
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase transition and electronic properties of Co–As binary compounds at high pressure †
Ao Zhang ab,
Yaqian Dana,
Han Liuab,
Siyuan Liuc,
Jincheng Yuea,
Junda Lia,
Yanping Huanga,
Yanhui Liu*a and
Tian Cui*a
ab,
Yaqian Dana,
Han Liuab,
Siyuan Liuc,
Jincheng Yuea,
Junda Lia,
Yanping Huanga,
Yanhui Liu*a and
Tian Cui*a
aInstitute of High Pressure Physics, School of Physical Science and Technology, Ningbo University, Ningbo 315211, China. E-mail: liuyanhui@nbu.edu.cn; cuitian@nbu.edu.cn
bDepartment of Physics, College of Science, Yanbian University, Yanji 133000, China
cSchool of Physics, Southeast University, Nanjing 211189, China
First published on 21st June 2022
Abstract
New stable stoichiometries in the Co–As system are investigated up to 100 GPa by the CALYPSO structure prediction method. In particular, we found three novel stable compounds of Co2As-Pnma, CoAs2-Pnnm, and CoAs3-C2/m at high pressure. According to the theoretical electronic band structures, the structures of Co2As-Pnma, CoAs2-Pnnm, and CoAs3-C2/m have metallic characters, and a pressure-induced electronic topological transition was found in CoAs3-C2/m, which is not shown in other stoichiometries of the Co–As system. The calculated results of the electron localization function show that there are polar covalent bond interactions between Co atoms and As atoms in CoAs2-Pnnm and CoAs3-C2/m. The present results can be helpful for understanding diverse structures and properties of Co–As binary compounds under high pressure.
1 Introduction
With several decades of effort, great achievements have been made in the field of condensed-matter physics. However, materials with better properties under extreme conditions are still being sought and studied. High pressure can provide a good way to search for new materials with new crystal phases, nonconventional stoichiometry, and outstanding properties.1–5 For instance, unexpected stable stoichiometry of some binary systems were predicted under pressure.6,7 Furthermore, the pressure-induced electronic topological transition in the structure of IrP has been uncovered by Ma by first-principles calculations.8 The study shows that pressure has a strong influence in tuning the electronic properties and can also enhance the properties of materials.9–11Compared with main group V compounds, the arsenide has been focused and researched under pressure since it exhibits unique structural features. For example, the arsenide is an important member of topological superconductors. Pressure-induced topological superconducting phases are observed in Cd3As2 and SrAs3.12,13 A non-monotonic pressure dependence of thermal conductivity was noticed in BAs due to the pressure-induced competing responses of three-phonon and four-phonon interactions, which has never been predicted or experimentally observed for any other materials.14,15 To the best of our knowledge, the Co–As system has rarely been reported under pressure. On the basis of the interesting properties of above arsenide, it is necessary to explore the structure characters and chemical bonding of the Co–As binary compounds under pressure.
In the present paper, by means of first-principles swarm-intelligence structure search, we explored the binary Co–As phase diagram at 0–100 GPa. The previously reported structures of Co2As-P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2m, CoAs-Pnam, CoAs2-P21/c, and CoAs3-Im
2m, CoAs-Pnam, CoAs2-P21/c, and CoAs3-Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) were successfully reproduced at ambient conditions. Furthermore, three novel stable compounds (Co2As-Pnma, CoAs2-Pnnm, and CoAs3-C2/m) were predicted under high pressure. At the same time, the details of electronic properties exhibit different bonding characters in these compounds under pressure. Interestingly, pressure-induced electronic topology transitions in CoAs3-C2/m phase have been confirmed. Our result represents a significant step toward understanding structural and electrical behaviour of the binary Co–As system under extreme condition.
were successfully reproduced at ambient conditions. Furthermore, three novel stable compounds (Co2As-Pnma, CoAs2-Pnnm, and CoAs3-C2/m) were predicted under high pressure. At the same time, the details of electronic properties exhibit different bonding characters in these compounds under pressure. Interestingly, pressure-induced electronic topology transitions in CoAs3-C2/m phase have been confirmed. Our result represents a significant step toward understanding structural and electrical behaviour of the binary Co–As system under extreme condition.
