
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free reductive cyclization of bis(2-aminophenyl) disulfide with CO2 in the presence of BH3NH3 to synthesize 2-unsubstituted benzothiazole derivatives†
Xiao Liabc,
Zhenbao Liuabc,
Hailong Hongabc,
Limin Han*abcd and
Ning Zhu *abc
*abc
aCollege of Chemical Engineering, Inner Mongolia University of Technology, Hohhot, 010051, China. E-mail: hanlimin_442@hotmail.com; zhuning@imut.edu.cn
bKey Laboratory of CO2 Resource Utilization at Universities of Inner Mongolia Autonomous Region, Hohhot, 010051, China
cInner Mongolia Engineering Research Center for CO2 Capture and Utilization, Hohhot, 010051, China
dWu Hai Vocational &Technical College, Wu Hai, 010070, China
First published on 20th June 2022
Abstract
An efficient and catalyst-free methodology for the reductive cyclization of various disulfides using BH3NH3 as a reductant and CO2 as a C1 resource was developed. The desired 2-unsubstituted benzothiazole derivatives were obtained in good to excellent yields. Moreover, mechanism investigation demonstrated that BH3NH3 played an important role in the formation of benzothiazole. As a reducing agent, BH3NH3 reduced CO2 and cleaved the S–S bond of the disulfide efficiently. In addition, the N–H bond of the amino group was also activated by BH3NH3. To the best of our knowledge, this is an unprecedented catalyst-free protocol for the synthesis of 2-unsubstituted benzothiazole from bis(2-aminophenyl) disulfide and CO2.
1 Introduction
CO2 is a low-cost, sustainable, and abundant C1 resource which has been widely employed in the synthesis of value-added chemicals.1–3 Among the various reactions reported for the transformations of CO2, reductive functionalization of amines with CO2 in the presence of a reducing reagent has attracted extensive attention.4–7 Significantly, new bonds such as C–N, C–O, C–C or C–S bonds were formed in the process of reductive functionalization by reducing CO2 with different functionalization reagents.8–12 Among these, formations of C–N and C–S bonds are important transformations in organic synthesis.One of the common compounds containing both C–N and C–S bonds is benzothiazole, a bicyclic compound in which a thiazole ring is fused with a benzene ring. It is an important synthetic intermediate and an important privileged scaffold of many natural compounds and/or bioactive compounds.13 Benzothiazole is used in the synthesis of pharmaceutical compounds such as ethoxzolamide, riluzole and florapronol (Fig. 1). In particular, 2-unsubstituted benzothiazole is the core structure that can be used to synthesize a variety of valuable 2-substituted benzothiazoles.
 | ||
| Fig. 1 Representative bioactive benzothiazoles. | ||
There are many methods to prepare 2-unsubstituted benzothiazole derivatives.14 It is worth noting that various raw materials have been utilized via different reaction routes in the synthesis of 2-unsubstituted benzothiazole. Among these raw materials, 2-aminothiophenol has attracted extensive attention of researchers. The most commonly used method is the condensation reaction of 2-aminothiophenol with formic acid,15 formamide,16 or formaldehyde17 in the presence of a catalyst. Recently, the synthesis of 2-unsubstituted benzothiazole using CO2 as the carbon source has been reported, in the presence of various reductants and catalysts (Scheme 1, route a). With H2 as the reductant, 2-unsubstituted benzothiazole could be obtained from the reaction of CO2 and 2-aminothiophenol catalyzed by CoF2/PP3/CsF.18 It should be noted that both H2 and CO2 need to be activated. Therefore, high temperatures and high pressures are needed in the reaction. With hydrosilanes as the reductant, 2-unsubstituted benzothiazole can be obtained from the reaction of CO2 and 2-aminothiophenol catalyzed by 1,5-diazabicyclo [4.3.0] non-5-ene (DBN),19 1,5,7-triazabicyclo [4.4.0] dec-5-ene (TBD),20 acetate-based ionic liquids [Bmim][OAc]21 or biomass-derived γ-valerolactone.22
Although these synthetic routes for preparing 2-unsubstituted benzothiazole using efficient, non-toxic, and renewable catalysts have been reported, the exploration of catalyst-free and atom-economic methods under mild conditions is of great significance. Notably, 2-aminothiophenol is unstable and easily oxidized to form disulfide,23,24 and 2-aminothiophenols containing various substituents are difficult to be prepared, which limits the application of 2-aminothiophenol as a substrate. Therefore, the method of synthesizing 2-unsubstituted benzothiazole compounds should be investigated using stable and readily available disulfides as raw materials. One of the key steps in the synthesis of benzothiazole from disulfide is the cleavage of S–S bond. It is found that the S–S bond can be cleaved by free radical reagents,25,26 metal catalysts,27,28 nucleophilic reagents,23,29 reducing reagents,30–32 thiol-disulfide exchange reaction33–35 and the disulfide-metal sulfide dynamic interchange reaction.36,37 Therefore, 2-unsubstituted benzothiazole is synthesized by reductive cyclization of disulfide and CO2, a crucial reducing reagent which could reduce CO2 and the S–S bond of disulfide is required (Scheme 1, route b).
 | ||
| Scheme 1 The synthesis of 2-unsubstituted benzothiazole. | ||
Recently, BH3NH3, a high hydrogen storage reagent, has been reported to reduce CO2 into formamides under mild conditions without any catalyst.20,38 Subsequently, we have prepared 1H-benzimidazole by the cascade reaction of CO2 and o-phenylenediamine under two-electron reduction process.39 As part of our ongoing work, we investigated BH3NH3 as a reductant to synthesize 2-unsubstituted benzothiazole derivatives from bis(2-aminophenyl) disulfide and CO2, in which C–N and C–S bonds were constructed simultaneously. This simple and effective method could be applicated in the synthesis of many 2-unsubstituted benzothiazole compounds.
