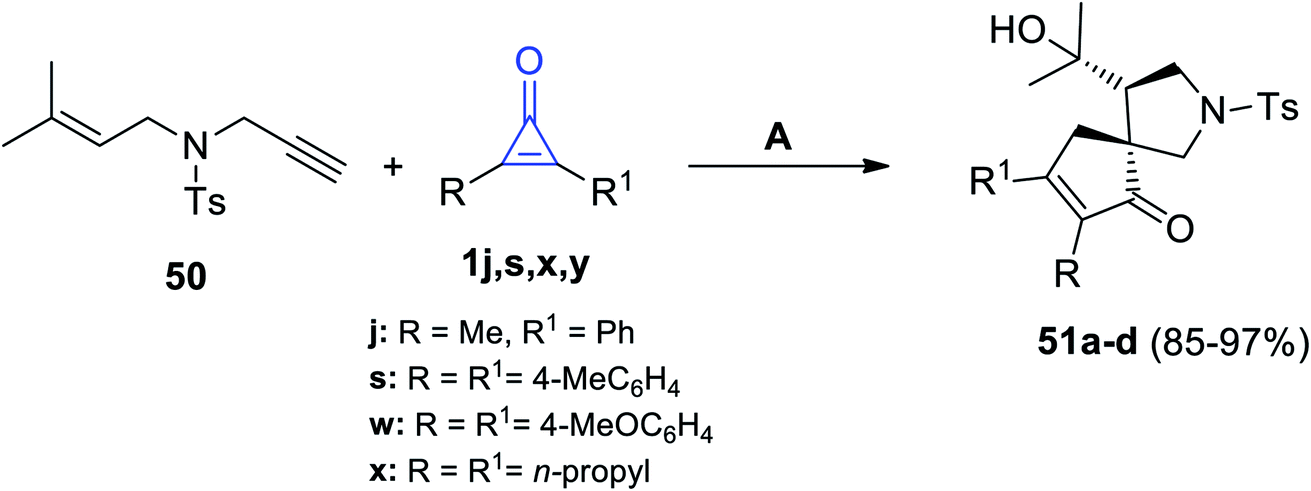DOI:
10.1039/D2RA03011J
(Review Article)
RSC Adv., 2022,
12, 18615-18645
Heterocycles from cyclopropenones
Received
12th May 2022
, Accepted 2nd June 2022
First published on 24th June 2022
Abstract
Great attention has been paid to cyclopropenones as they are present in many natural sources. Various synthesized cyclopropenone derivatives also show a wide range of biological activities. The cyclopropenone derivatives undergo a variety of reactions such as ring-opening reactions, isomerization reactions, C–C coupling reactions, C–H activation, cycloaddition reactions, thermal and photo-irradiation reactions, and acid–base-catalyzed reactions under the influence of various chemical reagents (electrophiles, nucleophiles, radicals, and organometallics) and external forces (heat and light). Many previous reviews have dealt with the chemistry and reactions of cyclopropenones. However and to the best of our knowledge, the utility of cyclopropenones in the synthesis of heterocycles has not been reported before. Therefore, it would be interesting to shed light on this new topic. The present review article provides, for the first time, a comprehensive compilation of synthetic methods for the synthesis of various heterocyclic ring systems, as a significant family in the field of organic chemistry.
1 Introduction
Cyclopropenone is an important building block for the construction of a large number of skeletons.1 Very recently, new developments have been made using cyclopropenone and its heteroanalogues, such as [3 + n] annulation reactions, acylation, organocatalytic reactions, metal catalytic reactions, base-mediated reactions, nucleophilic substitution reactions, light-induced reactions, σ-bond cross-exchange reactions and C–H activation reactions for the synthesis of diverse heterocycles. Besides, they are used as catalysts in a few reactions. The activation of C–C bonds is a powerful concept for the reorganization or coupling of organic scaffolds, yet it is a relatively challenging process to achieve in the context of synthetic methodology because of their inherent stability.2 In order to enable such methods, one can use C–C-strained, often cyclic, building blocks that are consequently spring-loaded for C–C bond activation.3–22
This review highlights the recent applications of cyclopropenone ring-expansion reactions aiming to synthesize various products (essentially various classes of heterocycles). Since there is no extensive comprehensive review concerned with the chemistry of cyclopropenone derivatives in the construction of heterocycles, the present study would be of great interest.
2 Chemistry
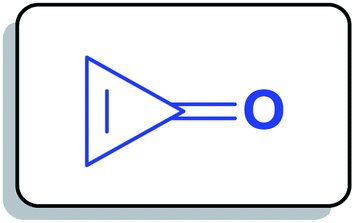
Cyclopropenone (1a) (Fig. 1) is a cyclic organic ketone with the molecular formula C3H2O composed of a cyclopropene with a ketone functional group. Cyclopropenone (1a) is an aromatic compound that polymerizes at room temperature.23
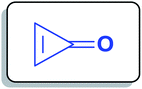 |
| | Fig. 1 Structure of cyclopropenone (1a). | |
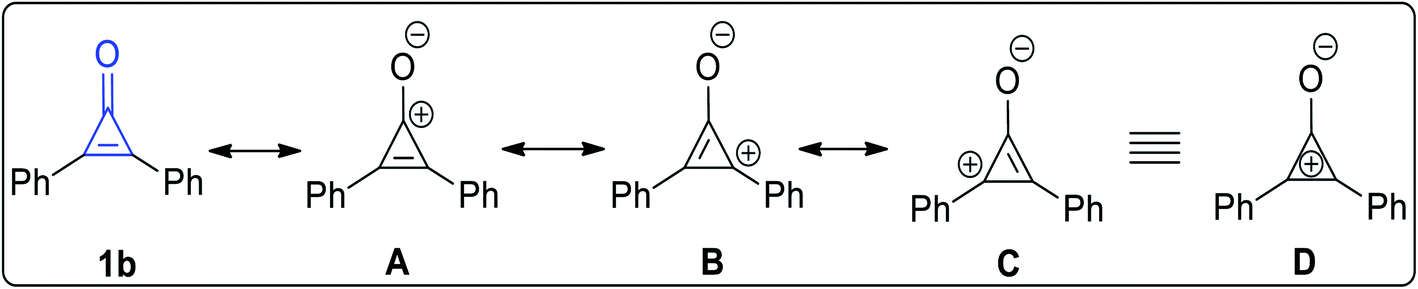
The stability of 2,3-diphenylcyclopropenone (1b) increases when the substituents are aryl groups. The possible resonance structures of 2,3-diphenylcyclopropenone (1b) (Fig. 2) are shown in A–C (equivalent to D), which contain a three-membered ring of sp2 carbon coupled to the electron-donor phenyl group in order to stabilize these structures (Fig. 2).24,25
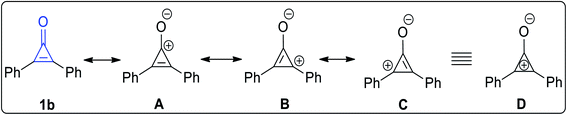 |
| | Fig. 2 Resonance structures of 2,3-diphenylcyclopropenone (1b). | |
Since the first synthesis of cyclopropenone (1) was done by Breslow,26 organic chemists have been quite interested in it. Cyclopropenones are commonly employed as electrophile-trapping agents.27,28 Due to their high strain29,30, cyclopropenones easily participate in cycloaddition,31 ring-opening,32–34 and ring-enlargement35–37 reactions. They are also used as organocatalysts in the conversion of aldoximes to nitriles.38 In addition, they are used as Lewis bases for the organocatalytic transformation of alcohols into alkyl chlorides.39 The effect of UV irradiation on cyclopropenones and their analogues results in efficient decarbonylation and the generation of the corresponding alkynes.40–45 Visible light causes cyclopropenones to undergo decarbonylation and form alkynes.46 Cyclopropenones can form metal complexes via chelation with transition metals at the oxygen center or at the double bond.47
2.1. Natural products containing cyclopropenone (1) and biological investigation of cyclopropenones
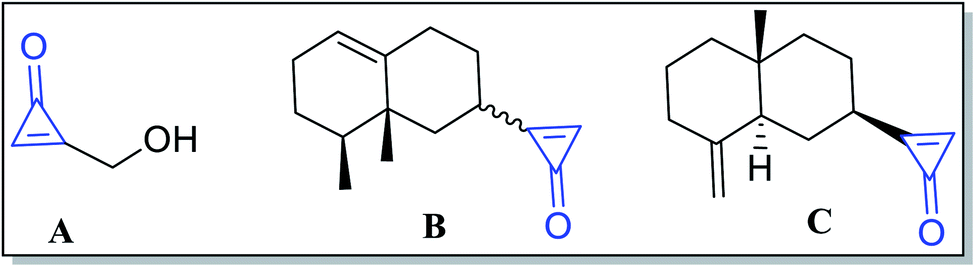
Numerous extracted natural products have cyclopropenone moieties such as 2-(hydroxymethyl)-cycloprop-2-enone (penitricin) (A),48 2-((8S,8aR)-8,8a-dimethyl-1,2,3,4,6,7,8,8a-octahydronaphthalen-2-yl)cycloprop-2-enone (B) and 2-((2R,4aR,8aS)-4a-methyl-8-methylenedecahydronaphthalen-2-yl)cycloprop-2-enone (C) (Fig. 3).49 Compound A was extracted from Penicillium aculeatum, whereas compounds B and C were extracted from plant sources. It was found that penitricin (A) is the only one that showed biological activity as an antibiotic agent.50
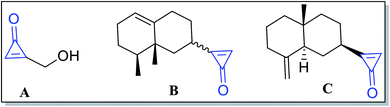 |
| | Fig. 3 Naturally occurring compounds A–C containing cyclopropenone molecules. | |
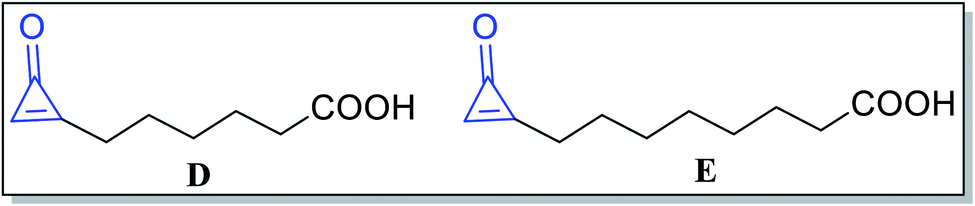
Besides, alutacenoic acid A (D) and alutacenoic acid B (E) (Fig. 4) are naturally occurring molecules that contain cyclopropenone structures. They were isolated from common fungi such as Eupenicillium alutaceum.51
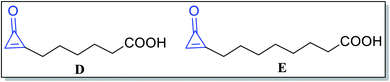 |
| | Fig. 4 Naturally occurring acids containing cyclopropenone structures. | |
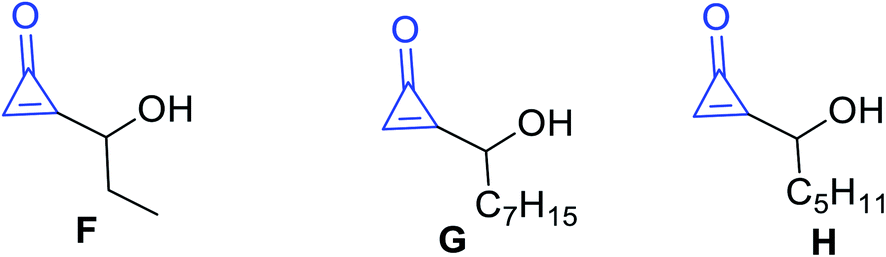
Other types of isolated naturally occurring products F, G and H (Fig. 5) having cyclopropenone moieties are 2-(1-hydroxypropyl)cycloprop-2-enone (F), 2-(1-hydroxyoctyl)-cycloprop-2-enone (G), and 2-(1-hydroxyhexyl)cycloprop-2-enone (H) (Scheme 5). Such compounds showed antibacterial activity more than penitricin (A).52
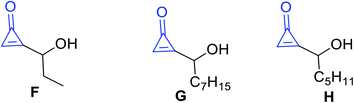 |
| | Fig. 5 Structures of naturally occurring compounds F–H. | |
Some studies reported in the literature illustrated the utility of diphenylcyclopropenone derivatives for dermatological treatments such as alopecia areata, alopecia totalis, and alopecia universalis.53,54 Moreover they were used for the treatment of recalcitrant warts,55 and others showed antitumor activities in the treatment of B16 melanoma.56,57
2.2. Synthesis of cyclopropenone (1) and its derivatives
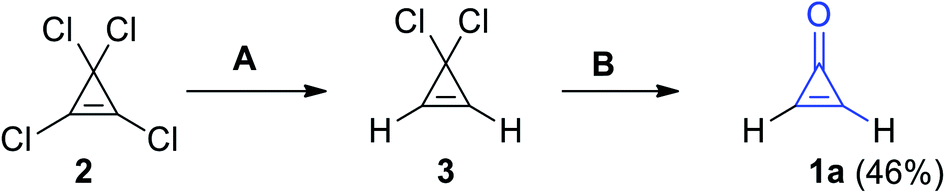
2.2.1. From acetals or their formation. Synthesis of cyclopropenone (1a) was achieved via selective dechlorination using (C4H9)3SnH on perchlorocycloprop-1-ene (2) to give 3,3-dichloro-cycloprop-1-ene (3). Hydrolysis of 3 led to the formation of compound 1a in 46% yield (Scheme 1).58
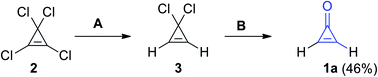 |
| | Scheme 1 Synthesis of cyclopropenone (1a) from perchlorocycloprop-1-ene (2). Reagents and conditions: A = (C4H9)3SnH, B = H2O. | |
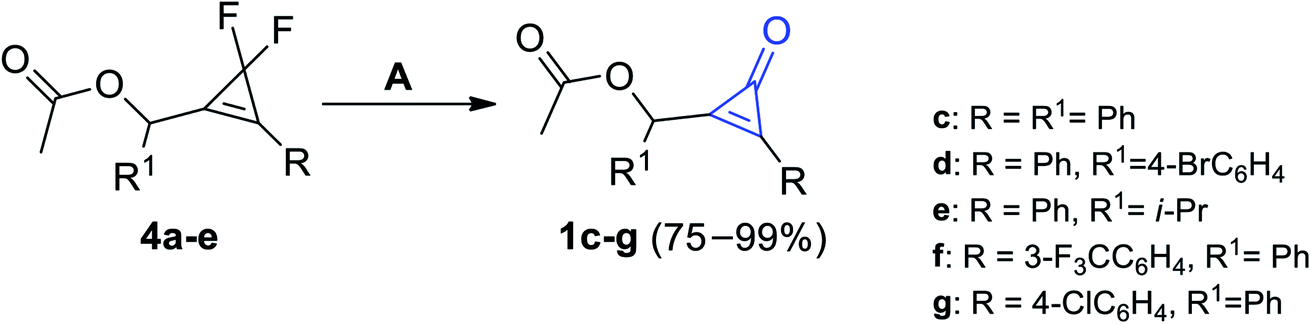
The most effective procedure for the synthesis of cyclopropenones was found during the treatment of compounds 4a–e with equal amounts of boron trifluoride (BF3) as a Lewis acid in ether to yield substituted cyclopropenones 1c–g in moderate to excellent yields (Scheme 2).59
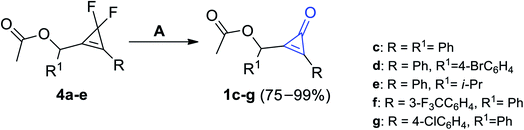 |
| | Scheme 2 Synthesis of substituted cyclopropenones 1c–g using BF3. Reagents and conditions: A = BF3, Et2O. | |
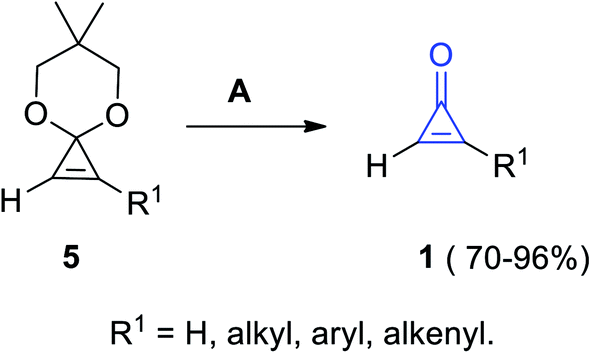
Another convenient method was known to obtain substituted cyclopropenones 1 in 70–96% yield, via the hydrolysis of 6,6-dimethyl-4,8-dioxaspiro[2.5]oct-1-ene derivatives 5 (cyclopropenone acetals) using Amberlyst-15 in acetone or in aqueous tetrahydrofuran (THF) at room temperature (Scheme 3).60
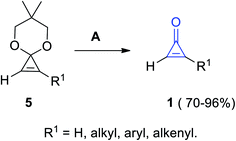 |
| | Scheme 3 Hydrolysis of 6,6-dimethyl-4,8-dioxaspiro[2.5]oct-1-ene derivatives 5 into 1. Reagents and conditions: A = Amberlyst-15, MeCOMe or aq. THF, rt. | |
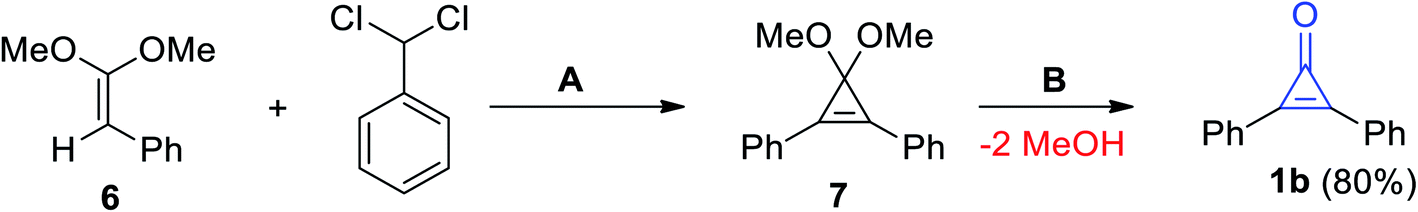
Breslow et al. also reported on the synthesis of diphenylcyclopropenone (1b) by treatment of (2,2-dimethoxyvinyl)benzene (6) with (dichloromethyl)benzene in the presence of potassium tert-butoxide (KO-t-Bu) to obtain 3,3-dimethoxy-1,2-diphenylcyclopropene (7) as an intermediate, which was converted by hydrolysis to give 1b in 80% yield (Scheme 4).25
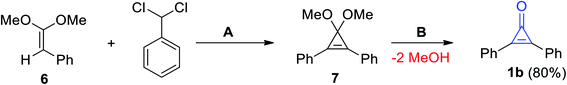 |
| | Scheme 4 Synthesis of 2,3-diphenylcyclopropenone (1b). Reagents and conditions: A = KO-t-Bu, B = H2O/H+. | |
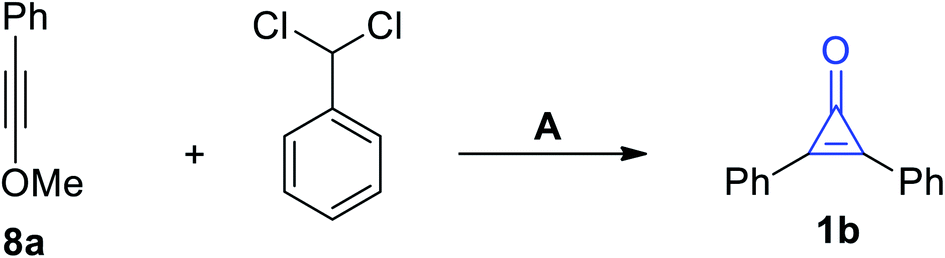
An alternative method for the synthesis of 1b was applied during the cycloaddition of phenylmethoxy-acetylene (8a) with (dichloromethyl)benzene in the presence of KO-t-Bu (Scheme 5).61
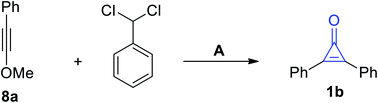 |
| | Scheme 5 Synthesis of 1b from the cycloaddition of phenylmethoxy-acetylene (8a) with (dichloromethyl)benzene. Reagents and conditions: A = (i) KO-t-Bu, (ii) H2O/H+. | |
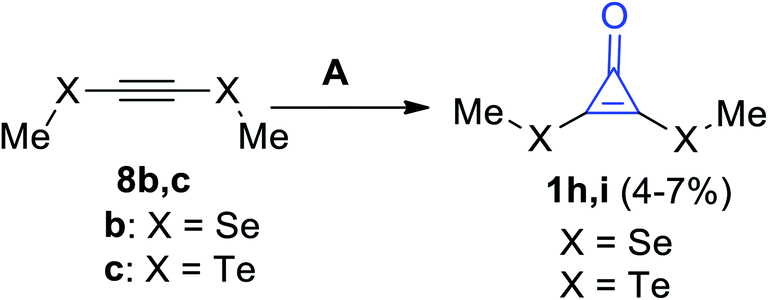
Cyclopropenones 1h,i were formed in a low yield (4–7%) via the reaction between acetylenes 8b,c and sodium trichloroacetate (Cl3COONa) using dimethylethane (DME), as shown in Scheme 6.62
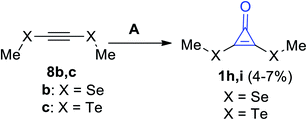 |
| | Scheme 6 Formation of cyclopropenone 1h,i via the reaction of acetylenes 8b,c with Cl3COONa. Reagents and conditions: A = (i) Cl3CCOONa, DME, reflux, (ii) H2O. | |
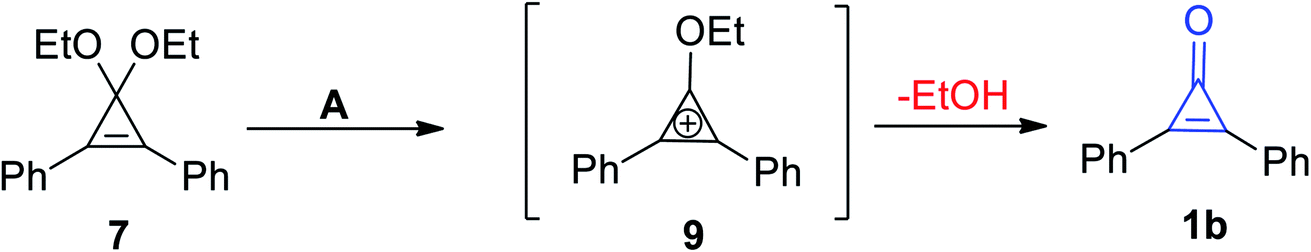
McGarrity et al. used a rapid injection NMR technique to observe the formation of the cyclopropenium intermediate (9) via hydrolysis of 1,1-diethoxy-2,3-diphenylcycloprop-2-ene (7) in slightly acidic aqueous acetone to obtain diphenylcyclopropenone (1b) (Scheme 7).63
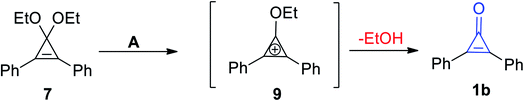 |
| | Scheme 7 Synthesis of cyclopropenone 1b. Reagents and conditions: A = MeCOMe, H2O/H3O+. | |
2.2.2. Bromination and/or elimination reaction of bromo-ketonic compounds. Various substituted cyclopropenones 1j were synthesized during the reaction of 1-phenyl-2-butanone (10) with bromine in DCM and Et3N. As an example, 2-methyl-3-phenylcycloprop-2-enone (1) was obtained in 45% yield (Scheme 8).64
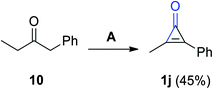 |
| | Scheme 8 Synthesis of 2-methyl-3-phenylcycloprop-2-enone (1j). Reagents and conditions: A = Br2, CH2Cl2, Et3N, 0 °C. | |
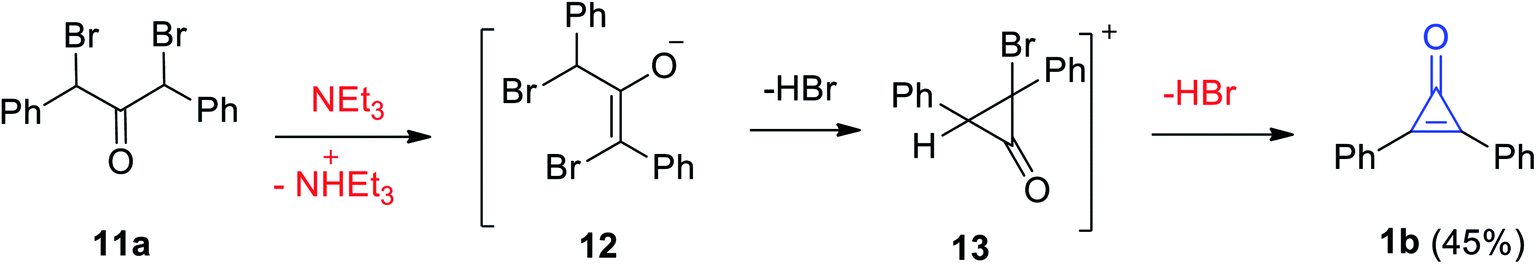
The Favorskii reaction was also used to synthesize 2,3-diphenylcyclopropenone (1b) in 45% yield during elimination of HBr by the action of Et3N on dibromodibenzyl ketone (11a). The reaction mechanism was the formation of intermediates 12 and 13 (Scheme 9).65
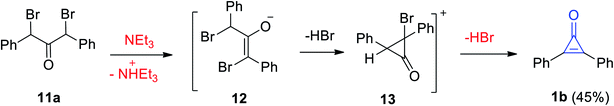 |
| | Scheme 9 Synthesis of diphenylcyclopropenone (1b) from dibromodibenzyl ketone (11a). | |
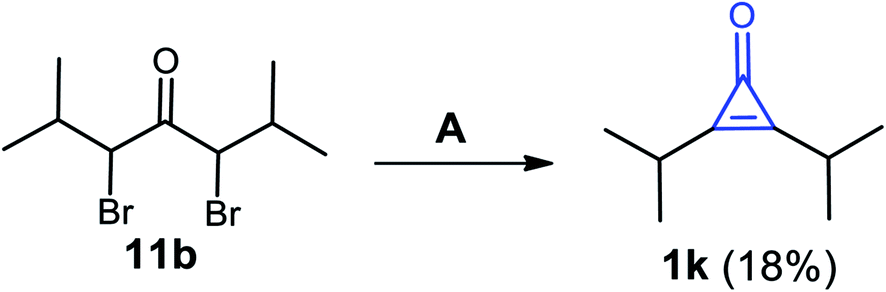
Previously, Curnow et al. reported that 2,3-diisopropylcycloprop-2-enone (1k) was also obtained in a low yield (18%), via dehydrobromination of 3,5-dibromo-2,6-dimethylheptan-4-one (11b) using NaH in THF as a solvent, followed by treatment with aqueous HCl (Scheme 10).66
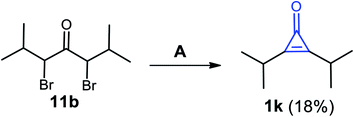 |
| | Scheme 10 Synthesis of 2,3-diisopropylcycloprop-2-enone (1k). Reagents and conditions: A = (i) NaH, THF, (overnight) and (ii) HCl/H2O. | |
2.2.3. Different methods of preparation. 2,3-Bis(methyl-(phenyl)-amino)cycloprop-2-enone (1l) was formed in 22% yield (Scheme 11) via the hydrolysis of N-(2,3-bis(methyl(phenyl)amino)cyclopropylidene)-N-methylbenzenaminium (14) using aqueous KOH. The reaction was proceeded via the formation of intermediate 15.67
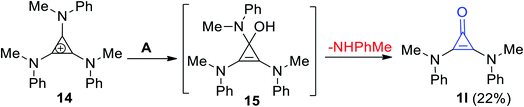 |
| | Scheme 11 Formation of 2,3-bis(methyl(phenyl)-amino)cycloprop-2-enone (1l). Reagents and conditions: A = KOH/H2O, MeOH, rt, 40 h. | |
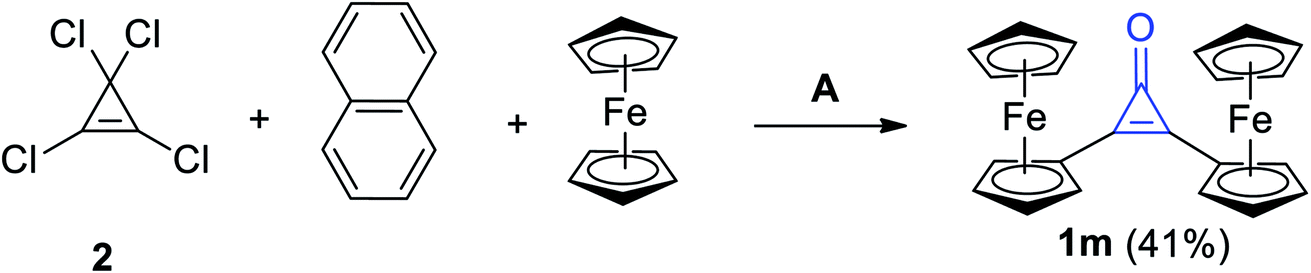
The reaction mixture of 2 with naphthalene and ferrocene in DCM in the presence of aluminum chloride (AlCl3) afforded among other products, diferrocenylcyclopropenone (1m) in 41% yield (Scheme 12).68
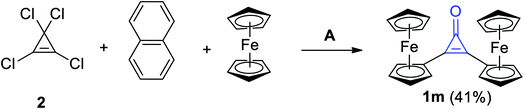 |
| | Scheme 12 Synthesis of diferrocenylcyclopropenone (1m). Reagents and conditions: A = AlCl3, CH2Cl2, H2O. | |
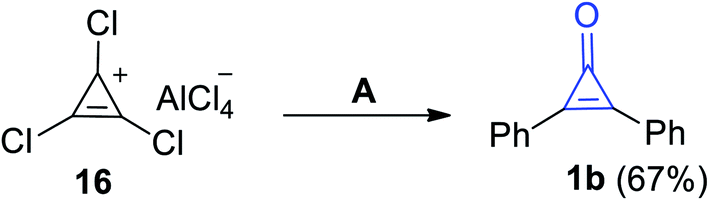
During heating of 1,2,3-trichlorocycloprop-2-en-1-ylium aluminum(III) chloride (16) in a mixture of benzene/H2O, 2,3-diphenylcyclopropenone (1b) was obtained in 67% yield (Scheme 13).69 As compound 16 underwent an electrophilic aromatic substitution (Friedel–Crafts alkylation) during reaction with benzene, which upon heating, gem-dichlorodiphenylcyclopropene was obtained. Then, the formed intermediate underwent hydrolysis (during workup) to afford 2,3-diphenylcyclopropenone (1b).
