
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVibrational spectroscopy investigation of defects in Zr- and Hf-UiO-66†
Brandon T. Yost *a,
Bradley Gibbonsb,
Addison Wilsona,
Amanda J. Morrisb and
L. E. McNeila
*a,
Bradley Gibbonsb,
Addison Wilsona,
Amanda J. Morrisb and
L. E. McNeila
aDepartment of Physics and Astronomy, University of North Carolina, Chapel Hill, North Carolina 27599, USA. E-mail: btyost@live.unc.edu
bDepartment of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA
First published on 11th August 2022
Abstract
Defect engineering in metal–organic framework compounds has allowed for improvements in catalysis-based functionalities, gas sensing, and gas storage. Metal–organic framework UiO-66 compounds with Zr- and Hf-based metal secondary building units were studied with Raman and infrared vibrational spectroscopy. Missing linker and missing cluster defects were engineered into the crystal structure via a modulated synthesis technique. Missing cluster defects in Hf-UiO-66 are first characterized by powder X-ray diffraction (PXRD) whereby two low-angle peaks were fit to extract the relative quantity of reo topology in four defective samples. A monotonic red-shift of the Raman-active Hf–O coordination bond vibration is interpreted as a signature of missing cluster defects, resulting from less-rigid charge-balancing monocarboxylate formate ions replacing the dicarboxylate linker molecule. This signature is hypothesized to be independent of the topology in which the defects appear. Missing linker defects in Zr-UiO-66 are characterized by infrared absorption spectroscopy by the quenching of C–C and C–H vibrational modes confined to the linker molecule. Together, Raman and infrared vibrational spectroscopies coupled with standard characterization techniques are employed to directly probe the nature of defects as well as offer new characterization tools for missing cluster defects in UiO-66.
Introduction
Metal–organic framework compounds (MOFs) are a class of nanoporous crystals that have been studied for applications in gas sensing, storage, and separation;1–3 drug delivery;4–6 and decontamination of chemical warfare agents (CWAs)7–9 among others. One of the more popular applications is in gas storage whereby the porous nature of the MOF allows for uptake of small molecules.1 To understand MOF–adsorbate interactions, spectroscopic techniques such as in situ infrared absorption or Raman spectroscopy are often employed to relate spectroscopic changes observed during uptake to bonding of a guest molecule to the framework.10–12 One of the most well-studied MOFs is Zr-UiO-66, a 12-connected Zr-based MOF with Zr6(μ3-O)4(μ3-OH)4 secondary building units (SBU) connected by 1,4-benzene dicarboxylic acid (BDC) ligands known as linker molecules.13 Zr-UiO-66 is typically able to hydrolyze organophosphorus nerve agents and their simulants only on the surface of the crystal14 where the agent or simulant can access open Zr sites. This limitation in Zr-UiO-66 is largely due to the small dicarboxylate linker in combination with the full 12-connected structure of Zr-UiO-66 that creates small (∼8 Å) pore apertures4 that do not allow the nerve agents or simulants to penetrate the pores to reach the catalytically-active Lewis-acidic metal nodes. One way to overcome this shortcoming is to introduce defects into the structure, on average creating larger pores and apertures.15,16The relevant defects in UiO-66 are of two types: missing linker and missing cluster defects.16 As the name suggests, a missing linker defect is created by the replacement of one or more dicarboxylate linker molecules per SBU with a charge-balancing capping agent, the nature of which depends on conditions during synthesis; typically formate ions from the dimethyl formamide (DMF) used in synthesis cap the defect sites, but also any additional modulator used during synthesis. Assuming no missing cluster defects are present, missing linker defects have been quantified using thermogravimetric analysis (TGA) by monitoring weight loss while slowly ramping the temperature of the MOF as organic linkers and excess solvent are burned off.17 Missing cluster defects are created by the absence of one or more SBUs in the crystal structure. Interestingly, ordered missing cluster defects can create a short-range deviation from the fcu topology of the pristine structure, introducing nanoregions of reo topology.18 It has been shown that ordered regions of reo topology introduce two peaks in the X-ray diffraction (XRD) pattern at around 5°, thus allowing XRD to be used to characterize UiO-66 by monitoring the emergence of reo topology.18 However, since these features will only show up in XRD if the missing cluster defects are ordered in small regions of reo topology, XRD is not sensitive to randomly-dispersed missing cluster defects, which makes direct quantification of missing cluster defects in a crystal challenging.
We employ Raman spectroscopy as well as FTIR absorption spectroscopy to directly probe the vibrational signature of each defect type in Zr-UiO-66 and Hf-UiO-66, importantly finding a spectroscopic signature of missing cluster defects in Hf-UiO-66.
Experimental
Synthetic procedures
Pristine UiO-66 was synthesized following previously-reported procedures.19 Pristine Hf-UiO-66 was synthesized following the same procedure, with 519 mg HfCl4 instead of ZrCl4. Defective UiO-66 samples containing missing linkers were synthesized following previously reported procedures.20 Briefly, 378.1 mg ZrCl4 and 539 mg of terephthalic acid were added to 10 mL of dimethylformamide (DMF) in a 6 dr glass vial. Modulator was added (6.3 mL acetic acid, 2.8 mL formic acid, or 4.2 mL formic acid) to produce defective Zr-UiO-66 with 14%, 22%, and 25% missing linkers respectively. The vial was capped and heated at 120 °C for 3 days. Defective Hf-UiO-66 containing missing cluster defects was synthesized by modifying procedures previously reported in the literature.18 The density of missing clusters was adjusted by changing the Hf![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :terephthalic acid ratio or by increasing the concentration of formic acid modulator. For all samples, 960 mg HfCl4 was added to 40 mL of DMF in a 100 mL Pyrex jar. Terephthalic acid and formic acid were added for each sample as follows. 5% reo: 332 mg terephthalic acid, 10 mL formic acid; 10% reo: 415 mg terephthalic acid, 15 mL formic acid; 17% reo: 415 mg terephthalic acid, 20 mL formic acid; 32% reo: 166 mg terephthalic acid, 10 mL formic acid.