2 Results and discussion
The model reaction of bis(2-aminophenyl) disulfide (1a, 0.5 mmol) and CO2 to synthesize benzothiazole (2a) was initially explored. First, the reaction was carried out in NMP at 1 MPa of CO2 with 3 equiv. of BH3NH3 at 100 °C in 24 h, and the desired product 2a was obtained in 32% yield (Table 1, entry 1). Notably, a large amount of disulfide was recovered, which demonstrated that the S–S bond of disulfide was not easily cleaved under this reaction condition and the raw material was not completely consumed. To improve the yield of target product, the reaction temperature was increased from 100 °C to 120 °C, which increased the yield of 2a from 32% to 85% (Table 1, entries 1–3). These experimental results indicated that higher temperature is beneficial to the cleavage of S–S bond. However, as the reaction temperature was increased to 140 °C, the yield of target product decreased to 46% (Table 1, entries 4–5), possibly because the higher reaction temperature decreased the solubility of CO2 and further reduced CO2 into methylation by-products.40 Consequently, the reaction temperature would be 120 °C in subsequent experiments.
|
||||||
|---|---|---|---|---|---|---|
| Entry | BH3NH3 (mmol) | Solvent (mL) | PCO2 (MPa) | T (°C) | t (h) | Yieldb [%] |
| a Reaction conditions: bis(2-aminophenyl) disulfide (0.1242 g, 0.5 mmol), solvent (1 mL).b Isolated yield.c No CO2. | ||||||
| 1 | 1.5 | NMP | 1 | 100 | 24 | 32 |
| 2 | 1.5 | NMP | 1 | 110 | 24 | 68 |
| 3 | 1.5 | NMP | 1 | 120 | 24 | 85 |
| 4 | 1.5 | NMP | 1 | 130 | 24 | 75 |
| 5 | 1.5 | NMP | 1 | 140 | 24 | 46 |
| 6 | 1.25 | NMP | 1 | 120 | 24 | 93 |
| 7 | 1 | NMP | 1 | 120 | 24 | 68 |
| 8 | 0.5 | NMP | 1 | 120 | 24 | 30 |
| 9 | 2 | NMP | 1 | 120 | 24 | 70 |
| 10 | 1.25 | NMP | 0.1 | 120 | 24 | Trace |
| 11 | 1.25 | NMP | 0.5 | 120 | 24 | 46 |
| 12 | 1.25 | NMP | 2 | 120 | 24 | 62 |
| 13 | 1.25 | 1,4-Dioxane | 1 | 120 | 24 | 13% |
| 14 | 1.25 | CH3CN | 1 | 120 | 24 | 25% |
| 15 | 1.25 | THF | 1 | 120 | 24 | 18% |
| 16 | 1.25 | DMSO | 1 | 120 | 24 | 26% |
| 17 | 1.25 | DMF | 1 | 120 | 24 | 80% |
| 18c | 1.25 | DMF | 0 | 120 | 24 | 75% |
| 19 | 1.25 | H2O | 1 | 120 | 24 | Trace |
| 20 | 1.25 | NMP | 1 | 120 | 18 | 83 |
| 21 | 1.25 | NMP | 1 | 120 | 12 | 60 |
| 22 | 1.25 | NMP | 1 | 120 | 6 | 25 |
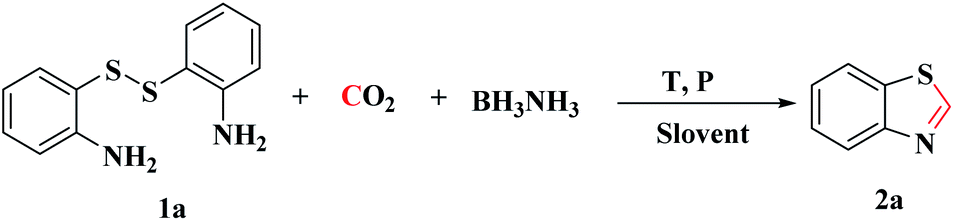
To further improve the yield of 2a, the amount of BH3NH3 and the pressure of CO2 were optimized (Table 1, entries 3, 6–9). Increasing the amount of BH3NH3 reduced the yield of the target product, which demonstrated that the amount of BH3NH3 was critical to the reaction. The excessive BH3NH3 might further reduce CO2 into methylated by-products.41 When the amount of BH3NH3 was 2.5 equiv. of 1a, the yield of 2a reached 93% (Table 1, entry 6). However, when the amount of BH3NH3 was further reduced, the yield of 2a was also reduced (Table 1, entries 7 and 8), suggesting that the amount of reducing agent is insufficient. Therefore, 2.5 equivalents of reducing agent were used in the subsequent reactions of 1a and CO2. Furthermore, the effect of CO2 pressure on the reaction yield was also examined (Table 1, entries 6, 10–12). Only trace amount of 2a was found at 0.1 MPa CO2 (Table 1, entry 10), indicating that higher concentration of CO2 was needed in the reaction of 1a and CO2. Increasing the pressure of CO2 to 1 MPa led to a higher yield of 2a (Table 1, entry 6). When the CO2 pressure was further increased to 2 MPa, the yield of 2a decreased significantly (Table 1, entry 12). These experimental results indicated that BH3NH3 is consumed by excess CO2, resulting in insufficient amounts of BH3NH3 to break the S–S bond. Therefore, the balanced optimization of the reaction temperature, the amount of BH3NH3 and CO2 pressure was critical for the high-yield formation of 2a from 1a and CO2.