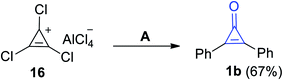 |
| | Scheme 13 Synthesis of 2,3-diphenylcyclopropenone (1b) from (16). Reagents and conditions: A = benzene, H2O. | |
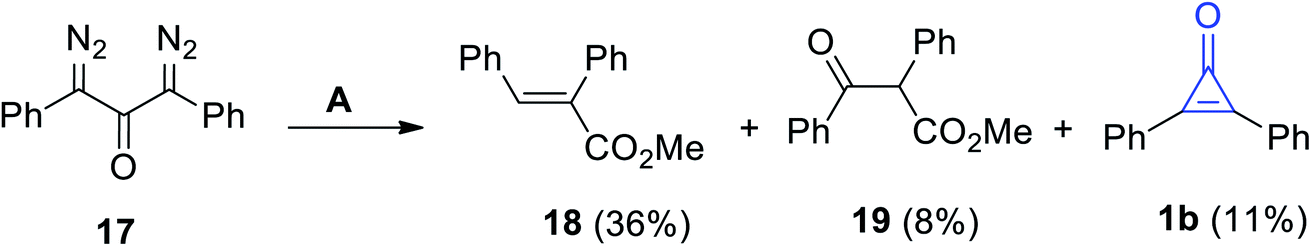
Decomposition of 1,3-bis(diazo)-1,3-diphenylpropan-2-one (17) in methanol in the presence of Ag2O yielded compounds 18 and 19, in addition to 2,3-diphenylcyclopropenone (1b), in a 11% yield (Scheme 14).70
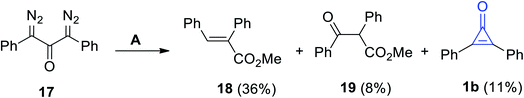 |
| | Scheme 14 Oxidation of 17 into diphenylcyclopropenone (1b), 18 and 19. Reagents and conditions: A = MeOH, Ag2O. | |
2.3. Utility of cyclopropenone derivatives in the synthesis of various heterocyclic compounds
2.3.1. Synthesis of four-membered rings with one heteroatom. An example of heterocyclic ring containing an heteroatom was reported.71 For example, the cyclization reaction of 2-(2-hydroxypropan-2-yl)-3-methylcycloprop-2-enone (1n) in CD3OD and triphenylphosphine (PPh3) yielded 3-ethylidene-4,4-dimethyloxetan-2-one (20) in 60% yield, whereas substituted furanone 21 was also formed as a side product with 39% yield (Scheme 15).71
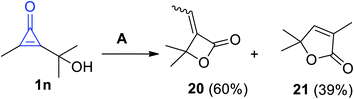 |
| | Scheme 15 Synthesis of 3-ethylidene-4,4-dimethyloxetan-2-one (20). Reagents and conditions: A = PPh3 (5 mol%), CD3OD, 25 °C. | |
2.3.2. Synthesis of five-membered rings with one heteroatom.
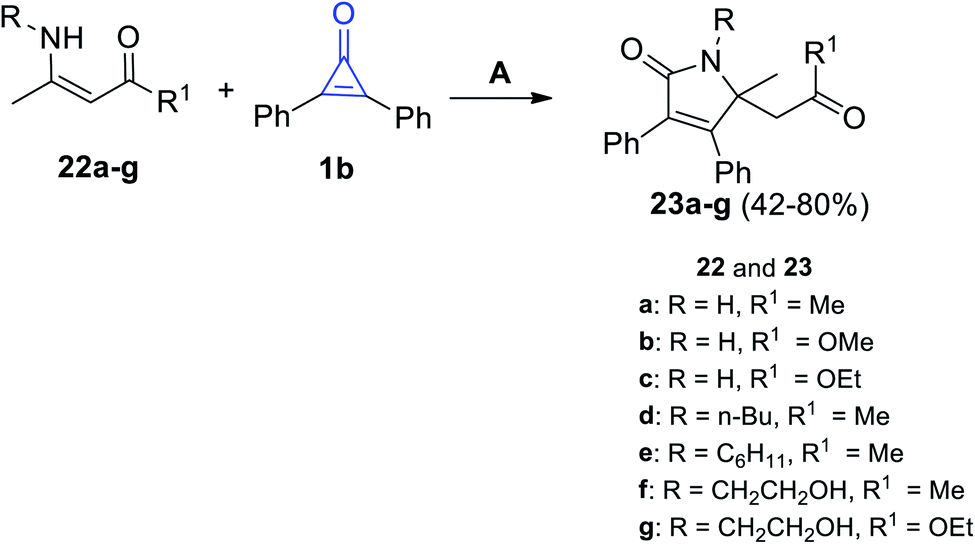
2.3.2.1. Synthesis of pyrrolones. Under microwave (MW) irradiation, the reaction of primary enaminone derivatives 22a–g with 1b catalyzed by Bi(NO3)3·5H2O afforded the corresponding 2-pyrrolinone derivatives 23a–g in 42–80% yield. The reaction was performed in toluene in the presence of bismuth nitrate Bi(NO3)3 as a catalyst (Scheme 16).72
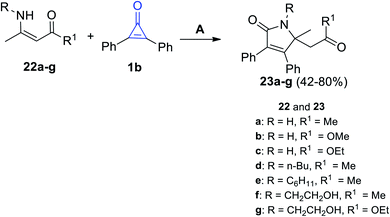 |
| | Scheme 16 Synthesis of 2-pyrrolinones 23a–g. Reagents and conditions: A = MW, toluene, Bi(NO3)3·5H2O, 30–90 min. | |
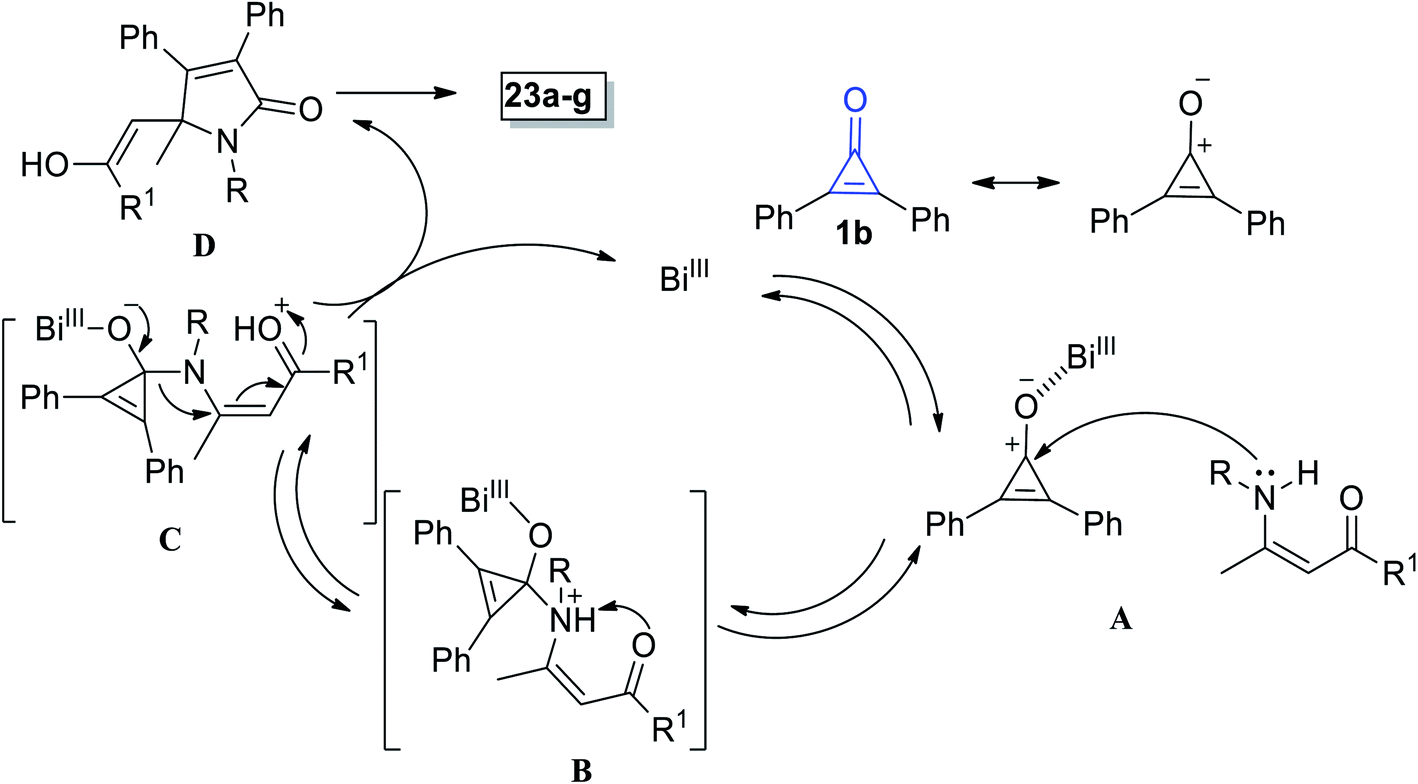
The suggested mechanism involves the formation of pyrrolinones 23a–g initiated via coordination between Bi(III) and the oxygen atom in 1b (Scheme 17). Then, the nitrogen atom of enaminone 22 would attack on carbonyl carbon of (1) via hard–hard interaction A.72 Proton shift to adduct B then occurred to yield intermediate C. Thereafter, adduct C underwent simultaneous ring expansion and Michael reaction, resulting in the formation of enol D, which finally gave 2-pyrrolinones 23a–g (Scheme 17).72
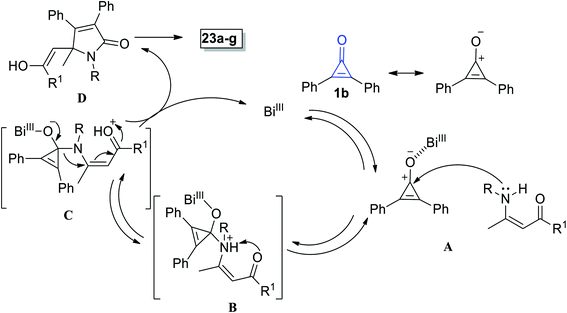 |
| | Scheme 17 Mechanism of the formation of 2-pyrrolinones 23a–g. | |
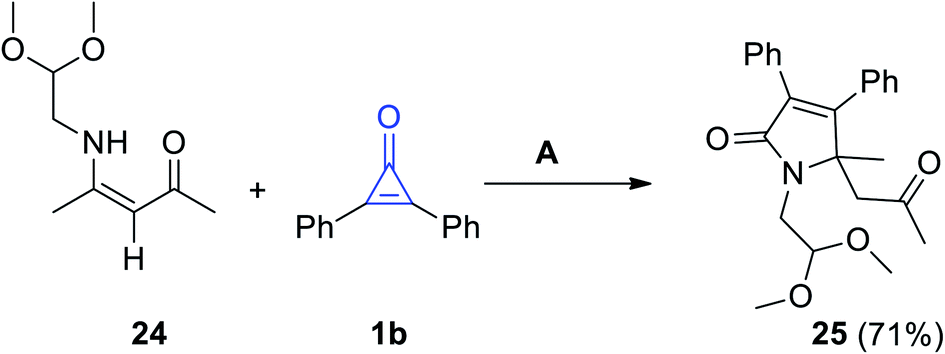
Reaction of (Z)-4-((2,2-dimethoxyethyl)amino)pent-3-en-2-one (24) and 1b in refluxing toluene for 6 d, proceeded to give 1-(2,2-dimethoxyethyl)-5-methyl-5-(2-oxopropyl)-3,4-diphenyl-1H-pyrrol-2(5H)-one (25) in 71% yield (Scheme 18).73
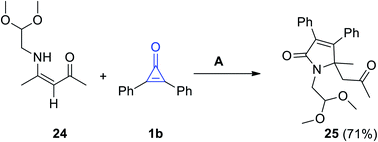 |
| | Scheme 18 Synthesis of compound 25. Reagents and conditions: A = toluene, reflux, 6 d. | |
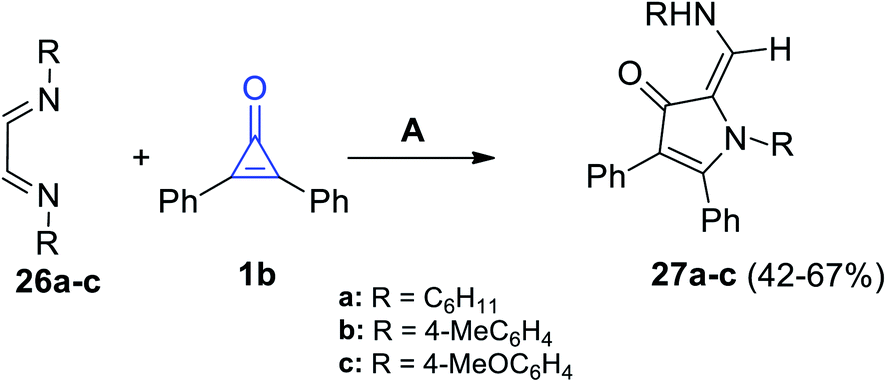
3-Pyrrolinone derivatives 27a–c were successfully synthesized in a moderate yield of 42–67%, via the reaction of diimines 26a–c with 2,3-diphenylcyclopropenone (1b) in dry ethanol for 2–5 h (Scheme 19).74
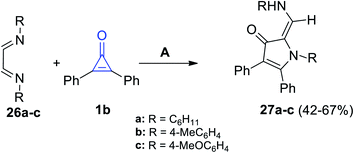 |
| | Scheme 19 Synthesis of 3-pyrrolinones 27a–c. Reagents and conditions: A = EtOH, 2–5 h. | |
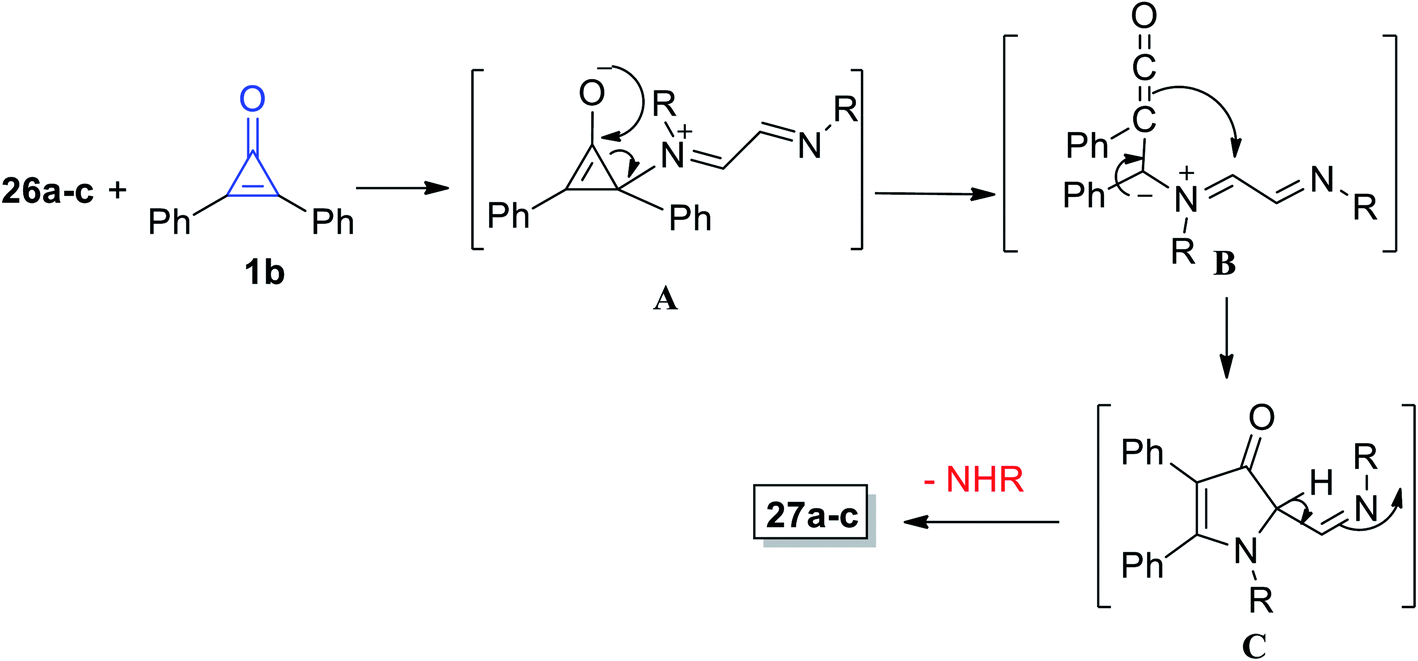
The mechanism of the formation of 3-pyrrolinone 27a–c is proposed and illustrated in Scheme 20. First, one of the imino nitrogen atoms of 26a–c was added to C-2 of 1b to obtain the intermediate (A). The ketene intermediate (B) was then obtained after ring opening, and then imines (C) were formed by cyclization via the ketene attack on iminium function, and finally, 3-pyrrolinones 27a–c were formed.74
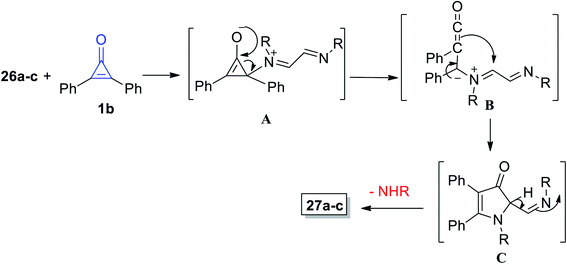 |
| | Scheme 20 Mechanism of the formation of 3-pyrrolinone derivatives 27a–c. | |
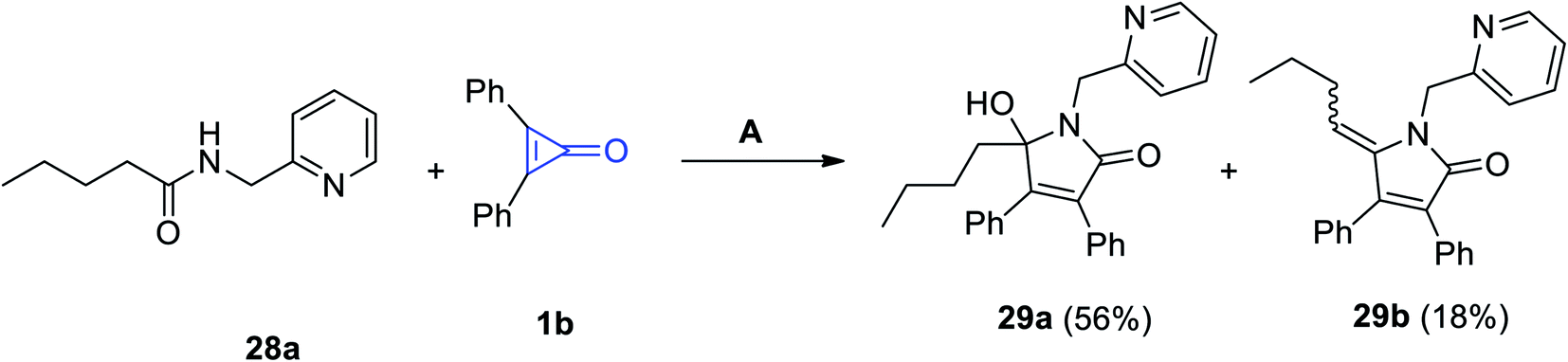
Haito and Chatani used [Rh(OAc)(cod)]2 as a catalyst in the reaction between N-(pyridin-2-ylmethyl)pentanamide (28a) and 2,3-diphenylcyclopropenone (1b). The reaction was performed in toluene in the presence of 2-phenylbenzoic acid (2-PhC6H4COOH) to produce 5-butyl-5-hydroxy-1-(pyridin-2-ylmethyl)-1H-pyrrol-2(5H)-one (29a) as a major product and also gave 5-butylidene-1-(pyridin-2-ylmethyl)-1H-pyrrol-2(5H)-one (29b) as a side product (Scheme 21).75
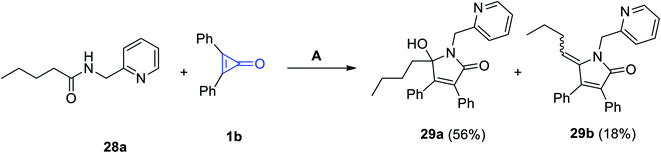 |
| | Scheme 21 Synthesis of N-pyridymethyl-pyrrol-3-ones 29a and 29b. Reagents and conditions: A = [Rh(OAc)(cod)]2 (5 mol%), toluene, 140 °C, 12 h. | |
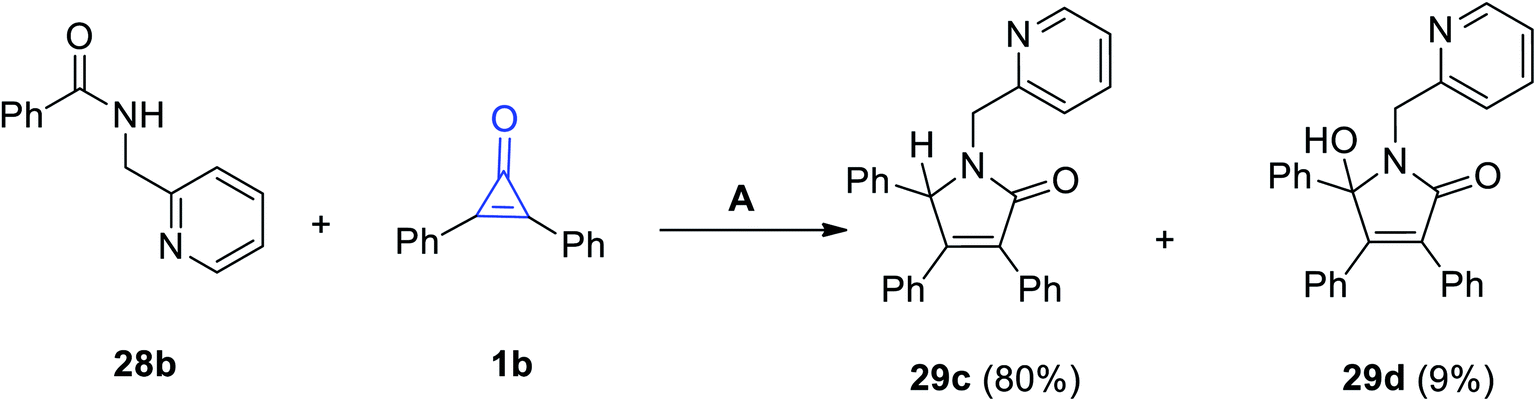
In continuation to the methods dealing with the synthesis of five-membered rings with one heteroatom, using cyclopropenone (1), Haito and Naoto Chatani reacted N-(pyridin-2-yl-methyl)benzamide (28b) with diphenylcyclopropenone (1b) in the presence of [Rh(OAc)(cod)]2 as a catalyst. 3,4,5-Triphenyl-1-(pyridin-2-ylmethyl)-1H-pyrrol-2(5H)-one (29c) and 5-hydroxy-3,4,5-triphenyl-1-(pyridin-2-ylmethyl)-1H-pyrrol-2(5H)-one (29d) were obtained in 80% and 9% yields, respectively (Scheme 22).75
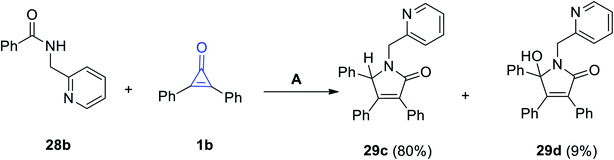 |
| | Scheme 22 Synthesis of pyrrol-2-ones 29c,d. Reagents and conditions: A = [Rh(OAc)(cod)]2 (10 mol%), toluene 2 ml, 140 °C, 1 h. | |
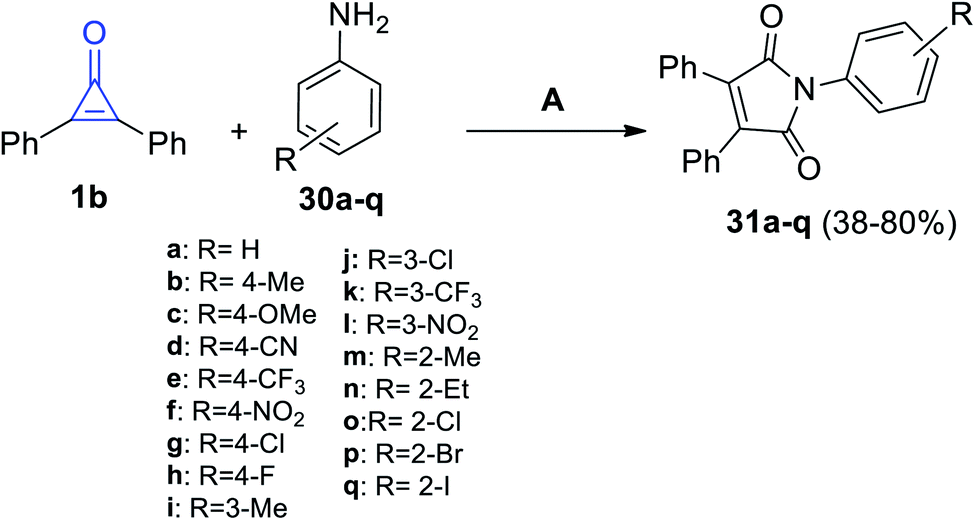
In 2020, Nanda et al. have reported that the reaction of 1b with various anilines 30a–q in the presence of palladium acetate as the catalyst, tetrabutylammonium bromide as an additive, and sodium acetate as the base at 120 °C for 12 h in DMF (0.25 M) gave substituted pyrroles 31a–q in 38–80% yield (Scheme 23).76
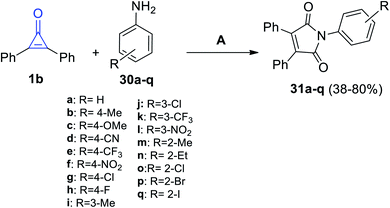 |
| | Scheme 23 Palladium-catalyzed synthesis of maleimides 31a–q. Reagents and conditions: A = Pd(OAc)2 (15 mol%), KOAc (4 equiv.), Bu4NBr (1 equiv.), K2S2O8 (1.5 equiv.), DMF (0.25 M), 120 °C, 1 h. | |
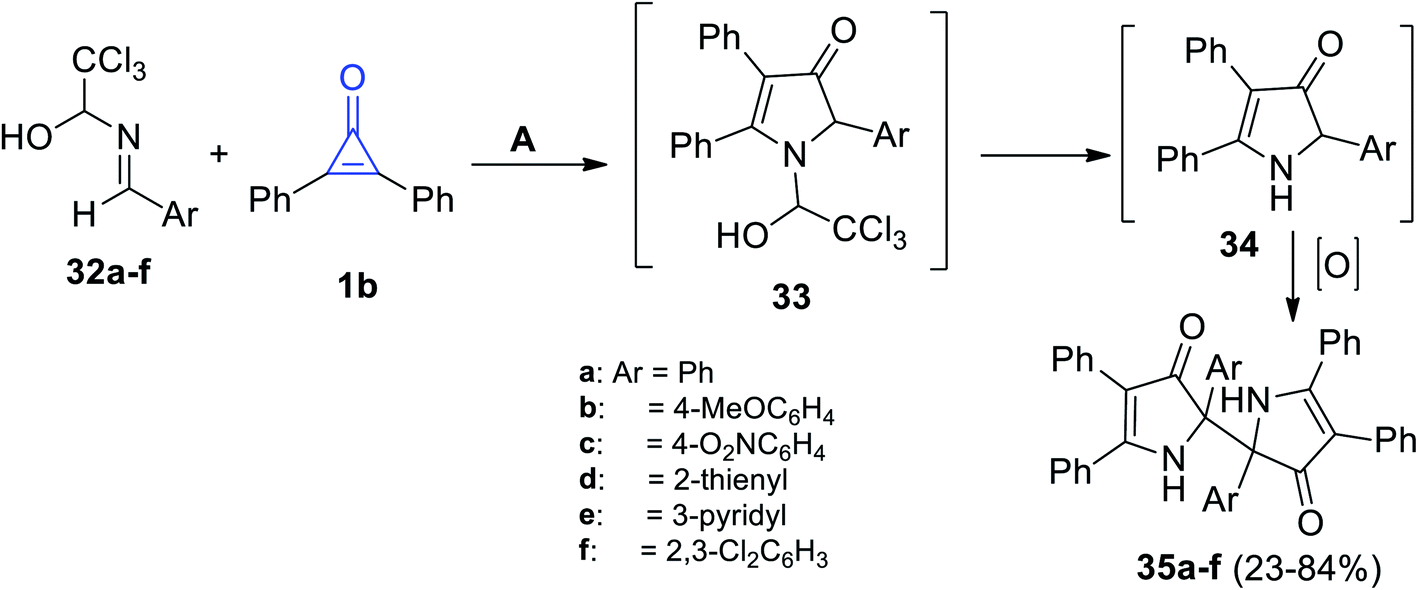
When 1-arylideneamino-2,2,2-trichloroethanols 32a–f were subjected to 1b at room temperature in methanol, the reaction produced 2,2′-diaryl-4,4′,5,5′-tetraphenyl-1,1′,2,2′-tetrahydro-3H,3′H-2,2′-bipyrrole-3,3′-diones 35a–f in 23–84% yield (Scheme 24), through the formation of intermediates 33 and 34, respectively.77
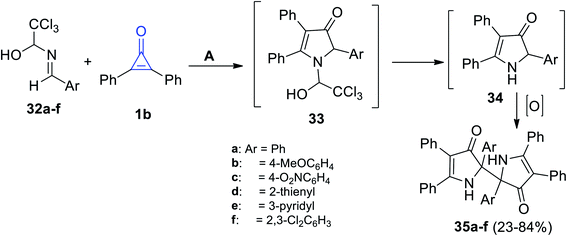 |
| | Scheme 24 Synthesis and mechanism describing the formation of compounds 35a–f. Reagents and conditions: A = MeOH, rt. | |
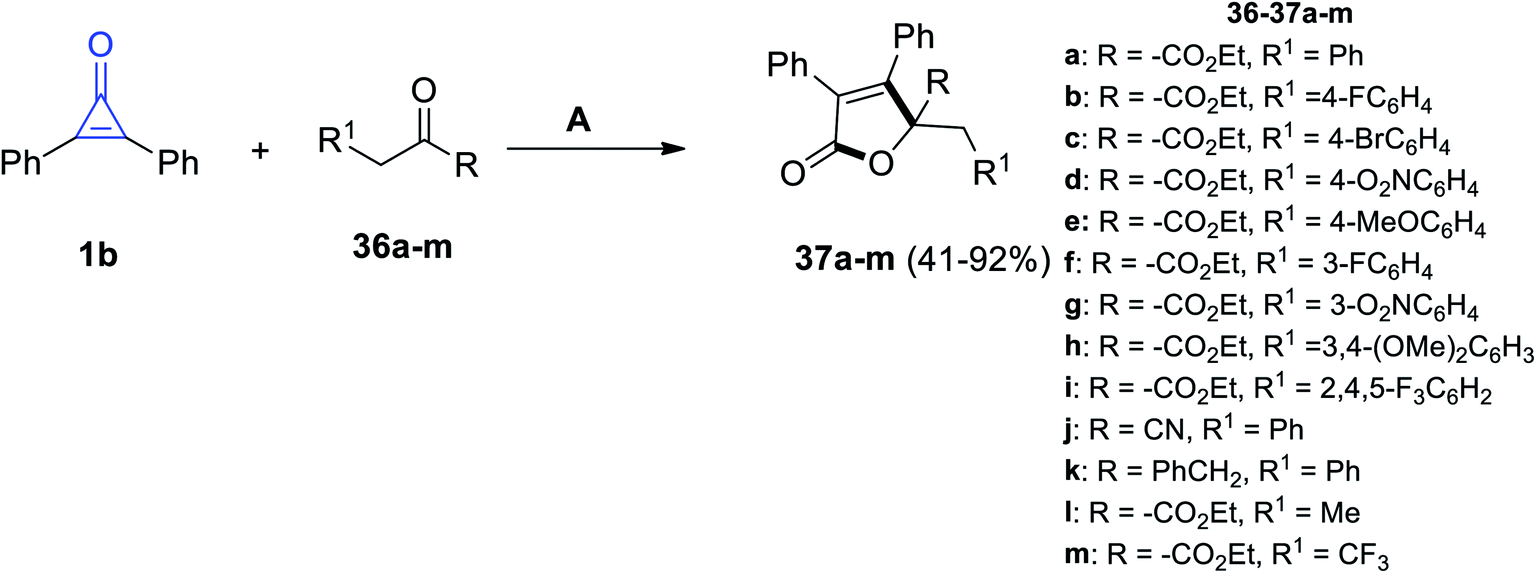
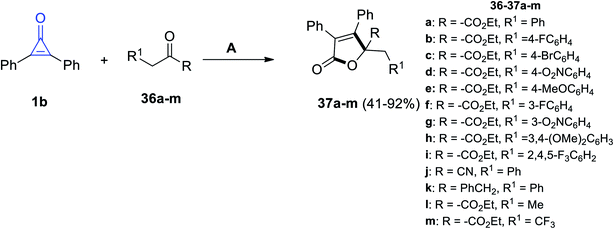 |
| | Scheme 25 Synthesis of substituted butanolides 37a–m. Reagents and conditions: A = DBU (20 mol%), DME, rt, 24 h. | |
