
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of a “Branch-Fruit” structure chitosan nanofiber physical hydrogels with high mechanical strength and pH-responsive controlled drug release properties†
Ying Wena,
Xiaofeng Li *ac,
Sihan Zhanga,
Chong Xiea,
Wei Maa,
Lun Liangb,
Zhenqiang Heb,
Hao Duanb,
Yonggao Mou*b and
Guanglei Zhao*a
*ac,
Sihan Zhanga,
Chong Xiea,
Wei Maa,
Lun Liangb,
Zhenqiang Heb,
Hao Duanb,
Yonggao Mou*b and
Guanglei Zhao*a
aState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510641, China. E-mail: glzhao@scut.edu.cn; Fax: +86-20-87111770; Tel: +86-20-87111770
bDepartment of Neurosurgery/Neuro-oncology, Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou 510000, China. E-mail: mouyg@sysucc.org.cn
cSchool of Food Sciences and Engineering, South China University of Technology, Wushan Road 381, Guangzhou 510641, China. E-mail: xflibio@scut.edu.cn
First published on 10th June 2022
Abstract
The poor mechanical properties of chitosan physical hydrogels seriously hinder their application in the biomedical field. Inspired by the structure of cell tissues, a novel chitosan nanofiber (CSNF)/Hyaluronic acid (HA)/β-glycerophosphate disodium (β-GP) drug-loaded hydrogel was prepared by micro-dissolution and physical crosslinking. The hydrogel has a “Branch-Fruit” structure and exhibits excellent mechanical properties, good biocompatibility and cell-adhesion properties. Human cancer cells (HeLa) can adhere to the hydrogel surface, which might facilitate tumor site-specific administration of drugs. This material also exhibits high pH sensitivity, with which drug release can be triggered under acidic conditions at pH 4.00. The mechanical strength and drug release behavior of this hydrogel can be easily adjusted by varying the CSNF content.
1. Introduction
At present, cancer has the second incidence rate in China and the first incidence rate in developed countries all over the world (Fig. 1).1Among various new treatment strategies, implantable hydrogels that deliver drugs locally and continuously are becoming a promising solution and a hotspot in research. Hydrogels are widely used for wound repair due to their controlled pore structure, size and shape and high similarity to the extracellular matrix. In addition, certain nanoparticles,2,3 peptides,4–6 ionic polymers7–9 and other microbial small molecules10 can be introduced into hydrogels to give them various functions that are more conducive to wound healing.10
In the past decades, researchers have been working on improving the mechanical strength and toughness of hydrogels, such as adding polymer particles,11 inorganic clays12 or carbon nanotubes13 to the gel systems. Other methods, such as forming interpenetrating polymer networks (IPN), semi-interpenetrating polymer networks14 or dual network structures,15 were also reported. Nanocomposite hydrogels (NPH) prepared by adding nanostructures show extremely high mechanical properties.16 However, most of the nanofillers currently used do not have long-term biocompatibility and controllable biodegradability, although some natural nanofillers with good biodegradability and biocompatibility have been applied in NPH.17,18 Till now, there are no reports of directly using nanofibers to prepare self-reinforced hydrogels.
Recently, various anticancer drugs such as doxorubicin,19 resveratrol,20 berberine,20 5-fluorouracil,21 etc. have been used to treat cancer diseases. The numbers of these anticancer drugs loaded on the fibers carriers during in vitro and in vivo cancer therapy. Also, among different fibrous carriers, chitosan nanofibers (CSNFs) have attracted great interest due to their macromolecular cationic properties, large specific surface area to volume ratio, inherent biological functions, and biocompatibility.22,23 Typically, CSNFs are fabricated by electrostatic spinning techniques.16,24 In our previous work, CSNFs was prepared by beating/refining papermaking treatment combined with high-pressure homogeneous phase post-treatment.25 Chitosan fibers (CSFs) with a size (diameter) of 15–60 μm are first treated with beating/refining equipment in the papermaking process. The beating process produces strong shearing, extrusion and other forces, making the CSFs possess micron or even nanometerized filaments, and different degrees of micro/nanosized pulp can be prepared according to requirements. Chitosan micro/nano fiber pulp can be further processed by deep beating/centrifugation separation or homogenization according to different experimental conditions and purposes.
The chitosan/hyaluronic acid (HA)/β-glycerol phosphate (β-GP) system is representative in the composition of pH-responsive drug-loaded hydrogels. Moreover, the undetermined carboxyl groups in HA are similar to pH buffers, which can alleviate the drug burst release caused by the sudden protonation of a large number of amine groups in CS in an acidic environment.26 However, the poor mechanical strength and long gel time of this system hinder its application in wound dressings after cancer surgery.
 | ||
Fig. 1 Chitosan nanofibers obtained by beating 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 revolutions and homogenizing for 5 times (a) SEM image, (b) TEM image. 000 revolutions and homogenizing for 5 times (a) SEM image, (b) TEM image. | ||
In this study, the traditional chitosan powder was replaced by CSNFs prepared by swelling-beating-homogenization method, and new drug-loaded chitosan composite hydrogels were prepared by micro-dissolution crosslinking method. The effects of CSNFs content on the mechanical properties, biocompatibility, and pH responsiveness of hydrogels were investigated. The drug release under normal physiological environment (pH = 7.4) and living environment for cancer cells (pH 5.6–6.8) were also studied to find out their pH sensitivity, as well as their potential application as a wound dressing after tumor resection surgery for cancer treatment.
2. Materials and methods
2.1 Materials
Hydrogels were prepared from CSNF, hyaluronic acid (purity > 97%, Macklin), and β-glycerophosphate disodium (purity > 95%, Macklin). Chitosan nanofibers (CSNFs) were prepared in lab according to the research of Zhang25 of this research group. The water used in the preparation process was obtained by a pure water machine (Purelab, ELGA). The culture media include DMEM basic (DMEM, Dulbecco's Modified Eagle Medium, Gibco), MEM α basic (α-MEM, minimum essential medium, Gibco), fetal bovine serum (FBS, Gibco), 0.25% trypsin-EDTA (Gibco), pen strep (PS, penicillin/streptomycin, Gibco), phosphate-buffered saline (PBS, pH 7.4 basic, Gibco), a cell counting kit-8 (CCK-8, Biosharp Life Sciences) and Calcein acetoxymethyl ester (Calcein-AM, Sigma). Other reagents used, such as acetic acid and ethanol, were commercially available and of analytical grade.2.2 Preparation of chitosan nanofibers (CSNFs)
Chitosan nanofibers (CSNFs) were prepared according to the method recently reported by Zhang.25 First, the chitosan regenerated fibers (CSFs, ≥95% deacetylated, ≥1000 mPa s viscosity) were cut into a length of 3–5 mm, and the CSFs were cut using a cutting mill (CM200, England) to maintain a uniform appearance during the subsequent processing. Take 30 g of milled CSFs (dry weight) and soak it in deionized (DI) water for swelling, and then concentrate the solid content of the chitosan slurry to 10% (w/v). The CSFs slurry was beaten with a PFI refiner (Hamjern Maskin 621, Norway) under a pressure of 3.33 N mm−1, and the slurry was beaten at 30000 revolutions. Finally, the slurry concentration was adjusted to 0.5% (w/v) and homogenized five times using a microflow nanohomogenizer (Nano BDe BEE Next Generation Homogenizers) to obtain a large number of relatively uniform CSNFs with a diameter 50–300 nm and a length bout 3–15 μm.