:terephthalic acid ratio or by increasing the concentration of formic acid modulator. For all samples, 960 mg HfCl4 was added to 40 mL of DMF in a 100 mL Pyrex jar. Terephthalic acid and formic acid were added for each sample as follows. 5% reo: 332 mg terephthalic acid, 10 mL formic acid; 10% reo: 415 mg terephthalic acid, 15 mL formic acid; 17% reo: 415 mg terephthalic acid, 20 mL formic acid; 32% reo: 166 mg terephthalic acid, 10 mL formic acid.
Methods
Powder X-ray diffraction (PXRD) for Zr-UiO-66 and Hf-UiO-66 samples was collected on a Rigaku MiniFlex benchtop diffractometer with Cu Kα radiation (λ = 1.5418 Å) with a 0.1° resolution and 15° min−1 scan rate. UiO-66 particle size and morphology were measured by scanning electron microscopy using a LEO/Zeiss 1550 field emission scanning electron microscope. Samples were prepared by drop casting a MOF suspension in ethanol onto fluorine-doped tin oxide (FTO). Missing linker defects were characterized by thermogravimetric analysis (TGA) on a TA TGA-5500 under an air flow from 30–650 °C with a 10 °C min−1 ramp rate as per ref. 20. N2 isotherms were collected on a Micromeritics 3Flex at 77 K. The samples were activated at 150 °C under vacuum overnight to remove excess moisture prior to analysis. From the N2 isotherm, a calculated pore size distribution was obtained and used to identify samples with missing cluster defects, since these samples will contain an additional pore 16 Å in diameter.Raman spectra were obtained using a Dilor Triple XY Raman spectrometer coupled to an optical microscope. A 473 nm laser excitation source with 100 μm spot size illuminated the powder sample on a glass slide on a microscope stage open to air. Scattered light collected by the microscope objective is dispersed across a liquid nitrogen cooled CCD detector after encountering a series of three gratings. Each of the three gratings are 1800 mm−1 holographic gratings blazed at 500 nm. Scans were obtained at frequencies no lower than approximately 100–150 cm−1 to avoid saturating the detector with Rayleigh-scattered laser light, and no higher than approximately 3000 cm−1 where the spectra are obscured by fluorescent backgrounds. However, in Zr-UiO-66, there are very few Raman-active modes at high frequencies and data collected in this region will not be shown. Fourier transform infrared absorption spectra were obtained in the range of 400 to 4000 cm−1 using a Nicolet Nexus 670 spectrometer. For all infrared data, each powder sample was crushed together with KBr that had been dried in an oven and then pressed into a pellet of 13 mm diameter and a thickness of 0.54 mm with a maximum pressure of approximately 10 tons using a hydraulic press. The pellet was mounted by placing it over a small hole in a stainless-steel plate and securing it with a magnet to hold the pellet in place. The interior volume of the spectrometer was flushed with nitrogen gas for the duration of the scans to avoid effects due to moisture in the ambient air.
Results and discussion
Raman spectroscopy
Raman spectroscopy and infrared absorption were used to characterize pristine Zr-UiO-66 and the results were compared to density functional theory calculations published by Valenzano, et al. to properly assign peaks in the spectrum to specific vibrational modes.17 However, perfect assignment of peaks is very difficult. This can be understood in a few ways: first, the UiO-66 structure is large and complex. As such, most of the vibrations are not pure motions of some well-defined species. For example, modes that appear dominated by motion on the Zr–O coordination bond often also have contributions from the motion of the benzene ring on the linker or the μ3-OH and μ3-O species in the metal SBU. Also, with the large number of atoms in a MOF crystal, there is a correspondingly large number of Raman- and IR-active modes, many of which are very closely spaced in frequency with slightly different atomic motions. Therefore, making an exact assignment of any peak in the spectrum is nearly impossible with such a complex system. Despite many of these closely-spaced modes having different specific motions, most modes within a small frequency region have atomic motions that typically occur on the same species. For example, Raman-active modes with calculated frequencies between 425 and 450 cm−1 are largely dominated by motion of the μ3-OH and its bond with neighboring Zr atoms. However, each mode in this range has slightly different motions, each with different contributions from the linker molecule: some appear to be pure Zr-μ3-OH wagging whereas others are a combination of this motion with more complex contributions from the carboxylate tails of the linker molecule as well as C–H wagging of the benzene ring. Therefore, rather than giving an explicit assignment for each peak, we will discuss our results in terms of different families of modes: those with significant Zr–O coordination bond contributions, modes dominated by motion of Zr-μ3-OH or Zr-μ3-O, and motion primarily confined to the linkers with little motion of the metal atoms or within the SBU.To study the effect of missing cluster defects in Hf-UiO-66, we first compare the vibrational spectra of pristine Zr-UiO-66 to those of pristine Hf-UiO-66. Importantly, the structure and topology (fcu) of the two pristine structures are nearly identical; the only difference being a one-to-one replacement of Zr in favor of Hf. Therefore, the assignment of vibrational modes is carried out in a two-step process: first by assigning relevant peaks in pristine Zr-UiO-66 as discussed above, then by comparing the vibrational spectrum of pristine Zr-UiO-66 to that of pristine Hf-UiO-66. Each spectrum is internally normalized to a peak at 865 cm−1 corresponding primarily to C–H motion confined to the linker molecule. During this comparison, we note that the Raman spectra between 400 and 1600 cm−1 are nearly identical (Fig. 1a) to one another. In this range of frequencies, most Raman-active modes are dominated by motions confined to the linker molecules. Since both pristine Hf-UiO-66 and pristine Zr-UiO-66 are synthesized in the same topology with the same linker molecule, it is unsurprising that the linker modes remain unchanged. This also suggests that the strength of the metal-linker coordination bond is similar in the two structures. Within the “fingerprint region” (150–400 cm−1) (Fig. 1b), the differences between the spectra of the two structures are noticeable. In this frequency range, the discrepancy between Zr-UiO-66 and Hf-UiO-66 suggests that most Raman-active modes have significant contributions from the metal atoms. With no full DFT calculation of Raman-active modes in Hf-UiO-66 similar to the calculation published by Valenzano, et al.17 band assignments in Hf-UiO-66 are made by first matching each peak to the corresponding peak in Zr-UiO-66. Then, the Zr-UiO-66 mode is assigned via the Valenzano calculation and the Hf-UiO-66 assignment is inferred by replacing Zr with Hf where applicable.