The effect of solvent on the reaction yield was also investigated. When weakly polar solvents such as 1,4-dioxane, CH3CN and THF were used (Table 1, entries 13–15), 2a was obtained in lower yields. When DMSO was used (Table 1, entry 16), a lower yield of 2a was obtained, likely due to the oxidizing property of DMSO to convert thiol into disulfide.42 When DMF was used (Table 1, entries 17 and 18), 2a was obtained in 80% yield. However, 2a could also be obtained in a yield of 75% in the absence of CO2, because DMF can serve as the C1 source to produce the target product in the presence of BH3NH3.16,43 Water was not suitable for the reaction system (Table 1, entry 19), because the formed carbonic acid promoted the hydrolysis of BH3NH3.44 To our delight, an excellent yield of 2a was obtained in NMP; therefore, NMP was chosen as the solvent in the following experiments. Finally, the reaction time was also investigated (Table 1, entries 6, 20–22), and the highest yield was obtained in 24 h. Therefore, 1 equiv. of 1a reacted with 1 MPa of CO2 in the presence of 2.5 equiv. BH3NH3 in 120 °C for 24 h to give the highest yield of the target product.
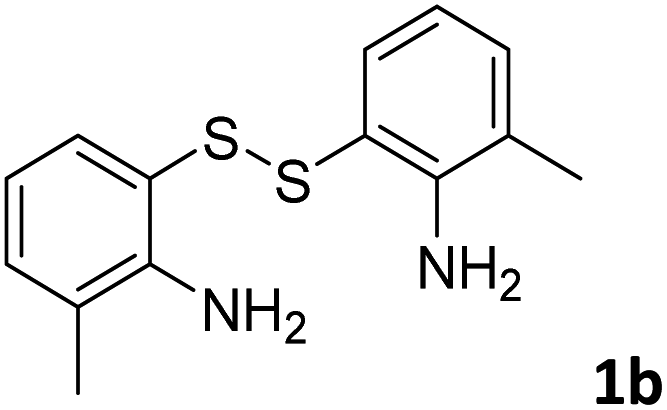
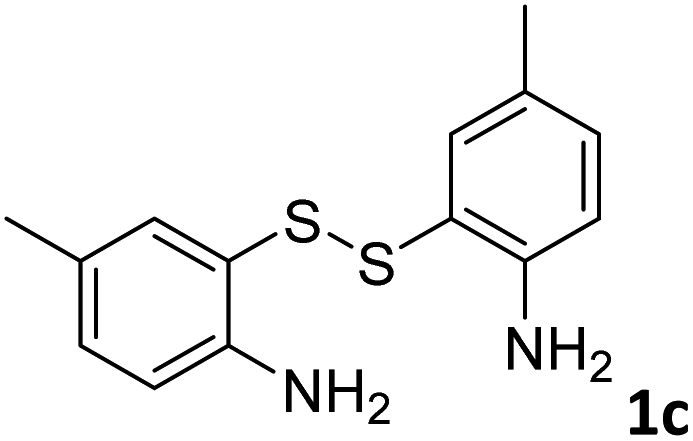
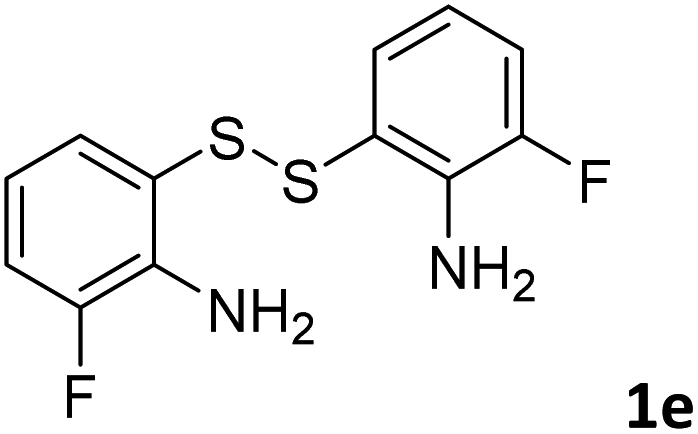
With the optimized reaction conditions in hand, the reaction scope was investigated with a range of differently substituted bis(2-aminophenyl) disulfide (Table 2). We found that all the substrates reacted with CO2 and BH3NH3 to produce the corresponding target products in good to excellent yields. The reaction of 1a with CO2 and BH3NH3 provided 2a in an isolated yield of 93% under the optimized reaction conditions (Table 2, entry 1). The substrates with electron-donating groups like methyl and methoxy displayed higher reactivity, and the corresponding products were obtained in excellent yields (86–95%, Table 2, entries 2–4). The substrates with electro-withdrawing groups including –F, –Cl, –Br and –CF3 displayed good reactivity, and the corresponding products were obtained in good yields (63–85%, Table 2, entries 5–10). Furthermore, bis(2-aminophenyl) disulfides bearing functional groups at the C4 position showed higher reactivity than those at the C6 position, (Table 2, entries 2 vs. 3; entries 6 vs. 8) which presumably attributed to the steric effects of substituent groups. When the substituent group was –SO2CH3, a strong electron-withdrawing group, corresponding 6-(methylsulfonyl)-benzothiazole was obtained in a yield of 55% (Table 2, entry 11). In addition, naphtho[2,3]-thiazole could be obtained in 66% yield from the corresponding disulfide (Table 2, entry 12). To the best of our knowledge, this is the first example of successfully synthesizing 6-(methylsulfonyl)-benzothiazole and naphtho[2,3]-thiazole using CO2 as the carbon source.