2 Computational methods
CALYPSO method is the swarm-intelligence based particle swarm optimization (PSO) algorithm,16–18 on which we predicted Co–As binary compounds under pressure of 0–100 GPa. In the framework of density functional theory, the structural optimizations, electronic structure and phonon calculations were performed. The generalized gradient approximation Perdew–Burke–Ernzerhof (GGA-PBE)19 was employed to treat exchange-correlation energy as implemented in the Vienna ab initio simulation package (VASP) code,20 and projector-augmented-wave method21 was employed for cobalt and arsenic with 3d84s1 and 4s24p3 as valence states, respectively. The cutoff energy of 800 eV and Monkhorst–Pack scheme with 2π × 0.04 Å−1 were chosen to ensure that all of the enthalpy calculations were well converged to better than 1 meV per atom.22,23 The full potential linearized augmented plane-wave approach developed in the WIEN2k package (Fig. S0†) is used to assess the correctness of the generalized gradient approximation Perdew–Burke–Ernzerhof utilized at high pressure.24 The DFT + U method was used to evaluate the effect of the U value for the transition metal element Co(ESI Fig. 6. and ESI Table 2†), the result proved that U had less impact on the Co–As system. The chemical stability of the Co–As system is examined by using the ab initio molecular dynamics (AIMD) simulations in the constant-temperature and constant-volume ensemble. The temperature is controlled at 300 K. The time step is set as 1 fs and each simulation lasts 5 ps. We used electron localization function (ELF)25 to investigate the bonding characteristics between atoms, and calculated phonons by supercell approach of PHONOPY program to determine dynamic stability of structure.26,273 Results and discussion
3.1. Crystal structure
The variable-cell simulation with cell sizes of 1–4 formula units (f.u.) was performed structure prediction for CoAsx (x = 1/2; 1; 2; 3) at 0–100 GPa. We calculated the enthalpy of formation (ΔHf) of each stoichiometric compound, as shown in Fig. 1, to study thermodynamic stability of predicted Co–As compounds. The relative thermodynamic stability of different Co–As compounds with respect to elemental solids Co and As is calculated according to following equation: ΔHf(CoAsx) = [H(CoAsx) − H(Co) − xH(As)]/(x + 1). In the formula, H(M) represents the enthalpy of structure at corresponding pressure (M = CoAsx, Co, As). In Fig. 1, solid star indicates thermodynamically stable phases and unstable or metastable phases are expressed by hollow star. | ||
| Fig. 1 Relative enthalpies of formation of Co–As phases with respect to elemental cobalt and arsenic solids. The convex hulls connecting stable phases (solid star) are shown by solid lines. Metastable phases are shown by open star. | ||
The built pressure-composition phase diagram of the Co–As system is shown in Fig. 2, with stable structures identified using colours and space groups. The metallic and non-metallic phases are represented by cyan and red colours, respectively. As can be seen in the Fig. 2, all predicted stable structures are metal phase under pressure. The structural parameters and atomic coordinates of predicted structures are presented in Table 1. The predicted stable structures at ambient pressure are in good agreement with the experimental results,28–31 validating the applicability to the Co–As system of our adopted structure searching method and PBE functional.