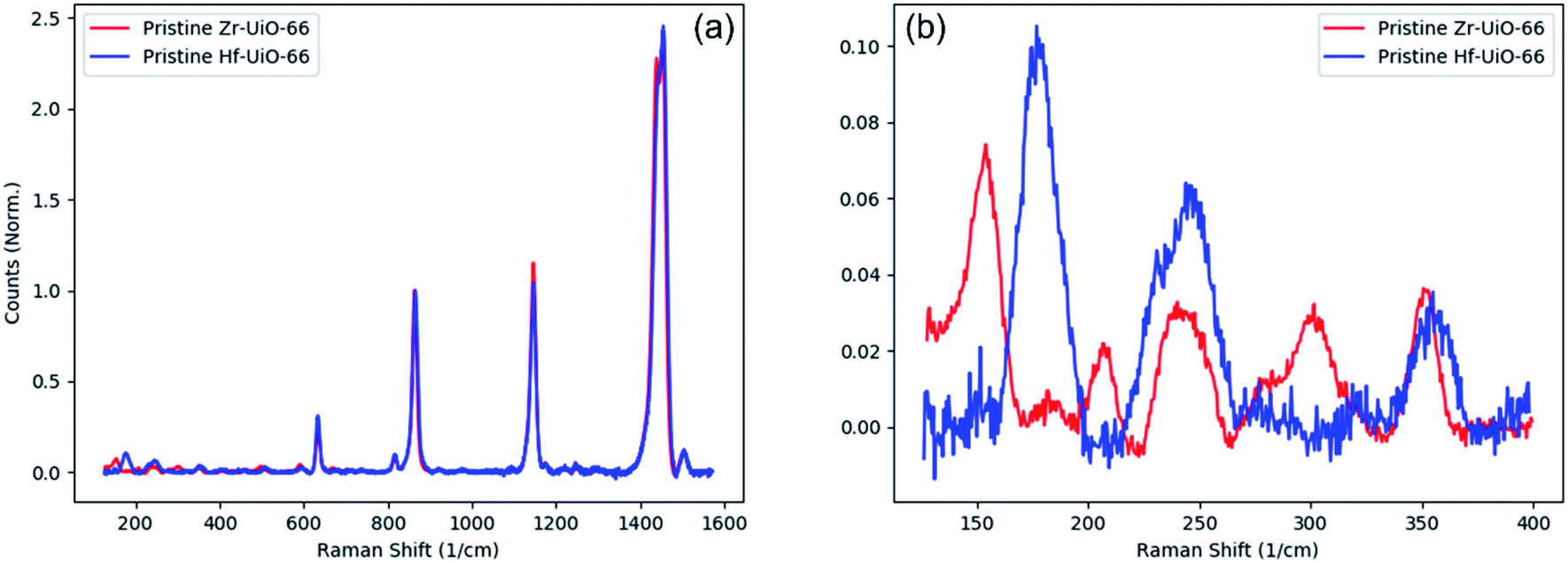 | ||
| Fig. 1 Zr-UiO-66 and Hf-UiO-66 comparison. (a) Full Raman spectra of Zr-UiO-66 and Hf-UiO-66 highlighting near-identical vibrational signatures of the two crystals. (b) Fingerprint region: differences between Zr- and Hf-UiO-66 are seen in modes corresponding primarily to vibrations of metal atoms. | ||
With properly-assigned Raman-active modes in Hf-UiO-66, we then begin to study the effects of missing cluster defects. Hf-UiO-66 was synthesized and first characterized by XRD as well as by a pore size distribution analysis (Fig. 2). The appearance and growth of two low-angle peaks in the XRD (see Fig. S1†) allowed the determination of the fraction of the sample in the reo topology across all of the defective samples as discussed above. The pristine Hf-UiO-66 showed neither of these two low-angle peaks and so is treated as defect-free. The treatment as defect-free is also corroborated by the pore size distribution analysis in Fig. 2 as well as TGA analysis. Above 400 cm−1, the spectra of the Hf-UiO-66 samples exhibit no change as a function of defect level. Although the samples being compared differ in density of missing cluster defects, it is nonetheless unsurprising that most of the spectra are identical. If a metal SBU is removed from the structure, the 12 linkers are either left “dangling” (i.e., only coordinated to one SBU rather than two) or the linkers are removed along with the SBU and replaced with charge-balancing capping agents, the latter being more likely. As such, the total number of linkers may be reduced as missing cluster defect density increases, but a large quantity of linker molecules will still be sampled with each measurement. Thus, the modes assigned primarily to linker motion (400–1600 cm−1) will still be present in each sample and located at the same frequency.