2.3 Preparation of CSNF/HA/β-GP composite hydrogel
The CSNFs suspension of 1.0%, 1.5% and 2.0% (w/v) was added with an acetic acid solution (0.03 mol L−1). After stirring for 30 minutes, it was dissolved in deionized water with 1.5% (w/v) HA and 10% (w/v) β-GP, stirred for 24 h, to form a HA/β-GP mixed solution. Then 0.8 mL of HA/β-GP mixed solution was added to 3.2 mL of acid-dissolved CSNFs solution in an ice bath to form CSNF/HA/β-GP solutions. After standing for 5 min, gels will be formed, denoted as HG1, HG2, HG3. HG0 is prepared with 2.0% chitosan powder (CSP), and the rest of the steps are the same. The composition of the composite hydrogels is shown in Table S1 (ESI†).2.4 Gelling time determination
The test tube inverting method27 was used to determine the time of sol–gel transition. The CSNF/HA/β-GP mixture was prepared according to the method described in Section 2.3 and stored at 4 °C. Briefly, 4 mL of CSNF/HA/β-GP mixture was added to a volume of 10 mL screw-top vial at low temperature, and then the vial was placed in a constant temperature water bath at 25 °C. The gel conditions were observed every 5 s. The gelling time is defined as the time when the mixture does not flow when inverted.2.5 Scanning electron microscopic observation
The hydrogels (HG0, HG1, HG2, HG3) were frozen in liquid nitrogen and then fractured to obtained the cross-section. The obtained sample was immediately transferred to a freeze drier (Christ ALPHA1-4 LD plus, German) and then lyophilized for 24 h. The cross-section of the freeze-dried sample was cut into 1 mm thick slices and pasted on the conductive tape. After coated with gold on an EMS 300 T sputter coater (Quorum Technologies), the surface morphology of the hydrogels was examined on a scanning electron microscopy (FESEM, Merlin, Germany) under a voltage of 5 kv.2.6 Dynamic rheological test
The rheological properties of hydrogels were determined by measuring their energy storage modulus (G′) and loss modulus (G′′) by a rotational rheometer (TA Instruments, New Castle, American) with a set parallel plate (diameter 40 mm, gap 0.5 mm). In order to reduce the damage to the gelling system, the hydrogel was measured in dynamic oscillation mode at a strain of 0.5%. According to the data in Fig. S1 (ESI†), the strain belongs to the linear viscoelastic interval. In dynamic frequency scanning, the temperature is preheated to 37 °C, and the scanning frequency is 0.1–100 HZ. Dynamic temperature sweep: The frequency is set to 1 HZ, and the temperature is increased from 25 °C to 70 °C at a rate of 5 °C min−1. In all experiments, the sample was equilibrated for 30 s before measurement, and a layer of low viscosity silicone oil was added around the sample to prevent evaporation of water.2.7 Texture analysis
Texture properties of gels were determined by TPA test (Texture profile analysis) on texture analyzer (TA XT. plus, USA). After keeping 24 h in 37 °C, samples placed at a mounted fixed table were conducted under a cylindrical probe (P/36R) with a speed of 1 mm s−1. During the TPA test, 30% deformation, a control force of 0.5 g and 5 s lapse existed between the two compressions processes. All operations were automatically controlled by the software of the texture analyzer.2.8 In vitro drug release
Adriamycin (DOX, red powder, 2 mg) was added into a vial containing 4 mL CSNF/HA/β-GP solution, and treated with ultrasound at low temperature (0 °C) for 1 h until DOX was completely dissolved. It was then left to rest at room temperature (25 °C) for 5 minutes until hydrogel formed. Subsequently, it remained at 37 °C. Then 10 mL of phosphate buffer solutions (PBS, pH 4.0, pH 6.5 and pH 7.4) were added to the vial containing the DOX loaded hydrogel. At a predetermined time point, 4 mL of medium was taken from the sample vial and the vial was refilled with 4 mL of fresh PBS solution to maintain a constant volume. The absorbance of this medium at 480 nm was measured using a UV-vis-NIR spectrometer (UV-1900, Shimadzu, Japan) and the experiment was repeated three times to take the mean value. The calibration curves of DOX in PBS solutions at different pHs was established prior to quantitative description of drug release, see Fig. S2 (ESI†).2.9 Adhesion of cancer cell to the hydrogel
Human cervical cancer cells (HeLa) were used to detect the adhesion of cancer cells to the hydrogel. HeLa cells were cultured in a high glucose medium (DMEM, Gibco) containing 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (PS, Gibco). The culture flasks containing HeLa cells were placed in an incubator at 5% CO2, 37 °C and saturated humidity. Prior to in vitro study, the hydrogels (HG0, HG3) were soaked in ethanol for 1 h, and then sterilized under UV light for 1 h. Subsequently, logarithmic growth phase HeLa cells (the growth curve of HeLa cells was shown in Fig. S3a of ESI†) were taken, digested with trypsin, centrifuged and resuspended in medium at a density of 1 × 105 cells per mL. The cells were seeded on the surface of the hydrogel with 1 mL cell suspension per well in a 24-well plate, and incubated in an incubator with 5% CO2 and 37 °C for 1 h and 4 h. The morphology and adhesion of HeLa cells were observed using scanning electron microscope and fluorescence microscope (FM, Carl Zeiss Axio vert. A1, Germany).In SEM observation, after 1 h and 4 h incubation of HeLa cells, the medium was aspirated and the unadhered cells were washed 3 times with PBS solution to remove the unadhered cells. The cells were fixed at 4 °C in 2.5% glutaraldehyde solution for 4 h. The cells were then sequentially dehydrated with a gradient of ethanol concentrations of 30%, 50%, 75%, 90%, 100% and 100% for 15 min each. Finally, the hydrogels co-incubated with the cells were freeze-dried as described in Section 2.5 and the cell morphology was observed on SEM.