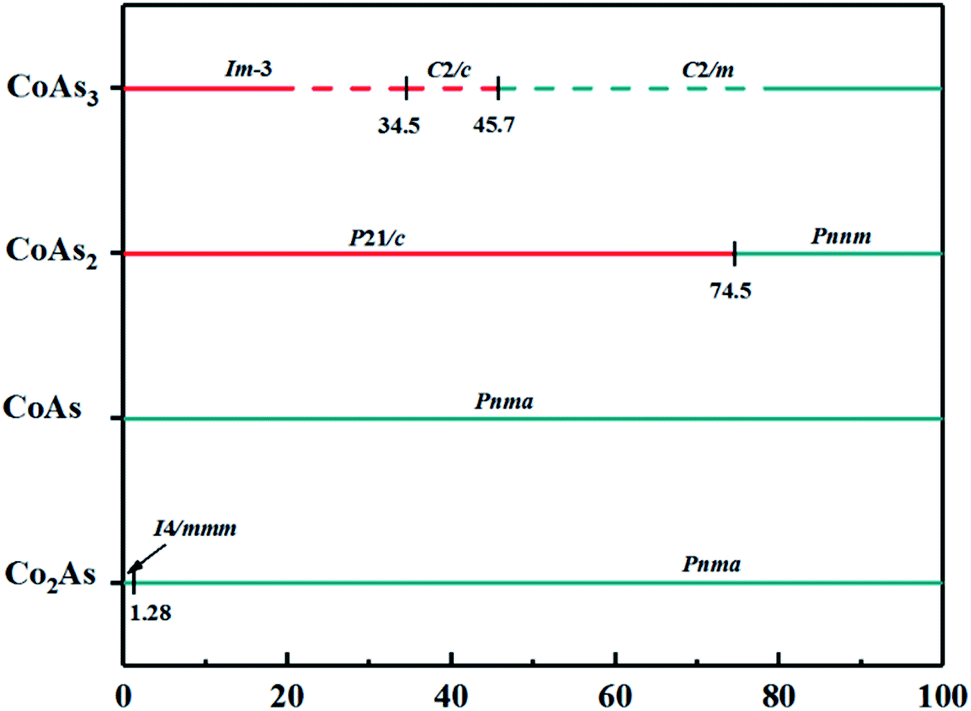 | ||
| Fig. 2 Predicted pressure-composition phase diagram of Co–As crystal phases. Cyan and red colours represent the metallic and non-metallic phases, respectively. The solid (dash) lines represent stable (metastable) phases. | ||
| Space group pressure | Lattice parameters | Atomic coordinates (fractional) | Sites | |||
|---|---|---|---|---|---|---|
| Pnma-Co2As 1.28 GPa | a = 5.611 Å | Co1 | 0.039 | 0.250 | 0.317 | 4c |
| b = 3.693 Å | Co2 | 0.648 | 0.250 | 0.565 | 4c | |
| c = 5.883 Å | As1 | 0.739 | 0.250 | 0.882 | 4c | |
| α = β = γ = 90.0° | ||||||
| Pnnm-CoAs2 74.5 GPa | a = 4.867 Å | Co1 | 0.500 | 0.000 | 0.500 | 2c |
| b = 5.433 Å | As1 | 0.823 | 0.139 | 1.000 | 4g | |
| c = 2.636 Å | ||||||
| α = β = γ = 90.0° | ||||||
| C2/m-CoAs3 45.7 GPa | a = 8.882 Å | Co1 | 0.176 | 0.000 | 0.923 | 4i |
| b = 4.348 Å | As1 | 0.055 | 0.000 | 0.288 | 4i | |
| c = 5.404 Å | As2 | 0.108 | 0.500 | 0.851 | 4i | |
| α = γ = 90.0° | As3 | 0.827 | 0.500 | 0.596 | 4i | |
| β = 92.06° | ||||||
Pressure exhibits unique advantages in finding new materials,32–36 thus the crystal structure and electron properties of these structures under high pressure will be examined in detail next. The full information of the initial pressure structures and phase diagrams of stable structures at different pressures are shown in ESI Fig. 1, 2, 3, and ESI Table 1,† respectively. The morphologies of predicted structures are presented in Fig. 3. The stoichiometric Co2As has the lowest-enthalpy structure with I4/mmm, then at 1.28 GPa, it gives to the Pnma phase. Simultaneously, the distance between Co atoms and As atoms changes from 2.346 Å to 2.370 Å. Our calculation indicates that Co2As-Pnma will be stable up until 100 GPa. Turning to CoAs, CoAs2, and CoAs3, Co atoms and As atoms in all predicted crystal structures form a Co–As octahedral environmental configuration, in which Co atoms occupy the center of the body and As atoms occupy the vertices. In the CoAs2-Pnnm, there are As–As bonds with bond lengths is 2.292 Å. CoAs3 is crystallized in a skutterudite-type structure at ambient pressure. The bond length of Co–As is 2.363 Å, and As atoms formed rectangular As4 ring with As–As distances of 2.655 Å and 2.529 Å. Our calculations show that the cubic CoAs3-Im will be stabilized up to 34.5 GPa. With the increase of pressure, the structure changes from Im phase to metastable C2/c phase, and in the C2/c phase, the As atoms are in the form of chains with the distance of 2.359 Å, and the distance between Co atoms and As