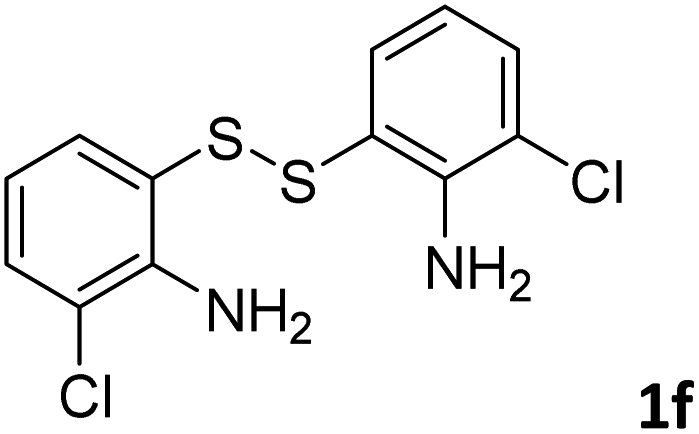
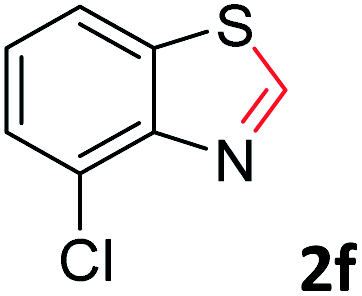
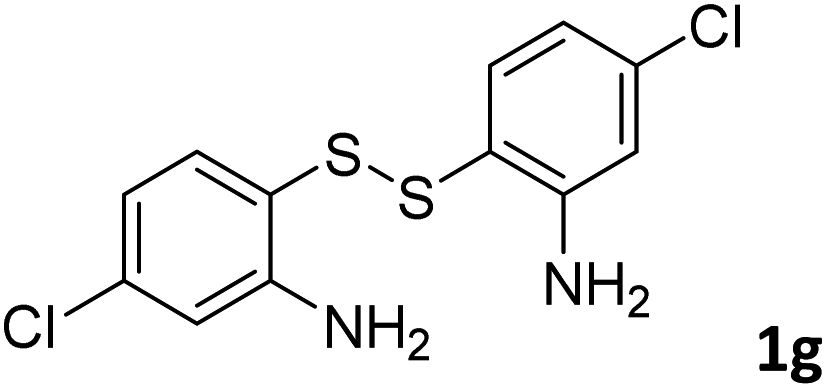






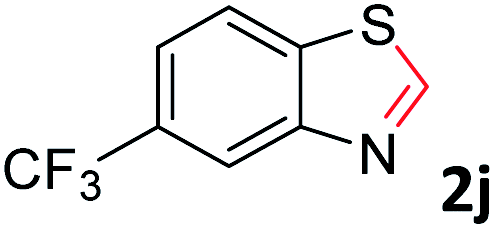


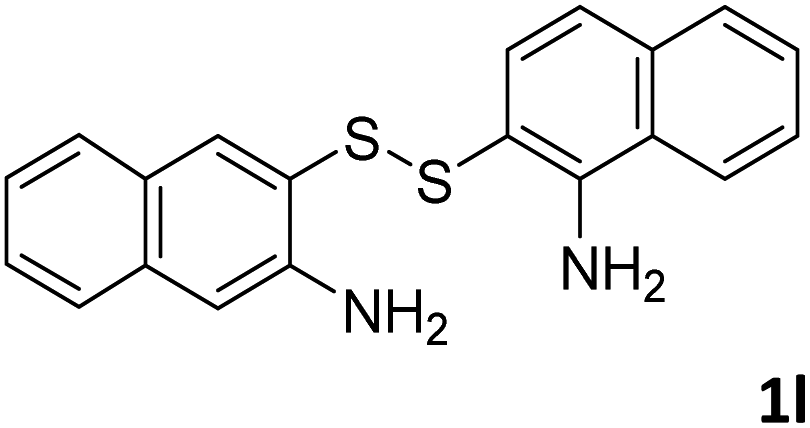
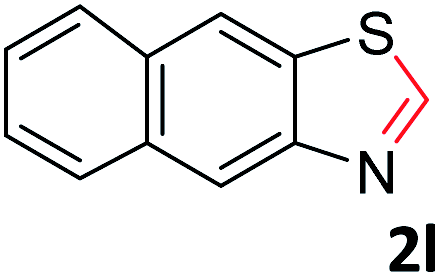
To verify the applicability of the developed methodology, the reductive cyclization of disulfide with CO2 to synthesize benzothiazole was carried out on the gram scale (Scheme 2). The desired product was obtained in 80% isolated yield, demonstrating that this methodology can be used in gram-scale synthesis.
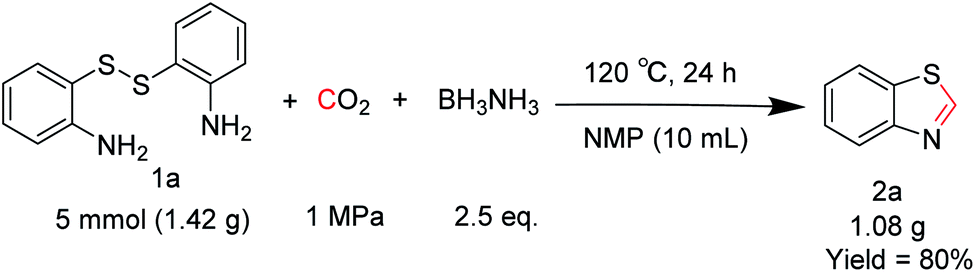 | ||
| Scheme 2 The gram-scale reaction. | ||
To explore the reaction mechanism, several control experiments were performed under the optimized conditions. As shown in Scheme 3, no desired product was detected in the absence of BH3NH3 or CO2, which indicated that both BH3NH3 and CO2 were indispensable (Scheme 3).
 | ||
| Scheme 3 Control experiments. | ||
When BH3NH3 and CO2 were mixed in NMP, it was interesting to find that the formate borohydride species BH3−n(OCOH)nNH3 (I) was formed, which was confirmed by 1H NMR (8.31 ppm, 8.04 ppm), 13C NMR (166.38 ppm), and11 B NMR (19.39 ppm) (Fig. S1†) analysis. The experimental results are similar with the data reported in the literature.41,45–47 This finding suggested that BH3−n(OCOH)nNH3 (I) was an intermediate for the synthesis of benzothiazole.