In fluorescence microscopy observation, the cells co-cultured with the hydrogels were washed 3 times with PBS solution to remove unadhered cells and then stained with Calcein-AM (Macklin) and incubated for 15 min in a 5% CO2, 37 °C incubator protected from light. Incubation of HeLa cells on hydrogels was observed under a fluorescence microscope (FM). Cell numbers were counted using Image-Pro Plus 6.0 software.
2.10 Cytotoxicity of the hydrogel
Mouse osteoblasts (MC3T3-E1) were used to assay the cytotoxicity of the hydrogels. MC3T3-E1 cells were cultured in low glucose medium (MEM, Gibco) containing 10% FBS and 1% PS. The culture flasks containing MC3T3-E1 cells were also placed in an incubator with saturated humidity, 5% CO2 and a temperature of 37 °C. Prior to in vitro studies, hydrogels (HG0, HG3) were soaked in ethanol for 1 h and then sterilised under UV light for 1 h. Subsequently, logarithmic growth phase (the growth curve of MC3T3-E1 cells was shown in Fig. S3b of ESI†) MC3T3-E1 cells were taken, digested with trypsin, centrifuged and resuspended in medium at a density of 1 × 105 cell per mL. The cells were seeded on the surface of the hydrogel and 96-well plates were prepared with 50 μL of cell suspension per well. After 1 d, 3 d and 5 d of incubation, 5 μL of CCK-8 solution was added to each well and placed in the incubator for 2–4 h before the absorbance value at 450 nm was detected by a microplate reader (DNM-9602). The control group was cells cultured in 96-well plates without hydrogel material. The culture medium was changed every two days.2.11 Statistical analysis
All quantitative results were obtained from at least three samples and three independent experiments. Data were presented as means ± SD.3. Results and discussion
3.1 Gel time of the chitosan nanofiber-based hydrogels
The gelation of CSNF/HA/β-GP is shown in Fig. S4 (ESI†). The hydrogel at 0 °C was a flowing liquid solution that flowed easily. As the temperature increased, gelation occurred and a change in turbidity (clear to cloudy white) was found. The change in turbidity of the hydrogel is considered to be a criterion for the sol–gel transition of the hydrogels during gelation. During gelation, all hydrogels showed the same sol–gel state change. Among them, the hydrogel HG0 could not be inverted due to its high water content. Table 1 shows that the gelation time decreases as the concentration of CSNFs increases, from around 547 s to 43 s.| Hydrogel name | Time (s) |
|---|---|
| HG0 | >3600 |
| HG1 | 546.673 ± 10.40 |
| HG2 | 63.33 ± 10.40 |
| HG3 | 43.33 ± 2.88 |
3.2 Dynamic rheological properties and texture attributes of the hydrogels
The energy storage modulus (G′) and the loss modulus (G′′) of the hydrogel are important indicators of the sol–gel transition, which are monitored during the formation process to characterize its internal structure. As HG0 is not a complete block gel and its fragility during movement makes it impossible to detect. Therefore it is not involved in the discussion. Fig. 2 shows a graph of the dynamic rheological properties of the hydrogel. Fig. 3a is the hydrogel frequency sweep rheological curve. When the frequency increased from 0.1 HZ to 100 HZ, the G′ values of the hydrogels HG1, HG2 and HG3 were always greater than G′′ and remained basically unchanged, indicating that the gels have a stable internal structure that is unchanged within the tested frequency range. The curve of G′′ fluctuates slightly, which might be caused by water seepage in the gel during compression in the test. In terms of intensity, the G′ values of this hydrogel are much higher than that of a typical chitosan physical hydrogel,26 and increases with increasing concentration of CSNFs. This phenomenon suggests that CSNFs in hydrogels have similar reinforcing effects to other nanofillers (such as carbon nanotubes, nanoclays and other polymeric nanoparticles).28 Generally, nanofibers play a very important role in the reinforcement process through good dispersion and interfacial adhesion for effective load transfer.18,29 Compared with chitosan particles, the addition of CSNFs results in a more compact hydrogel interior, as well as an electrostatic interaction between the carboxyl groups on hyaluronic acid and the ammonium groups on chitosan, allowing the hyaluronic acid molecular chains to intersperse in the network of CSNFs, which together with the CSNFs form the internal support structure of the hydrogel.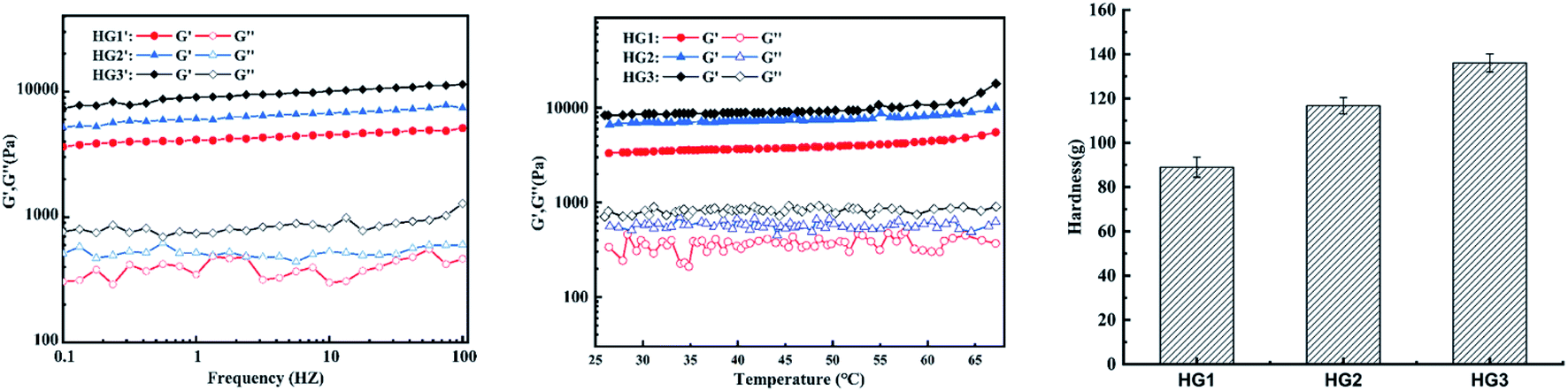 | ||
| Fig. 2 The rheological curves of the hydrogels HG1, HG2 and HG3 in the (a) frequency and (b) temperature mode. (c) Hardness of HG1, HG2 and HG3. | ||
 | ||
| Fig. 3 SEM images focused on the cross-section of hydrogels originating from gelling system with varying CSNFs content: (a) HG0; (b) HG1; (c) HG2; (d)–(f) HG3. (a) to (d) Scale bars represent 10 μm. (e) Scale bars represent 1 μm and (f) scale bars represent 500 nm. | ||
The effect of temperature on the G′ and G′′ values of the hydrogels is shown in Fig. 2b. The G′ values of all tested hydrogels were consistently greater than G′′ at temperatures ranged from 25 °C to 70 °C, indicating that CSNFs hydrogels have good thermal stability within the temperature range and might maintain their physical properties unchanged in human body. The G′ values of the hydrogels tended to increase as the temperature rose from 45 °C to 70 °C. This was due to that much higher temperatures may increase the hydrogen bond breakage and increase the hydrophobicity of the chitosan nanofibers, resulting in tighter entanglement and a rise in G′.
