CrossRef CAS.
- N. Shahabadi and R. Farhadi, Multispectroscopic and molecular docking studies on DNA binding of guaifenesin drug, Nucleosides, Nucleotides Nucleic Acids, 2021, 40, 317–335 CrossRef CAS PubMed.
- M. A. Khajeh, G. Dehghan, S. Dastmalchi, M. Shaghaghi and M. Iranshahi, Spectroscopic profiling and computational study of the binding of tschimgine: a natural monoterpene derivative, with calf thymus DNA, Spectrochim. Acta, Part A, 2018, 192, 384–392 CrossRef CAS PubMed.
- N. Shahabadi, M. Falsafi and M. Maghsudi, DNA-binding study of anticancer drug cytarabine by spectroscopic and molecular docking techniques, Nucleosides, Nucleotides Nucleic Acids, 2017, 36, 49–65 CrossRef CAS PubMed.
- M. W. Weatherburn, Phenol-hypochlorite reaction for determination of ammonia, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- A. Mumtaz, J. Arshad, A. Saeed, M. A. H. Nawaz and J. Iqbal, Synthesis, characterization and urease inhibition studies of transition metal complexes of thioureas bearing ibuprofen moiety, J. Chil. Chem. Soc., 2018, 63, 3934–3940 CrossRef CAS.
- B. Bano, K. M. Khan, A. Lodhi, U. Salar, F. Begum, M. Ali, M. Taha and S. Perveen, Synthesis, in vitro urease inhibitory activity, and molecular docking studies of thiourea and urea derivatives, Bioorg. Chem., 2018, 80, 129–144 CrossRef CAS PubMed.
- S. Wahid, S. Jahangir, M. A. Versiani, K. M. Khan, U. Salar, M. Ashraf, U. Farzand, A. Wadood, M. Taha and S. Perveen, Atenolol thiourea hybrid as potent urease inhibitors: design, biology-oriented drug synthesis, inhibitory activity screening, and molecular docking studies, Bioorg. Chem., 2020, 94, 103359 CrossRef CAS PubMed.
- W.-Y. Li, W.-W. Ni, Y.-X. Ye, H.-L. Fang, X.-M. Pan, J.-L. He, T.-L. Zhou, J. Yi, S.-S. Liu, M. Zhou, Z.-P. Xiao and H.-L. Zhu, N-Monoarylacetothioureas as potent urease inhibitors: synthesis, SAR, and biological evaluation, J. Enzyme Inhib. Med. Chem., 2020, 35, 404–413 CrossRef CAS PubMed.
- C. V. K. Reddy, D. Sreeramulu and M. Raghunath, Antioxidant activity of fresh and dry fruits commonly consumed in India, Food Res. Int., 2010, 43, 285–288 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: CIF of crystal & IC50 data files in a zip folder as supplementary material. CCDC 2149670. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra03160d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Pervaiz Ali Channar
*b,
Pervaiz Ali Channar