atoms turns into 2.242 Å. As pressure increases to 45.7 GPa, the structure changes to the C2/m phase, and distances between Co–As and As–As change to 2.286 Å and 2.510 Å, respectively. To judge their stability, the phonon dispersion curves for Co2As-Pnma, CoAs2-Pnnm, and CoAs3–C2/m are calculated, as presented in Fig. 4. All above phases are dynamically stable in their accessible pressures since no imaginary vibrational modes are observed in the Brillouin zone.37–41 To confirm the thermodynamic stability of the predicted structure, the AIMD at finite temperatures for Co2As-Pnma, CoAs2-Pnnm, and CoAs3–C2/m are calculated, as presented in ESI Fig. 4.†42,43
 | ||
| Fig. 3 Structures of predicted Co–As system for (a) Co2As-Pnma at 1.28 GPa; (b) CoAs2-Pnnm at 74.5 GPa, and (c) CoAs3-C2/m at 45.7 GPa. The blue and pink spheres are Co atoms and As atoms, respectively. | ||
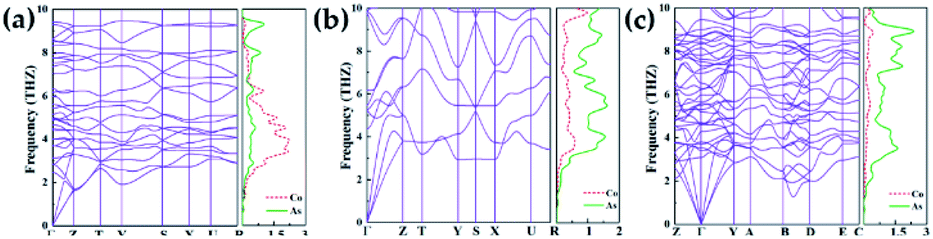 | ||
| Fig. 4 Phonon dispersion relations and phonon density of states projected on Co atoms and As atoms for (a) Co2As-Pnma at 1.28 GPa (b) CoAs2-Pnnm at 74.5 GPa, and (c) CoAs3-C2/m at 45.7 GPa. | ||
3.2. Electronic properties
As shown in Fig. 5, we calculated band structures and the partial density of states (DOS) to investigate electronic properties of various Co–As compounds. Under high pressure, predicted structures exhibit metallic feature by evidence of overlap between conduction bands and valence bands at the Fermi level. In the structures of Co2As-Pnma, the majority of the occupied states come from Co-d states near Fermi level (Fig. 5(a)), while in CoAs2-Pnnm and CoAs3-C2/m, there exist strong hybridization between Co-d and As-p states (−2 to 2 eV). In CoAs2 and CoAs3, the pressure-induced transition from non-metal to metal was recognized. As is shown in ESI Fig. 5,† compared with the typical arsenide GaAs under the high pressure, all the novel phases of high pressure exhibit similar metallic properties. Due to the pressure, the electrons become unlocalized, making the material exhibit metallic properties.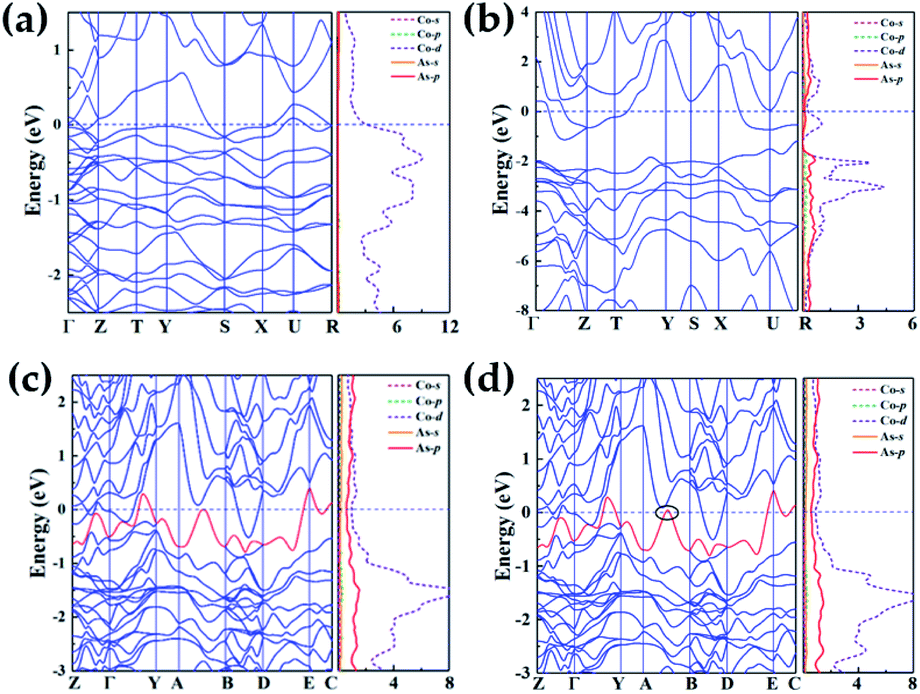 | ||
| Fig. 5 Electronic band structure and the projected electronic DOS on Co atoms and As atoms for (a) Co2As-Pnma at 1.28 GPa; (b) CoAs2-Pnnm at 74.5 GPa; (c) CoAs3-C2/m at 45.7 GPa, and (d) CoAs3-C2/m at 48 GPa. Note that Zero energy is at the Fermi level. | ||