The characteristic parameters of the hardness texture of the hydrogel can be obtained from the texture curve. The hardness is the force required to deform the sample and is represented on the characteristic curve as the maximum peak during the first compression cycle. It can reflect the mechanical properties of the hydrogel. Fig. 2c clearly shows the increase in hardness values of the hydrogels as the concentrations of CSNFs are increased. That is because the CSNFs in the hydrogel contribute to its mechanical strength. This result is also consistent with the rheological properties results.
3.3 Morphologies of the hydrogels after freeze-drying
Freeze-drying technology has been widely used to characterize the morphology of hydrogels.26,29 SEM images of the cross-sections of the freeze-dried gels (Fig. 3) showed that, all the gels are three-dimensional porous reticular structures after freeze-drying. As the concentration of CSNFs increase, the porous gels gradually changes from a filamentary pore structure to a lamellar pore structure with a significantly-reduced pore size, and its surface also become rough from smooth (Fig. 3a–d). The SEM image of the HG3 cross-section magnified (Fig. 3e and f) showed the typical structure of chitosan nanofiber on the surface of the pore wall. The pore size can vary with the concentration of CSNFs. The smaller pore size is beneficial for loading small molecule drugs, and the rough pore wall is beneficial for cell adhesion.30 And the uniform and continuous porous structure can improve the mechanical strength of the soft material.30 Hence, our findings may serve to the application of chitosan nanofiber-reinforced hydrogels in the biomedical fields.3.4 Formation process of the hydrogels
As shown in Fig. 4, the gelation process consists of three steps. The CSNFs are slightly acid-hydrolyzed, and the CSNF/HA/β-GP solution is stored at a low temperature to form mechanical method are dendritic, with increased specific surface charge and good dispersibility in water.25 In slightly acidic aqueous medium, the amino groups on the molecular chain of CSNFs will be protonated. The strong electrostatic repulsive force partially dissolves the surface of CSNFs (Fig. 4b). When the mixed solution of β-GP and HA is introduced, the carboxyl and phosphate groups in the HA molecules will partially neutralize the protonated amino groups, reducing the repulsive force between the CS molecules. However, CSNF/HA/β-GP molecules are rich in polar groups and hydrophilic groups, such as amino, hydroxyl, carbonyl and ether, which can form hydrogen bonds with water molecules. Make the mixed solution form a temporary metastable state (Fig. 4c). As the temperature rises, the hydrogen bonds between water molecules and CSNFs and HA are broken. At the same time, the electrostatic attraction between NH3+ on CSNFs and OPO32− on β-GP and COO− on HA increases, thereby promoting the entanglement of the CSNFs chain itself and the association with the HA chain.31 In addition, an increase in temperature will reduce the proton dissociation constant (pKa) of CSNF, resulting in a decrease in the ionization effect of CSNF. Under the combined influence of these effects, the sol transforms into a hydrogel (Fig. 4d).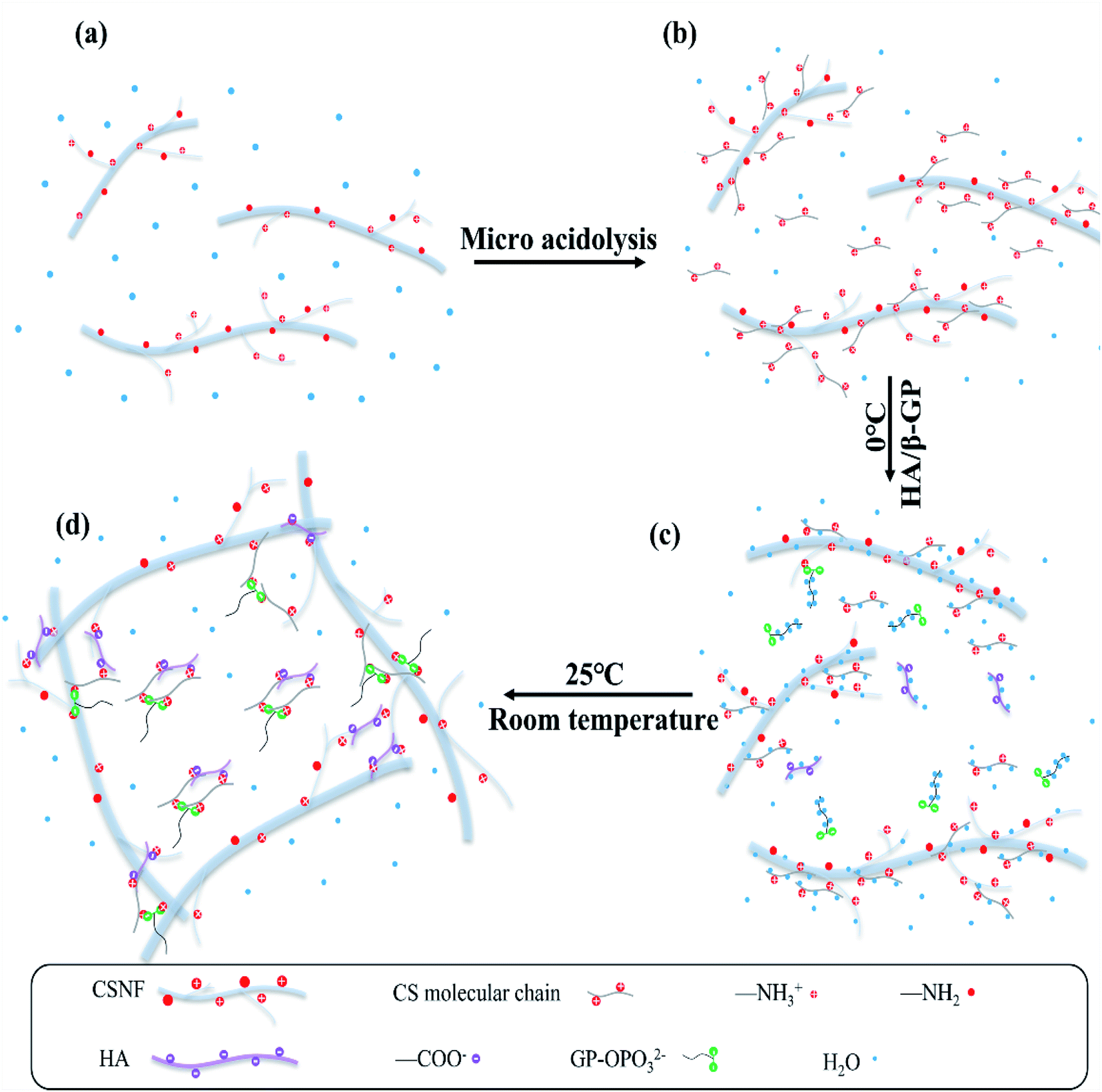 | ||
| Fig. 4 Representation of the gelation mechanism of CSNF/HA/β-GP precursor solution. | ||
3.5 In vitro drug release