 | ||
| Fig. 2 Pore-size distribution of pristine and defective Hf-UiO-66. Peaks near 6 angstroms and 8 angstroms represent tetrahedral and octahedral pores respectively while the peaks between 15–20 angstroms represent missing cluster defects. | ||
Vibrational modes involving the Hf atoms are likely to be more dependent on the density of missing cluster defects than are the linker modes. Within the fingerprint region, there are five peaks of interest centered at approximately 176, 231, 245, 320, and 355 cm−1 (Fig. 3). These modes most commonly involve either motion of the Hf–O coordination bond or the Hf-μ3O, however, the mode at 320 cm−1 cannot accurately be assigned due to the lack of a clear corresponding peak in the pristine Zr-UiO-66 Raman spectrum. Also, the mode at 176 cm−1 is the most complex vibration, involving motion of Hf–O and Hf-μ3O in the SBU as well as C–H motion on the linker molecule. With each spectrum internally normalized to a peak at 865 cm−1 as discussed above, all of the modes within the fingerprint region that have been assigned to motion involving the Hf atom have lower intensities in defective samples than in the pristine sample, likely due to the Raman intensity scaling with the number of scattering species. However, a few important patterns begin to emerge upon closer inspection. First, the mode centered at 245 cm−1 has very similar intensity in all samples regardless of defect density, and second, the neighboring vibrational mode centered at 231 cm−1 has been quenched in all defective samples. Attempting to assign these peaks to specific atomic motions is difficult for reasons discussed above. However, within the fingerprint region this difficulty is exacerbated by the large quantity of Raman-active modes calculated in this frequency range. With this in mind, there are two reasonable interpretations of these two modes: either the 231 cm−1 mode involves the Hf-μ3O while the 245 cm−1 mode involves the Hf–O coordination bond, or the reverse.
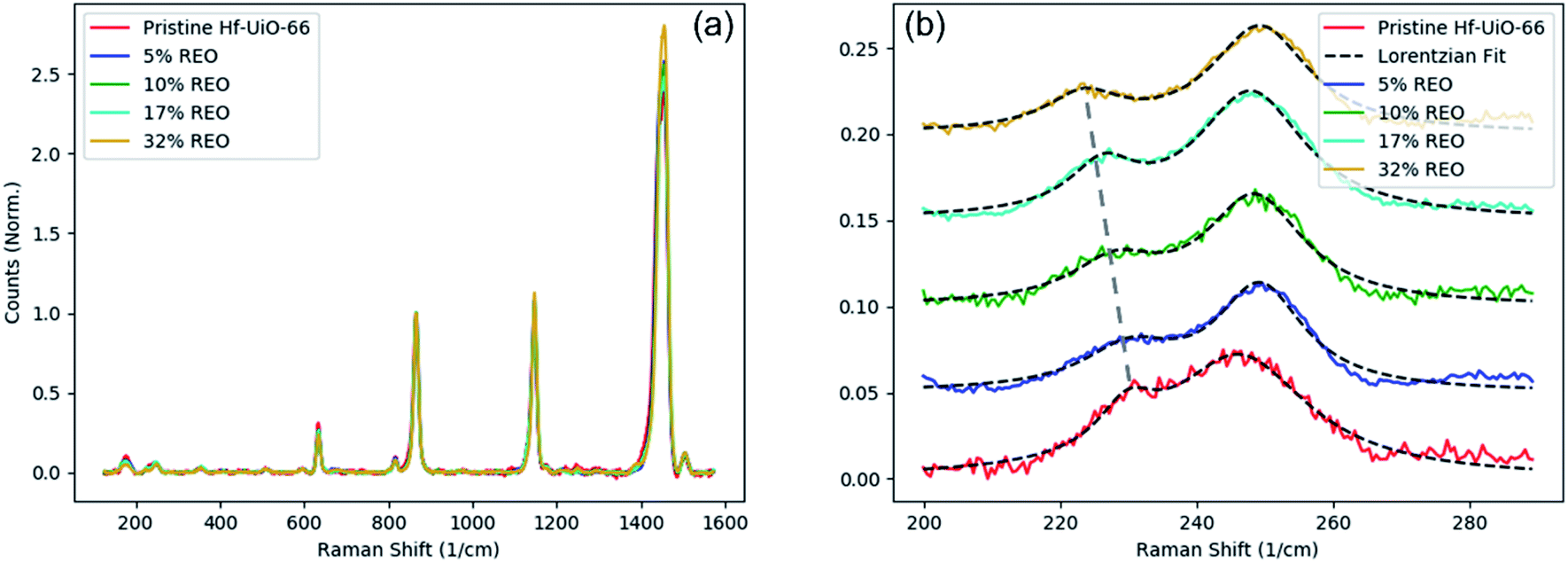 | ||
| Fig. 3 Missing cluster defects in Hf-UiO-66. (a) Full Raman spectra of Hf-UiO-66 defect series, highlighting the near-perfect agreement among all defective samples. (b) Fingerprint region showing mode quenching as a function of defect density in Hf-UiO-66. The 231 cm−1 vibrational mode is monotonically red-shifted as a function of defect density. | ||
Regardless of how these two Raman-active modes are assigned, there is a clear trend that represents missing cluster defects. First, the mode centered at 231 cm−1 in pristine Hf-UiO-66 is largely quenched compared to all defective samples regardless of defect density. Also, this mode is monotonically and roughly linearly red-shifted as a function of defect density (Fig. 4). Next, the mode centered at 245 cm−1 in pristine Hf-UiO-66 remains largely unchanged regardless of defect density; the intensities of this peak in the defective samples are lower than that of the pristine sample, but the measured quenching is significantly less strong than that of the 231 cm−1 mode. Due to the measured shift of the 231 cm−1 mode, we therefore hypothesize that the 231 cm−1 mode is due to vibrations of the Hf–O coordination and the 245 cm−1 is due to vibrations of the Hf-μ3O species. The quenching of both of these modes can be understood by an average decrease of scattering species through the removal of metal SBUs and the corresponding removal of linker molecules. We conclude that the 231 cm−1 mode corresponds to the Hf–O coordination because it experiences a monotonic red-shift as well as a quenching of the intensity relative to the pristine Hf-UiO-66. At higher defect densities, the metal SBU adjacent to a defect site is capped by formate ions that are less massive than the linker molecule. Therefore, the more defective samples contain more Hf–O species in which the oxygen atom is part of the formate capping group rather than the heavier linker molecule, causing the observed frequency shift. Mass differences between the formate and linker are not sufficient to describe the observed red shift, as a decrease in the mass would act to increase the vibrational