Another organocatalyzed synthesis of substituted 2-furanones was achieved by Reitel et al. As the chiral compound 39 was used as a catalyst in the reaction between ethyl-3-oxo-3-phenyl-propaneperoxoate (38) and 1b to yield 5-((ethylperoxy)-methyl)-3,4,5-triphenylfuran-2(5H)-one (40) in 60% yield (Scheme 26).79
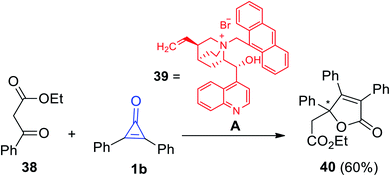 |
| | Scheme 26 Synthesis of 5-((ethylperoxy)-methyl)-3,4,5-triphenylfuran-2(5H)-one (40). Reagents and conditions: A = CH2Cl2, 50% aq KOH (1 equiv.), rt, 1 h, 39 (20 mol%). | |
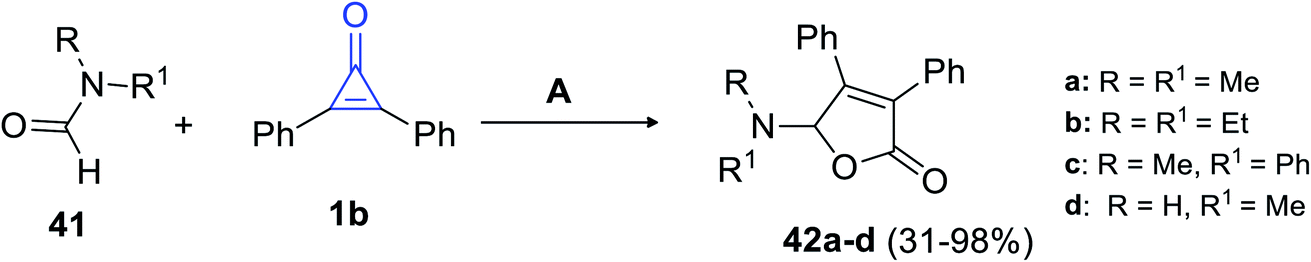
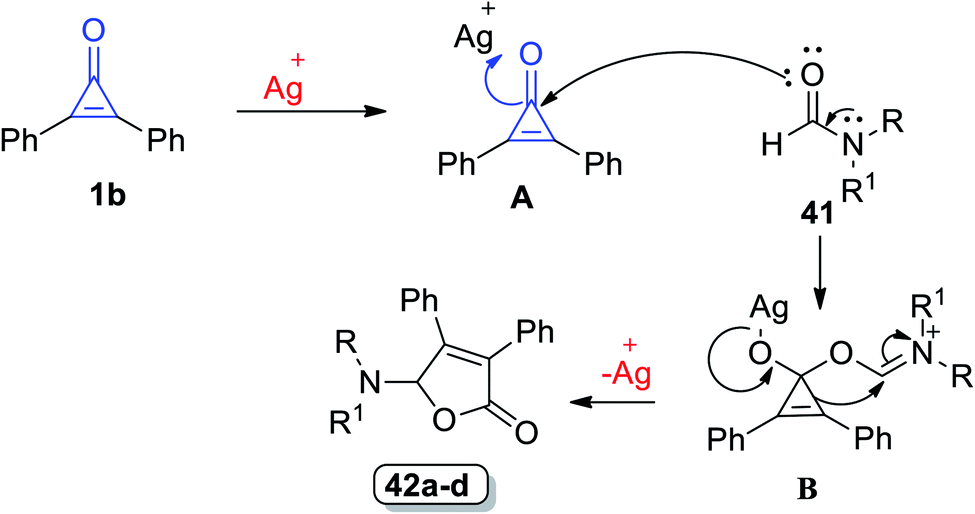
In 2018, Matsuda et al. succeeded in synthesizing trisubstituted 2-furanones by using another convenient method. The reaction between formamides 41 and 1b (10–20 equiv.) was performed for refluxing chlorobenzene, which was catalyzed with silver trifluoromethane-sulfonate (AgTOf) to give diphenylfuranone derivatives 42a–d in 31–98% yield (Scheme 27).80
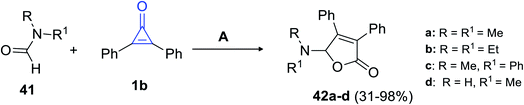 |
| | Scheme 27 Synthesis of substituted diphenylfuranones 42a–d. Reagents and conditions: A = AgTOf (10 mol%), PhCl, 130 °C, 2 h. | |
The suggested mechanism is illustrated as follows: diphenylcyclopropenone underwent ring-opening [3 + 2] annulation and the corresponding formamides 42 were formed (Scheme 28). The carbonyl oxygen atom of 1 was coordinated to Ag+ to form A, C-1 atom of A was attacked by the carbonyl oxygen atom of 41 and then the intermediate B was obtained. The final step afforded the furane ring via ring expansion and regenerated Ag+.80
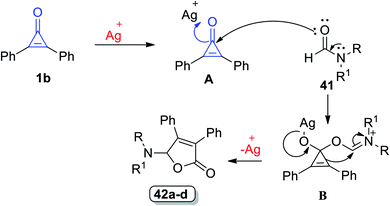 |
| | Scheme 28 Mechanism of the formation of diphenylfuranones 42a–d. Reagents and conditions: A = AgTOf (10 mol%), C6H5Cl, 130 °C, 2 h. | |
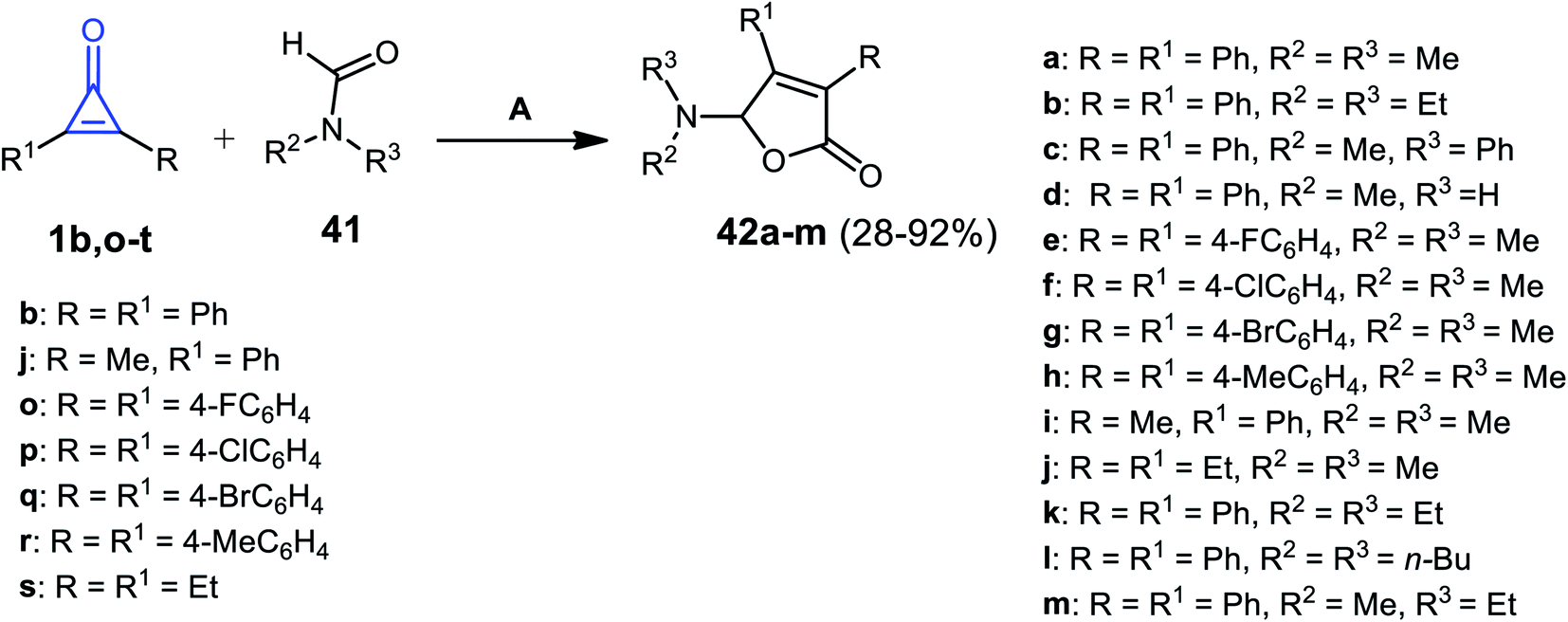
Ren et al. in 2018, used silver catalysis to afford other furanone derivatives 42a–m in 28–92% yield, via the reaction between cyclopropenone derivatives 1b,j,o–s and formamides 41 using silver hexafluoroantimonate(V) AgSbF6 at 80 °C (Scheme 29).81
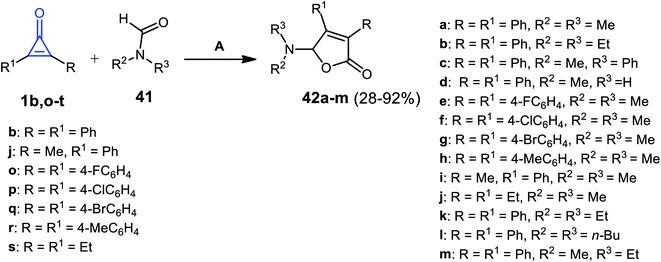 |
| | Scheme 29 Synthesis of compound 42a–m. Reagent and conditions: A = AgSbF6 (10 mol%), 80 °C, 20 h. | |
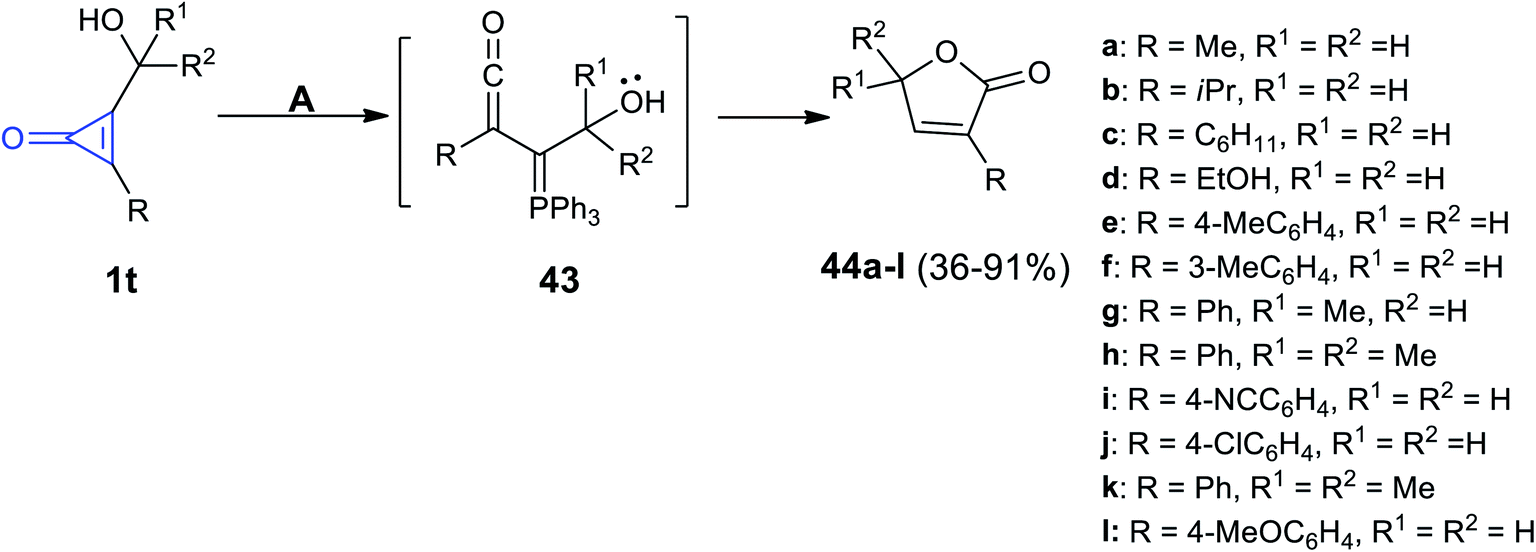
Triphenylphosphine (TPP) mediated the reaction developed by Nguyen et al. The reaction occurred between the substituted cyclopropenones 1t in methanol and TPP at 23 °C to yield butenolides 44a–l in 36–91% yield via the formation of triphenylphosphine ylidene intermediate 43 (Scheme 30).71
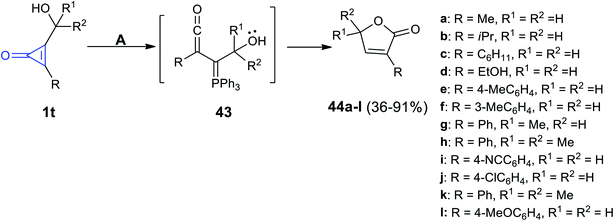 |
| | Scheme 30 TPP mediated the synthesis of 44a–l. Reagents and conditions: A = PPh3 (5 mol%), MeOH, 23 °C. | |
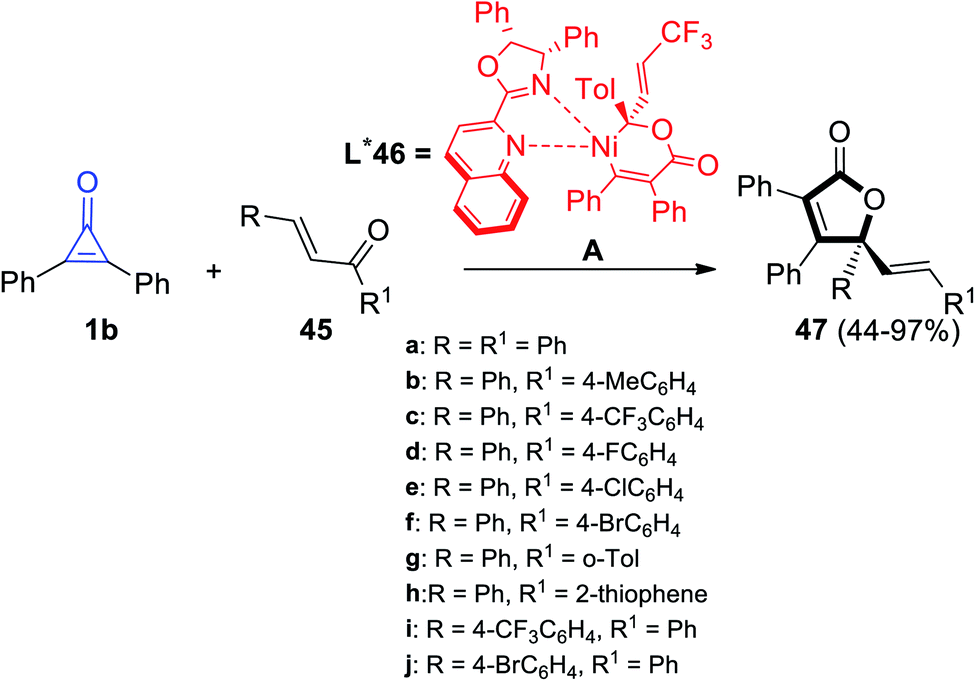
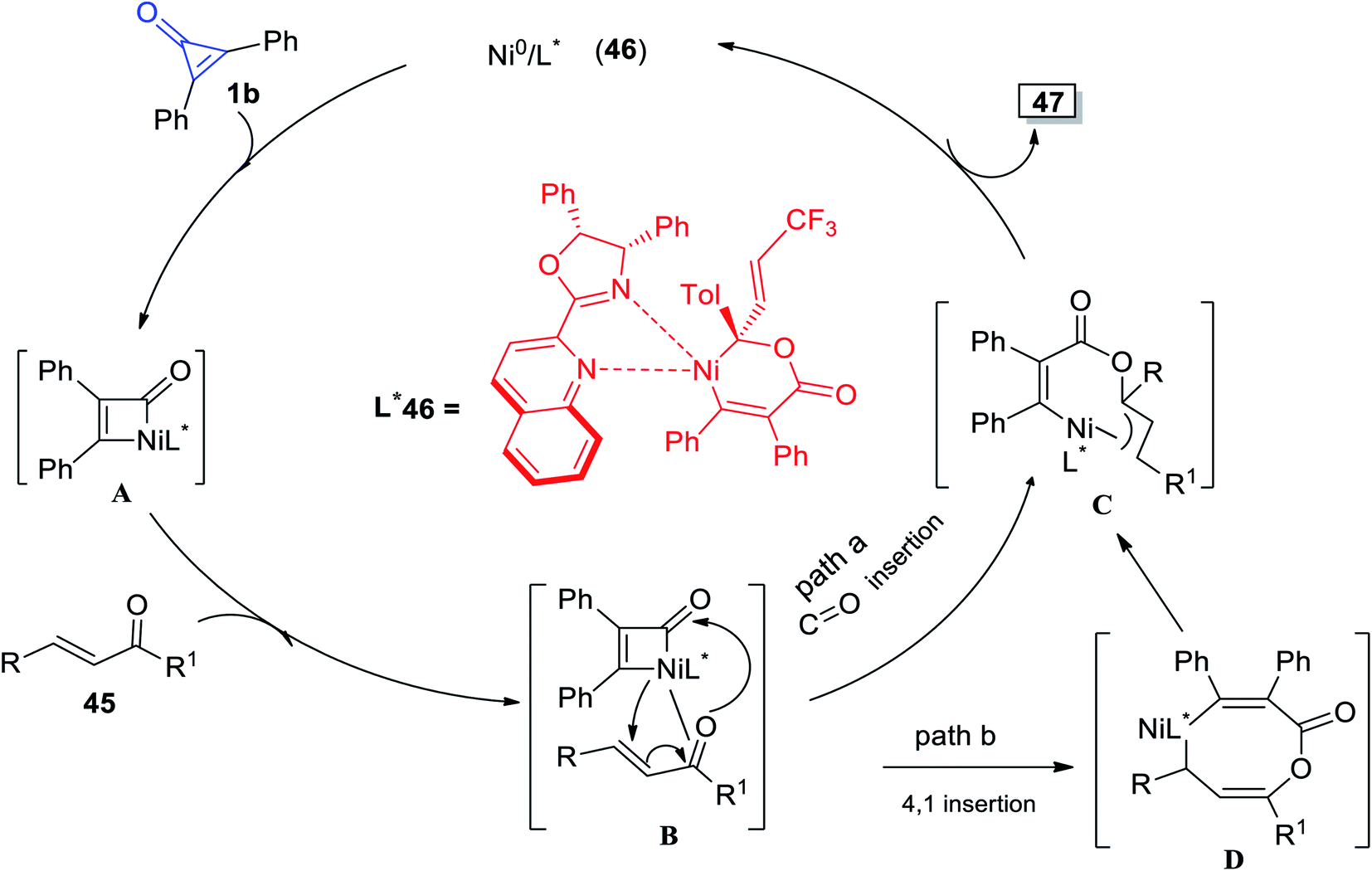
γ-Alkenylbutenolide 47 was synthesized by Bai et al. via cycloaddition reaction between 1b and enones 45 at room temperature in the presence of Ni complex 46 as an organic catalyst (Scheme 31).82
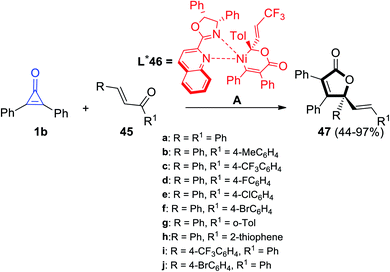 |
| | Scheme 31 Synthesis of γ-alkenylbutenolide 47 from enones 45 and 1b. Reagents and conditions: A = Ni(cod)2 (1–2 mol%), L* 46 (2–3 mol%), toluene, rt, 1–24 h. | |
The plausible reaction mechanism was proposed in Scheme 32, as intermediate A was obtained by oxidative addition of 1b to Ni0. The intermediate A was then thought to migrate into the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond via intermediate B and enantioselective Ni–C(acyl) migratory insertion occurred (path a). The Ni2–allyl intermediate C that resulted reductively removed the final product 47, allowing the catalyst to be regenerated. Alternatively, or even more probable, the intermediate B may go through a concerted 4,1-insertion of the Ni–acyl into the enone, yielding an 8-membered nickelacycle D that was a direct predecessor of C (path b). The olefin unit is involved in both pathways to allow for allyl stabilization (Scheme 32).82
O bond via intermediate B and enantioselective Ni–C(acyl) migratory insertion occurred (path a). The Ni2–allyl intermediate C that resulted reductively removed the final product 47, allowing the catalyst to be regenerated. Alternatively, or even more probable, the intermediate B may go through a concerted 4,1-insertion of the Ni–acyl into the enone, yielding an 8-membered nickelacycle D that was a direct predecessor of C (path b). The olefin unit is involved in both pathways to allow for allyl stabilization (Scheme 32).82
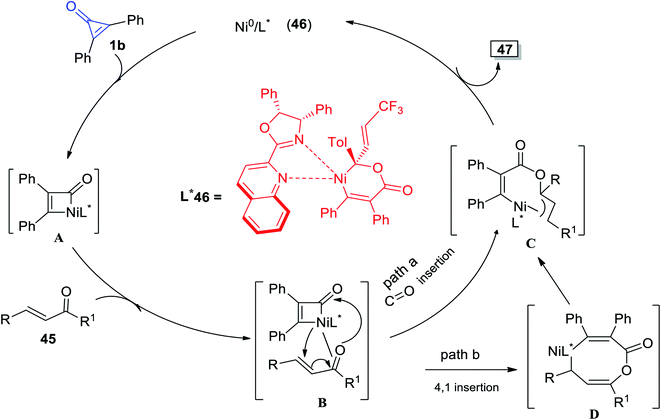 |
| | Scheme 32 Mechanism describing the synthesis of γ-alkenylbutenolide 47. | |
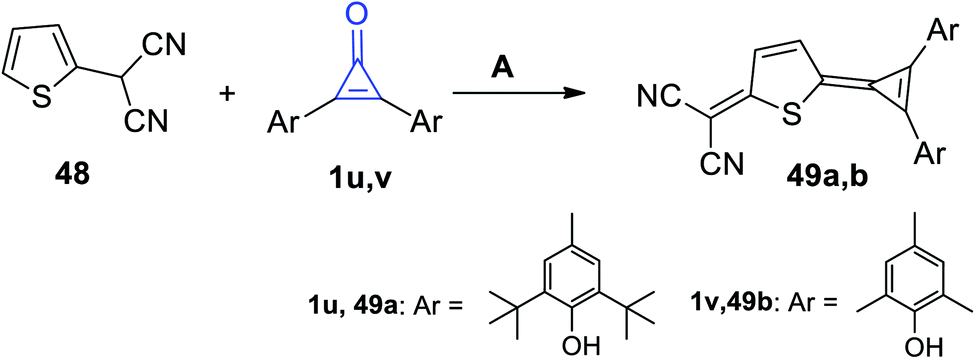
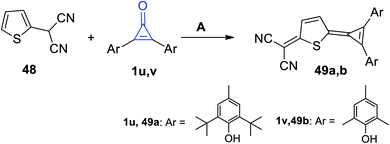 |
| | Scheme 33 Reaction of 2-(thiophen-2-yl)malononitrile (48) with diarylcyclopropenones 1u,v. Reagents and conditions: A = Ac2O, reflux. | |
Matsuda and Sakurai used gold catalysis in their reaction between 4-methyl-N-(3-methylbut-2-en-1-yl)-N-(prop-2-yn-1-yl)benzene-sulfonamide (50) and cyclopropenone 1j,s,w,x. The former reaction was carried out in DCM, at room temperature, and in the presence of (IPr)AuNTf2 to give the spiro compounds 51a–d in 85–97% yield, as shown in Scheme 34.84
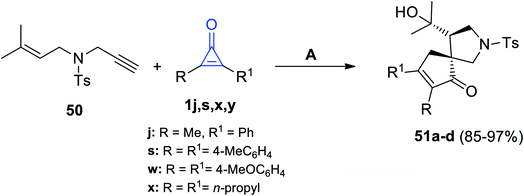 |
| | Scheme 34 (IPr)AuNTf2-catalyzed synthesis of compounds 51a–d. Reagents and conditions: A = (IPr)AuNTf2 (2 mol), DCM, rt. | |
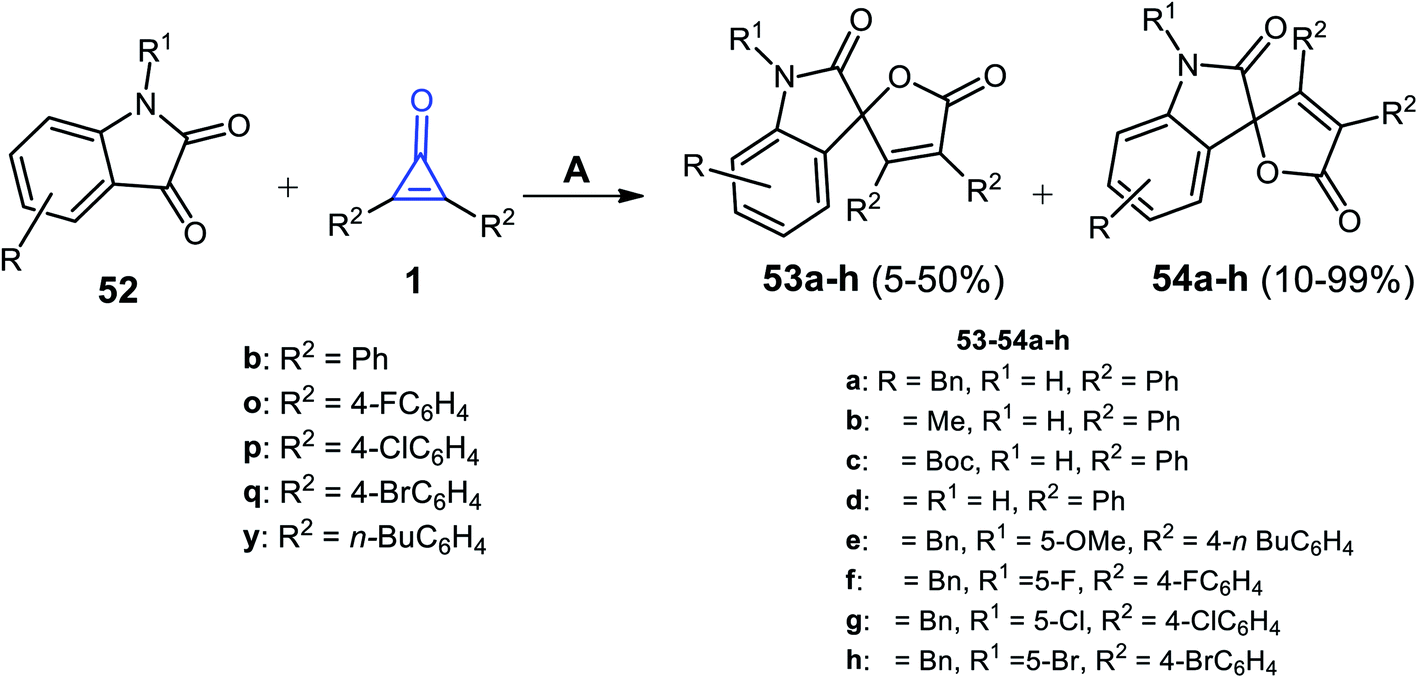
Xu et al. succeeded to synthesize the spiro heterocyclic compounds 53 and 54 from the reaction between cyclopropenone derivatives 1b,o,p,q,y and isatines 52 (Scheme 35). The reaction was carried out in toluene and 4-dimethylaminopyridine (DMAP) as a catalyst at 50 °C. The two isomers of spiro furano-indolinone 53a–h and 54a–h were formed in 5–50% and 10–99% yield, respectively.85
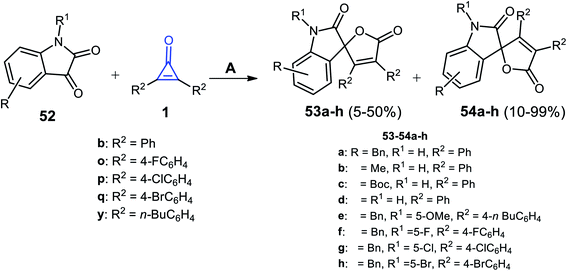 |
| | Scheme 35 Synthesis of spiro furano-indolinone 53a–h and 54a–h. Reagents and conditions: A = DMAP (20 mol%), toluene, 50 °C, 6 h. | |
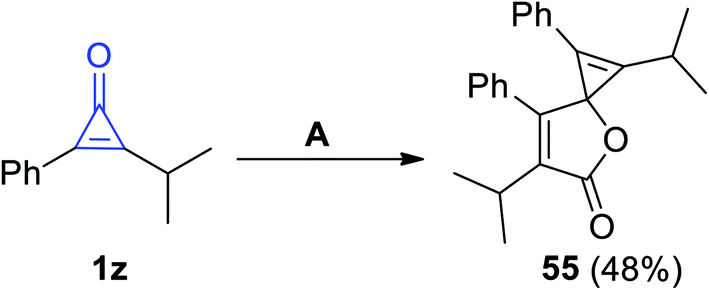
Previously, Cunha and Rocha had reported that 1,6-diisopropyl-2,7-diphenyl-4-oxaspiro-[2.4]hepta-1,6-dien-5-one (55) was synthesized in 48% yield, via refluxing 2-isopropyl-3-phenyl-cycloprop-2-en-1-one (1z) in dioxane in the presence of copper chloride (CuCl) for 12 h, as shown in Scheme 36.86
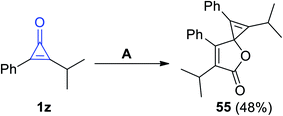 |
| | Scheme 36 Synthesis of the spiro compound 55. Reagents and conditions: A = dioxane, CuCl, 12 h. | |
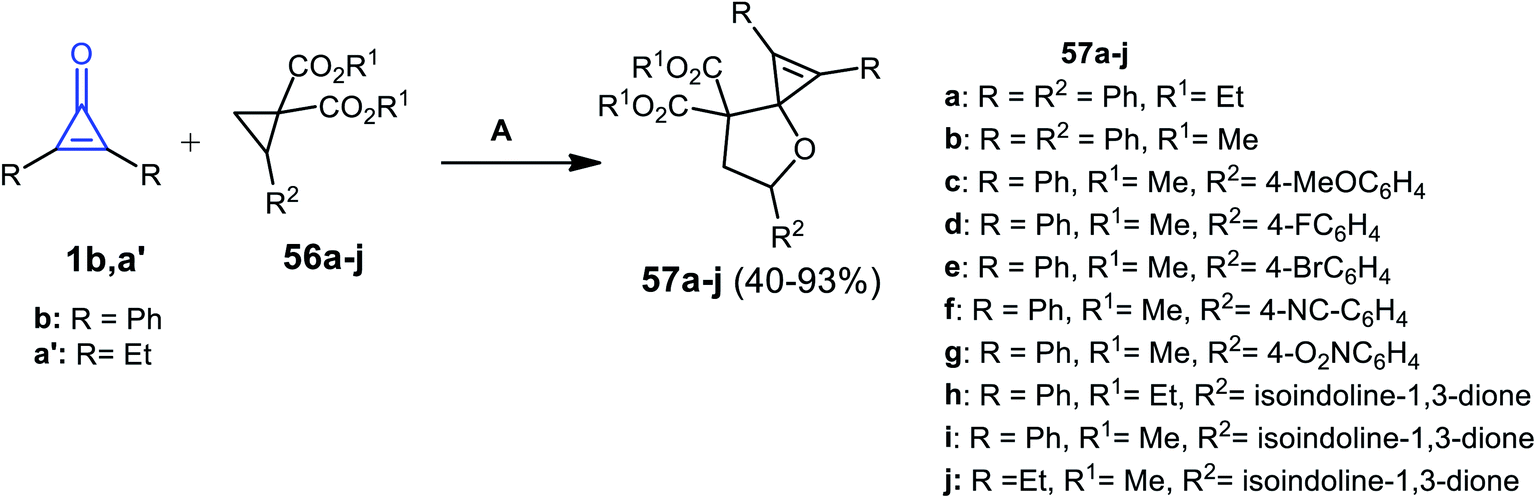
In 2014, Rivero et al. have reported that the reaction between substituted cyclopropenones 1b,a′ and trisubstituted cyclopropanes 56a–j in DCM catalyzed by scandium(III) trifluoromethane-sulfonate Sc(OTf)3 for 4 h gave the spiro heterocyclic compounds 57a–j (Scheme 37).87