Using DOX as an anti-cancer drug, the cumulative release of DOX in the drug-loaded hydrogel at 37 °C and different pHs (7.4, 6.5, 4.0) was evaluated. Fig. 5a shows that the cumulative release of DOX in the hydrogel is obviously dependent on the pH. As the pH of the environment decreases, the cumulative release of DOX gradually increases. At pH 7.4, all drug-loaded hydrogels were released suddenly in the first 6 h, and the cumulative release amount was close to 30%, with no significant increase thereafter. After 72 h, the cumulative release amount of all drug-loaded hydrogels was about 30%. When the pH dropped to 6.5, all drug-loaded hydrogels were released suddenly in the first 15 h, and the cumulative release amount was increased as compared with that at pH 7.4. The drug-release was completed within 40 h. After 40 h, little change in the cumulative release amount was observed. When the pH continued to decrease to 4.0, all drug-loaded hydrogels were released suddenly in the first 15 h, and slowly released during 15–40 h with no significant increase afterwards. After 72 h, the cumulative release of all drug-loaded hydrogels reached 88%. Among them, the cumulative release of HG1 reached 98%, being almost completely released. Compared with previously reported drug-loaded hydrogels prepared with CSP, the cumulative release is at least 20% higher and up to 40% higher.26 The intelligent pH response of drug-loaded hydrogels is mainly due to two aspects. Under acidic conditions, a large number of amine groups are protonated on CSNF, and the electron repulsion between CSNF increases, which promotes the release of DOX. Meanwhile, the pending carboxyl groups in HA are similar to pH buffers, which can alleviate the burst release of the drug caused by the sudden protonation of a large number of amine groups in CSNF. Another reason is the partial carboxylation on HA, which disrupts the electrostatic interaction between CSNF and HA and promotes the massive release of DOX.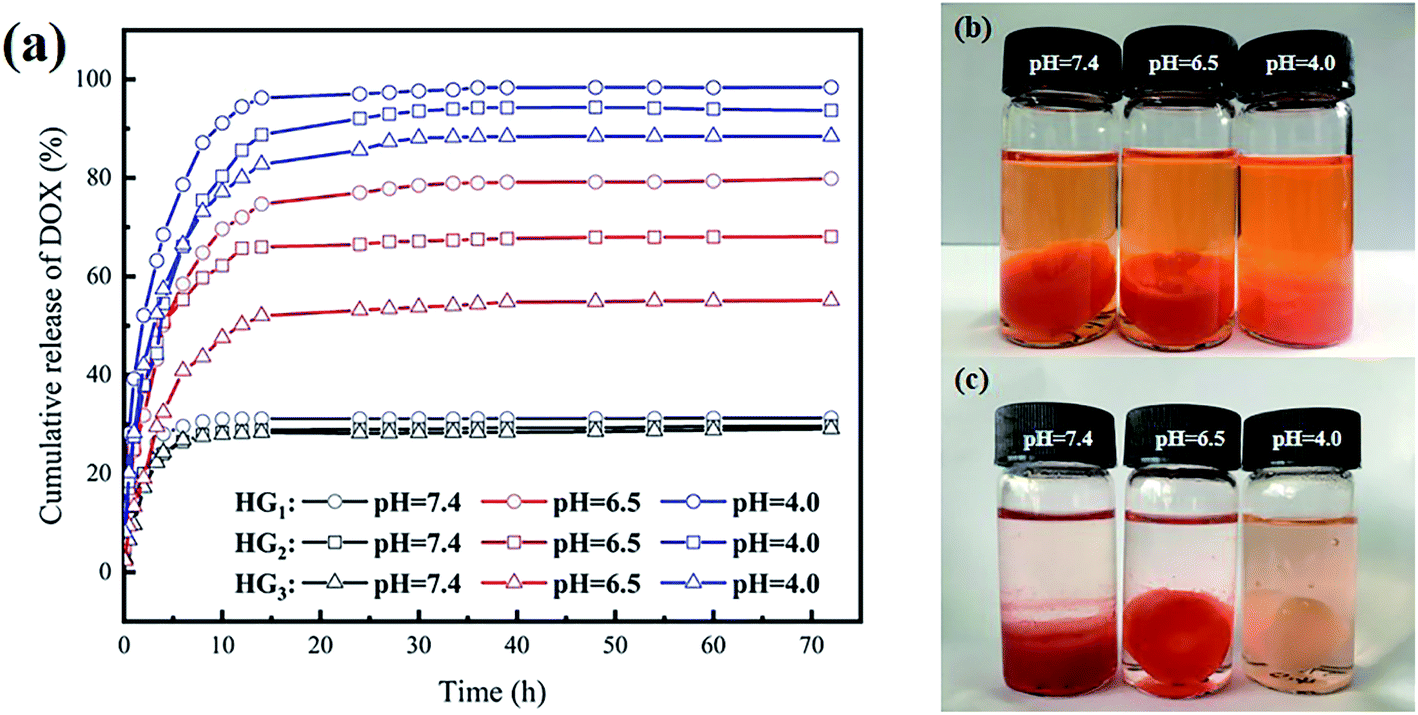 | ||
| Fig. 5 (a) The cumulative percentage release of DOX in PBS buffers of different pH; hydrogel photographs of DOX in HG3 released in PBS buffers of different pH for 72 h (b) before and (c) after. | ||
Analysis of CSNFs content shows that, the cumulative release of HG3 is always the lowest under the same conditions, followed by HG2, and the cumulative release of HG1 is always the highest. At pH 7.4, there were no significant difference in the cumulative release of the three drug-loaded hydrogels is not. As the pH decreased, evident differences can be found among the three drug-loaded hydrogels. The greatest difference was found at pH 6.5, which was mainly caused by the CSNFs content. There was a difference of approximately 12% for each carrier hydrogel, with cumulative release reaching 80% for HG1, 68.1% for HG2 and 55.2% for HG3. At pH 4.0, the difference approximately 5.0% was found for each carrier, with cumulative releases of 98.5%, 93.7% and 88.5% for HG1, HG2 and HG3, respectively. Fig. 5a shows that HG3 has the highest cumulative release under the same acidic conditions. This can be attributed to the fact that under acidic conditions the amine groups on CSNF are protonated and an electron repulsion is generated between the chitosan chains. The higher the CSNF content, the greater the electron repulsion, which facilitates the release of the drug from the hydrogel.