frequency. To explain the observed decrease in the vibrational frequency, there must also be a corresponding decrease of the spring constant of the metal–oxygen coordination bond. This is reasonable due to the nature of the molecules: the dicarboxylate linker molecule is coordinated to metal SBUs on both ends and is correspondingly more rigid than the monocarboxylate formate capping species. Therefore, the observed decrease in vibrational frequency is directly related to defect-induced rigidity change of the Hf–O coordination bond. The linear trend (Fig. 4b) between peak position and defect level extracted from PXRD fits suggests that the quantity of missing cluster defects – regardless of the topology in which they occur – increases with the increasing PXRD-derived defect level. These data also suggest that the monotonic decrease of the Hf–O coordination bond vibrational frequency implies that the larger the percentage of the crystal volume in the reo topology, the more highly defective the samples are, not simply that the same number of defects is increasingly well ordered.
 | ||
| Fig. 4 Signature of missing cluster defects. Correlation plot of peak position versus the percentage of reo topology as extracted from fitting PXRD peaks along with a linear fit. | ||
The above claim that the Raman spectroscopic fingerprint of missing clusters in Hf-UiO-66 is independent of the topology in which they occur is supported by a group theory analysis of the pristine fcu topology versus the defective reo topology. Atomic coordinate calculations were recently performed by Cliffe, et al.18 on optimized structures of fcu Zr-UiO-66, reo Zr-UiO-66, fcu Hf-UiO-66, and reo Hf-UiO-66. These structurally-optimized atomic coordinates were used to determine the number and symmetry of the allowed Raman-active modes by means of the Bilbao Crystallographic Server.21–24 Comparison of the modes of the various structures to one another shows that the reo Hf-UiO-66 has more than twice as many Raman-active modes as does the fcu Hf-UiO-66 regardless of mode symmetry (A1, E, or T2). However, in the experimental spectra, when comparing defective Hf-UiO-66 to pristine Hf-UiO-66, there is a one-to-one correspondence between all modes across the entire measured range. We would expect additional peaks to be observed if the defective samples were to have large regions of reo topology, so it is likely that either the differences in the experimental Raman spectra are due to randomly-dispersed missing cluster defects or that the regions of the sample that adopt the reo topology are not sufficiently large and ordered to be detected by Raman spectroscopy. The other possibility is that our pristine Hf-UiO-66 sample in fcu topology is not truly pristine and has missing clusters in the reo phase already, thus obscuring the differences between pristine and defective Hf-UiO-66. However, both the pore size distribution (Fig. 2) of the pristine Hf-UiO-66 as well as the agreement between the Raman spectra of pristine Hf-UiO-66 and pristine Zr-UiO-66 suggest that this is unlikely.
This analysis was applied to a series of Zr-UiO-66 samples synthesized with formic acid as a modulator (Fig. 5). Three samples with varying concentrations of formic acid modulator (labeled FA100, FA200, and FA300) were compared to pristine Zr-UiO-66 with Raman spectroscopy. In pristine Zr-UiO-66, the Raman band at 300 cm−1 is attributed to motion of the Zr–O coordination bond coupled with motion of the aromatic C–H of the BDC linker. Comparing pristine Zr-UiO-66 to FA100 and FA200, a similar monotonic decrease as was observed in Hf-UiO-66 is also observed with increasing formic acid modulator concentration. However, FA300, the sample with the highest concentration of formic acid during synthesis does not appear to follow this trend. This discrepancy highlights the difficulty in defect quantification within MOFs, and we relate this discrepancy to missing linker defects. In Hf-UiO-66, it is suggested that most defects are missing cluster defects and the sharpness of the two low-angle PXRD reflections suggests very well-ordered missing cluster defects in the reo topology.