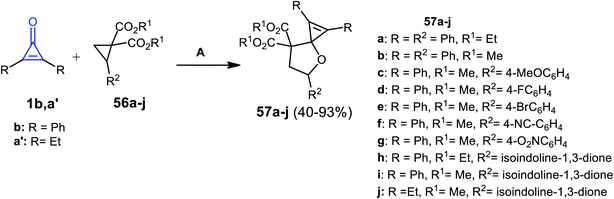 |
| | Scheme 37 Sc(OTf)3 mediated the synthesis of 57a–j. Reagents and conditions: A = Sc(OTf)3 (10 mol%), DCM, 25 °C 4 h. | |
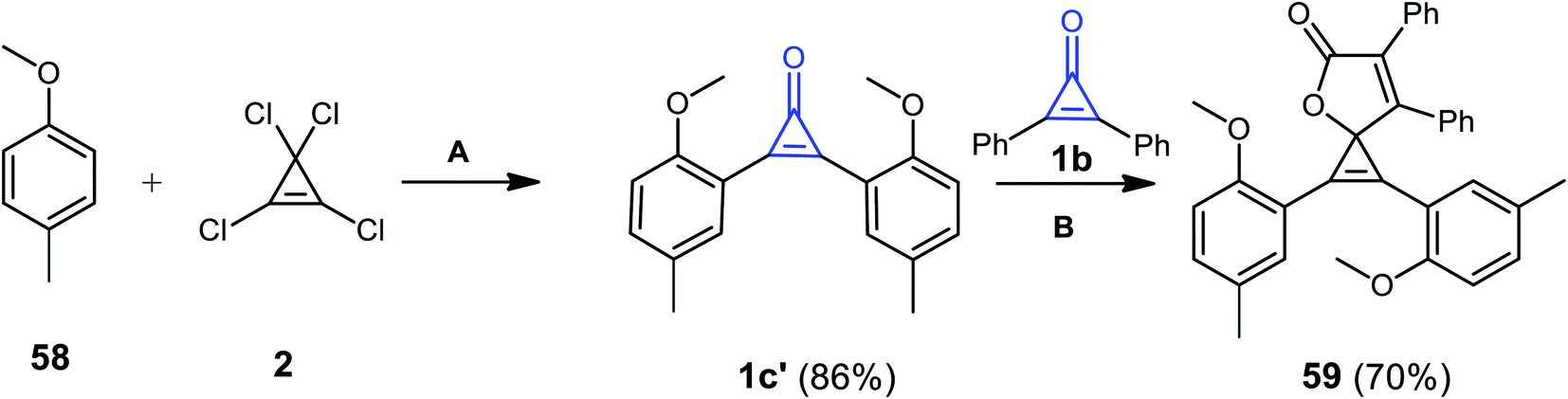
1,2-Bis(2-methoxy-5-methylphenyl)-6,7-diphenyl-4-oxaspiro[2.4]hepta-1,6-dien-5-one (59) was obtained in 70% yield via the reaction of two derivatives of cyclopropenones 1b and 1b′ together with compound 2 in the presence of CuBr as a catalyst. One of these cyclopropenones was synthesized from the reaction of 58 with 2 in AlCl3/CH2Cl2 at −20 °C (Scheme 38).88 The reaction was due to the formation of 1c′ (Scheme 38).88
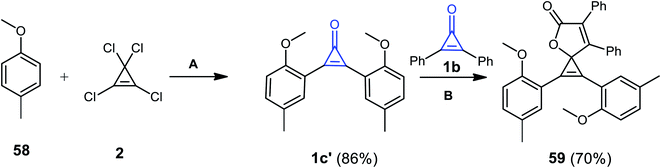 |
| | Scheme 38 Synthesis of 1,2-bis(2-methoxy-5-methylphenyl)-6,7-diphenyl-4-oxaspiro[2.4]hepta-1,6-dien-5-one (59). Reagents and conditions: A = (i) AlCl3, CH2Cl2, −20 °C, 45 min; (ii) H2O; B = CuBr, CH2Cl2, 70 °C, 12 h. | |
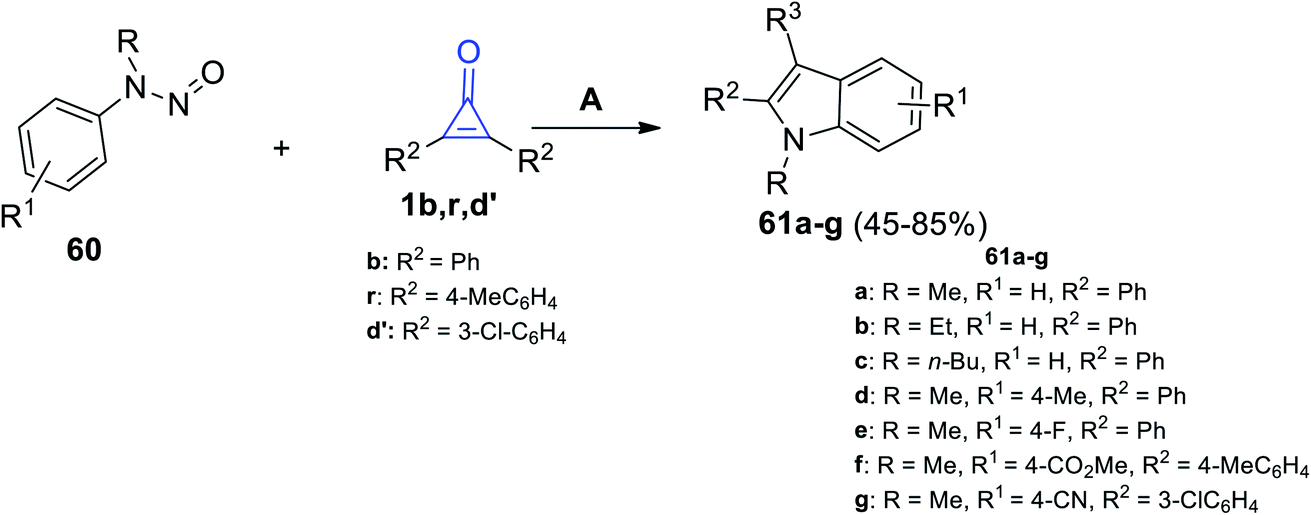
Then, we proceeded to the formation of fused compounds such as indoles 61a–g, which were obtained in 45–85% yield, via the reaction between N-nitrosoanilines 60 and cyclopropenones 1b,r,d′ in the presence of [RhCp*(OAc)2] and AgNTf2 in DCE at 120 °C for 24 h (Scheme 39).89
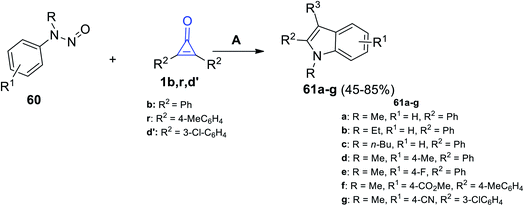 |
| | Scheme 39 Synthesis of indoles 61a–g. Reagents and conditions: A = [RhCp*(OAc)2], AgNTf2, DCE, 120 °C, 24 h. | |
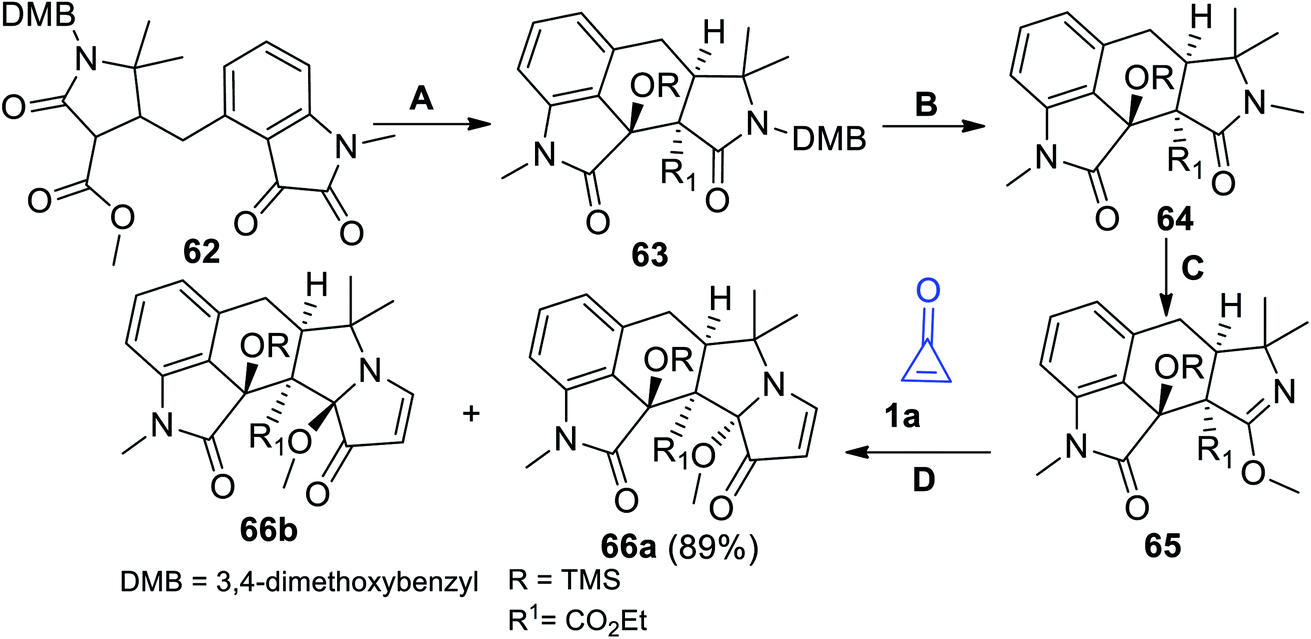
The interesting approach to prepare compounds containing indole moieties is outlined in Scheme 40. The strategy started with treatment compound 62 with trimethylsilyl trifluoromethanesulfonate (TMS-OTf), Et3N and TiCl4 (method A). Oxidation of 63 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in CHCl3 (method B) afforded compound 64. Rearrangement of 64 using methyl trifilate (MeOTf) (method C) gave the corresponding compound 65 (Scheme 40).90 Upon heating 65 with cyclopropenone (1a) in MeCN (method D), the reaction gave the target products 66a in 89% yield together with its diastereomer 66b (Scheme 40). The two diastereomers 66a,b were used as precursors in the synthesis of (±) Aspergilline A.90
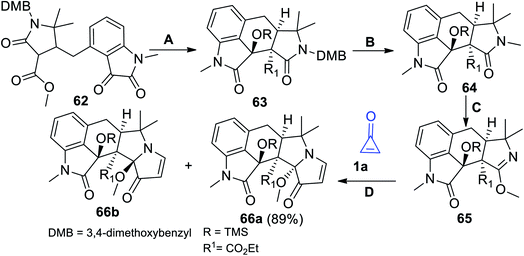 |
| | Scheme 40 Synthesis of compounds 66a,b. Reagents and conditions: A = TMS-OTf, Et3N, DCM, TiCl4, 0–35 °C; B = CHCl3, DDQ, H2O, 70 °C; C = MeOTf, CH2Cl2; D = MeCN, 50 °C. | |
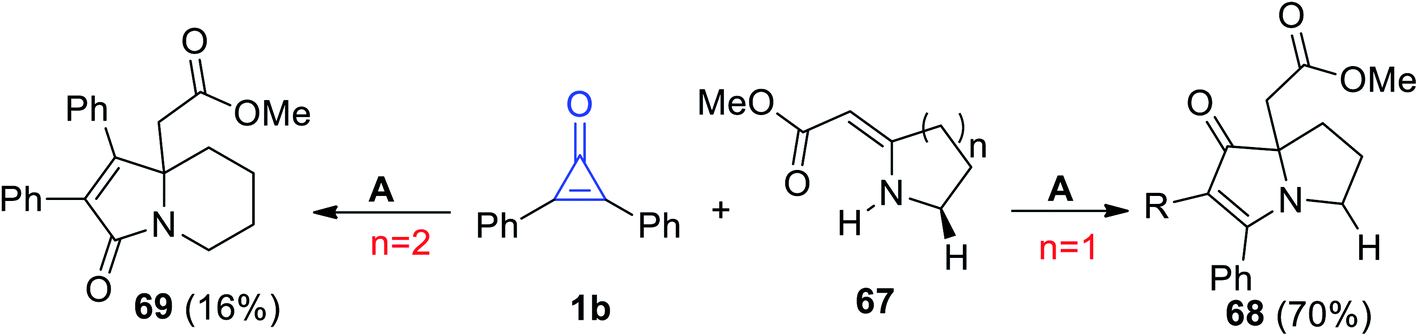
Cunha et al. reported on a direct path to obtain pyrrolizidine 68 and indolizidine 69 by the reaction of 67 with 1b in refluxing toluene (Scheme 41).91
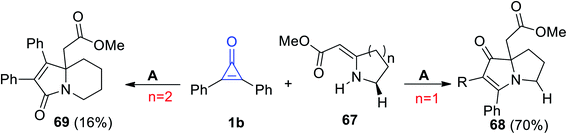 |
| | Scheme 41 Synthesis of pyrrolizidine 68 and indolizidine 69. Reagents and conditions: A = toluene, reflux. | |
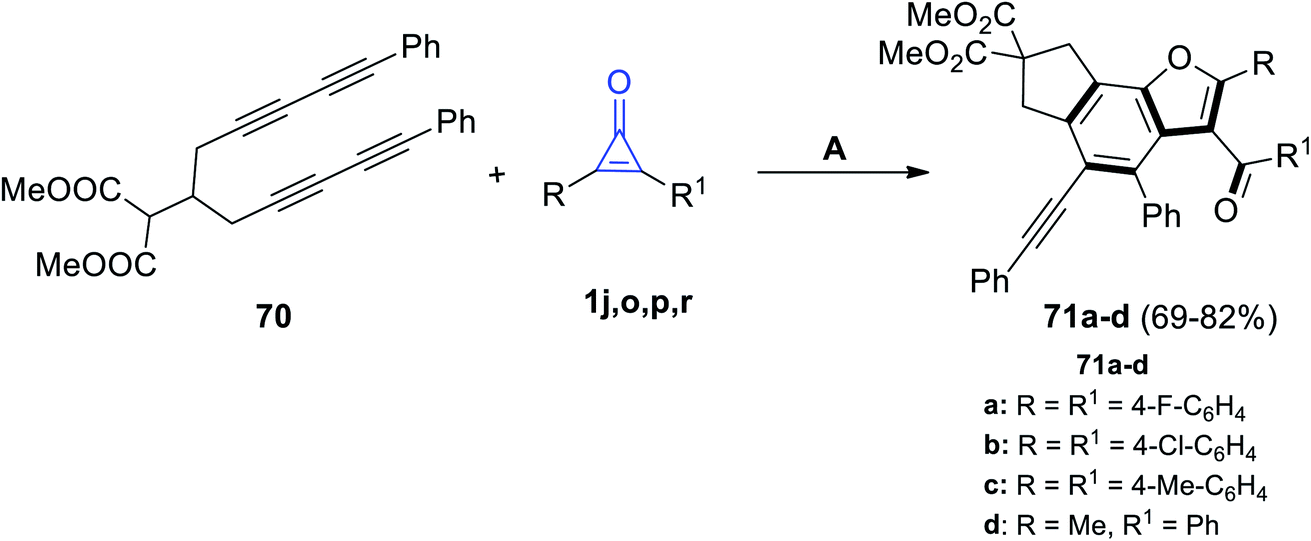
Recently, in 2021, Yao et al. reacted dimethyl 2-(1,11-diphenylundeca-1,3,8,10-tetrayn-6-yl)malonate (70) with cyclopropenone derivatives 1j,o,p,r under the stream of O2 to give the benzo[b]furan derivatives 71a–d in 69–82% yield, as shown in Scheme 42.92
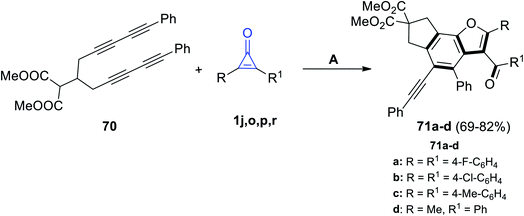 |
| | Scheme 42 Synthesis of benzo[b]furane derivatives 71a–d. Reagents and conditions: A = O2, MeCN, reflux, 12 h. | |
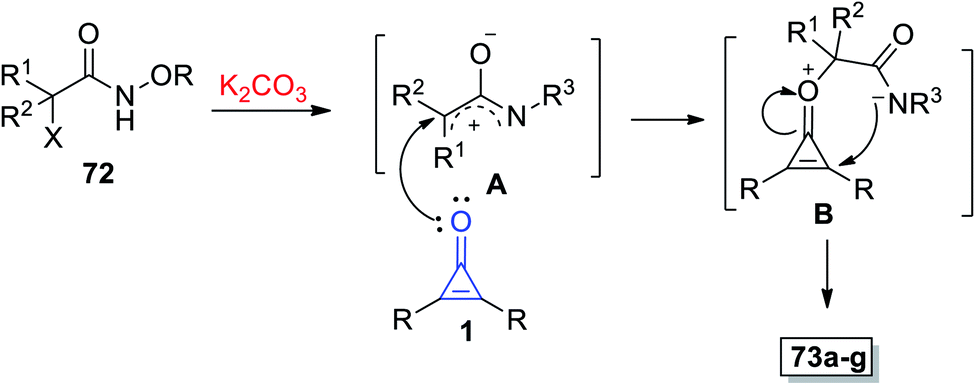
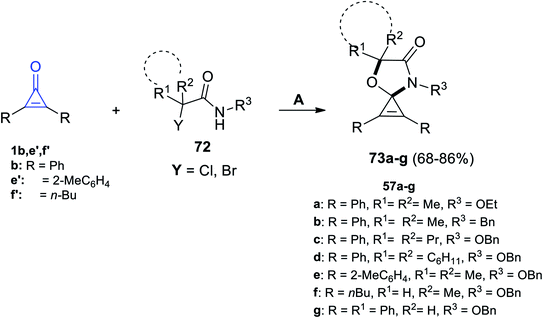 |
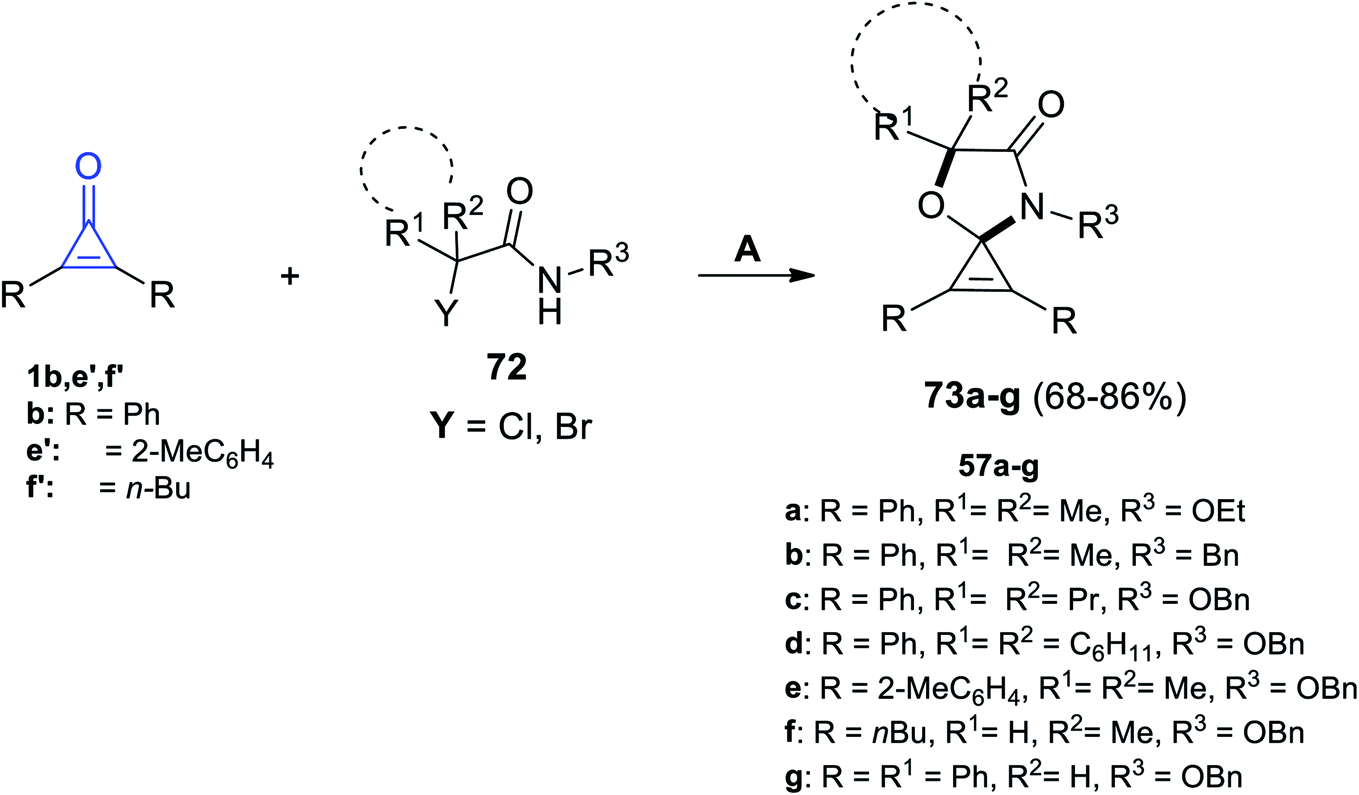
| | Scheme 43 Synthesis of spiro oxazoles 73a–g. Reagents and conditions: A = K2CO3 (0.8 mmol), HFIP (2 ml), 50 °C, 12 h. | |
The mechanism described the formation of 73a–g as an initiation step using K2CO3, and 72 was converted in situ into azaoxyallyl cation intermediate A. Thereafter, the azaoxyallyl cation A gave the zwitterionic intermediate B after addition of carbonyl oxygen in the cyclopropenone 1. Finally, the spirocyclic oxazoles 73 were obtained by intramolecular nucleophilic addition of B, as illustrated in Scheme 44.93
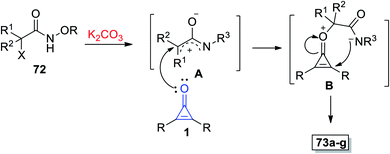 |
| | Scheme 44 Mechanism illustrating the formation of spiro oxazoles 73a–g. | |
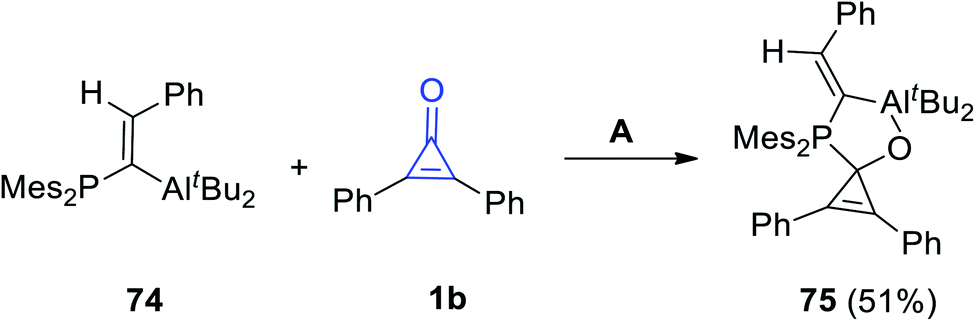
When (E)-di-t-butyl(1-(dimesitylphosphino)-2-phenylvinyl)aluminum (74) reacted with 1b in n-pentane at −40 °C, the spiro product 75 was produced in 51% yield (Scheme 45).94
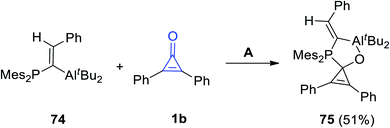 |
| | Scheme 45 Synthesis of spiro compound 75. Reagents and conditions: A = n pentane, −40 °C, overnight. | |
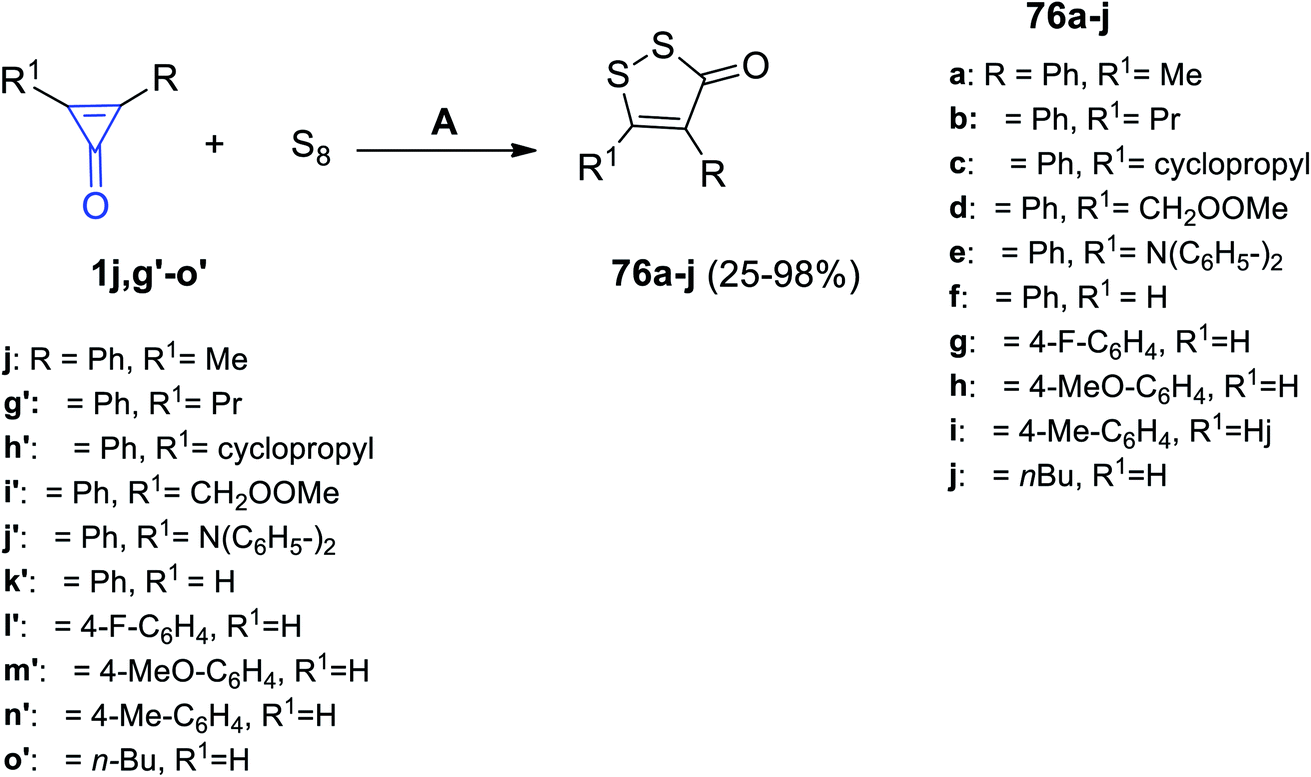
In 2020, Wu et al. had reported that the reaction between diaryl cyclopropenones 1j,g′–o′ and elemental sulfur in dimethylformamide (DMF) at room temperature for 12 h provided disubstituted dithiolone derivatives 76a–j in 25–98% yield, as shown in Scheme 46.95
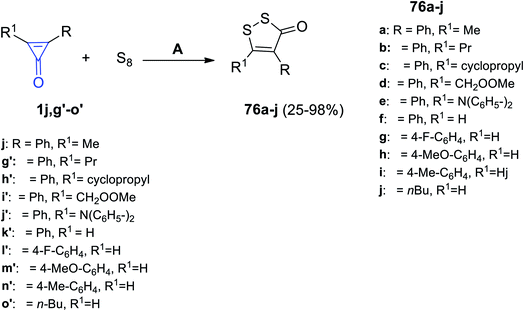 |
| | Scheme 46 Synthesis of dithiolone derivatives 76a–j. Reagents and conditions: A = KF, DMF, air, rt, 12 h. | |
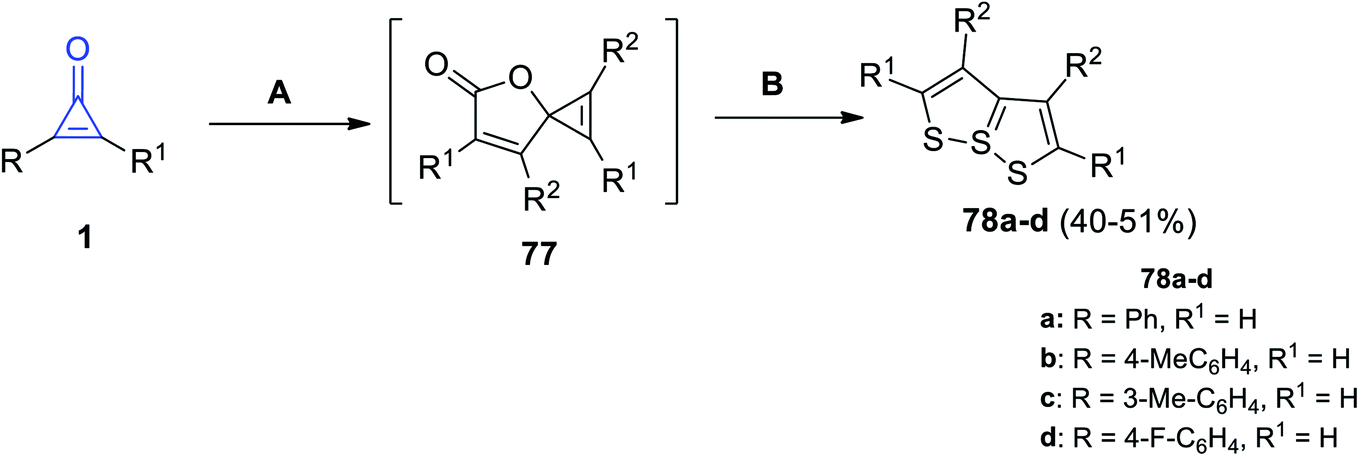
[1,2]Dithiolo[5,1-e][1,2]dithiole derivatives 78a–d were successfully synthesized in 40–51% yield, by refluxing two moles of cyclopropenones 1 in DCM at 70 °C in the presence of CuBr, the spironolactone intermediate 77 was formed and then reacted with elemental sulfur in DMF at 50 °C for 5 h to give the targeted compounds (Scheme 47).95
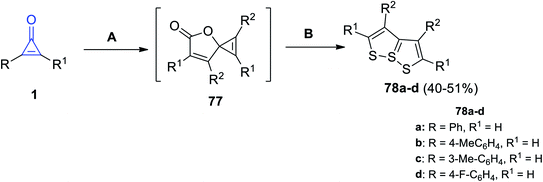 |
| | Scheme 47 Synthesis of [1,2]dithiolo[5,1-e][1,2]dithioles 78a–d. Reagents and conditions: A = CuBr (5 mol%), DCM, 75 °C, N2,12 h, B = KF (2 equiv.), S8, DMF, 50 °C, air, 5 h. | |
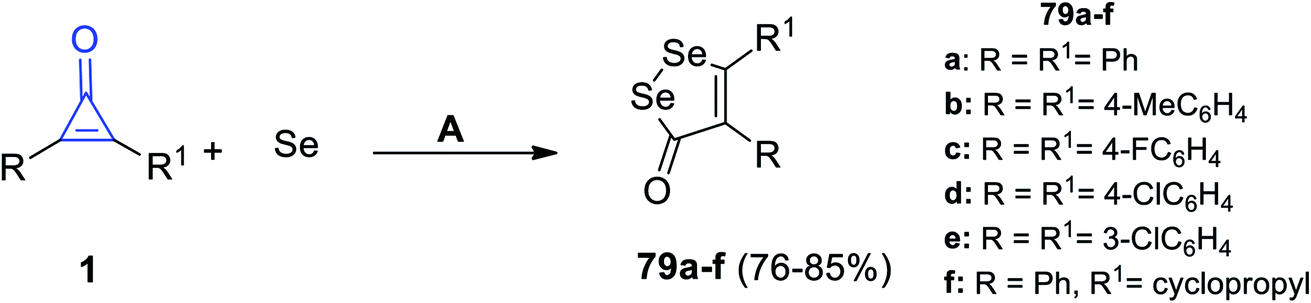
Similarly, Wu et al. also succeeded to obtain diselenolone derivatives 79a–f in 76–85% yield (Scheme 48), via the reaction between cyclopropenones 1 and elemental selenium in dimethylsulfoxide (DMSO) at 120 °C under N2 flow for 12 h.95
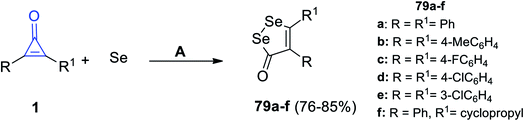 |
| | Scheme 48 Synthesis of diselenolone derivatives 79a–f. Reagents and conditions: A = DMSO, N2, 120 °C, 12 h. | |
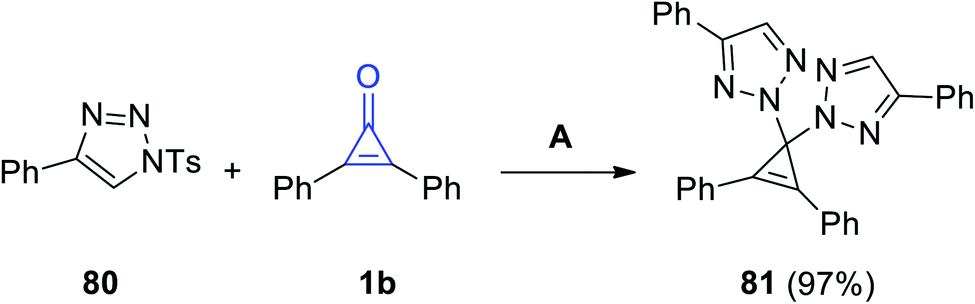
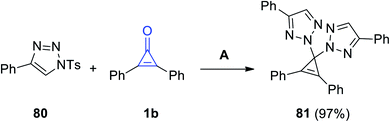 |
| | Scheme 49 Synthesis of 2,2′-(2,3-diphenylcycloprop-2-ene-1,1-diyl)bis(4-phenyl-2H-1,2,3-triazole) (81). Reagents and conditions: A = DCE, 80 °C, 2.5 h. | |
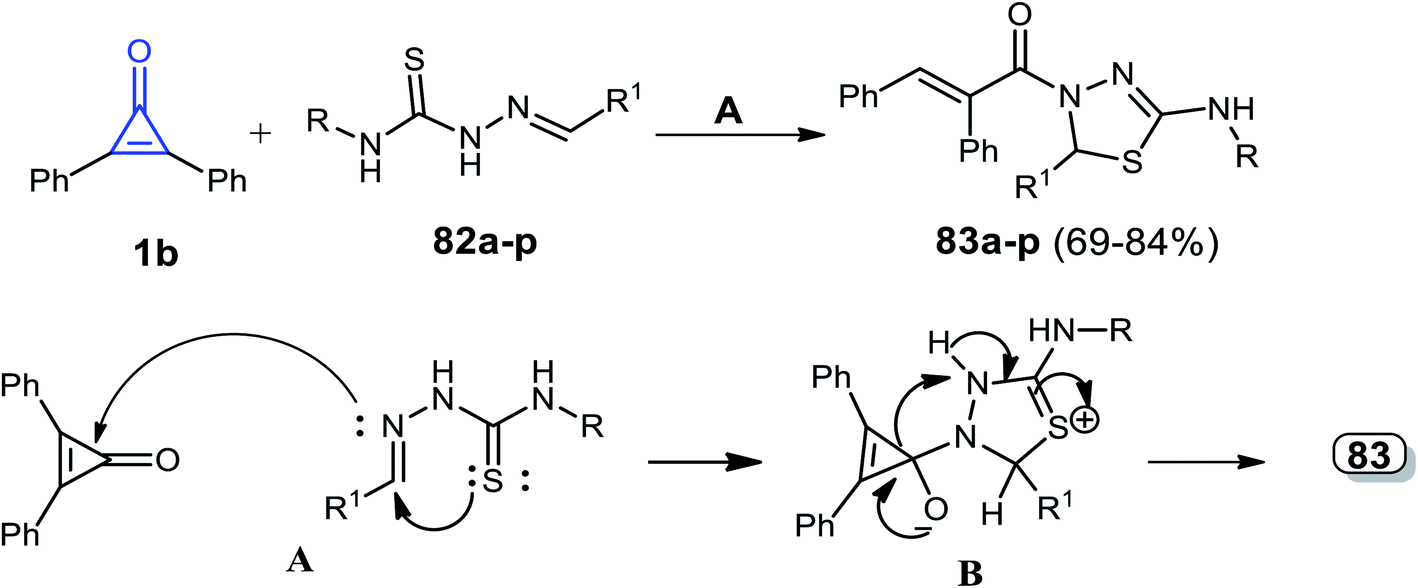
Hassan et al. reported that a series of thiadiazoles 83a–p were obtained in 69–84% yield, via a catalyst-free reaction between 1b and alkenylidene hydrazine carbothioamides 82a–p in dry ethanol.97