From Fig. 5b and c, it can be observed that after 72 h, the color of the drug-loaded hydrogel HG3 at pH 4.0 faded and was consistent with the cumulative release results.
3.6 Release kinetics model fitting of DOX in the hydrogels
To investigate the drug release mechanism, the cumulative drug release curves are fitted using the zero-order, first-order, Higuchi, and Ritger–Peppas models (shown in Fig. 5a) and the values of the fitted parameters are shown in Tables S2–S4 (ESI†). Among those parameters, k, b and n are the correlation coefficients of the model, and R2 is the determination coefficient. The fitted data is based on the principle of maximizing the coefficient of determination. Tables S2–S4† show that the Ritger–Peppas model has the best fitting effect on most DOX release curves, and n is less than 0.45. So, Fick diffusion dominated the release of DOX. However, the DOX release curves for HG1 and HG2 at pH 4 is most consistent with the first order model. It can be proposed from the first-order model that the diffusion dynamics are mainly driven by concentration differences, and that the dissolution of drugs from solid particles within liquid media is mainly a surface effect. The first-order model can be applied to describe the dissolution of water-soluble drugs in porous drug dosage forms. Due to the sparser structure of HG1 and HG2 relative to HG3, the amino groups on chitosan are deprotonated under acidic conditions, resulting in increased repulsion between the chitosan chains and the opening of the entangled chains, allowing the concentration difference to dominate the DOX efflux from the matrix.3.7 Cell adhesion and cytotoxicity of the hydrogel
Because human cervical cancer cells (HeLa) can express CD44,32 they can be used as proof of cancer cells. Therefore, this HeLa cell model was adopted in this experiment to assess the adhesion performance of cancer cells cultured on composite hydrogels for different time. As shown in Fig. 6a–d, comparing the FM images, it can be found that more cells adhere to the surface of HG3 than HG0. The statistical results of the cell number per unit area intuitively show that HG3 has better adhesion to cells (Fig. 6e). The hydrogel HG3 was selected for cytotoxicity experiments. From Fig. 6f, the absorbance values of cells on hydrogel HG3 were almost not significantly different from those cultured on well plates, and the relative proliferation rates (RGR) of cells were all above 90%. According to the US Pharmacopoeia class V toxicity rating criteria, this composite hydrogel has low cytotoxicity with a class I cytotoxicity rating.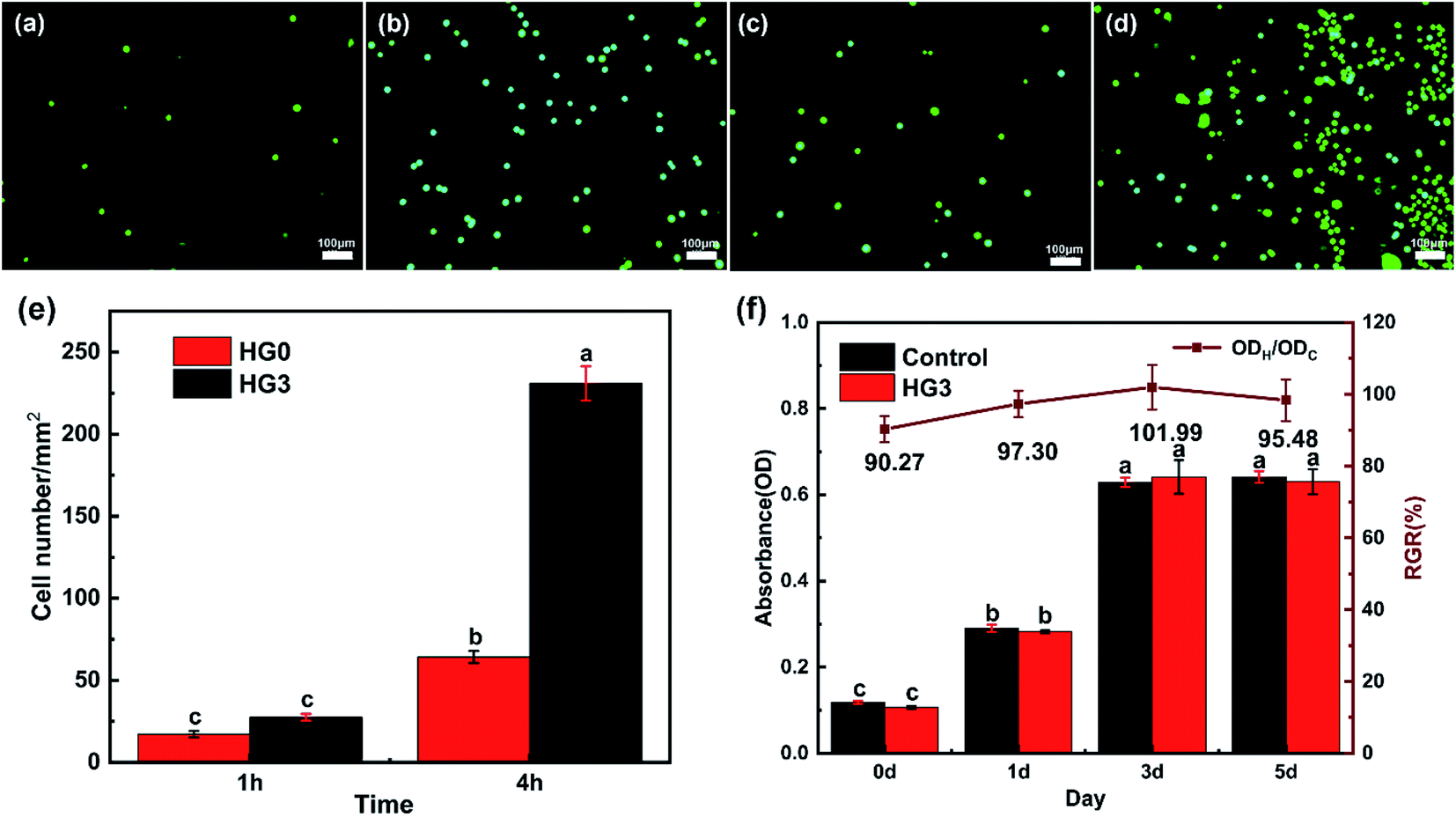 | ||