It is worth noting that the electronic topological transition has a significant impact on the corresponding physical and chemical properties and can occur when the Fermi surface of the electronic system is changed by doping, high pressure, temperature or other external reagents.44–46 Therefore, we have checked the potential pressure-induced electronic topological transition in Co–As system. Attractively, the electronic topological transition exists in CoAs3-C2/m as shown in Fig. 5(c), it is found that there is no band along the A–B direction at 45.7 GPa. The band moves down and approaches to Fermi level at A–B direction when pressure increases. Fig. 5(d) clearly shows that the red band along the A–B direction is not fully occupied at 48 GPa and will form a hole pocket near the Fermi surface. An electronic topological transition in CoAs3-C2/m structure is clearly observed since the noteworthy changes in the surface pro files Fermi at high pressure.
The electron localization function (ELF),47 as depicted in Fig. 6, is used to understand bonding character of the Co–As compounds. Generally speaking, the regions with higher tendency of electron pairing (such as cores, and lone pairs) have large ELF values (>0.5), which indicates the formation of covalent bonds. However, the non/less electron localization have small values (<0.5), which indicates that there are ionic/metallic bonds between atoms. There is a mixed behaviour of ionic and metallic bonds simultaneously exits in Co2As-Pnma phases. Note that in Fig. 6(b) and (c), the great ELF values about 0.8 between the Co and As atoms in CoAs2-Pnnm and CoAs3-C2/m structures, which suggests the covalent bonding character. The above statement is in good agreement with DOS. The DOS shows a large overlap between 3d states of Co and 4p states of As (Fig. 5(b) and (c)). Interestingly, in CoAs2-Pnnm and CoAs3-C2/m structure, it can be observed that there is electronic localization between As and As that does not favour either side, indicating the existence of nonpolar covalent bonds. It is more obvious that As atoms exist in chain form in CoAs3-C2/m. To quantify the amount of charge belonging to each atom at different stoichiometries, we use the Bader charge analysis, which offers a description of electron transfer.47
 | ||
| Fig. 6 Contours of the ELF for the structures of (a) Co2As-Pnma at 1.28 GPa; (b) CoAs2-Pnnm at 74.5 GPa, and (c) CoAs3-C2/m at 45.7 GPa with isosurface of 0.8. | ||
4 Conclusions