The reaction mechanism was explained by the attack of the azomethine (CHN) nitrogen atom of compound 82 to the carbonyl group of 1b. The mechanism of the reaction was explained by the formation of intermediate B via a spontaneous intramolecular nucleophilic attack of the sulfur atom lone pair on the CHN group. That was followed by cyclization, via the formation of intermediate B, which rearranged to the final product 83 (Scheme 50 and Table 1).97
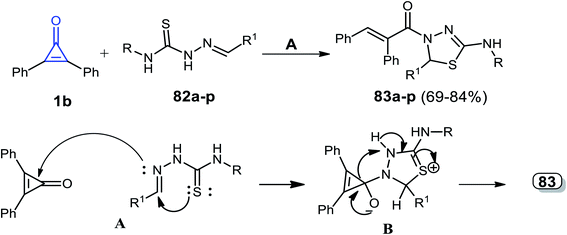 |
| | Scheme 50 Synthesis and mechanism of thiadiazoles 83a–p. Reagents and conditions: A = EtOH, 4–8 h. | |
Table 1 Substituents and the yield of thiadiazoles 83a–p
| |
R |
R1 |
Yield (%) |
| a |
Ph– |
C6H5–CHCH (E) |
82 |
| b |
Bn– |
C6H5–CHCH (E) |
75 |
| c |
CH2CH–CH2– |
C6H5–CHCH (E) |
70 |
| d |
Ph– |
2- MeO–C6H4–CHCH (E) |
72 |
| e |
Bn– |
2- MeO–C6H4–CHCH (E) |
76 |
| f |
CH2CH–CH2– |
2- MeO–C6H4–CHCH (E) |
68 |
| g |
Ph– |
C6H5–CHC–Me2 (E) |
84 |
| h |
Ph– |
Me–(CH2)2–CHCH– (E) |
83 |
| i |
Bn– |
Me–(CH2)2–CHCH– (E) |
65 |
| j |
CH2CH–CH2– |
Me–(CH2)2–CHCH– (E) |
71 |
| k |
Ph– |
Me–CHCH– (E) |
75 |
| l |
Bn– |
Me–CHCH– (E) |
71 |
| m |
CH2CH–CH2– |
Me–CHCH– (E) |
67 |
| n |
Ph– |
Me2–CH– |
76 |
| o |
Bn– |
Me2–CH– |
74 |
| p |
CH2CH–CH2– |
Me2–CH– |
69 |
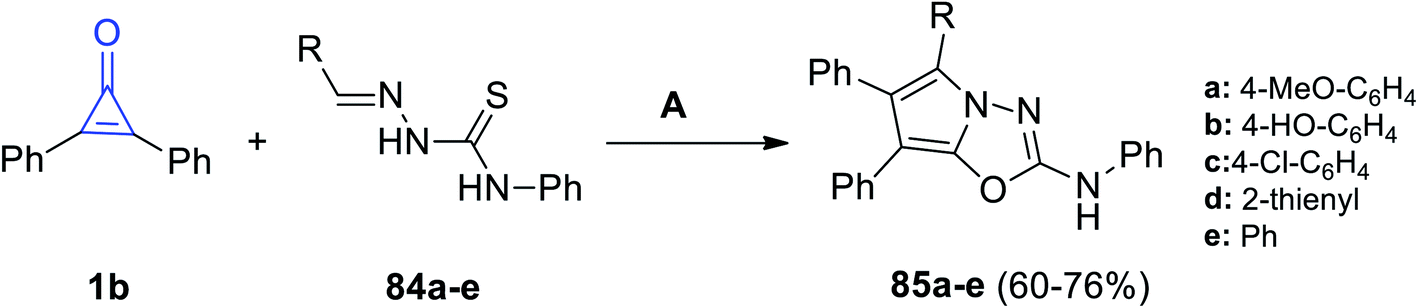
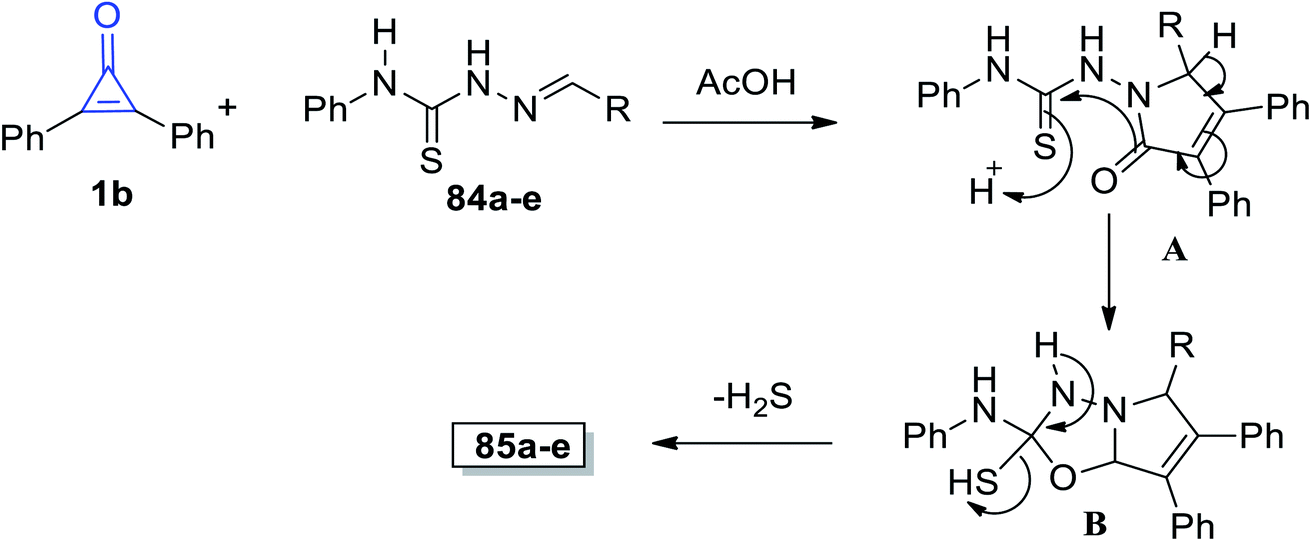
Aly et al. reported that the reaction mixture of 1b and ylidene-N-phenylhydrazine-carbothioamides 84a–e in glacial acetic acid at room temperature afforded 2,5,6,7-tetrasubstituted-pyrrolo[2,1-b](1,3,5-oxadiazolyl)-2-amines 85a–e in 60–76% yield (Scheme 51).98
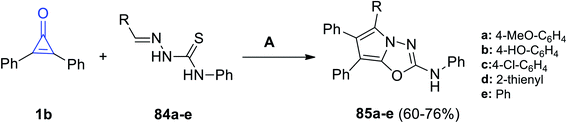 |
| | Scheme 51 Synthesis of pyrrolo[2,1-b](1,3,5-oxadiazolyl)-2-amines 85a–e. Reagents and conditions: A = AcOH, 4–8 h. | |
The suggested mechanism is explained as follows: the afforded product structures supported the formal [2 + 3] cycloaddition pathway proposed by Eicher to generate the adducts (A). This was followed by cyclization of intermediate (B) with aromatization of the pyrrole ring. Eventually, intermediate B lost a molecule of hydrogen sulfide and the final product 85 was formed, as shown in Scheme 52.98
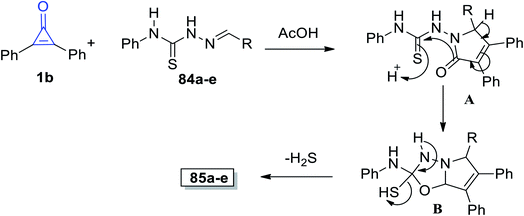 |
| | Scheme 52 Mechanism of the formation of pyrrolo[2,1-b](1,3,5-oxadiazolyl)-2-amines 85a–e. | |
2.3.3. Synthesis of six-membered rings.
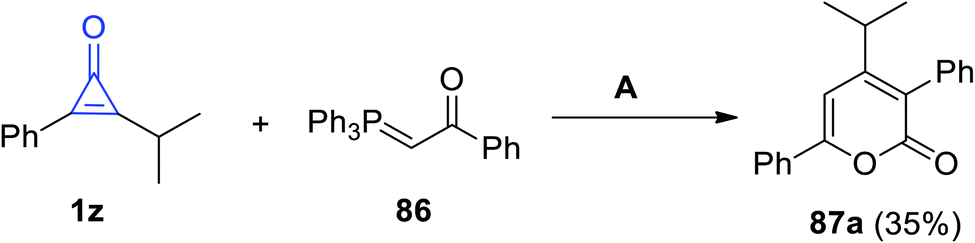
2.3.3.1. Six-membered rings with one heteroatom. On reacting 2-isopropyl-3-phenylcycloprop-2-en-1-one (1z) with 1-phenyl-2-(triphenyl-phosphoranylidene)ethanone (86) in benzene for 5 h, 4-isopropyl-3,6-diphenyl-2H-pyran-2-one (87a) was obtained in 35% yield (Scheme 53).86
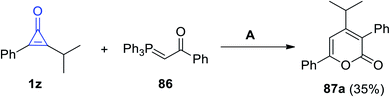 |
| | Scheme 53 Synthesis of 4-isopropyl-3,6-diphenyl-2H-pyran-2-one (87a). Reagents and conditions: A = benzene, reflux, 5 h. | |
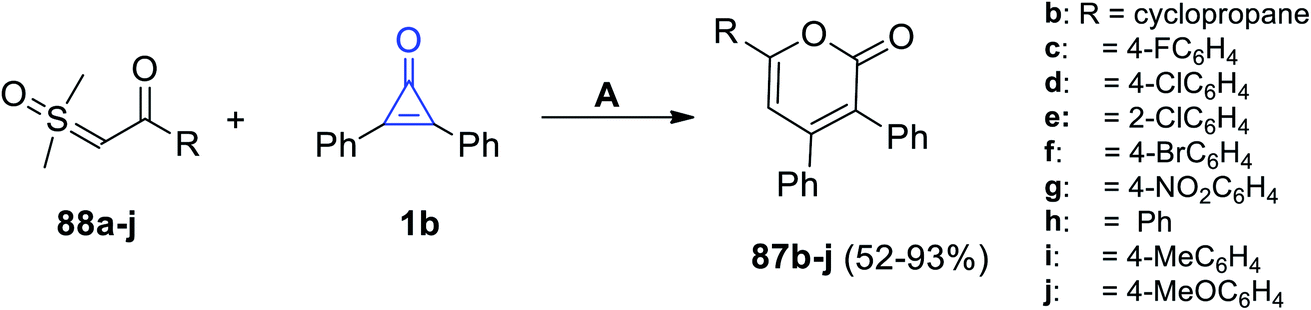
Furthermore, Zhou et al. reported on the synthesis of a series of 2-pyranone derivatives 87b–j in 52–93% yield, that was achieved by the reaction between β-ketosulfoxonium ylides 1b in MeCN at 100 °C for 12 h, and dichloro(pentamethyl-cyclopentadienyl)-rhodium(III) dimers (Cp*RhCl2)2 (Scheme 54).99
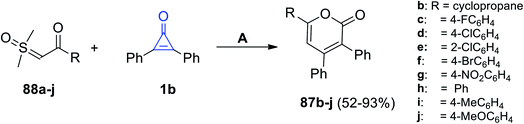 |
| | Scheme 54 Synthesis of 2-pyranone derivatives 87b–j. Reagents and conditions: A = (Cp*RhCl2)2, NaOAc, MeCN, 100 °C, 12 h. | |
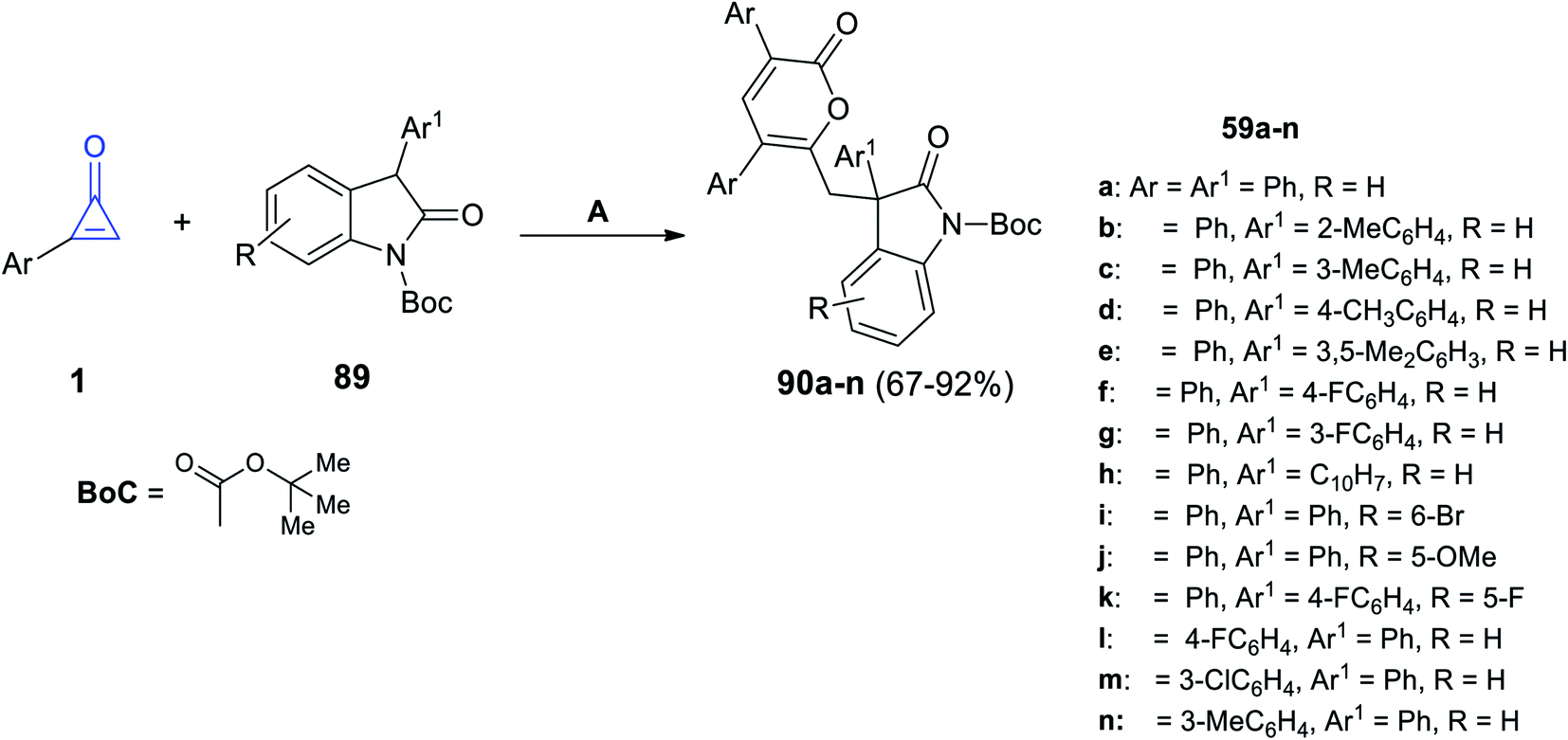
The reaction between 2-arylcycloprop-2-enones 1 and substituted 2-indolones 89 at 25 °C in acetonitrile for 1 h and DABCO was used as an organic catalyst to form compound 90a–n in 67–92% yield (Scheme 55).100
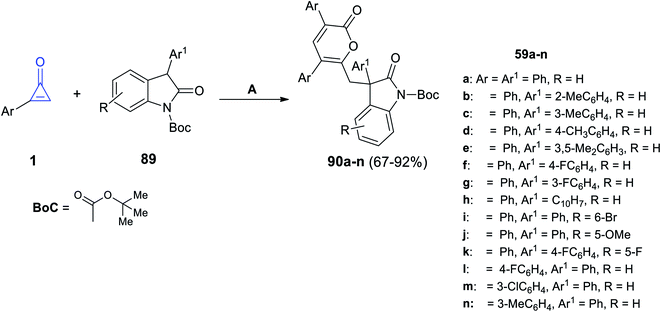 |
| | Scheme 55 Synthesis of compound 90a–n. Reagents and conditions: A = DABCO (20 mol%), MeCN, 25 °C, 1 h. | |
The mechanism was described as follows. First, DABCO subtracts one proton from two to form the nucleophile A, on the less sterically hindered side. Thereafter, 1,4-addition of the CC bond of 1 was initiated via nucleophile A, yielding the enolate intermediate B. The intermediate B was then added intermolecularly to another molecule of 1 to form intermediate C, which was converted to intermediate D by a concerted process after the ring-opening process and intramolecular nucleophilic addition; after that, it was passed through another ring-opening step to produce intermediate E, which after protonation gave the final product 90, and the catalyst was regenerated (Scheme 56).100
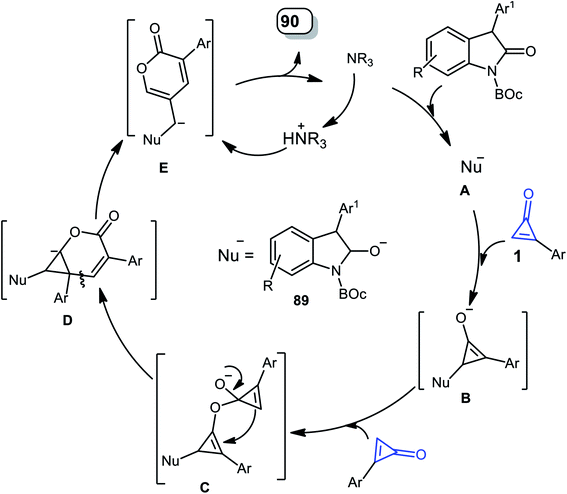 |
| | Scheme 56 Mechanism describing the formation of 90a–n. | |
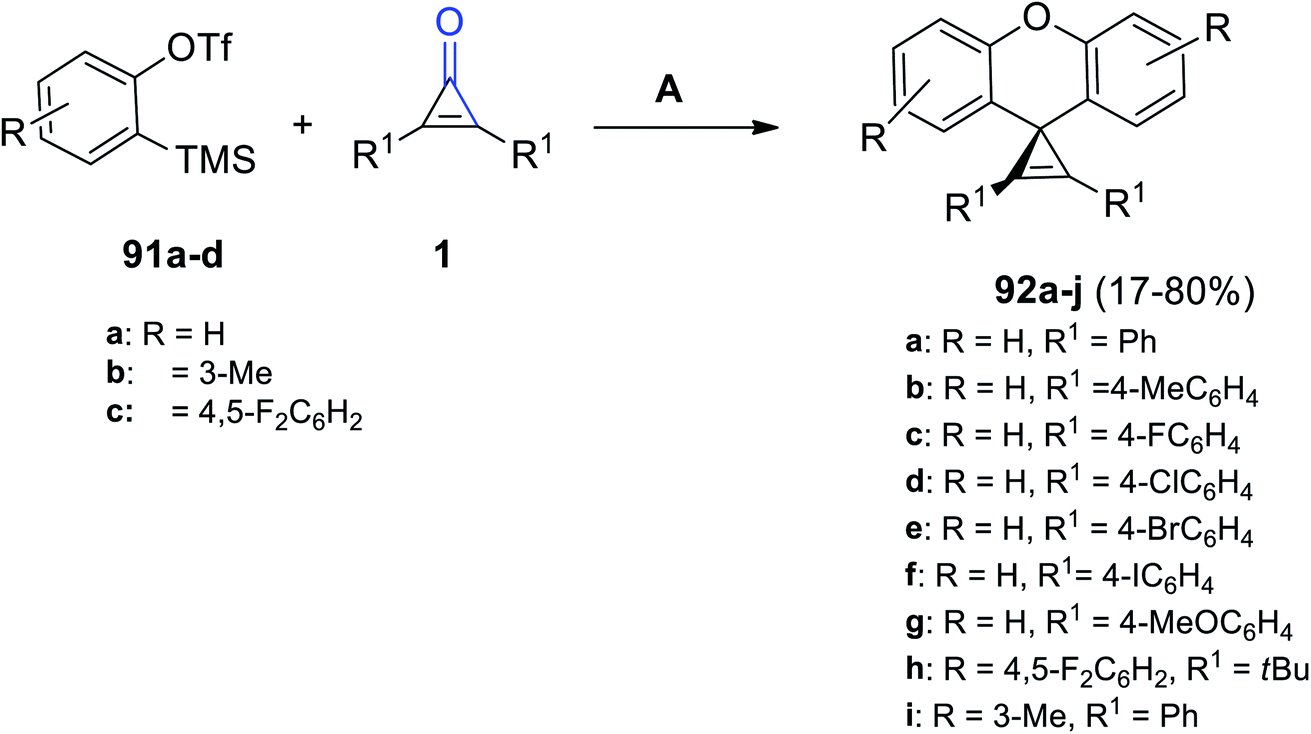
A series of substituted diaryl spiro[cycloprop[2]ene-1,9′-xanthene] derivatives 92a–j were successfully synthesized in 40–80% yield, via the reaction between compound 91a–d (2.5 equiv.) and diaryl cyclopropenones 1 (1 equiv.) (Scheme 57). The reaction was performed in MeCN using cesium fluoride (CsF) as a catalyst at 30 °C for 24 h.101
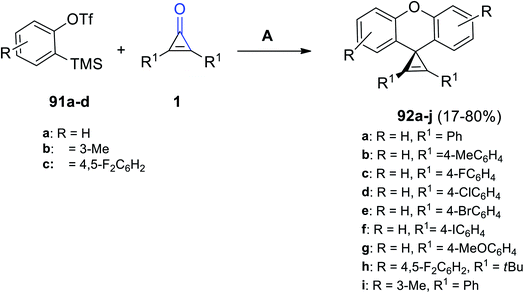 |
| | Scheme 57 Synthesis of substituted diaryl spiro[cycloprop[2]ene-1,9′-xanthene] derivatives 92a–j. Reagents and conditions: A = CsF, MeCN, 30 °C, 24 h. | |
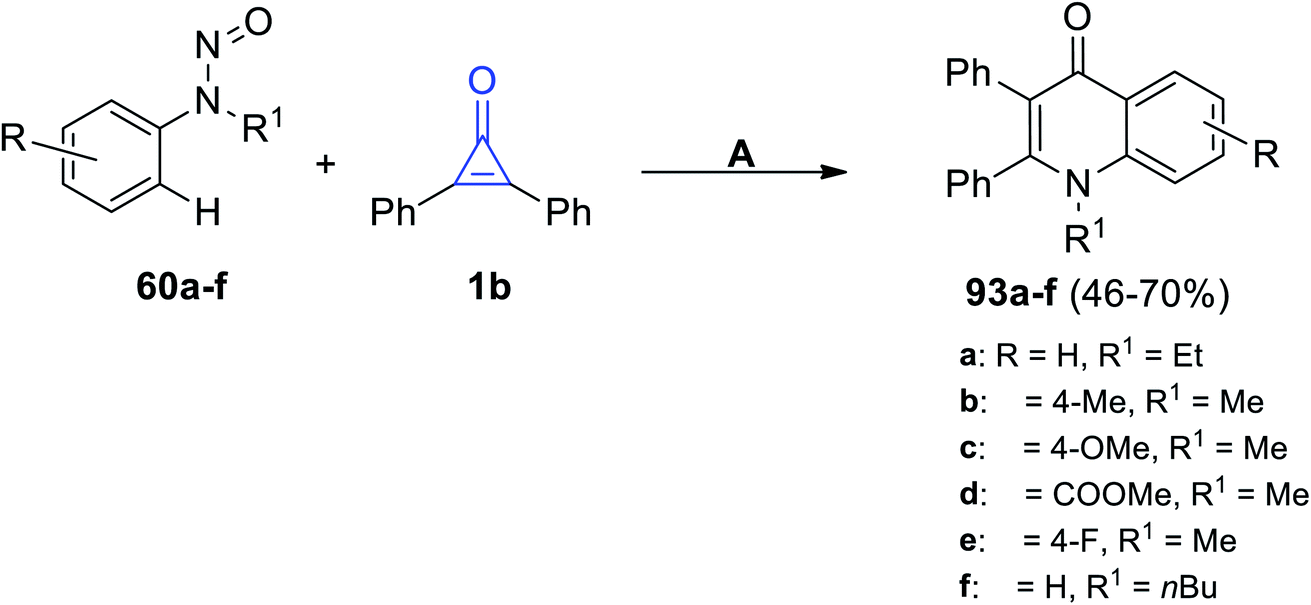
The rhodium-catalyzed reaction occurred between substituted N-nitrosoanilines 60 and 1b in the presence of AgBF4 and DCE at 100 °C for 12 h. The quinoline-4-one derivatives 93a–f were produced in 46–70% yield, as shown in Scheme 58.102
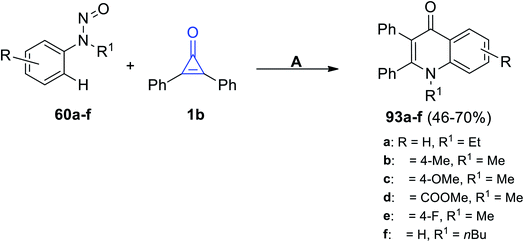 |
| | Scheme 58 Rhodium(III)-catalyzed synthesis of quinoline-4-one derivatives 93a–f. Reagents and conditions: A = (Cp*RhCl2)2 (2 mol%), AgBF4 (1.2 equiv.), NaF, DCE, 100 °C, 12 h. | |
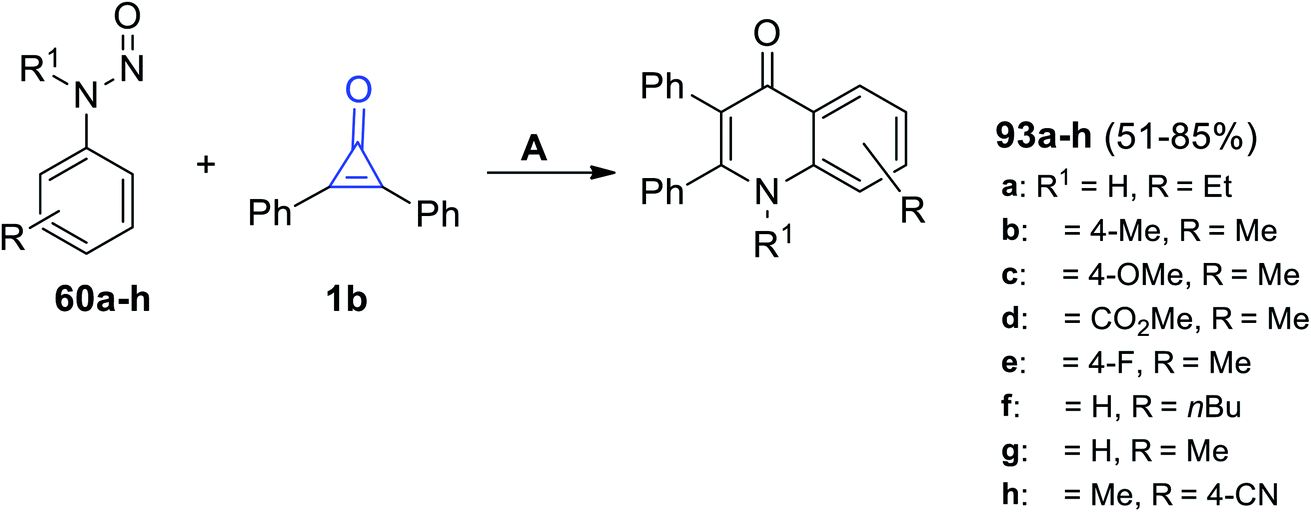
Moreover, in 2020, Shi et al. reacted with 1b with N-nitrosoanilines 60 using [RhCp*(OAc)2], [Rh(COD)Cl]2 and AgBF6 in DCM and yielded 4-quinolones 93a–h in 51–85% yield (Scheme 59).89
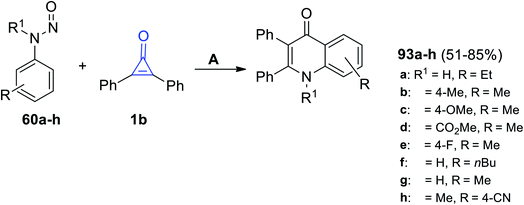 |
| | Scheme 59 Synthesis of 4-quinolones 93a–h. Reagents and conditions: A = [RhCp*(OAc)2] (0.5 mol%), [Rh(COD)Cl]2, AgBF6 (0.02 mmol), DCM, 120 °C, 36 h. | |
(3S,4aS,9bS)-Diethyl-3-methyl-4-oxo-4a-phenyl-1,4,4a,9b-tetrahydrobenzofuro[3,2-b]pyridine-2,2(3H)-dicarboxylates 96a–d were synthesized via the reaction of (E)-diethyl 2-((2-hydroxybenzylidene)amino)malonate derivatives (94) with 1j at room temperature in the presence of the catalyst 95 (Scheme 60).103
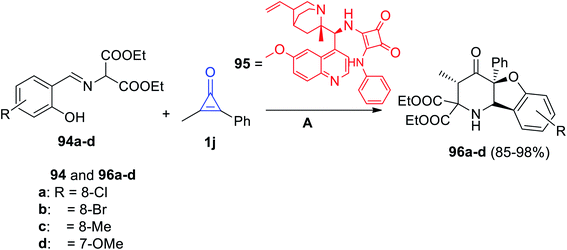 |
| | Scheme 60 Synthesis of compounds 95a–d. Reagents and conditions: A = mesitylene, rt, cat 96. | |
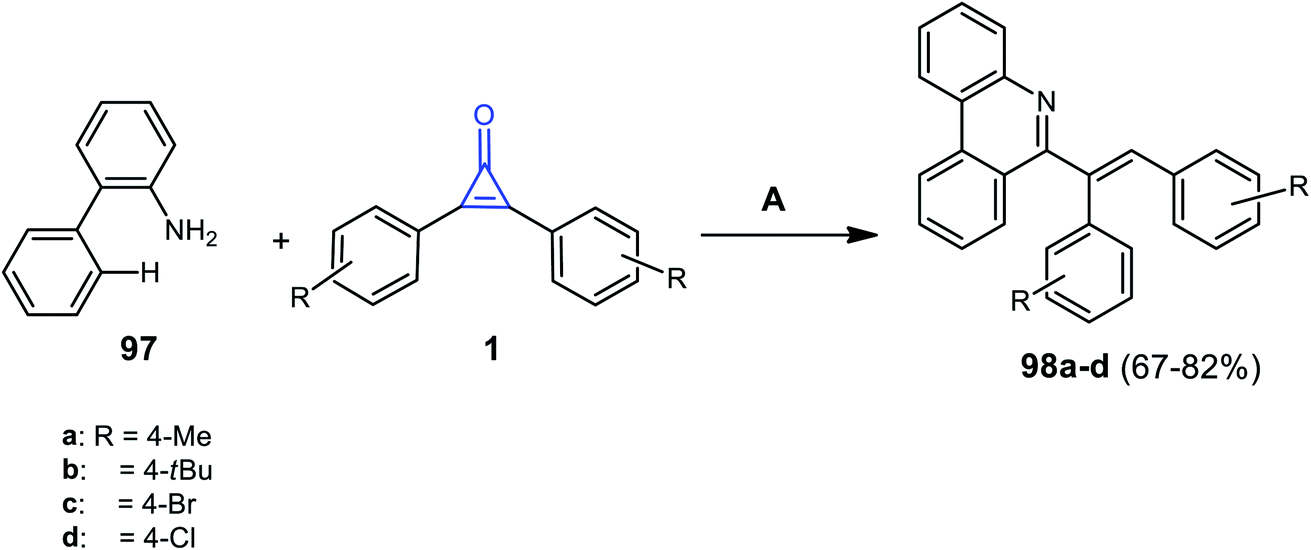
In 2020, it was reported that phenanthridine derivatives 98a–d were synthesized in 67–82% yield, via refluxing [1,1′-biphenyl]-2-amine (97) with substituted cyclopropenones 1 in DCM for 48 h and in the presence of [Ru(p-cymene)Cl2]2 and Ag2CO3 (Scheme 61).104