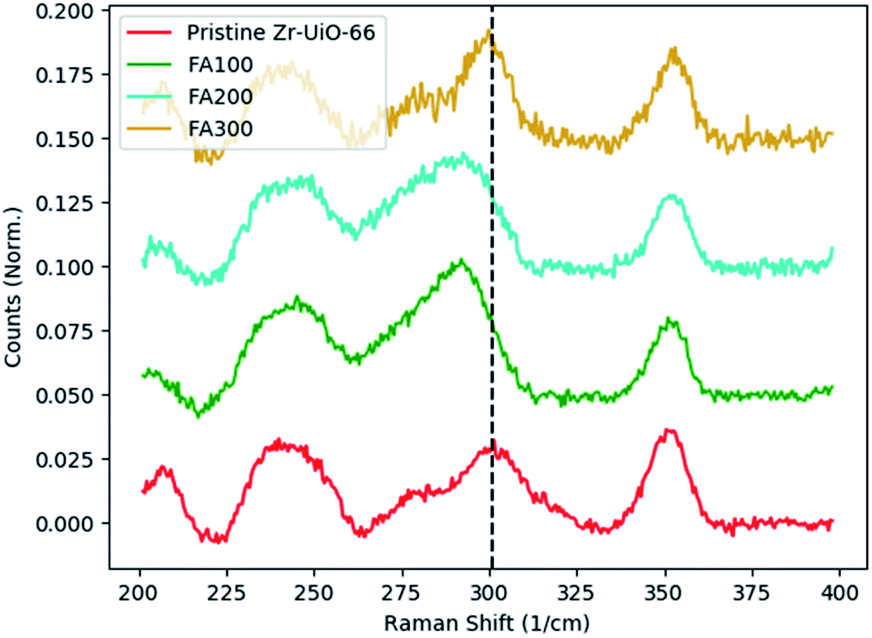 | ||
| Fig. 5 Missing clusters in Zr-UiO-66. Low concentrations of formic acid modulator show a monotonic decrease in the observed frequency of the Zr–O coordination vibrations. However, further increasing the concentration (FA300) results in a large quantity of missing linker defects. Despite the presence of missing cluster defects as confirmed by PXRD, the large quantity of missing linker defects overshadows the missing cluster defects and disrupts the observed monotonic decrease. The black dotted line depicts the center of the 300 cm−1 peak in pristine Hf-UiO-66 and FA300. | ||
However, in the Zr-UiO-66, TGA data suggests that there is a large quantity of missing linker defects, with overall defect level increasing with increased modulator; that is, FA300 is more defective than FA200 and FA100. As will be discussed below, missing linker defects are very challenging to study with Raman spectroscopy, with most defective samples showing near-perfect agreement across the entire measured range. Therefore, we hypothesize that the large number of missing linker defects present in the FA300 sample hinders the ability to study missing cluster defects with Raman spectroscopy and an accurate study of missing clusters requires first minimizing the quantity of randomly distributed missing linker defects.
To study missing linker defects, similar spectra were obtained for all Zr-UiO-66 samples containing missing linker defects and compared to the spectrum of the pristine counterpart (Fig. 6a). Each spectrum was internally normalized to the peak at 860 cm−1 consistent with the Hf-UiO-66 data presented above. Unsurprisingly, most of the region between 400 cm−1 to 1100 cm−1 in the Raman spectra remains unchanged regardless of defect level, though some deviations from the spectrum of pristine Zr-UiO-66 can be seen. This can be understood by noting that most of the vibrational modes in this region are dominated by motion of the linker molecules. Regardless of defect level, each sample contains identical BDC linker molecules that contribute to the measured spectrum. Any defect-induced changes in linker-dominated modes are expected to be very slight and likely involve an intensity change due to an average decrease in the number of scattering species as a function of defect density rather than a change of the vibrational frequency. Unlike the Hf-UiO-66 samples synthesized with missing cluster defects, most of the fingerprint region is very similar in pristine and defective Zr-UiO-66 regardless of defect density. This is not very surprising, however as these samples fabricated with between 13% and 25% defect density only correspond to between approximately 0.78 and 1.5 linkers removed per SBU (12 linker molecules shared between 2 SBUs). That is, on average even the most highly defective samples have structures that are extremely similar to the pristine counterpart. Combined with the large crystal volume sampled in these measurements, this causes all samples to appear extremely similar to one another. This differs from the case of missing cluster defects as the removal of one SBU is likely to remove all 12 linker molecules that were coordinated to the metal SBU. Despite the inability to find trends indicating the presence and nature of missing linker defects like those found for missing cluster defects in Hf-UiO-66, Raman spectroscopy still proves to be a useful characterization tool. In the 22% and 25% defective Zr-UiO-66 samples, dimethylformamide (DMF) used in synthesis as well as a formic acid modulator imply the presence of charge-balancing formate ions at the defect sites. In the 13% defective Zr-UiO-66, the modulator employed was acetic acid. Therefore, there is competition between formate capping groups from DMF and acetate ion capping groups from acetic acid. Direct vibrational signatures of the acetate capping agents are noticeable in the Raman spectrum of the 13% defective sample. As seen in Fig. 6b, the spectrum of the acetic acid-modulated sample also shows a very noticeable new peak at approximately 660 cm−1. Published Raman spectra on acetic acid show features at 603 and 621 cm−1 with no additional features near 660 cm−1.25 Therefore, we hypothesize that the side peak located at 660 cm−1 in the 13% defective sample is due not to free acetic acid in the pores left over from synthesis, but rather to the acetate ions capping the defect sites.