Furthermore, 1H NMR analysis was employed to identify the cleavage of S–S bond of disulfide by BH3NH3 during the reaction process (Fig. 2). As illustrated in Fig. 2c, a peak pattern similar to that of 2-aminothiophenol appeared when disulfide reacted with BH3NH3 under an inert atmosphere at 120 °C in deuterated DMF (Fig. 2c red rectangle). However, the aromatic hydrogen shifted to upfield in 2-aminothiophenol after reduced by BH3NH3, which might be resulted from the coordination of boron atom of BH3NH3 with the nitrogen atom of 2-aminothiophenol.48 In addition, the 1H NMR spectra of reaction mixture (Fig. 2c) showed that the –NH2 group hydrogen in 2-aminothiophenol formed from disulfide reduction shifted downfield from 4.87 to 5.19 (Fig. 2c blue rectangle), indicating that the N–H bond of 2-aminothiophenol was activated. These results demonstrated that BH3NH3 reduced disulfide to 2-aminothiophenol and activated the N–H bond of amino group, which increased the nucleophilicity of the amino group in 2-aminothiophenol.49
 | ||
| Fig. 2 The reaction of disulfide with BH3NH3 detected by 1H NMR in deuterated DMF. (a) Disulfide 1a, (b) the mixture of disulfide 1a and BH3NH3, (c) the reaction of disulfide with BH3NH3 for 1 h, (d) 2-aminothiophenol. | ||
Based on our experimental results and previous reports,18,21,50 a possible reductive cyclization reaction mechanism was proposed (Scheme 4). Firstly, BH3NH3 reacts with CO2 to produce the intermediate BH3−n(OCOH)nNH3 (I). Meanwhile, the S–S bond of the disulfide is cleaved by BH3NH3 to form complex (II) of 2-aminothiophenol with BH3. Subsequently, the nucleophilic N atom of 2-aminothiophenol attacks the carbon atom of BH3−n(OCOH)nNH3 (I) to form intermediate III. Finally, intermediate IV is formed through the intramolecular nucleophilic cyclization of intermediate III, followed by dehydration to yield the target product benzothiazole.
 | ||
| Scheme 4 The proposed mechanism of reductive cyclization of CO2 with disulfide. | ||
3 Experimental
3.1 General information
All reagents and solvents were purchased from commercial suppliers and used without further purification. All reactions were monitored by TLC with GF254 silica gel coated plates. Purification of reaction products were carried out by chromatography using silica gel (200–300 mesh). The 1H, 13C and 11B NMR spectra were recorded on an Agilent 500 MHz DD2 spectrometer at 500 MHz (1H), 126 MHz (13C) and 160 MHz (11B) in d6-DMSO, or CDCl3 or D2O using tetramethylsilane (TMS) or solvent residue as internal standard. All chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. Melting points were measured with SGC X-4 microscopic melting point meter and were uncorrected. Molecular weights were obtained using Shimadzu LCMS-2020 (ESI) instrument.3.2 General procedure for the synthesis of benzothiazoles (2a–2l)
A stainless-steel autoclave reactor equipped with a magnetic stirrer was charged with bis(2-aminophenyl) disulfide (0.5 mmol), BH3NH3 (1.25 mmol) and NMP (1 mL). Then the stainless-steel autoclave was sealed and pressurized with 1 MPa of CO2 after air was exchanged by CO2 at ambient temperature. And then it was heated and stirred at 120 °C for 24 h. When the reaction was completed, it was cooled down to room temperature and the excessive CO2 was released slowly. Subsequently, the reaction mixture solution was quenched by brine water and extracted with ethyl acetate three times. The combined organic layers were dried over anhydrous MgSO4 and evaporated under reduced pressure. The desired products were obtained in good to excellent yields after purification by column chromatography on silica gel using petroleum ether/ethyl acetate as the eluent. All the desired products were identified through comparisons with the corresponding 1H NMR, 13C NMR data reported in the literature.3.3 Mechanistic study
3.4 Characterization (NMR) of the products