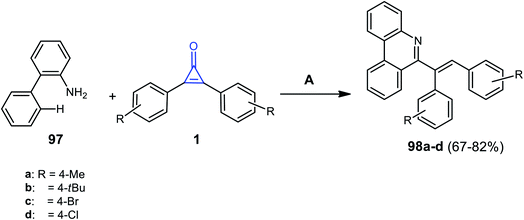 |
| | Scheme 61 Synthesis of phenanthridine derivatives 98a–d. Reagents and conditions: A = [Ru(p-cymene)Cl2]2, Ag2CO3, H3BO3, DCM, reflux, 48 h. | |
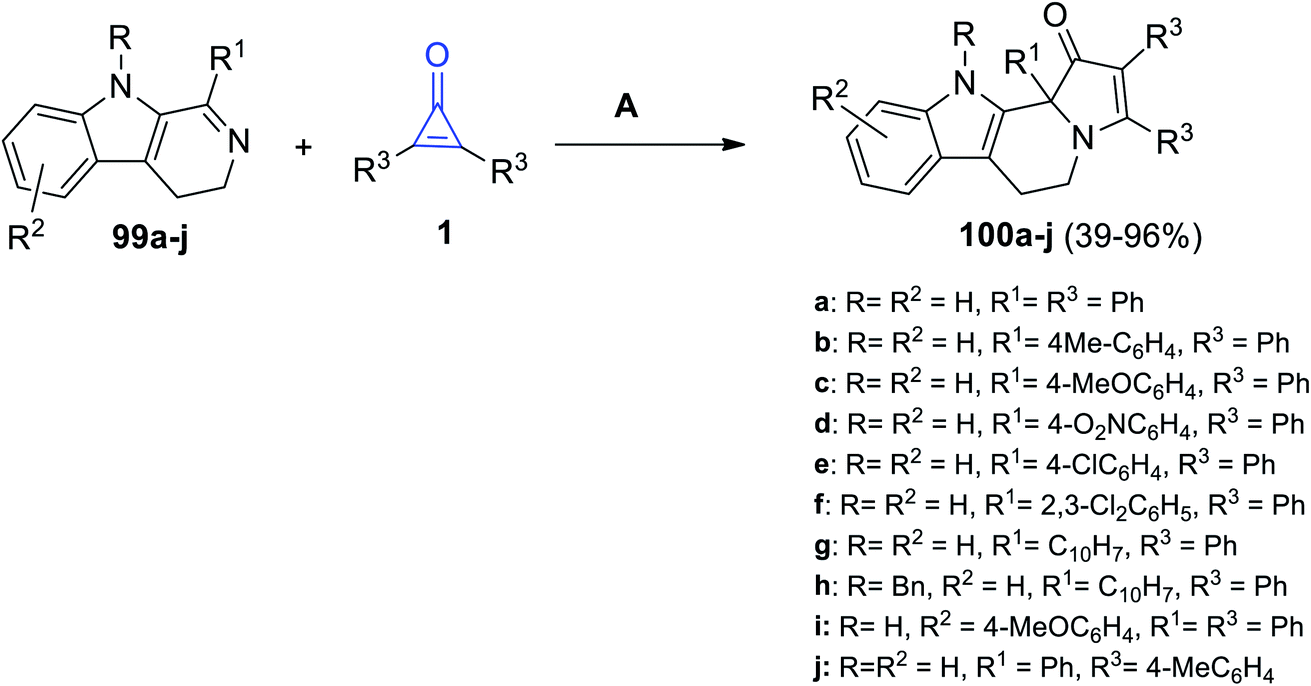
Substituted pyrido[3,4-b]indoles 99a–j could react easily with cyclopropenone derivatives 1 at room temperature in absolute ethanol for 5–50 h. The reaction yielded compounds 100a–j in 39–93% yield (Scheme 62).105
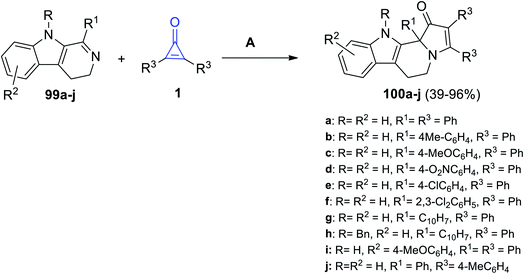 |
| | Scheme 62 Synthesis of 100a–j. Reagents and conditions: A = EtOH, rt, 5–50 h. | |
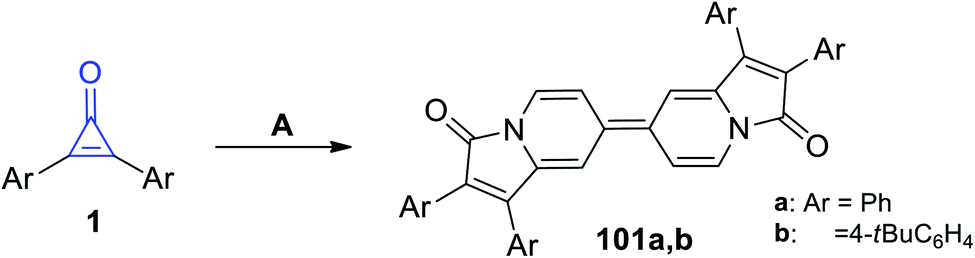
Refluxing diarylcyclopropenones 1 in pyridine at 100 °C in the presence of cupric acetate (Cu(OAc)2) afforded biindolizines 101a–b, as shown in Scheme 63.106
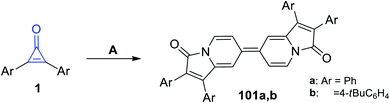 |
| | Scheme 63 Synthesis of biindolizines 101a,b. Reagents and conditions: A = pyridine, Cu(OAc)2, 15 min. | |
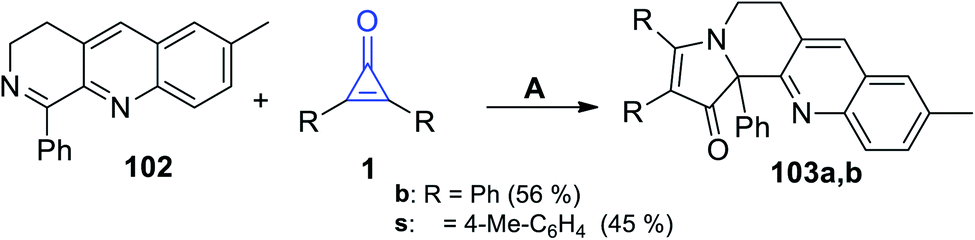
7-Methyl-1-phenyl-3,4-dihydrobenzo[b][1,7]naphthyridine (102) easily reacted with cyclopropenones 1 without using any catalyst at room temperature in EtOH to afford 2,3-disubstituted-9-methyl-12b-phenyl-5,6-dihydrobenzo[b]pyrrolo[1,2-h][1,7]naphthyridin-1(12bH)-ones 103a–b in 45–56% yield, as shown in Scheme 64.105
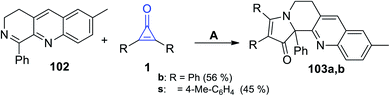 |
| | Scheme 64 Synthesis of the fused heterocyclic compounds 103a,b. Reagents and conditions: A = EtOH, rt. | |
Carbonylation of symmetrical cyclopropenones 1 with alkynes 8c–e occurred in refluxing toluene for 20 h. The reaction was mediated by triruthenium dodecacarbonyl Ru3(CO)12 and yielded pyranopyrandiones 104a–c in 54–82% yield (Scheme 65).107
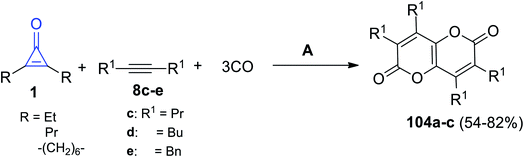 |
| | Scheme 65 Synthesis of pyranopyrandiones 102a–c. Reagents and conditions: A = toluene, Ru3(CO)12, Et3N, 150 °C, 20 h. | |
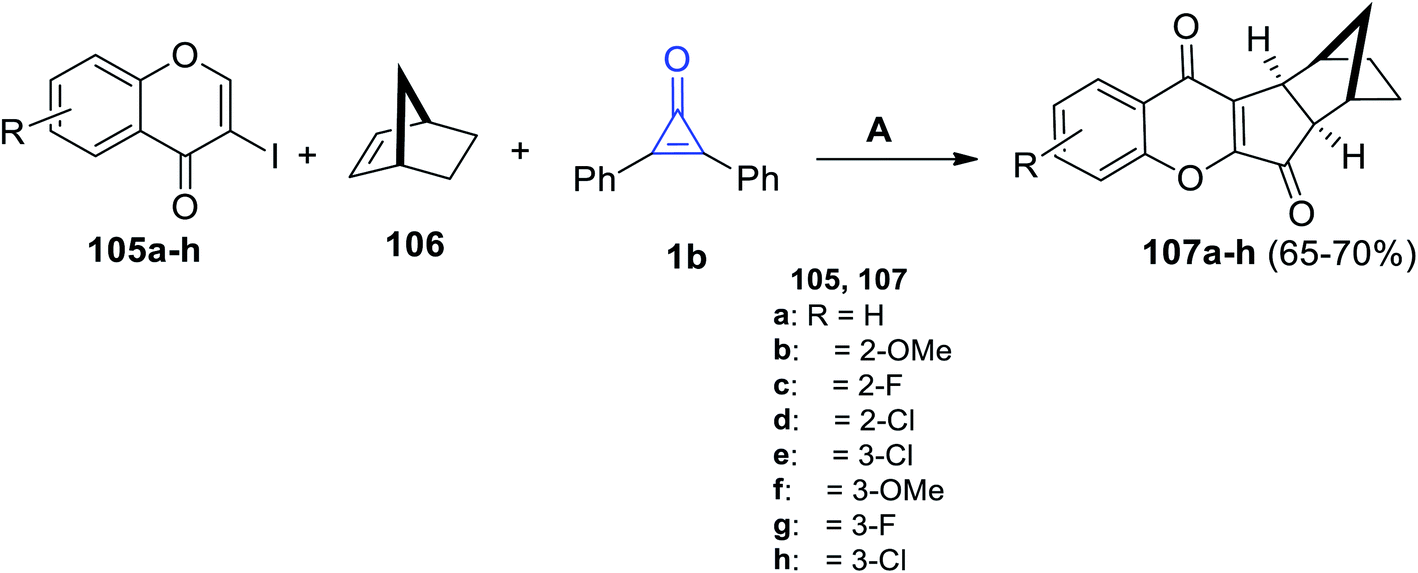
Zhu et al. in 2021, have developed a palladium-catalyzed three-component reaction of substituted iodochromen-4-ones 105a–h, (1R,4S)-bicyclo[2.2.1]hept-2-ene (106) and 1b in fluorobenzene in the presence of tris-(4-trifluoromethyl-phenyl)phosphine P(4-CF3-C6H4)3 at 100 °C for 24 h. (6aR,7S,10R,10aS)-6a,7,8,9,10,10a-Hexahydro-7,10-methanoindeno[2,1-b]chromene-6,11-diones 107a–h were obtained in 65–70% yield (Scheme 66).108
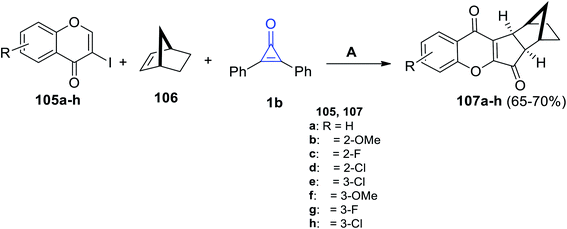 |
| | Scheme 66 Synthesis of compounds 107a–h. Reagents and conditions: A = PdCl2, P(4-CF3-C6H4)3 (20 mol%), Cs2CO3, C6H5F, 100 °C, 24 h. | |
In 2021, Chen et al. successfully synthesized a series of tetrasubstituted pyrano[2,3-b]indol-2(9H)-ones 109a–l in 65–91% yield via the reaction between substituted isatines 40 and diaryl cyclopropenones 1 at 110 °C in toluene and in the presence of lanthanide amides [(Me3Si)2N]3La(μ-Cl)Li(THF)3 and ligand 108 as a catalyst (Scheme 67).109
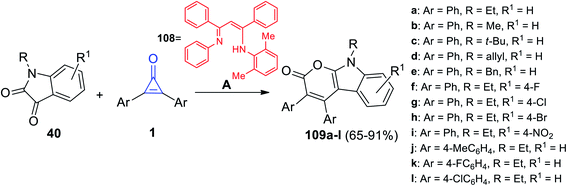 |
| | Scheme 67 Synthesis of tetrasubstituted pyrano[2,3-b]indol-2(9H)-ones (109a–l). Reagents and conditions: A = [(Me3Si)2N]3La(μ-Cl)Li(THF)3, toluene, 110 °C, 2.5 h, (108), HOP(OEt)2. | |
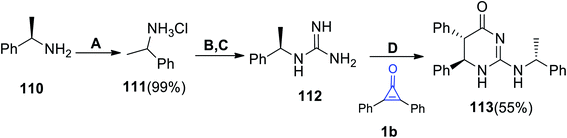 |
| | Scheme 68 Synthesis of 5,6-dihydropyrimidine 113. Reagents and conditions: A = conc. HCl, 1,4 dioxane, 20 °C; B = NH2CN, pH 8–9, H2O, reflux, 5 h; C = passed through Amberlite IRA-401 (hydroxide form); D = benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (1:1), rt, 18 h. :EtOH (1:1), rt, 18 h. | |
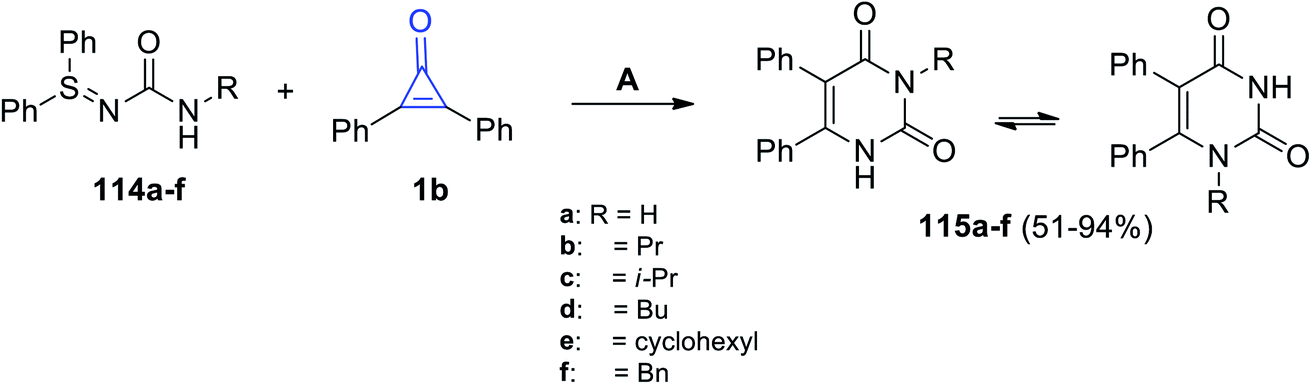
A catalyst-free reaction between N-carbamoyl sulfilimines 114a–f and 1b in toluene at 110 °C for 13 h afforded the two isomers of diphenyl pyrimidinediones 115a–f in 51–94% yields, as shown in Scheme 69.111
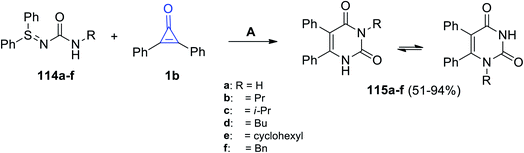 |
| | Scheme 69 Synthesis of diphenyl pyrimidinediones 115a–f. Reagents and conditions: A = toluene, 110 °C, 13 h. | |
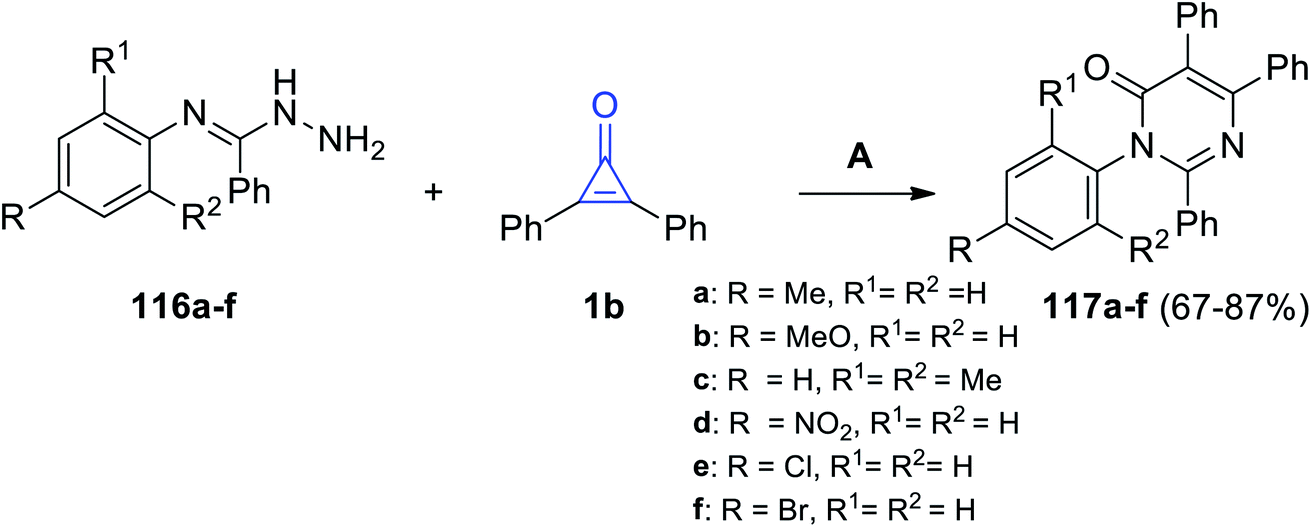
Aly et al. in 2016, successfully synthesized pyrimidines 117a–f in 67–87% yield, via the reaction between 116a–f and 1b in EtOH and Et3N (Scheme 70).112
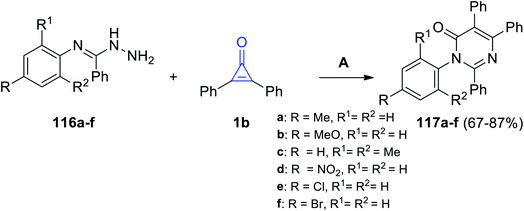 |
| | Scheme 70 Synthesis of pyrimidines 117a–f. Reagents and conditions: A = EtOH, Et3N, reflux 6–10 h. | |
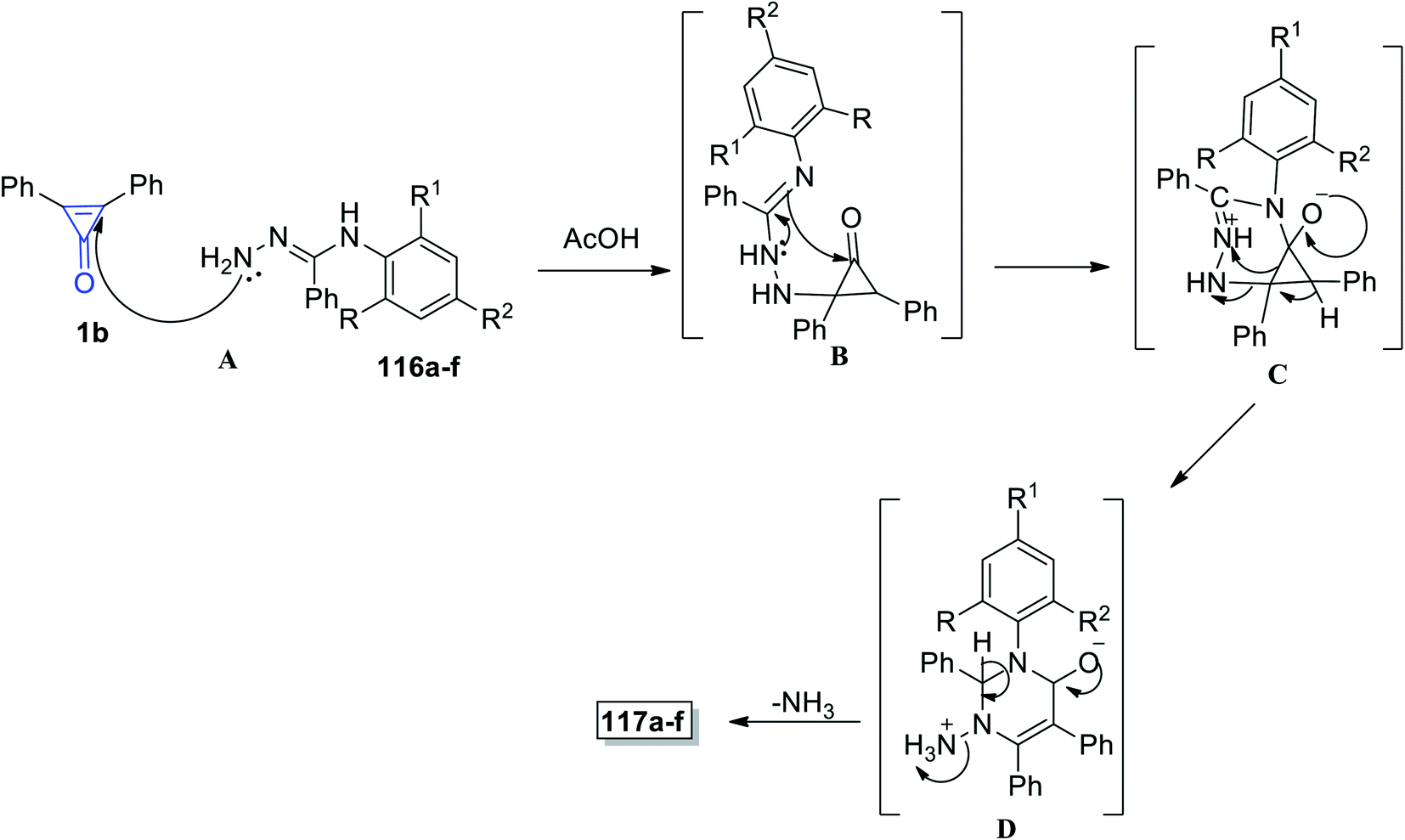
The proposed reaction mechanism is illustrated in Scheme 71; the carbonyl group of cyclopropenone was attacked by the hydrazine nitrogen atom yielding intermediate B, following which an amidine-like reaction of N-3 to carbonyl may occur to obtain salt C. Nucleophilic addition to positively charged nitrogen via ring opening followed by proton transfer would afford D, and the final product 117 was obtained by losing ammonia from D (Scheme 71).112
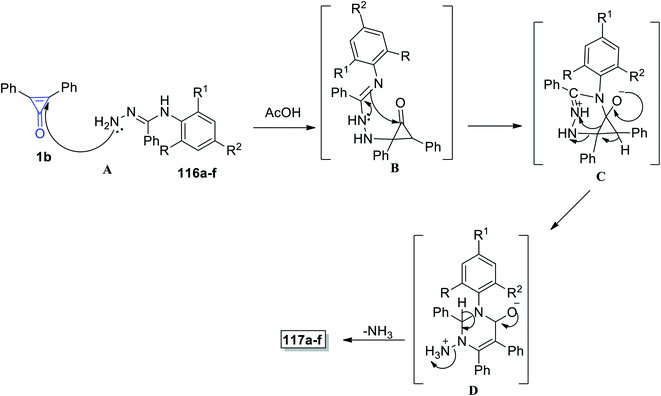 |
| | Scheme 71 Mechanism of the formation of pyrimidines 117a–f. | |
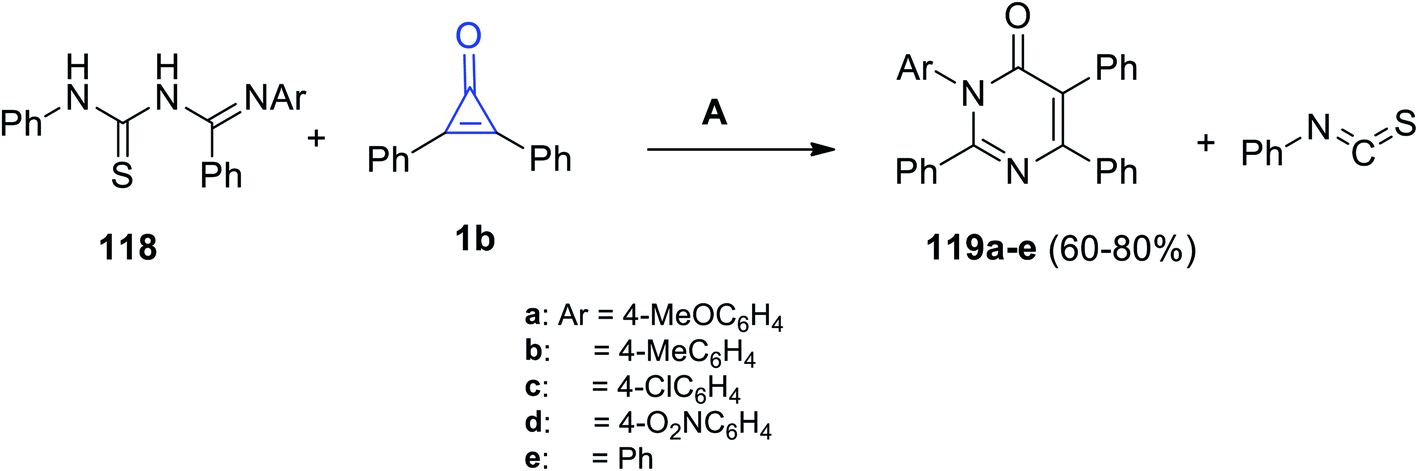
Aly et al. also reported that the reaction between (E)-N′-aryl-N-(phenylcarbamothioyl)-benzimidamides 118 and 1b in EtOH produced substituted 3-aryl-2,5,6-triphenylpyrimidin-4(3H)-ones 119a–e, as shown in Scheme 72.113
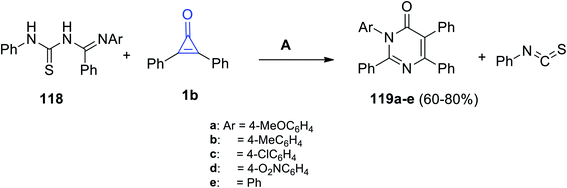 |
| | Scheme 72 Synthesis of substituted 3-aryl-2,5,6-triphenylpyrimidin-4(3H)-ones (119a–e). Reagents and conditions: A = EtOH, reflux, 10–16 h. | |
Mohan and Jose, in 2017, have reported that reaction between (E)-disubstituted diazene-1,2-dicarboxylates 120a–j and diarylcyclopropenone 1 in DCM at room temperature. The reaction was catalyzed by PPh3 to afford substituted 1,3-oxazin-6-one 121a–j in 50–70% yield (Scheme 73).114
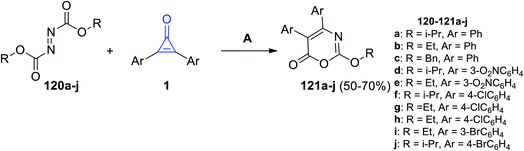 |
| | Scheme 73 Synthesis of substituted 1,3-oxazin-6-ones 121a–j. Reagents and conditions: A = PPh3 (1 equiv.), DCM, rt, 15 min. | |
The reaction was initiated via PPh3 attack on 120 to give A and Huisgen zwitterion's nucleophilic attack on cyclopropenone (1) in step B; the intermediate (C) was formed followed by internal cyclization of D to generate 1,3-oxazin-6-ones 121a–j, as illustrated in Scheme 74.114
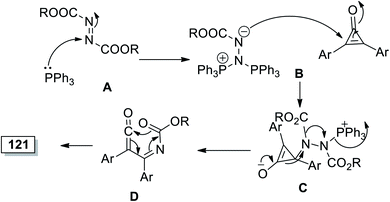 |
| | Scheme 74 Mechanism of the formation of substituted 1,3-oxazin-6-ones 121a–j. | |
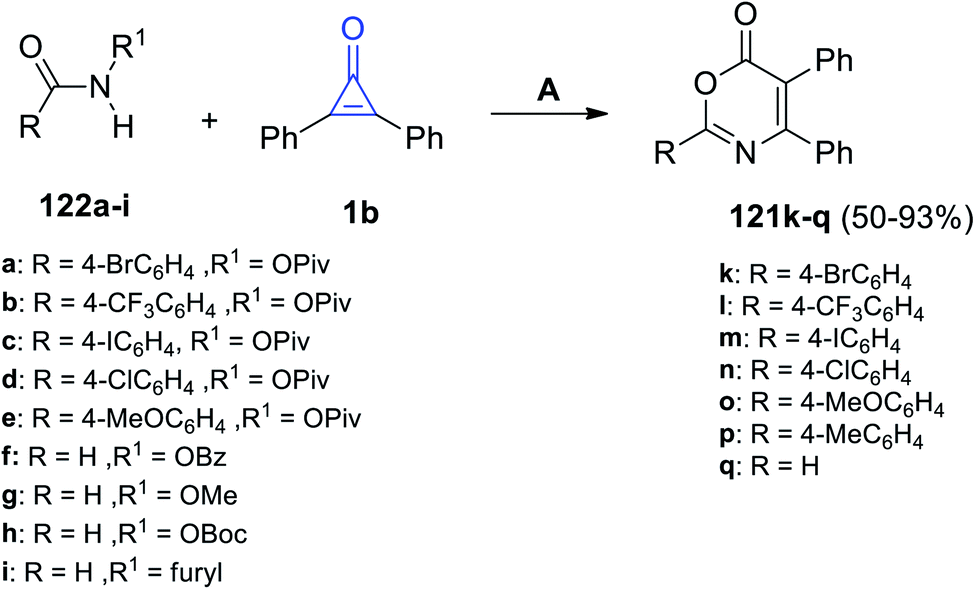
Via the reaction of equal amounts of amide derivatives 124a–i with 2,3-diphenylcyclopropenones (1) in DCE using CsOAc as a catalyst, the substituted 1,3-oxazin-6-ones 121k–q were obtained in 50–93% yield (Scheme 75).115
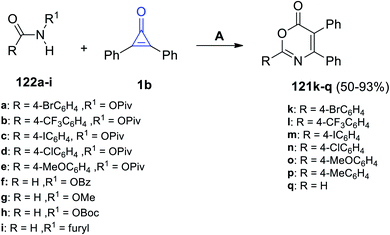 |
| | Scheme 75 Scandium-catalyzed synthesis of substituted 1,3-oxazin-6-ones 121k–q. Reagent and conditions: A = CsOAc (1 equiv.), DCE, rt, 6 h. | |
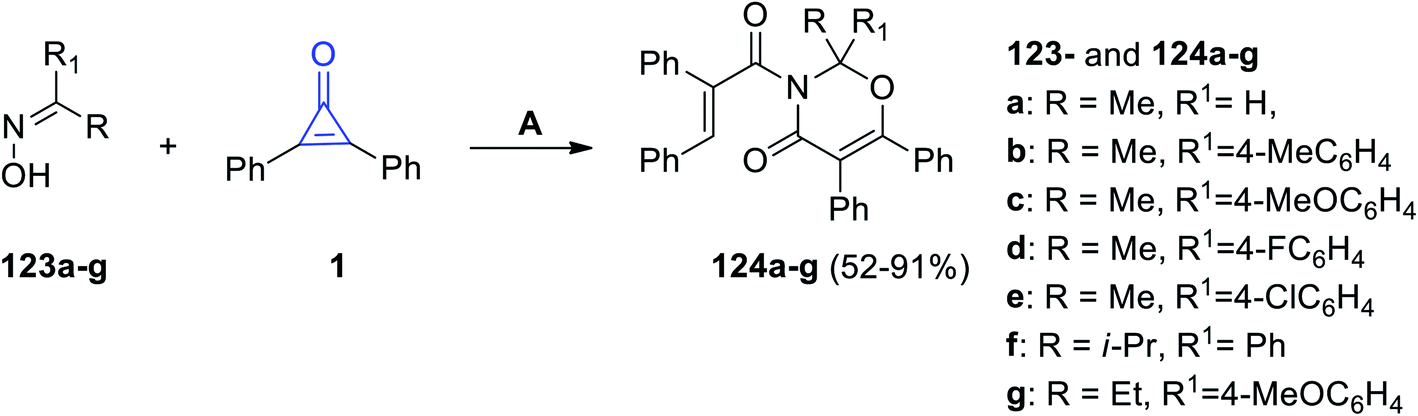