 | ||
| Fig. 6 Missing linkers in Zr-UiO-66. (a) Full Raman spectra of Zr-UiO-66 defect series, highlighting the near-perfect agreement among all defective samples. (b) Emergence of a new peak centered at 660 cm−1 in the acetic acid-modulated sample suggesting the presence of acetate-capped defect sites. | ||
FT-IR analysis
While Raman spectroscopy has proven very useful for identifying and understanding the nature of missing cluster defects in Hf-UiO-66 due to the accessibility of the low-wavenumber fingerprint region, infrared absorption spectroscopy allows for a more complete characterization of the vibrational modes of UiO-66 as well as signatures of missing linker defects in Zr-UiO-66, implying that the modes observed in the infrared spectrum are either not Raman-active or are only very weakly so. This trend highlights the importance of a comprehensive approach using both Raman and IR vibrational spectroscopy to ensure a full and accurate characterization of complex systems. As seen in Fig. 7, the spectra of the various Zr-UiO-66 samples show near-perfect agreement in IR-active modes regardless of defect density. As discussed in other literature, most of the IR-active modes within the scan range of 400 to 4000 cm−1 are linker-dominated with a few of the lowest-wavenumber modes involving motion of Zr-(OC) or μ3-O.26,27 Coupled with the relatively small number of linkers missing per SBU in defective samples, it is unsurprising that changes in the defect density do not greatly alter any of these modes. Slight differences between pristine and defective Zr-UiO-66 are seen nonetheless, namely around 795 cm−1 and 1085 cm−1. In each case, the intensities of the modes in defective Zr-UiO-66 are strongly quenched relative to those seen in pristine Zr-UiO-66. The vibrational mode at 795 cm−1 is assigned to motion of the μ3-OH coupled with a C–C bending and C–H wagging of the linker molecule. The observed quenching is likely due to the removal of many of the C–C bonds in defective samples, where the C–C bond is replaced by a C–H due to the formate-capping of the defect sites. Interestingly, the peak at 1085 cm−1 corresponds nearly entirely to C–H wagging on the benzene ring of the linker molecule, and this mode is quenched in all of the defective samples in a similar fashion to the mode at 795 cm−1. Again, this could be due simply to fewer scatterers present; a higher density of missing linker defects implies a larger number of benzene rings (and thus C–H bonds) missing from the structure as compared to the pristine crystal. However, as discussed previously, the formate capping groups are terminated by a C–H, though the symmetry of the C–H modes will differ due to the geometric change of the defect-capping C–H groups. These data suggest that the symmetric C–H mode at 1085 cm−1 is quenched whereas the antisymmetric C–H mode at 1105 cm−1 is largely unchanged with increasing defect density.Further IR investigations of the Hf-UiO-66 sample corroborate the hypothesis that most of the measured IR-active modes represent predominantly linker motion (Fig. 8). As with the Raman data, the IR spectra for pristine Hf-UiO-66 and pristine Zr-UiO-66 show a one-to-one match of peak positions, suggesting that very few of the IR-active modes in this frequency range directly involve the metal atoms.
 | ||
| Fig. 7 FT-IR of missing linkers in Zr-UiO-66. (a) All FT-IR spectra of defective and pristine Zr-UiO-66 show very close agreement to one another. The spectra were internally normalized to the peak near 1000 cm−1 shown by the dotted line. (b) Quenching of the 795 cm−1 mode assigned to motion of the μ3-OH coupled with a C–C bending and C–H wagging of the linker molecule. (c) Quenching of the 1085 cm−1 mode assigned to the benzene ring C–H wagging. | ||
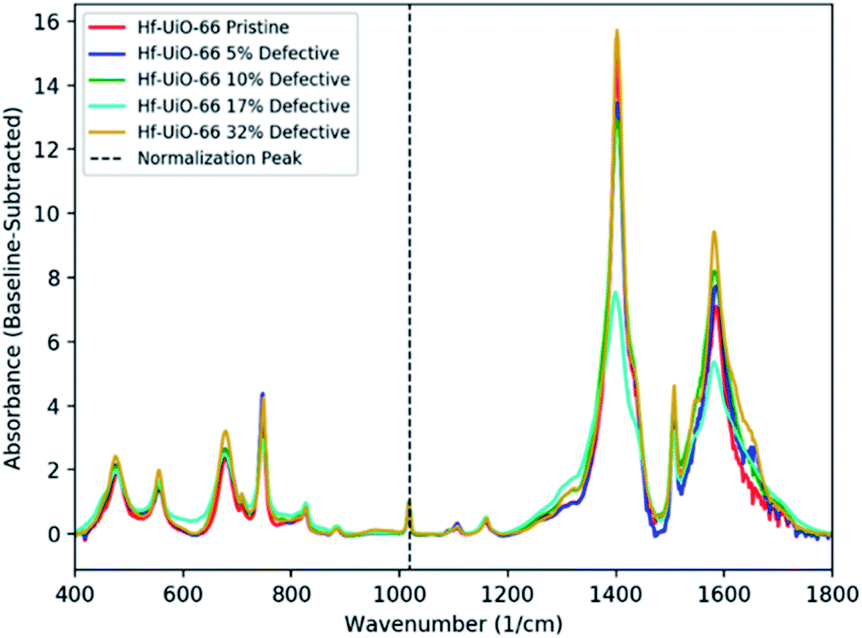 | ||
| Fig. 8 FTIR of Hf-UiO-66. Full FTIR spectra of pristine and defective Hf-UiO-66 internally normalized to the peak at the dotted line show near-perfect agreement between all defective samples and the pristine counterpart. It is very likely that any signatures of missing cluster defects occur at frequencies too low to be accessed in this FTIR measurement. | ||
As discussed above, we also see weak quenching of 1085 cm−1 modes in Hf-UiO-66 samples in the same fashion as indicated missing linkers in Zr-UiO-66. Therefore, we hypothesized the presence of missing linker defects in Hf-UiO-66, which is corroborated by the feature between 1.1 and 1.2 nm in the pore-size distribution analysis (Fig. 2). In contrast to the previously-discussed Raman spectroscopy data, FTIR appears not to be sensitive to missing cluster defects in Hf-UiO-66, primarily due to the inaccessibility of the low-wavenumber fingerprint region with this instrument.