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.126 g, 93%); 1H NMR (500 MHz, chloroform-d) δ 9.00 (s, 1H), 8.14 (d, J = 8.1 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.54–7.49 (m, 1H), 7.44 (t, J = 7.5 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 153.91, 153.21, 133.70, 126.17, 125.54, 123.62, 121.87. MS (ESI): m/z calcd for C7H6SN [M + H]+: 136.19, found 136.01.:1, 0.128 g, 86%); 1H NMR (500 MHz, chloroform-d) δ 8.98 (s, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.36–7.31 (m, 2H), 2.80 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 152.70, 152.53, 133.56, 133.45, 126.66, 125.47, 119.29, 18.37. MS (ESI): m/z calcd for C8H8NS [M + H]+: 150.21, found 150.05.:1, 0.141 g, 95%); 1H NMR (500 MHz, chloroform-d) δ 8.91 (s, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.33 (d, J = 9.9 Hz, 1H), 2.51 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 152.83, 151.35, 135.69, 133.86, 127.82, 123.03, 121.51, 21.50. MS (ESI): m/z calcd for C8H8NS [M + H]+: 150.21, found 150.05.:1, 0.149 g, 90%); mp: 71–73 °C; 1H NMR (500 MHz, chloroform-d) δ 8.84 (s, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.40 (s, 1H), 7.13 (dd, J = 9.0, 3.0 Hz, 1H), 3.89 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 158.06, 151.44, 147.71, 135.12, 123.95, 115.89, 104.00, 55.80. MS (ESI): m/z calcd for C8H8NOS [M + H]+: 166.21, found 166.00.:1, 0.096 g, 63%); 1H NMR (500 MHz, chloroform-d) δ 9.00 (s, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.42 (td, J = 2.01,1.13 Hz, 1H), 7.25–7.21 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 156.30 (d, J = 257.79 Hz), 154.37, 142.26 (d, J = 13.73 Hz), 136.60 (d, J = 3.4 Hz), 126.45 (d, J = 7.18 Hz), 117.55 (d, J = 4.41 Hz), 111.81 (d, J = 17.77 Hz). MS (ESI): m/z calcd for C7H5FNS [M + H]+: 154.17, found 154.00.:1, 0.119 g, 70%); mp: 50–52 °C; 1H NMR (500 MHz, chloroform-d) δ 9.08 (s, 1H), 7.87 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 154.84, 150.25, 135.30, 128.67, 126.48, 126.21, 120.49. MS (ESI): m/z calcd for C7H5ClNS [M + H]+: 170.63, found 169.95.:1, 0.131 g, 77%); mp: 104–106 °C; 1H NMR (500 MHz, chloroform-d) δ 9.04 (s, 1H), 8.14 (s, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 10.2 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 155.72, 154.02, 132.41, 131.99, 126.22, 123.46, 122.58. MS (ESI): m/z calcd for C7H5ClNS [M + H]+: 170.63, found 170.00.:1, 0.137 g, 80%); mp: 41–42 °C; 1H NMR (500 MHz, chloroform-d) δ 8.99 (s, 1H), 8.06 (d, J = 8.7 Hz, 1H), 7.95 (s, 1H), 7.49 (d, J = 9.4 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 154.34, 151.71, 134.95, 131.73, 127.12, 124.33, 121.49. MS (ESI): m/z calcd for C7H5ClNS [M + H]+: 170.63, found 170.00.:1, 0.182 g, 85%); mp: 53–55 °C; 1H NMR (500 MHz, chloroform-d) δ 8.98 (d, J = 2.5 Hz, 1H), 8.11 (t, J = 2.1 Hz, 1H), 8.00 (dd, J = 8.7, 2.3 Hz, 1H), 7.65–7.61 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 154.32, 152.11, 135.44, 129.77, 124.72, 124.44, 119.37. MS (ESI): m/z calcd for C7H5BrNS [M + H]+: 215.08, found 213.90.:1, 0.148 g, 73%); mp: 63–65 °C; 1H NMR (500 MHz, chloroform-d) δ 9.13 (s, 1H), 8.43 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 155.87, 152.88, 137.23, 129.01 (q, J = 32.51 Hz), 124.11 (q, J = 272.79 Hz), 122.61, 121.99 (q, J = 3.53 Hz), 120.97 (q, J = 4.16 Hz). MS (ESI): m/z calcd for C8H5F3NS [M + H]+: 204.18, found 204.00.:1, 0.117 g, 55%); mp: 99–102 °C; 1H NMR (500 MHz, chloroform-d) δ 9.26 (s, 1H), 8.64 (d, J = 1.8 Hz, 1H), 8.32 (d, J = 8.6 Hz, 1H), 8.07 (d, J = 6.8 Hz, 1H), 3.14 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 158.50, 156.19, 137.72, 134.45, 124.86, 124.67, 122.58, 44.88. MS (ESI): m/z calcd for C8H8NO2S2 [M + H]+: 214.27, found 213.95.:1, 0.122 g, 66%); mp: 48–51 °C; 1H NMR (500 MHz, chloroform-d) δ 9.11 (s, 1H), 8.85 (d, J = 8.2 Hz, 1H), 7.99–7.95 (m, 2H), 7.86 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 7.1 Hz, 1H), 7.60 (d, J = 7.0 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 152.52, 149.77, 131.91, 130.65, 128.79, 128.05, 127.17, 126.40, 126.26, 123.66, 118.97. MS (ESI): m/z calcd for C11H8NS [M + H]+: 186.24, found 186.00.
:1, 0.126 g, 93%); 1H NMR (500 MHz, chloroform-d) δ 9.00 (s, 1H), 8.14 (d, J = 8.1 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.54–7.49 (m, 1H), 7.44 (t, J = 7.5 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 153.91, 153.21, 133.70, 126.17, 125.54, 123.62, 121.87. MS (ESI): m/z calcd for C7H6SN [M + H]+: 136.19, found 136.01.:1, 0.128 g, 86%); 1H NMR (500 MHz, chloroform-d) δ 8.98 (s, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.36–7.31 (m, 2H), 2.80 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 152.70, 152.53, 133.56, 133.45, 126.66, 125.47, 119.29, 18.37. MS (ESI): m/z calcd for C8H8NS [M + H]+: 150.21, found 150.05.:1, 0.141 g, 95%); 1H NMR (500 MHz, chloroform-d) δ 8.91 (s, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.33 (d, J = 9.9 Hz, 1H), 2.51 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 152.83, 151.35, 135.69, 133.86, 127.82, 123.03, 