Recently, it has been reported that the oxime derivatives 123a–g reacted with 2,3-diphenyl-cyclopropenones (1) to give substituted 1,3-oxazine-4-ones 124a–g in 52–91% yield (Scheme 76). The reaction was performed at 80 °C in cyclohexane for 18 h, and Ag2O was also used as a catalyst.116
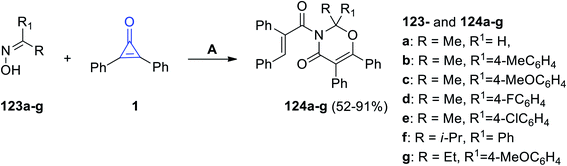 |
| | Scheme 76 Synthesis of 1,3-oxazine-4-ones 124a–g. Reagents and conditions: A = Ag2O, cyclohexane, 80 °C, 18 h. | |
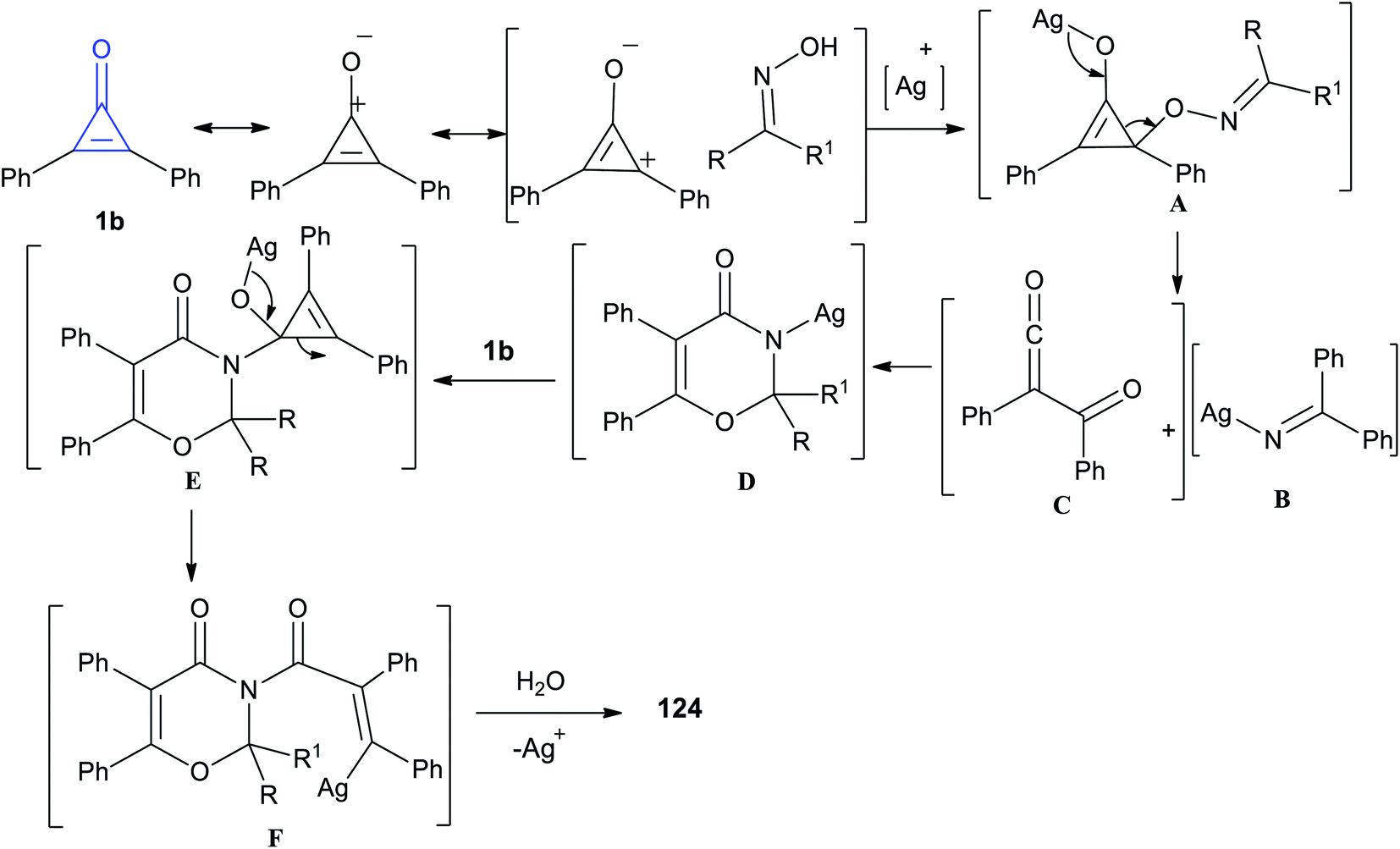
The reaction mechanism is illustrated in Scheme 77. At first, the resonance structure of cyclopropenone was nucleophilically attacked by oximes, yielding intermediate A. Subsequently the intermediate A was fragmented into intermediates B and C. Accordingly, the intermediate D was obtained via a [4 + 2] cycloaddition between B and C. Then, a reaction occurred between D and another cyclopropenone to generate intermediate E, and the intermediate F was subjected to a rearrangement/protonation process to form compound 124.116
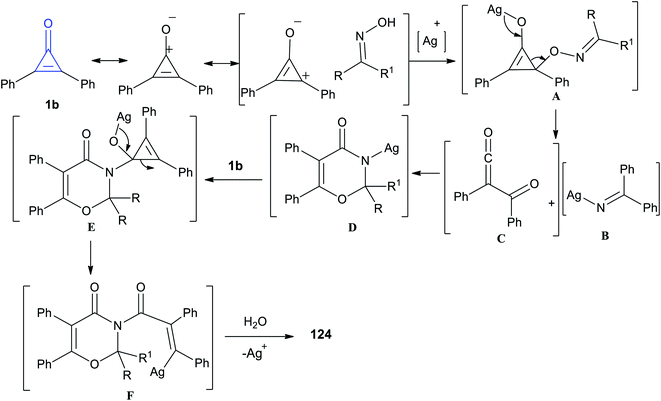 |
| | Scheme 77 Mechanism describing the formation of 1,3-oxazine-4-ones 124a–g. | |
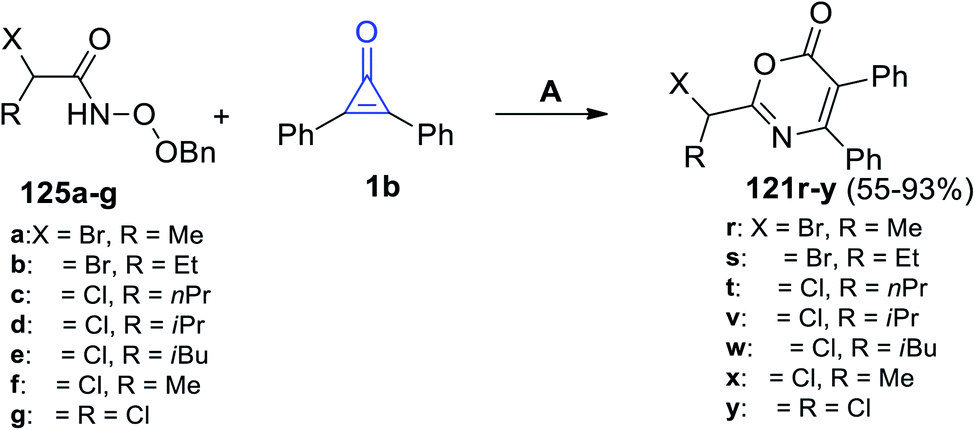
In 2021, Sizhan, et al. successfully synthesized substituted 1,3-oxazin-6-ones 121r–y in 55–93% yield, via the reaction between α-halogenated hydroxamates 125a–g and 1b in DCM and Et3N (Scheme 78).117
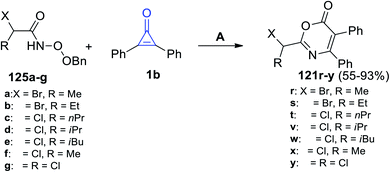 |
| | Scheme 78 Synthesis of substituted 1,3-oxazin-6-ones 123r–y. Reagents and conditions: A = DCM, reflux, Et3N, 2 h. | |
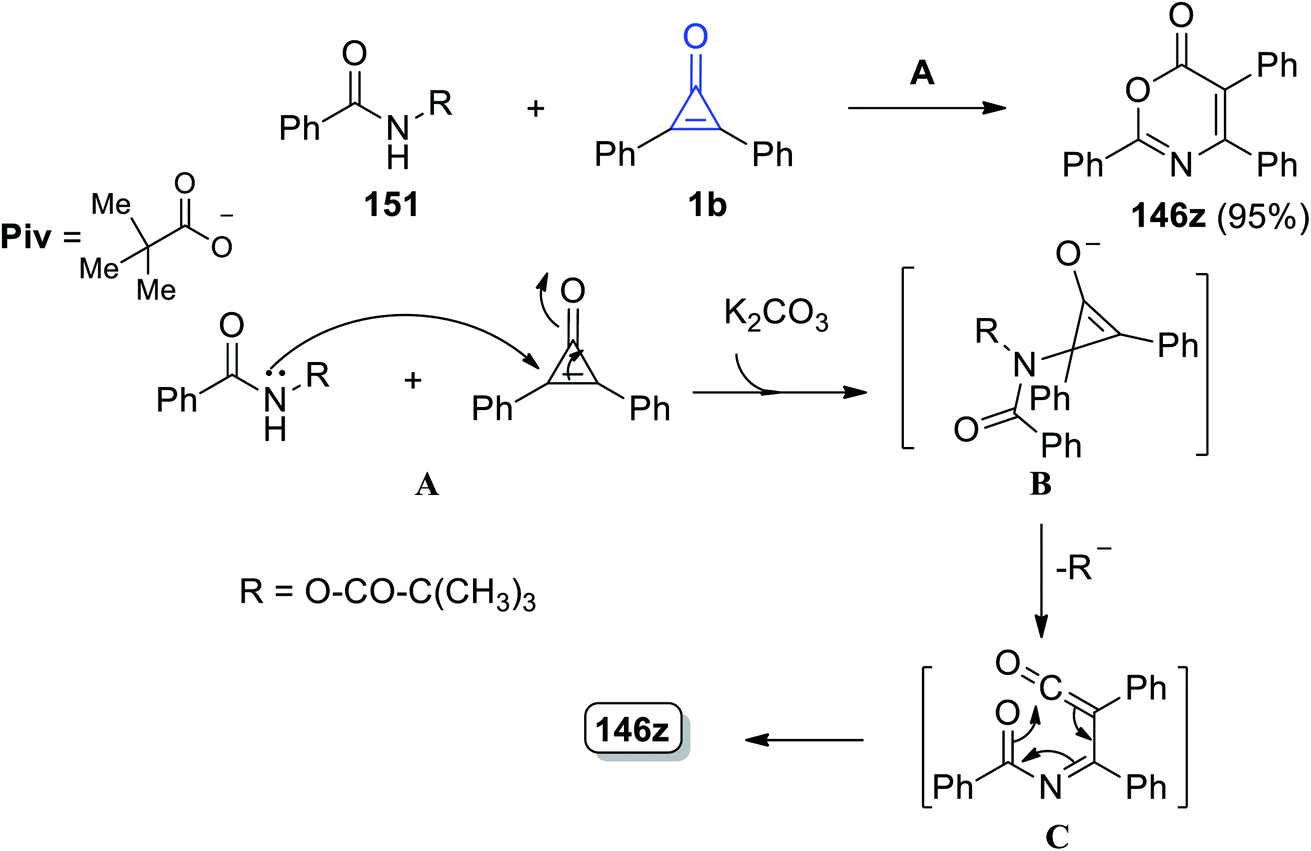
2,4,5-Triphenyl-6H-1,3-oxazin-6-one (121z) was obtained in 95% yield by reacting N-(pivaloyloxy)benzamide (126) with 2,3-diphenylcyclopropenone (1) at 60 °C in THF using K2CO3. The mechanism of the reaction was explained by the nucleophilic attack of the nitrogen atom of amide 26 on 1b and intermediate B was then formed in the presence of base. During the ring opening, B lost a pivalate anion to give ketene C, which rearranged to form the final structure (121z), via a 6π electrocyclization process, as shown in Scheme 79.118
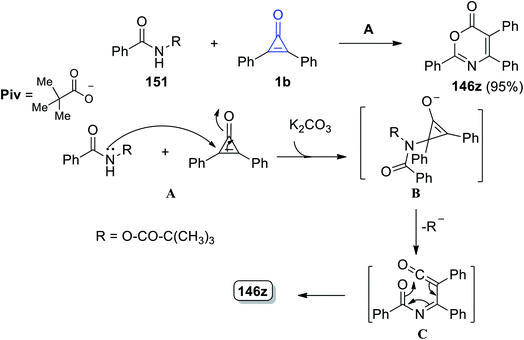 |
| | Scheme 79 Synthesis of 2,4,5-triphenyl-6H-1,3-oxazin-6-one (123z). Reagents and conditions: A = K2CO3 (0.5 equiv.), THF, 60 °C, 2 h. | |
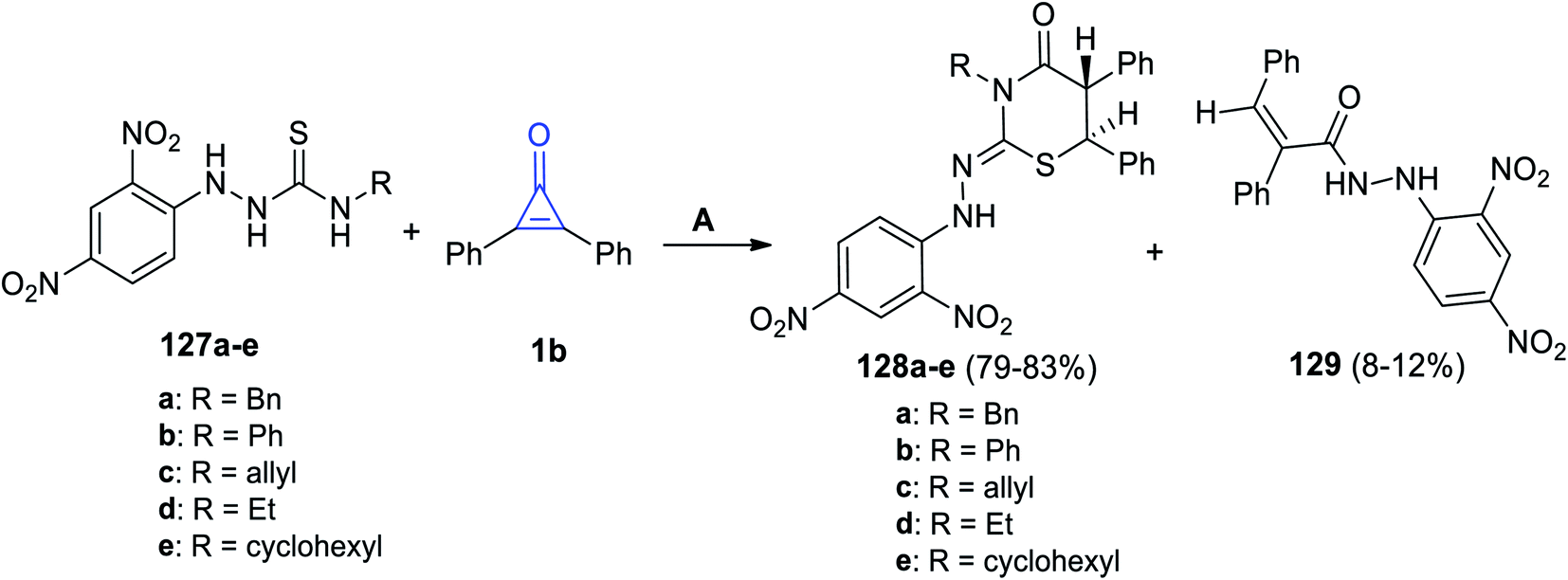
In 2019, substituted thiazinane derivatives 128a–e were successfully synthesized by Hassan et al. in 2019. It was achieved during the reaction of hydrazinecarbothioamide derivatives 127a–e with 1b in refluxing EtOH (Scheme 80). The reaction afforded compounds 128a–e, as well as a side product 129.119
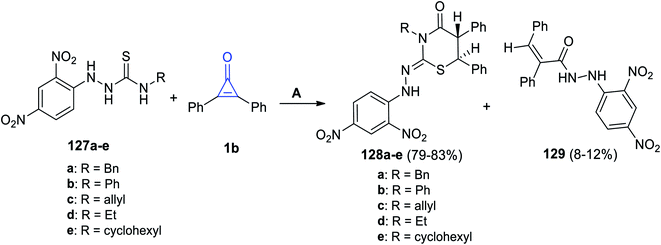 |
| | Scheme 80 Synthesis of thiazinanes 128a–e. Reagents and conditions: A = EtOH, reflux. | |
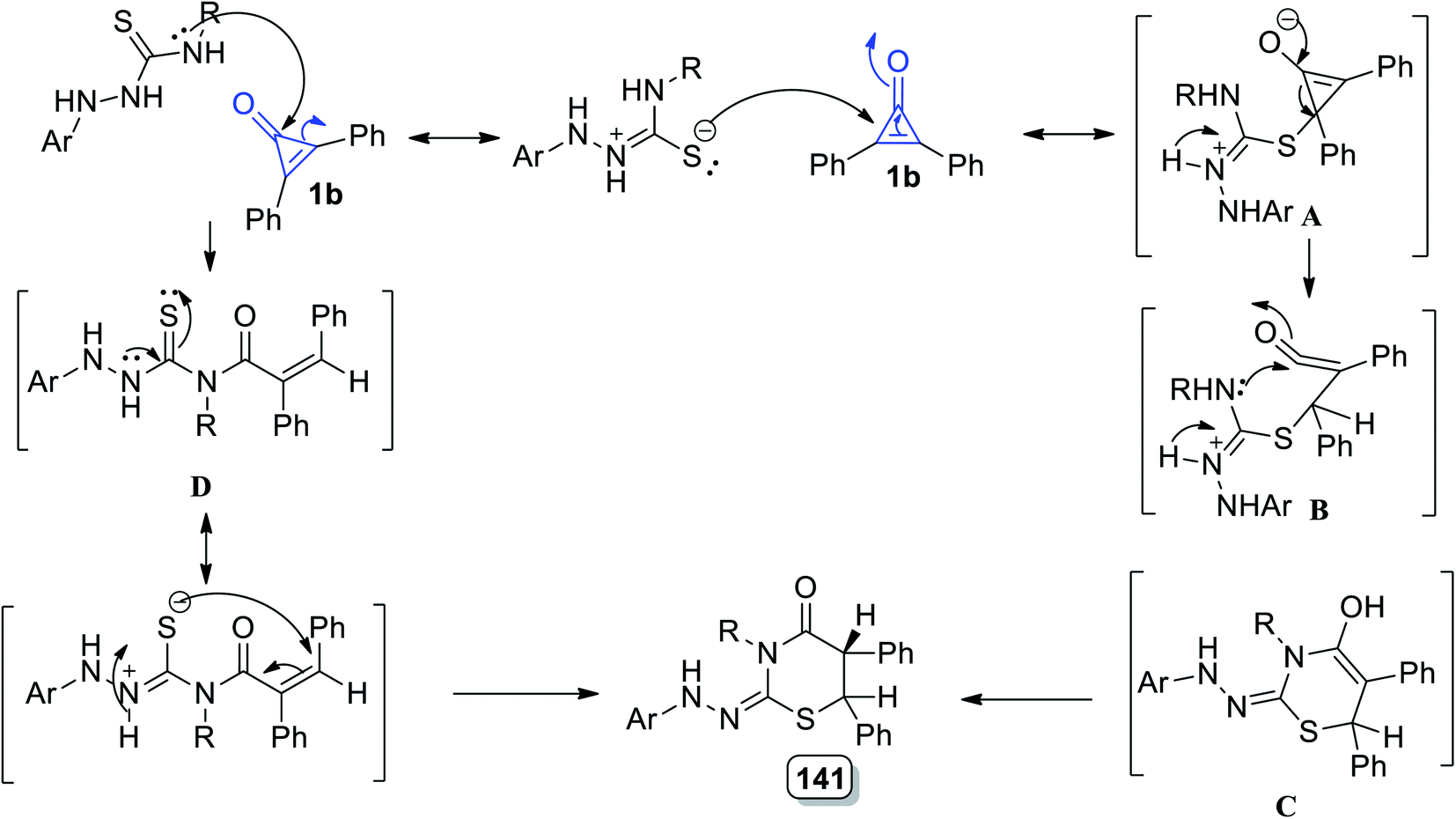
The reaction mechanism could be simply described as the conjugate double bond of 1b was attacked by the sulfur atom generating intermediate A. The intermediate B was formed via the intramolecular nucleophilic attack of N4–H on CO, which rearranged to generate 128. However, the carbonyl group of 1 was attacked by N4–H, resulting in the formation of the product 128 via intermediate D (Scheme 81).119
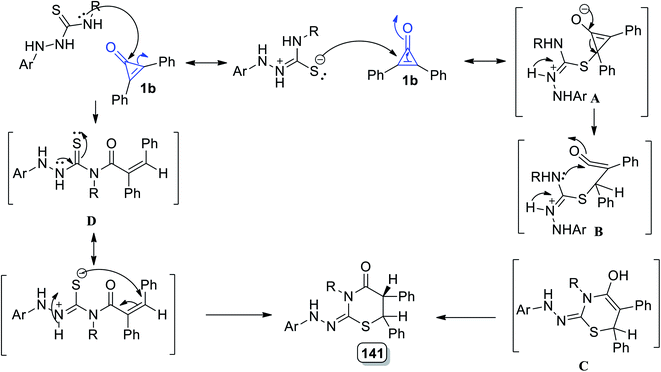 |
| | Scheme 81 Mechanism of the formation of thiazinanes 128a–e. | |
When pyrazolylthioureas 130a–c were subjected to 1b, the reaction proceeded to give compounds 131a–c in 75–90% yield (Scheme 82). The reaction was performed in ethanol for 4–7 h in the presence of DDQ as the oxidizing agent.120
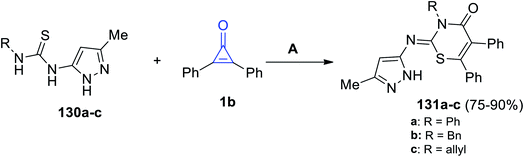 |
| | Scheme 82 Synthesis of thiazines 131a–c. Reagents and conditions: A = DDQ, EtOH, reflux, 4–7 h. | |
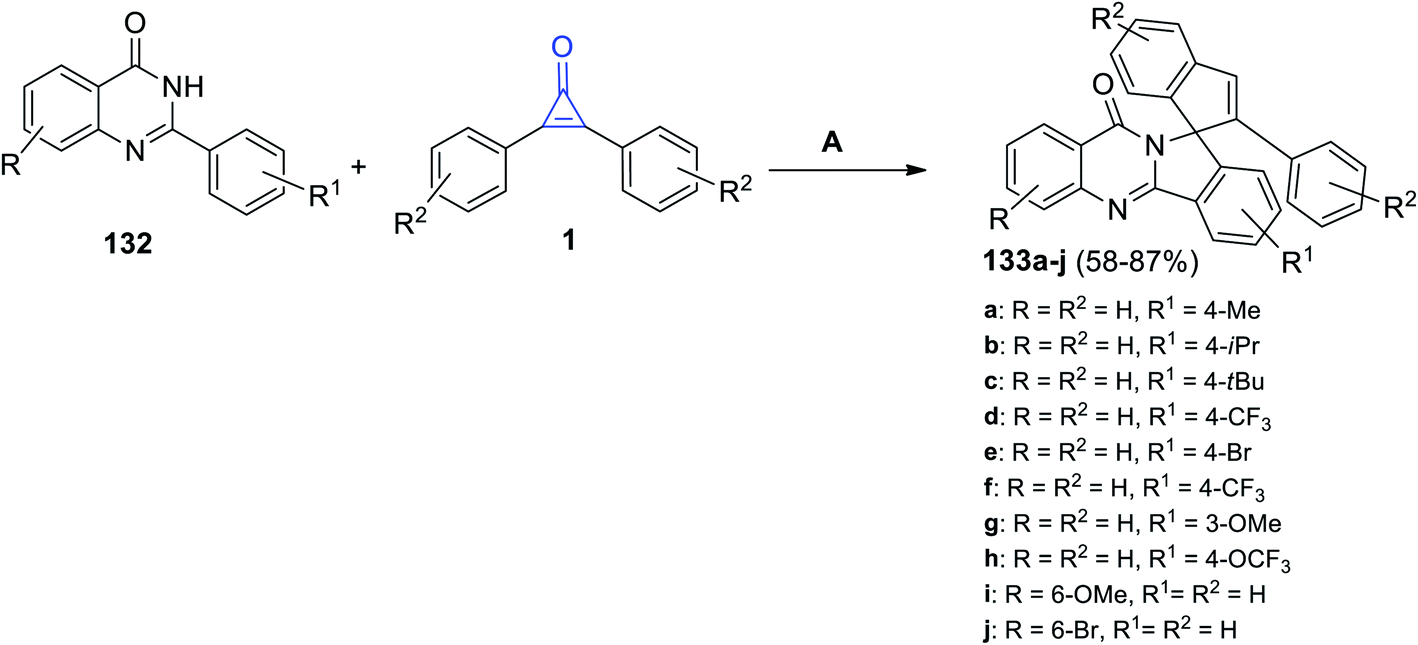
Moreover, in 2021, Shi et al. succeeded to synthesize the heterocyclic substituted 2-phenyl-10′H-spiro[indene-1,12′-isoindolo[1,2-b]quinazolin]-10′-ones 133a–j in 58–87% yield by reacting 2-phenylquinazolin-4-ones 132a–j with cyclopropenones 1 in refluxing DCE for 24 h, and a (cymene)-ruthenium dichloride dimer [Ru(p-cymene)Cl]2 was used as a metal catalyst (Scheme 83).121
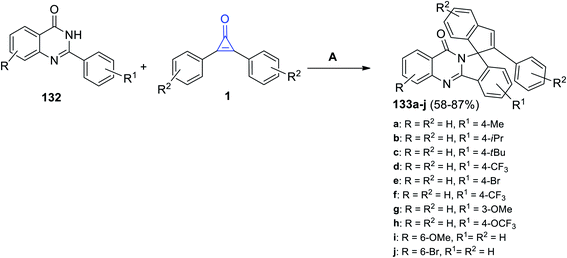 |
| | Scheme 83 Synthesis of substituted quinazolin-ones 133a–j. Reagents and conditions: A = [Ru(p-cymene)Cl]2, AgSbF6, AdCOOH, DCE, reflux, 24 h. | |
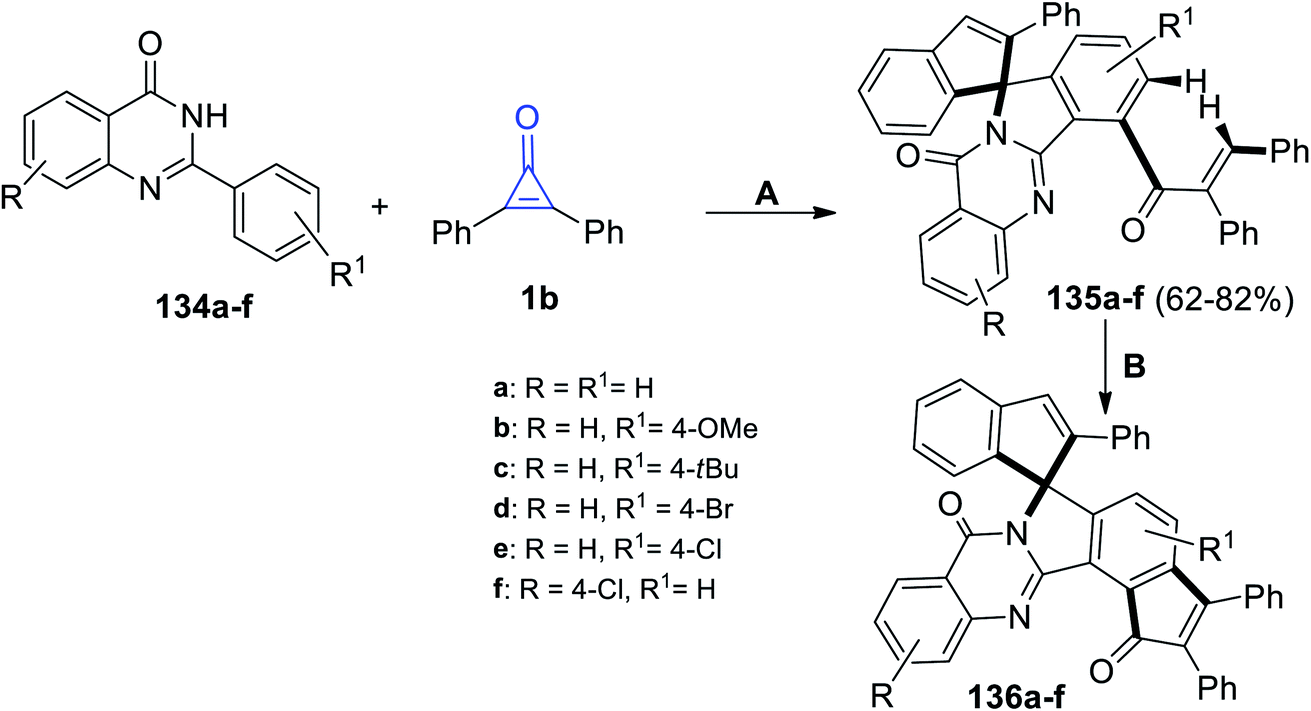
Shi et al. also reacted substituted 2-phenylquinazolin-4-ones 134a–f with 1b to obtain compounds 135a–f in 62–82% yield. The reaction was performed in trifluoroethanol (TFE) at 110 °C for 48 h. Subsequently, treatment of compound 135a–f with trifluoroacetic acid (TFA) at 140 °C and in the presence of Rh[Cp*(OAc)] and Ag2O yielded compounds 136a–f (Scheme 84).121
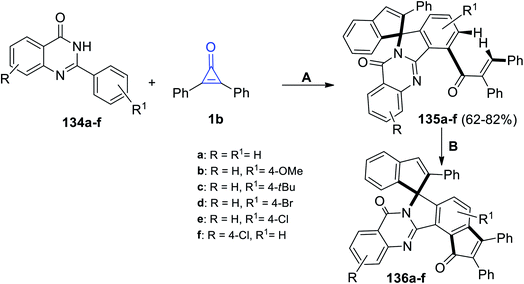 |
| | Scheme 84 Synthesis of compounds 135a–f and 136a–f. Reagents and conditions: A = Rh[Cp*(OAc)], TFE, 110 °C, 48 h; B = Pd(OAc)2, Ag2O, TFA, 140 °C, 24 h. | |
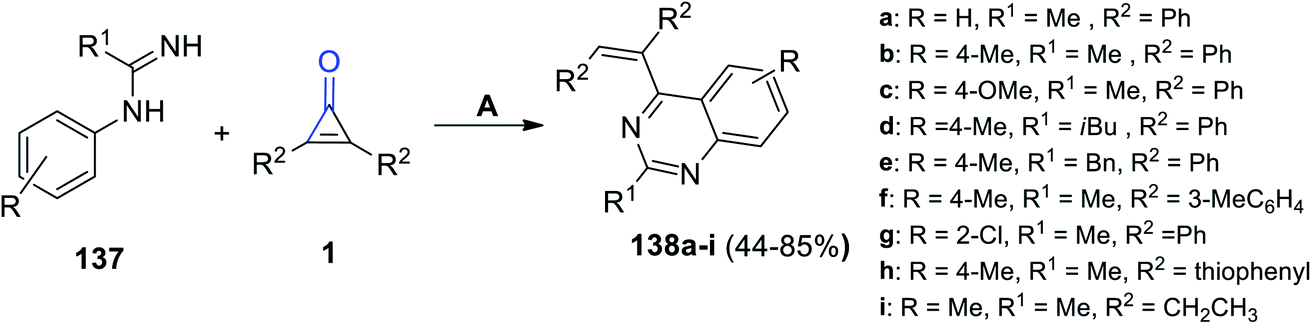
Refluxing 137 with 2,3-substituted cyclopropenones 1 in DCM yielded quinazoline derivatives 138a–i in 44–85% yield. The reaction was catalyzed using (Cp*RhCl2)2 and AgSbF6, as shown in Scheme 85.122
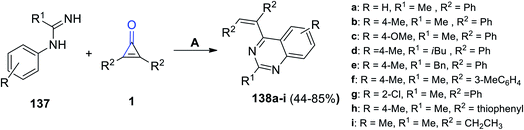 |
| | Scheme 85 Synthesis of quinazoline derivatives 138a–i. Reagents and conditions: A = (Cp*RhCl2)2, AgSbF6, DCM, reflux, 36 h, O2. | |
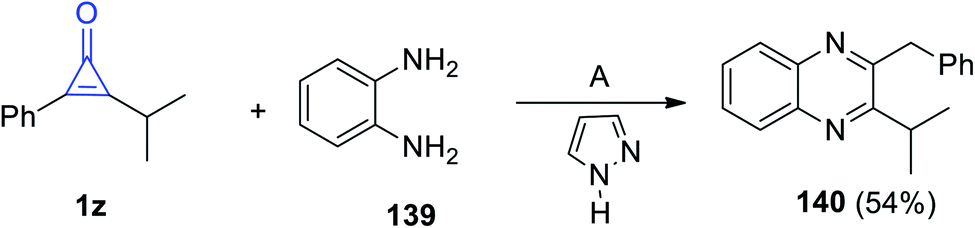
2-Benzyl-3-isopropylquinoxaline (140) was synthesized via reacting 2-isopropyl-3-phenylcycloprop-2-en-1-one (1z) with benzene-1,2-diamine (139) in diethyl ether for 8 h and in the presence of 1H-pyrazole as a catalyst (Scheme 86).86
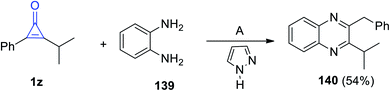 |
| | Scheme 86 Synthesis of 2-benzyl-3-isopropylquinoxaline (140). Reagents and conditions: A = Et2O, 8 h. | |
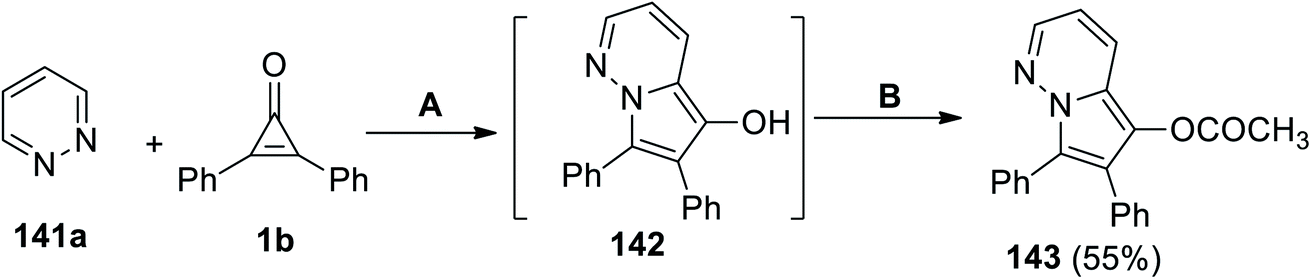
6,7-(Diphenylpyrrolo[1,2-b]pyridazin-5-yl)acetate (143) was successfully synthesized in 55% yield via a cyclization reaction between pyridazine (141a) and 1b in DCE at the reflux temperature. Subjecting the formed intermediate 142 with acetic anhydride and 4-(N,N-dimethylamino)pyridine (DMAP) as a base catalyst gave the final product 143 (Scheme 87).122
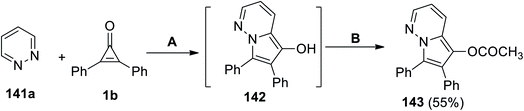 |
| | Scheme 87 Synthesis of 6,7-diphenylpyrrolo[1,2-b]pyridazin-5-yl acetate (143). Reagents and conditions: A = DCE, reflux, B = Ac2O, DMAP. | |
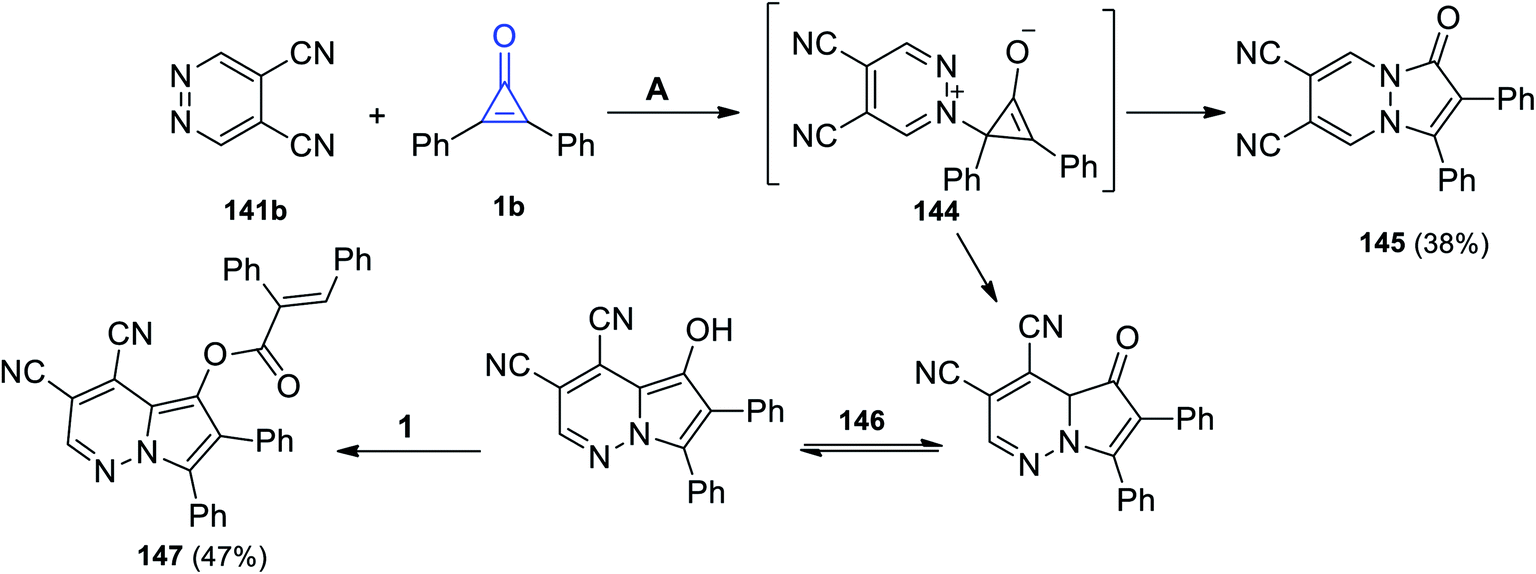