| Fig. 6 FM images of HeLa cells cultured on HG0 (a and b) and HG3 (c and d) for 1 h (a and c) and 4 h (b and d); (e) comparison between the number of cells attached to HG0 and HG3, (f) absorbance of MC3T3-E1 cells after 1, 3 and 5 days of incubation on well plates and HG3 respectively three repetitions with ± SD. | ||
Fig. 7 shows the SEM images of HeLa cells cultured on HG0 and HG3 for 1 h and 4 h. The adhesion performance of the cells on the hydrogel HG3 is better than the others, which are in good agreement with the FM images in Fig. 6. It can be seen from Fig. 7 that cells on the surface of HG3 produced a large number of filopodia and actin filaments and stretched along the surface of HG3 regardless of whether they were cultured for 1 h or 4 h. For HG0 prepared from chitosan powder, the cells only attached to the surface of the hydrogel and did not produce filopodia and actin filaments. This phenomenon may be due to the rough surface of the hydrogel prepared by chitosan nanofibers, which can specifically bind to CD44. This further verifies that HG3 prepared by CSNFs is more beneficial to cell adhesion.
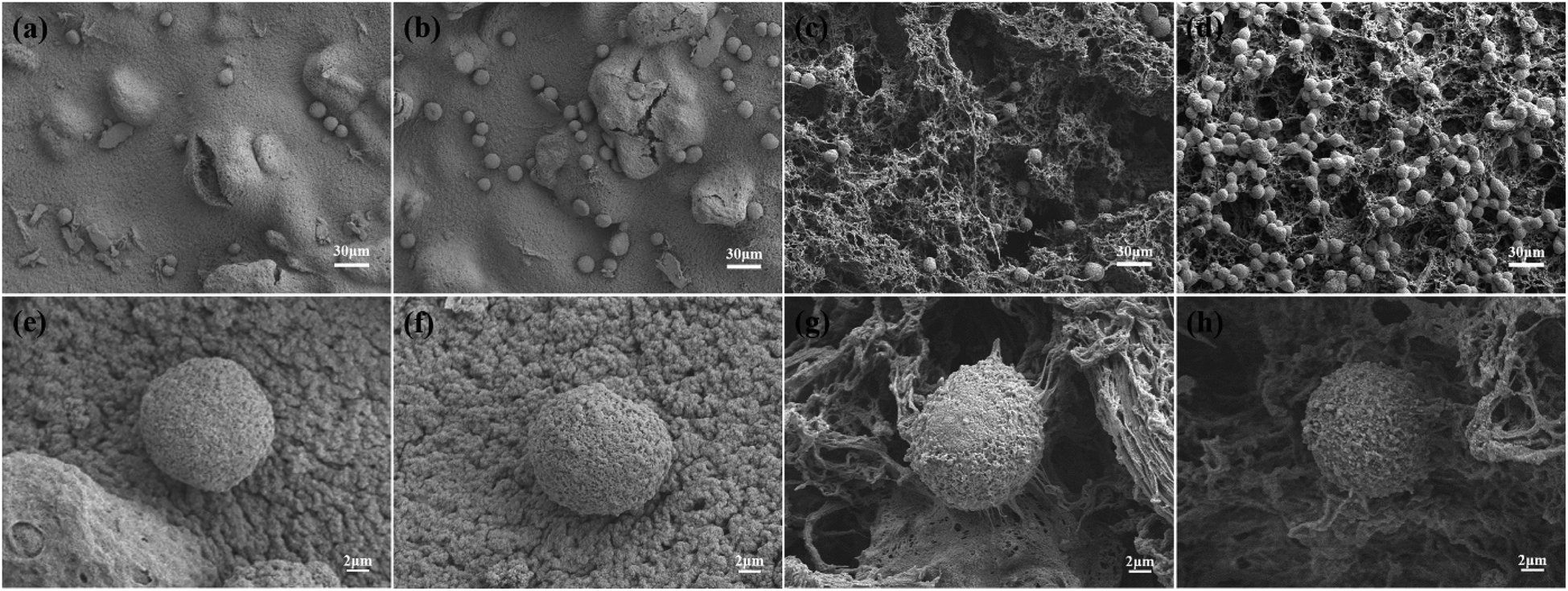 | ||
| Fig. 7 SEM images of HeLa cells cultured on HG0 (a and b) and HG3 (c and d) for 1 h (a and c) and 4 h (b and d), respectively. And (e)–(h) show details of the individual cells in (a)–(d) respectively. | ||
4 Conclusions
In this research, a novel of CSNF/HA/β-GP composite hydrogel was successfully prepared by micro-dissolution crosslinking method, which has excellent mechanical properties, better biocompatibility and good adhesion performance to cancer cells. The cells can attach to the surface of the hydrogel, due to the rough surface of the hydrogels that can specifically bind to CD44. Meanwhile, the anti-cancer drugs loaded in the hydrogel can be released as much as possible under acidic tumor conditions. These properties highlight the potential application of these novel hydrogels with the loaded anti-tumor drug as a wound dressing after tumor resection surgery, targeting to kill residual cancer cells and thus preventing cancer recurrence.Author contributions
Y. W. contributed to the design of the methodology, performing the main experiments, data analysis, and writing the original draft of the manuscript. S. Z. contributed by performing part of the experiments and analyzing data. C. X. and W. M. contributed to the validation of the experiments and the data analysis. X. L. contributed to the design of the methodology, review and revising of the writing, supervision, and funding acquisition. L. L., Z. H., H. D. and Y. M. contributed to the design of the methodology, review and revising of the writing. G. Z. contributed to the main conceptualization, design of the methodology, supervision, and funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21878107 and 21978101).Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed
.