121.51, 21.50. MS (ESI): m/z calcd for C8H8NS [M + H]+: 150.21, found 150.05.:1, 0.149 g, 90%); mp: 71–73 °C; 1H NMR (500 MHz, chloroform-d) δ 8.84 (s, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.40 (s, 1H), 7.13 (dd, J = 9.0, 3.0 Hz, 1H), 3.89 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 158.06, 151.44, 147.71, 135.12, 123.95, 115.89, 104.00, 55.80. MS (ESI): m/z calcd for C8H8NOS [M + H]+: 166.21, found 166.00.:1, 0.096 g, 63%); 1H NMR (500 MHz, chloroform-d) δ 9.00 (s, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.42 (td, J = 2.01,1.13 Hz, 1H), 7.25–7.21 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 156.30 (d, J = 257.79 Hz), 154.37, 142.26 (d, J = 13.73 Hz), 136.60 (d, J = 3.4 Hz), 126.45 (d, J = 7.18 Hz), 117.55 (d, J = 4.41 Hz), 111.81 (d, J = 17.77 Hz). MS (ESI): m/z calcd for C7H5FNS [M + H]+: 154.17, found 154.00.:1, 0.119 g, 70%); mp: 50–52 °C; 1H NMR (500 MHz, chloroform-d) δ 9.08 (s, 1H), 7.87 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 154.84, 150.25, 135.30, 128.67, 126.48, 126.21, 120.49. MS (ESI): m/z calcd for C7H5ClNS [M + H]+: 170.63, found 169.95.:1, 0.131 g, 77%); mp: 104–106 °C; 1H NMR (500 MHz, chloroform-d) δ 9.04 (s, 1H), 8.14 (s, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 10.2 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 155.72, 154.02, 132.41, 131.99, 126.22, 123.46, 122.58. MS (ESI): m/z calcd for C7H5ClNS [M + H]+: 170.63, found 170.00.:1, 0.137 g, 80%); mp: 41–42 °C; 1H NMR (500 MHz, chloroform-d) δ 8.99 (s, 1H), 8.06 (d, J = 8.7 Hz, 1H), 7.95 (s, 1H), 7.49 (d, J = 9.4 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 154.34, 151.71, 134.95, 131.73, 127.12, 124.33, 121.49. MS (ESI): m/z calcd for C7H5ClNS [M + H]+: 170.63, found 170.00.:1, 0.182 g, 85%); mp: 53–55 °C; 1H NMR (500 MHz, chloroform-d) δ 8.98 (d, J = 2.5 Hz, 1H), 8.11 (t, J = 2.1 Hz, 1H), 8.00 (dd, J = 8.7, 2.3 Hz, 1H), 7.65–7.61 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 154.32, 152.11, 135.44, 129.77, 124.72, 124.44, 119.37. MS (ESI): m/z calcd for C7H5BrNS [M + H]+: 215.08, found 213.90.:1, 0.148 g, 73%); mp: 63–65 °C; 1H NMR (500 MHz, chloroform-d) δ 9.13 (s, 1H), 8.43 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 155.87, 152.88, 137.23, 129.01 (q, J = 32.51 Hz), 124.11 (q, J = 272.79 Hz), 122.61, 121.99 (q, J = 3.53 Hz), 120.97 (q, J = 4.16 Hz). MS (ESI): m/z calcd for C8H5F3NS [M + H]+: 204.18, found 204.00.:1, 0.117 g, 55%); mp: 99–102 °C; 1H NMR (500 MHz, chloroform-d) δ 9.26 (s, 1H), 8.64 (d, J = 1.8 Hz, 1H), 8.32 (d, J = 8.6 Hz, 1H), 8.07 (d, J = 6.8 Hz, 1H), 3.14 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 158.50, 156.19, 137.72, 134.45, 124.86, 124.67, 122.58, 44.88. MS (ESI): m/z calcd for C8H8NO2S2 [M + H]+: 214.27, found 213.95.:1, 0.122 g, 66%); mp: 48–51 °C; 1H NMR (500 MHz, chloroform-d) δ 9.11 (s, 1H), 8.85 (d, J = 8.2 Hz, 1H), 7.99–7.95 (m, 2H), 7.86 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 7.1 Hz, 1H), 7.60 (d, J = 7.0 Hz, 1H). 13C NMR (126 MHz, chloroform-d) δ 152.52, 149.77, 131.91, 130.65, 128.79, 128.05, 127.17, 126.40, 126.26, 123.66, 118.97. MS (ESI): m/z calcd for C11H8NS [M + H]+: 186.24, found 186.00.4 Conclusions
In conclusion, a green, catalyst-free and efficient approach to synthesize 2-unsubstituted benzothiazole derivatives by the reductive cyclization of bis(2-aminophenyl) disulfide with CO2 was realized, which could be used to prepare a variety of 2-unsubstituted benzothiazole derivatives in good to excellent yields. The reaction mechanism investigation demonstrated that BH3NH3 plays an important role in the formation of benzothiazole, as an efficient reductant to reduce CO2, to cleave the S–S bond of disulfide and to activate the N–H bond of amino group. This simple and green route provides a new method for the utilization of CO2 in the synthesis of benzothiazole derivatives.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Program for Grassland Excellent Talents of Inner Mongolia Autonomous Region, the National Natural Science Foundation of China (21865020), the Natural Science Foundation of Inner Mongolia (No. 2019LH02009 and No. 2021ZD02), the Innovation Research Team of CO2 Resource Utilization in Universities of Inner Mongolia Autonomous Region (NMGIRT2212), and the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (2019).Notes and references
- A. H. Chowdhury, P. Bhanja, N. Salam, A. Bhaumik and S. M. Islam, Mol. Catal., 2018, 450, 46–54 CrossRef CAS
.