In summary, three novel stable compounds (Co2As-Pnma, CoAs2-Pnnm, and CoAs3-C2/m) of Co-As compounds are discovered under high pressure using unbiased structure searching combined with density functional theory calculations. The theoretical phonon dispersion curves suggest that all these phases are dynamically stable. The calculated electronic band structures and density of states suggest that the Co2As-Pnma, CoAs2-Pnnm, and CoAs3-C2/m phases are metallic. Moreover, with the increase of pressure, the CoAs2 and the CoAs3 realize the transition from semiconductor to metal at 74.5 GPa and 45.7 GPa, respectively. A pressure-induced electronic topological transition has been found in CoAs3-C2/m phase. The electron localization function results show that there are ionic bonds and metal bonds in the predicted structures. Further analysis of the bonding nature shows that, there are polar covalent bonds between Co atoms and As atoms in CoAs2-Pnnm, and CoAs3-C2/m. Our results are important for understanding the structures and properties of Co–As system under high pressure.Author contributions
Conceptualization, A. Z., H. L.; methodology, S. L., Y. D., Y. H. and A. Z.; validation, A. Z., J. Y. and J. L.; writing—original draft, A. Z.; writing—review and editing, A. Z., Y. L. and T. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the Natural Science Foundation of China (Grant No. 11764043).Notes and references
- Y. Ma, M. Eremets and A. R. Oganov, et al., Nature, 2009, 458(7235), 182–185 CrossRef CAS PubMed.
- M. Miao, Nat. Chem., 2013, 5(10), 846–852 CrossRef CAS PubMed.
- Y. Liu, R. Wang and Z. Wang, et al., Nat. Commun., 2022, 13(1), 1–7 Search PubMed.
- N. N. Patel, A. K. Verma and A. K. Mishra, et al., Phys. Chem. Chem. Phys., 2017, 19(11), 7996–8007 RSC.
- Q. Zeng, S. Yu and D. Li, et al., Phys. Chem. Chem. Phys., 2017, 19(12), 8236–8242 RSC.
- J. Shi, W. Cui and J. A. Flores-Livas, et al., Phys. Chem. Chem. Phys., 2016, 18(11), 8108–8114 RSC.
- Y. Liu, C. Wang, P. Lv, H. Sun and D. Duan, Chem.–Eur. J., 2018, 24, 11402–11406 CrossRef CAS PubMed.
- X. Ma, X. Li, D. Zhou, J. Xu, W. Gao and Y. Liu, J. Alloys Compd., 2019, 791, 1257–1262 CrossRef CAS.
- M. Jacobsen, S. Sinogeikin, R. Kumar and A. Cornelius, J. Phys. Chem. Solids, 2012, 73, 1154–1158 CrossRef CAS.
- S. V. Ovsyannikov and V. V. Shchennikov, Chem. Mater., 2010, 22, 635–647 CrossRef CAS.
- Y. Zhang, Y. Ma, A. Geng, C. Zhu, G. Liu, Q. Tao, F. Li, Q. Wang, Y. Li and X. Wang, et al., J. Alloys Compd., 2016, 685, 551–558 CrossRef CAS.
- E. Uykur, R. Sankar, D. Schmitz and C. A. Kuntscher, Phys. Rev. B, 2018, 97, 195134 CrossRef CAS.
- E. J. Cheng, W. Xia, X. B. Shi, Z. H. Yu, L. Wang, L. M. Yan, D. C. Peets, C. C. Zhu, H. Su, Y. Zhang, D. Z. Dai, X. Wang, Z. Q. Zou, N. Yu, X. F. Kou, W. W. Zhao, Y. F. Gao and S. Y. Li, npj Quantum Mater., 2020, 5, 38–45 CrossRef CAS.