- T. Dai, C. Wang, Y. Wang, W. Xu, J. Hu and Y. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 15163–15173 CrossRef CAS PubMed
- M. Zabara, B. Senturk, M. Gontsarik, Q. Ren, M. Rottmar, K. Maniura Weber, R. Mezzenga, S. Bolisetty and S. Salentinig, Adv. Funct. Mater., 2019, 29, 1904007 CrossRef
- S. Wei, P. Xu, Z. Yao, X. Cui, X. Lei, L. Li, Y. Dong, W. Zhu, R. Guo and B. Cheng, Acta Biomater., 2021, 124, 205–218 CrossRef CAS PubMed
- N. Rezaei, H. G. Hamidabadi, S. Khosravimelal, M. Zahiri, Z. A. Ahovan, M. N. Bojnordi, B. S. Eftekhari, A. Hashemi, F. Ganji, S. Darabi and M. Gholipourmalekabadi, Int. J. Biol. Macromol., 2020, 164, 855–862 CrossRef CAS PubMed
- O. Qianqian, K. Songzhi, H. Yongmei, J. Xianghong, L. Sidong, L. Puwang and L. Hui, Int. J. Biol. Macromol., 2021, 181, 369–377 CrossRef PubMed
- C. Zhou, G. W. N. Chia, J. C. S. Ho, T. Seviour, T. Sailov, B. Liedberg, S. Kjelleberg, J. Hinks and G. C. Bazan, Angew. Chem., Int. Ed., 2018, 57, 8069–8072 CrossRef CAS PubMed
- H. Xu, Z. Fang, W. Tian, Y. Wang, Q. Ye, L. Zhang and J. Cai, Adv. Mater., 2018, 30, 1801100 CrossRef PubMed
- B. Jin, G. Zhang, J. Lian, Q. Zhang, X. Zhan and F. Chen, J. Mater. Chem. A, 2019, 7, 12266–12275 RSC
- Y. Liang, X. Zhao, T. Hu, B. Chen, Z. Yin, P. X. Ma and B. Guo, Small, 2019, 15, 1900046 CrossRef PubMed
- C. Lu, C. Yu and Y. Yeh, Acta Biomater., 2021, 130, 66–79 CrossRef CAS PubMed
- C. Hu, M. S. Haider, L. Hahn, M. Yang and R. Luxenhofer, J. Mater. Chem. B, 2021, 9, 4535–4545 RSC
- M. Mihajlovic, M. Mihajlovic, P. Y. W. Dankers, R. Masereeuw and R. P. Sijbesma, Macromol. Biosci., 2019, 19, 1800173 CrossRef PubMed
- Z. Zou, B. Zhang, X. Nie, Y. Cheng, Z. Hu, M. Liao and S. Li, RSC Adv., 2020, 1, 33973–39722 Search PubMed
- Y. Yang, X. Wang, F. Yang, L. Wang and D. Wu, Adv. Mater., 2018, 30, 1707071 CrossRef PubMed
- L. Wang, H. Lv, L. Liu, Q. Zhang, P. Nakielski, Y. Si, J. Cao, X. Li, F. Pierini, J. Yu and B. Ding, J. Colloid Interface Sci., 2020, 565, 416–425 CrossRef CAS PubMed
- W. Xing and Y. Tang, Nano Mater. Sci., 2021, 1–14 Search PubMed
- H. Cui, W. Jiang, C. Wang, X. Ji, Y. Liu, G. Yang, J. Chen, G. Lyu and Y. Ni, Composites, Part B, 2021, 225, 109316 CrossRef CAS
- P. Abasian, M. Radmansouri, J. M. Habibi, M. V. Ghasemi, A. Mohammadi, M. Irani and F. S. Jazi, Int. J. Biol. Macromol., 2019, 121, 398–406 CrossRef CAS PubMed
- P. Balan, J. Indrakumar, P. Murali and P. S. Korrapati, Int. J. Biol. Macromol., 2020, 142, 201–211 CrossRef CAS PubMed
- J. J. Grant, S. C. Pillai, T. S. Perova, S. Hehir, S. J. Hinder, M. McAfee and A. Breen, Chemosensors, 2021, 9, 70 CrossRef CAS
- F. Rao, Y. Wang, D. Zhang, C. Lu, Z. Cao, J. Sui, M. Wu, Y. Zhang, W. Pi, B. Wang, Y. Kou, X. Wang, P. Zhang and B. Jiang, Theranostics, 2020, 10, 1590–1603 CrossRef CAS PubMed
- P. G. Shikhi-Abadi and M. Irani, Int. J. Biol. Macromol., 2021, 183, 790–810 CrossRef CAS PubMed
- C. Cui, S. Sun, S. Wu, S. Chen, J. Ma and F. Zhou, Engineered Regeneration, 2021, 2, 82–90 CrossRef
- S. Zhang, G. Zhao, J. Wang, C. Xie, W. Liang, K. Chen, Y. Wen and X. Li, ACS Appl. Mater. Interfaces, 2021, 13, 12347–12358 CrossRef CAS PubMed
- W. Zhang, X. Jin, H. Li, R. Zhang and C. Wu, Carbohydr. Polym., 2018, 186, 82–90 CrossRef CAS PubMed
- H. Y. Zhou, X. G. Chen, M. Kong, C. S. Liu, D. S. Cha and J. F. Kennedy, Carbohydr. Polym., 2008, 73, 265–273 CrossRef CAS
- B. Maharjan, J. Park, V. K. Kaliannagounder, G. P. Awasthi, M. K. Joshi, C. H. Park and C. S. Kim, Carbohydr. Polym., 2021, 251, 117023 CrossRef CAS PubMed
- S. Nitta, M. Akagi and H. Iwamoto, Polym. J., 2019, 51, 501–509 CrossRef CAS
- J. Zhang, B. J. Allardyce, R. Rajkhowa, Y. Zhao, R. J. Dilley, S. L. Redmond, X. Wang and X. Liu, ACS Biomater. Sci. Eng., 2018, 4, 3036–3046 CrossRef CAS PubMed
- X. Liu, Y. Chen, Q. Huang, W. He, Q. Feng and B. Yu, Carbohydr. Polym., 2014, 110, 62–69 CrossRef CAS PubMed
- X. Mo, F. Wu, Y. Li and X. Cai, Mater. Today Commun., 2021, 28, 102682 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: The data in this work are available in the manuscript or available from the corresponding author upon request. See https://doi.org/10.1039/d2ra01622b |
| This journal is © The Royal Society of Chemistry 2022 |