Conclusions
In conclusion, Raman spectroscopy and FTIR absorption spectroscopy were used to directly probe the nature of defects in the metal–organic frameworks Zr-UiO-66 and Hf-UiO-66. The Raman-active Hf–O coordination bond in Hf-UiO-66 was shown to occur at lower frequencies with increasing defect level. This was attributed to the less-rigid formate charge-balancing capping group replacing linkers at adjacent metal SBUs. The monotonic and nearly linear red-shift of the frequency of the Hf–O coordination bond suggests that Raman spectroscopy is sensitive to missing cluster defects in Hf-UiO-66 regardless of the topology in which they occur. In conjunction with the standard XRD technique for studying missing cluster defects, Raman spectroscopy is shown to add an extra layer of insight; allowing for the probing of missing cluster defects directly rather than small, ordered regions of reo topology within the larger fcu crystal. In FT-IR absorption spectra, missing linker defects result in a quenching of C–C and C–H modes on the carboxylate tail of the linker molecule and the benzene ring of the linker molecule respectively. This is due to the replacement of linkers by monocarboxylate capping agents such as formate and acetate in defective materials. While powerful in characterization, as shown in the aforementioned study of missing linker defects, FT-IR absorption spectroscopy alone cannot offer a complete understanding of defects in UiO-66. Due to the inaccessibility of the low-wavenumber region, standard FT-IR measurements are not sensitive to missing cluster defects that alter the Hf–O and Hf-μ3O vibrational modes. Together, these spectroscopic techniques provide a direct probe of defects within UiO-66, allowing the disentanglement of different defect types: the spectroscopic fingerprint of one type of defect is independent of the nature or quantity of the other defect.Author contributions
BTY, BG and AW are responsible for investigation; AJM and LEM are responsible for supervision and conceptualization. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was performed with the support of the Department of Defense Threat Reduction Agency under grants HDTRA11910008 (UNC-CH) and W911NF-19-2-0156 (Virginia Tech).Notes and references
- B. Li, H.-M. Wen, W. Zhou and B. Chen, J. Phys. Chem. Lett., 2014, 5, 3468–3479 CrossRef CAS.
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef CAS.
- Y. Li, A.-S. Xiao, B. Zou, H.-X. Zhang, K.-L. Yan and Y. Lin, Polyhedron, 2018, 154, 83–97 CrossRef CAS.
- M.-X. Wu and Y.-W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- S. Beg, M. Rahman, A. Jain, S. Saini, P. Midoux, C. Pichon, F. J. Ahmad and S. Akhter, Drug Discovery Today, 2017, 22(4), 625–637 CrossRef CAS.
- M. Nasrabadi, M. A. Ghasemzadeh and M. R. Z. Monfared, New J. Chem., 2019, 43, 16033 RSC.
- J. E. Mondloch, M. J. Katz, W. C. Isley III, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- S.-Y. Moon, Y. Liu, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 6795–6799 CrossRef CAS.
- K. Vellingiri, L. Philip and K.-H. Kim, Coord. Chem. Rev., 2017, 353, 159–179 CrossRef CAS.
- N. Nijem and Y. J. Chabal, Comments Inorg. Chem., 2014, 34, 78–102 CrossRef CAS.
- A. Centrone, D. Y. Siberio-Pérez, A. R. Milward, O. M. Yaghi, A. J. Matzger and G. Zerbi, Chem. Phys. Lett., 2005, 411, 516–519 CrossRef CAS.
- J. Ethiraj, F. Bonino, C. Lamberti and S. Bordiga, Microporous Mesoporous Mater., 2015, 207, 90–94 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- M. J. Katz, S.-Y. Moon, J. E. Mondloch, M. H. Beyzavi, C. J. Stephenson, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 2286 RSC.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- W. Xiang, Y. Zhang, Y. Chen, C.-J. Liu and X. Tu, J. Mater. Chem. A, 2020, 8, 21526 RSC.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- B. Gibbons, E. C. Bartlett, M. Cai, X. Yang, E. M. Johnson and A. J. Morris, Inorg. Chem., 2021, 60, 16378–16387 CrossRef CAS PubMed.
- M. I. Aroyo, J. M. Perez-Mato, D. Orobengoa, E. Tasci, G. de la Flo and A. Kirov, Bulg. Chem. Commun., 2011, 43(2), 183–197 CAS.
- M. I. Aroyo, J. M. Perez-Mato, C. Capillas, E. Kroumova, S. Ivantchev, G. Madariaga, A. Kirov and H. Wondratschek, Z. Kristallogr., 2006, 221(1), 15–27 CAS.
- M. I. Aroyo, A. Kirov, C. Capillas, J. M. Perez-Mato and H. Wondratschek, Acta Crystallogr., 2006, A62, 115–128 CrossRef CAS.
- E. Kroumova, M. I. Aroyo, J. M. Perez Mato, A. Kirov, C. Capillas, S. Ivantchev and H. Wondratschek, Phase Transitions, 2003, 76(1–2), 155–170 CrossRef CAS.
- F. Wan, L. Du, W. Chen, P. Wang, J. Wang and H. Shi, Energies, 2017, 10, 967 CrossRef.
- Y. Han, M. Liu, K. Li, Y. Zuo, Y. Wei, S. Xu, G. Zhang, C. Song, Z. Zhang and X. Guo, CrystEngComm, 2015, 17, 6434 RSC.
- Z. Jin and H. Yang, Nanoscale Res. Lett., 2017, 12, 539 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction patterns, scanning electron microscopy images, thermal gravimetric analyses, nitrogen adsorption isotherm, and pore size distributions. See https://doi.org/10.1039/d2ra03131k |
| This journal is © The Royal Society of Chemistry 2022 |