- N. K. Ravichandran and D. Broido, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- L. Wang, F. Tian, X. Liang, Y. Fu, X. Mu, J. Sun, X.-F. Zhou, K. Luo, Y. Zhang and Z. Zhao, et al., Phys. Rev. B, 2019, 99, 174104 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- H. Wang, Y. Wang, J. Lv, Q. Li, L. Zhang and Y. Ma, Comput. Mater. Sci., 2016, 112, 406–415 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B, 2010, 82, 094116 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953 CrossRef PubMed.
- D. Thirumalai, R. W. Hall and B. Berne, J. Chem. Phys., 1984, 81, 2523–2527 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- P. Blaha, K. Schwarz, P. Sorantin and S. B. Trickey, Comput. Phys. Commun., 1990, 59, 399 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B, 2008, 78, 134106 CrossRef.
- K. Parlinski, Z. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063 CrossRef CAS.
- P. S. Lyman and C. Prewitt, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 14–20 CrossRef.
- J. C. Quesnel and R. Heyding, Can. J. Chem., 1962, 40, 814–818 CrossRef CAS.
- J. Ackermann and A. Wold, J. Phys. Chem. Solids, 1977, 38, 1013–1016 CrossRef CAS.
- I. Lindeberg and Y. Andersson, J. Alloys Compd., 1991, 175, 163–169 CAS.
- Q. Zhu, D. Y. Jung and A. R. Oganov, et al., Nat. Chem., 2013, 5(1), 61–65 CrossRef CAS PubMed.
- X. Dong, A. R. Oganov and A. F. Goncharov, et al., Nat. Chem., 2017, 9(5), 440–445 CrossRef CAS PubMed.
- S. Wei, D. Li, Y. Lv, Z. Liu, C. Xu, F. Tian, D. Duan, B. Liu and T. Cui, Phys. Chem. Chem. Phys., 2016, 18, 18074–18080 RSC.
- X. Ren, X. Yan, Z. Yu, W. Li and L. Wang, J. Alloys Compd., 2017, 725, 941–945 CrossRef CAS.
- C. Xu, H. Yu, B. Kuo, S. Ma, X. Xiao, D. Li, D. Duan, X. Jin, B. Liu and T. Cui, Phys. Chem. Chem. Phys., 2018, 20, 6108–6115 RSC.
- Y. Pan, W. Guan and Y. Li, Phys. Chem. Chem. Phys., 2018, 20, 15863–15870 RSC.
- Y. Pan, J. Alloys Compd., 2019, 779, 813–820 CrossRef CAS.
- Y. Pan, P. Wang and C.-M. Zhang, Ceram. Int., 2018, 44, 12357–12362 CrossRef CAS.
- Y. Pan and W. Guan, Phys. Chem. Chem. Phys., 2017, 19, 19427–19433 RSC.
- Y. Pan, Mater. Res. Bull., 2017, 93, 56–62 CrossRef CAS.
- H. Huang, H. H. Wu, C. Chi, J. Zhu, B. Huang and T. Y. Zhang, Nanoscale, 2019, 11(40), 18758–18768 RSC.
- H. H. Wu, H. Huang, J. Zhong, S. Yu, Q. Zhang and X. C. Zeng, Nanoscale, 2019, 11(25), 12210–12219 RSC.
- H. Ghosh and S. Sen, J. Alloys Compd., 2016, 677, 245–251 CrossRef CAS.
- Y. Zhang, L. Song, X. Shao, Y. Li, P. Zhu, H. Xu and J. Yang, J. Alloys Compd., 2017, 715, 237–241 CrossRef CAS.
- Y. Zhang, X. Shao, Y. Zheng, L. Yan, P. Zhu, Y. Li and H. Xu, J. Alloys Compd., 2018, 732, 280–285 CrossRef CAS.
- D. Song, D. Wan, H. H. Wu, D. Xue, S. Ning, M. Wu, T. Venkatesan and S. J. Pennycook, Nanoscale, 2020, 12(12), 6844–6851 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02114e |
| This journal is © The Royal Society of Chemistry 2022 |