- X. F. Liu, X. Y. Li, C. Qiao, H. C. Fu and L. N. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS PubMed
- C. K. Ran, L. L. Liao, T. Y. Gao, Y. Y. Gui and D. G. Yu, Curr. Opin. Green Sustain. Chem., 2021, 32, 100525 CrossRef CAS
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed
- A. I. Ojeda-Amador, J. Munarriz, P. Alamán-Valtierra, V. Polo, R. Puerta-Oteo, M. V. Jiménez, F. J. Fernández-Alvarez and J. J. Pérez-Torrente, ChemCatChem, 2019, 11, 5524–5535 CrossRef CAS
- M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002–1017 CrossRef CAS PubMed
- W. F. Zhao, H. Li, Y. Li, J. X. Long, Y. F. Xu and S. Yang, Sustain. Chem. Pharm., 2020, 17, 100276 CrossRef
- Z. Q. Guo, B. Zhang, X. H. Wei and C. J. Xi, ChemSusChem, 2018, 11, 2296–2299 CrossRef CAS PubMed
- Z. Q. Guo, B. Zhang, X. H. Wei and C. J. Xi, Org. Lett., 2018, 20, 6678–6681 CrossRef CAS PubMed
- G. H. Jin, C. G. Werncke, Y. Escudiee, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed
- C. Shen, P. F. Zhang, Q. Sun, S. Q. Bai, T. S. Hor and X. G. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC
- G. Q. Yuan, C. R. Qi, W. Q. Wu and H. F. Jiang, Curr. Opin. Green Sustain. Chem., 2017, 3, 22–27 CrossRef
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed
- J. H. Zhu, Z. B. Zhang, C. X. Miao, W. Liu and W. Sun, Tetrahedron, 2017, 73, 3458–3462 CrossRef CAS
- J. O. Rathi and G. S. Shankarling, Sol. Energy, 2019, 189, 471–479 CrossRef CAS
- X. Gao, B. Yu, Q. Q. Mei, Z. Z. Yang, Y. F. Zhao, H. Y. Zhang, L. D. Hao and Z. M Liu, New J. Chem., 2016, 40, 8282–8287 RSC
- S. Dadwal, M. Kumar and V. Bhalla, J. Org. Chem., 2020, 85, 13906–13919 CrossRef CAS PubMed
- Z. G. Ke, B. Yu, H. Wang, J. F. Xiang, J. J. Han, Y. Y. Wu, Z. H. Liu, P. Yang and Z. M. Liu, Green Chem., 2019, 21, 1695–1701 RSC
- X. Gao, B. Yu, Y. F. Zhao, L. D. Hao and Z. M. Liu, RSC Adv., 2014, 4, 56957–56960 RSC
- B. Zhang, G. X. Du, W. Hang, S. Wang and C. J. Xi, Eur. J. Org. Chem., 2018, 2018, 1739–1743 CrossRef CAS
- X. Gao, B. Yu, Z. Z. Yang, Y. F. Zhao, H. Y. Zhang, L. D. Hao, B. X. Han and Z. M. Liu, ACS Catal., 2015, 5, 6648–6652 CrossRef CAS
- J. L. Song, B. W. Zhou, H. Z. Liu, C. Xie, Q. L. Meng, Z. R. Zhang and B. X. Han, Green Chem., 2016, 18, 3956–3961 RSC
- D. B. Lima, F. Penteado, M. M. Vieira, D. Alves, G. Perin, C. Santi and E. J. Lenardão, Eur. J. Org. Chem., 2017, 2017, 3830–3836 CrossRef CAS
- X. Liu and Z. B. Dong, Eur. J. Org. Chem., 2020, 2020, 408–419 CrossRef CAS
- R. Y. Tang, Y. X. Xie, Y. L. Xie, J. N. Xiang and J. H. Li, Chem. Commun., 2011, 47, 12867–12869 RSC
- D. Asakawa, H. Takahashi, S. Iwamoto and K. Tanaka, Phys. Chem. Chem. Phys., 2019, 21, 26049–26057 RSC
- M. Arisawa and M. Yamaguchi, Molecules, 2020, 25, 3595–3630 CrossRef CAS PubMed
- N. Taniguchi, J. Org. Chem., 2015, 80, 1764–1770 CrossRef CAS PubMed
- Y. Liu, X. Sun, X. Zhang, J. Liu and Y. G. Du, Org. Biomol. Chem., 2014, 12, 8453–8461 RSC
- W. Munbunjong, E. H. Lee, P. Ngernmaneerat, S. J. Kim, G. Singh, W. Chavasiri and D. O. Jang, Tetrahedron, 2009, 65, 2467–2471 CrossRef CAS
- J. McNulty, V. Krishnamoorthy, D. Amoroso and M. Moser, Bioorg. Med. Chem. Lett., 2015, 25, 4114–4117 CrossRef CAS PubMed
- F. Hofbauer and I. Frank, Chem.–Eur. J., 2010, 16, 5097–5101 CrossRef CAS PubMed
- R. Lu, S. Itabashi, N. Enjo and T. Miyakoshi, Curr. Org. Synth., 2014, 11, 295–300 CrossRef CAS
- N. Zhu, F. Zhang and G. Liu, J. Comb. Chem., 2010, 12, 531–540 CrossRef CAS PubMed
- B. H. Zhou, H. L. Hong, H. C. Wang, T. M. Zhang, L. M. Han and N. Zhu, Eur. J. Org. Chem., 2018, 2018, 6983–6990 CrossRef CAS
- C. Q. Lou, N. Zhu, R. H. Fan, H. L. Hong, L. M. Han, J. B. Zhang and Q. L. Suo, Green Chem., 2017, 19, 1102–1108 RSC
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed
- T. X. Zhao, G. W. Zhai, J. Liang, P. Li, X. B. Hu and Y. T. Wu, Chem. Commun., 2017, 53, 8046–8049 RSC
- X. Li, J. H. Zhang, Y. Yang, H. L. Hong, L. M. Han and N. Zhu, J. Organomet. Chem., 2021, 954–955, 122079 CrossRef CAS
- C. Fang, C. Lu, M. Liu, Y. Zhu, Y. Fu and B. L. Lin, ACS Catal., 2016, 6, 7876–7881 CrossRef CAS
- Q. Z. Zou, G. C. Long, T. X. Zhao and X. B. Hu, Green Chem., 2020, 22, 1134–1138 RSC
- K. S. Du and J. M. Huang, Green Chem., 2018, 20, 1405–1411 RSC
- B. Bhanage and D. Nale, Synlett, 2015, 26, 2835–2842 CrossRef
- F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746–749 CrossRef CAS PubMed
- I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef CAS
- B. Zhang, Z. N. Fan, Z. Q. Guo and C. J. Xi, J. Org. Chem., 2019, 84, 8661–8667 CrossRef CAS PubMed
- Q. Z. Zou, Y. Yi, T. X. Zhao, F. Liu, C. Kang and X. B. Hu, J. CO2 Util., 2021, 50, 101590 CrossRef CAS
- Q. Y. Zhao, R. D. Dewhurst, H. Braunschweig and X. N. Chen, Angew. Chem., Int. Ed., 2019, 58, 3268–3278 CrossRef CAS PubMed
- H. Lv, Q. Xing, C. T. Yue, Z. Q. Lei and F. W. Li, Chem. Commun., 2016, 52, 6545–6548 RSC
- S. Chun, S. Yang and Y. K. Chung, Tetrahedron, 2017, 73, 3438–3442 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03134e |
| This journal is © The Royal Society of Chemistry 2022 |