The reaction between pyridazine-4,5-dicarbonitrile (141b) and 1b in acetone at 110 °C afforded 1-oxo-2,3-diphenyl-1H-pyrazolo[1,2-a]pyridazine-6,7-dicarbonitrile (145) in 38% yield via the formation of intermediate 144 (Scheme 88). In addition to the formation of 145, [(E)-3,4-dicyano-6,7-diphenylpyrrolo[1,2-b]pyridazin-5-yl]-2,3-diphenylacrylate (147) was also obtained in 47% yield. The formation of 147 can be explained as due to the cyclization process of intermediate 144 into 146, which tautomerized and reacted with another molecule of 1 to give 147 (Scheme 88).123
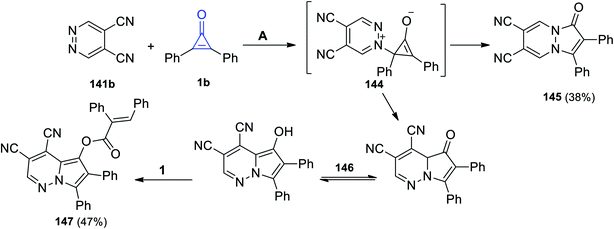 |
| | Scheme 88 Synthesis of pyridazines 145 and 147. Reagents and conditions: A = MeCOMe, 110 °C, 48 h. | |
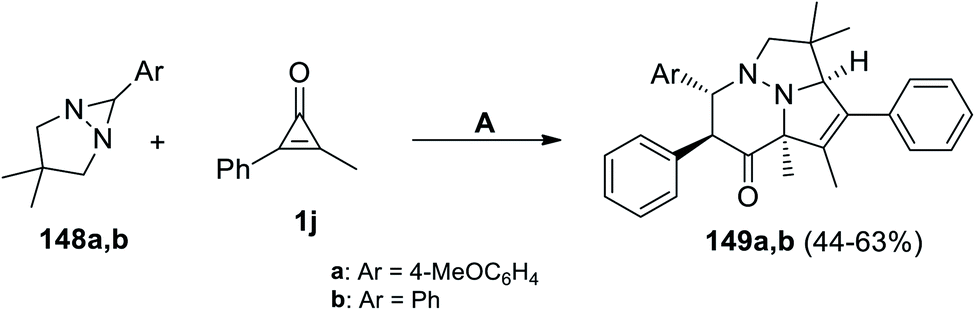
Molchanov et al. have developed on the synthesis of pyridazines 149a,b during refluxing a mixture of 6-aryl-3,3-dimethyl-1,5-diazabicyclo[3.1.0]hexanes 148a,b with 2-methyl-3-phenylcycloprop-2-enone (1j) in p-xylene for 20 min (Scheme 89).124
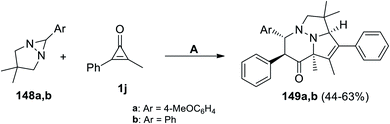 |
| | Scheme 89 Synthesis of pyridazines 149a,b. Reagents and conditions: A = p-xylene, reflux, 138 °C, 20 min. | |
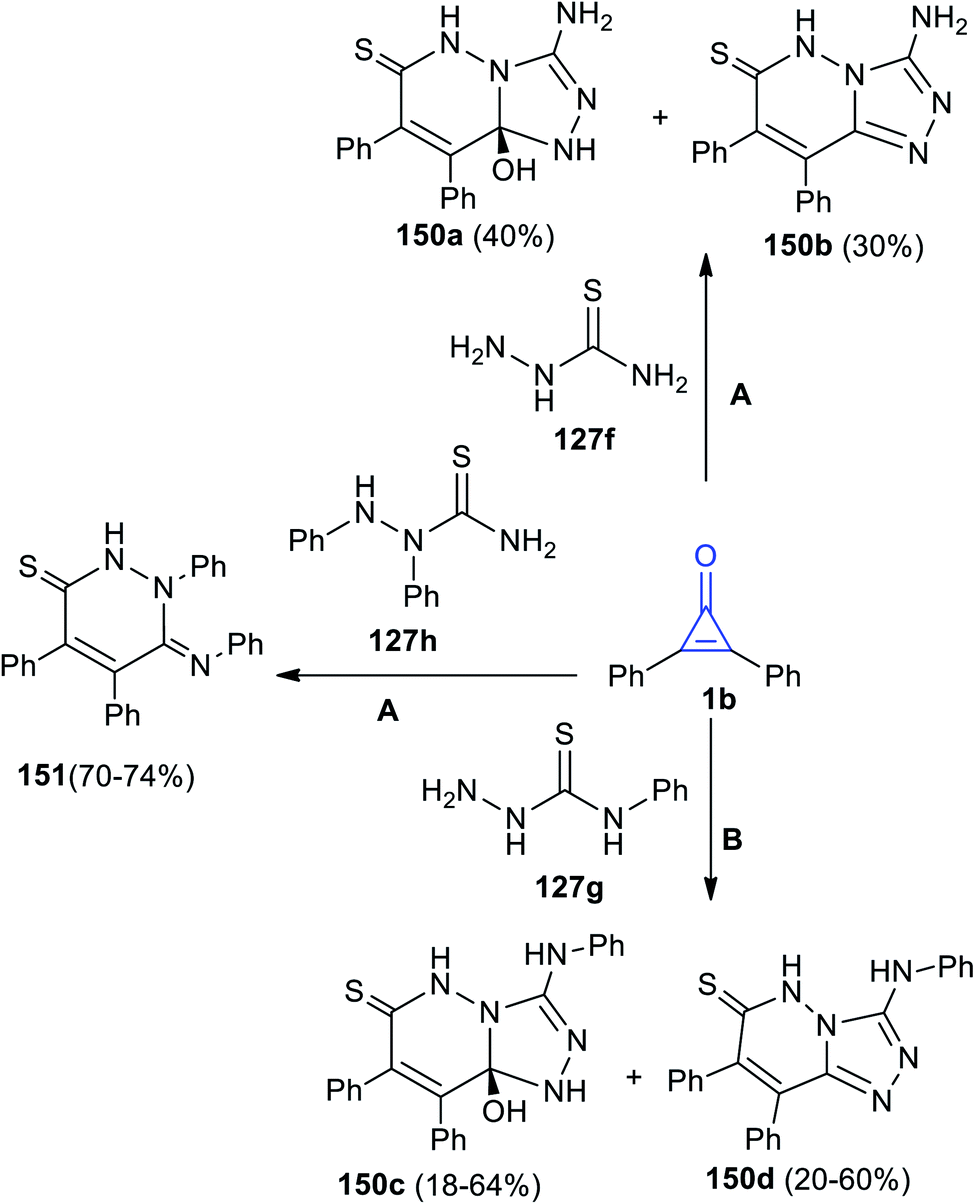
Aly's group reacted 1b with hydrazinecarbothioamides 138a–c in MeOH.125
The reaction yielded substituted triazolopyridazines 150a–d. However, when 1,2-diphenyl-hydrazinecarbothioamide (129h) was used, the reaction gave only (Z)-1,4,5-triphenyl-6-(phenyl-imino)-1,6-dihydropyridazine-3(2H)-thione (151) (Scheme 90).125
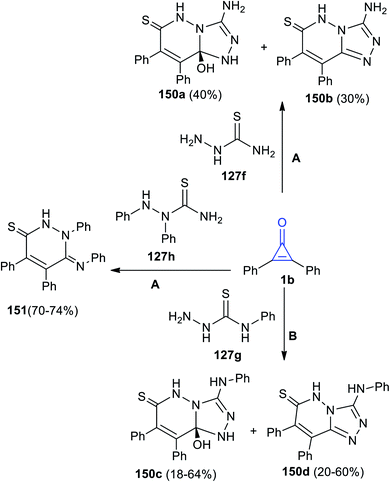 |
| | Scheme 90 Synthesis of pyridazines 150, 151. Reagents and conditions: A = MeOH, reflux, 48 h, B = MeOH, reflux, 6–12 h. | |
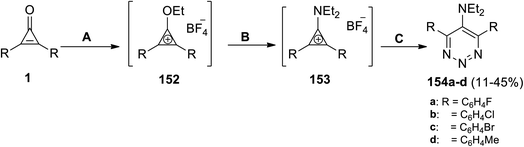 |
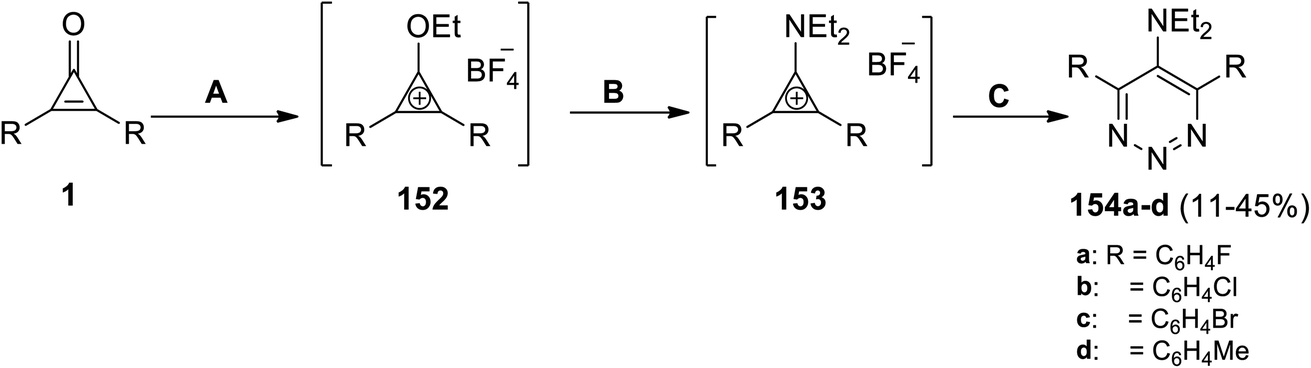
| | Scheme 91 Synthesis of triazines (154a–d). Reagents and conditions: A = Et3OBF4, CH2Cl2; B = Et2NH; C = NaN3, CH2Cl2, DMF. | |
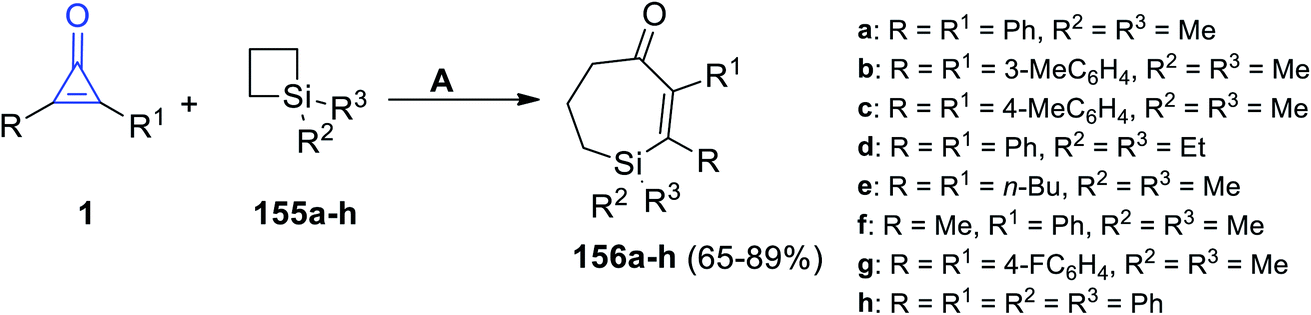
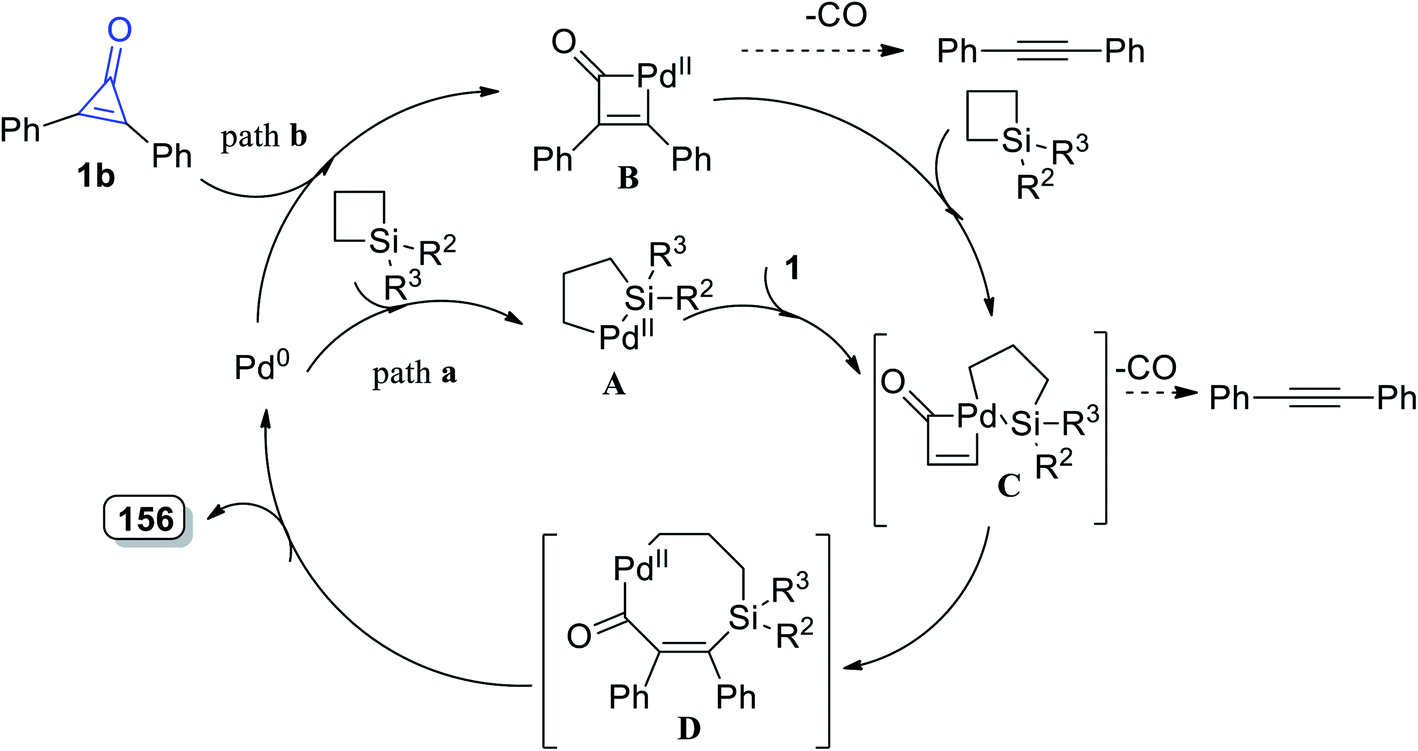
2.3.4. Synthesis of seven-membered-ring heterocycles. Tetrasubstituted-6,7-dihydro-1H-silepin-4(5H)-ones 156a–h were obtained in 65–89% yield by refluxing 1,1-disubstituted siletanes 155a–h with cyclopropenone derivatives 1 in toluene at room temperature for 48 h, and Pd(OAc)2 (Scheme 92).127 The reaction was initiated via bond cleavage of silacyclobutane (155): the C–Si bond of silacyclobutane was broken through an oxidative addition, which was proposed to give palladacycle A as the first step. Then, after a series of oxidative additions to the C–C bond of cyclopropenone 1b, intermediate C was formed (Scheme 93). The eight-membered palladacycle D was formed via the reductive elimination of intermediate C, which might go through a second reductive elimination step to produce the final product 156 and form the palladium (0) catalyst again. The possibility of path b could not be completely excluded, which would first involve the cleavage of the C–C σ-bond of cyclopropenone (1) (Scheme 93).127
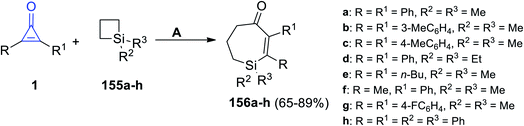 |
| | Scheme 92 Synthesis of tetrasubstituted-6,7-dihydro-1H-silepin-4(5H)-ones 156a–h. Reagents and conditions: A = (1 mol%), Pd(OAc)2 toluene, rt, 48 h. | |
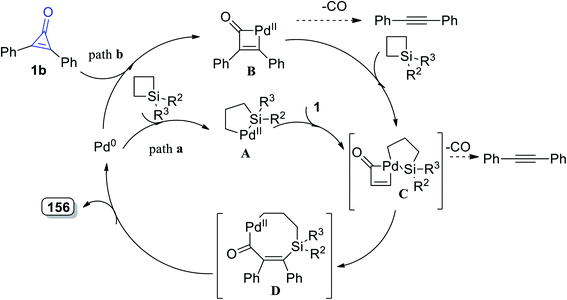 |
| | Scheme 93 Mechanism of the formation of 6,7-dihydro-1H-silepin-4(5H)-ones 157a–h. | |
2.3.5. Synthesis of nine-membered-ring heterocycles. When compounds 157a–c reacted with 2,3-diphenylcyclopropenone (1b) in absolute ethanol, intermediate 158 was formed followed by the formation of nine-membered-ring bicyclic products 159a–c in 51–62% yield (Scheme 94).128
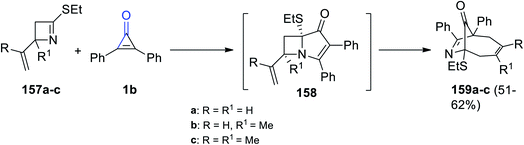 |
| | Scheme 94 Synthesis of compounds 159a–c. Reagents and conditions: A = EtOH, reflux. | |
3 Conclusion
In summary, the use of cyclopropane derivatives for the construction of various heterocycles can provide a practical alternative to traditional methods for the preparation of such compounds. Since cyclopropenones have high strain, they easily participated in various reactions and, therefore, in the construction of various organic molecules. In that review, we consider the utility of cyclopropenones in the synthesis of heterocycles, especially those of prospective biologically active compounds. The reactions of cyclopropenones are described as highly regioselective, often stereoselective, and permit the synthesis of a variety of heterocyclic systems, both saturated and unsaturated. The substrates employed in most cases are simple (and often commercially available), making the methods amenable to the rapid construction of diverse collections of compounds.
Author contributions
A. A. Aly (conceptualization, writing, editing, and submitting), Alaa A. Hassan, Sara M. Mostafa (supervision), Asmaa. H. Mohamed (editing of revision), Esraa M. Osman (methodology and writing a draft). A. A. Nayl (editing of revision). All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
References
- K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371–1427, DOI:10.1021/cr010011q.
- G. Dong, C-C Bond Activation, Springer, Berlin, 1st edn, 2014, vol. 346, pp. 1–258 Search PubMed.
- W. D. Jones, Nature, 1993, 364, 676–677, DOI:10.1038/364676a0.
- B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883, DOI:10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3.
- C. H. Jun, Chem. Soc. Rev., 2004, 33, 610–618, 10.1039/B308864M.
- C. Aissa, Synthesis, 2011, 21, 3389–3407, DOI:10.1055/s-0030-1260233.
- N. Cramer and T. Seiser, Synlett, 2011, 4, 449–460, DOI:10.1055/s-0030-1259536.
- M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100–1105, 10.1039/C0CC02566F.
- A. Korotvicka, D. Necas and M. Kotora, Curr. Org. Chem., 2012, 16, 1170–1214, DOI:10.2174/138527212800564213.
- A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567–581, 10.1039/C4QO00053F.
- I. Marek, A. Masarwa, P. O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414–429, DOI:10.1002/anie.201405067.
- L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464, DOI:10.1021/acs.chemrev.5b00138.
- M. Murakami and N. J. Ishida, Am. Chem. Soc., 2016, 138, 13759–13769, DOI:10.1021/jacs.6b01656.
- D. S. Kim, W. J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015, DOI:10.1021/acs.chemrev.6b00554.
- P. H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360, DOI:10.1021/acscatal.6b03210.
- Q. Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590–4627, 10.1039/C6CS00107F.
- B. Zhao, T. Rogge, L. Ackermann and Z. Shi, Chem. Soc. Rev., 2021, 50, 8903–8953, 10.1039/C9CS00571D.
- R. Vicente, Chem. Rev., 2021, 121, 162–226, DOI:10.1021/acs.chemrev.0c00151.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139, DOI:10.1021/acs.chemrev.0c00160.
- M. Murakami and N. Ishida, Chem. Rev., 2021, 121, 264–299, DOI:10.1021/acs.chemrev.0c00569.
- X. Wang, C. Yu, I. L. Atodiresei and F. W. Patureau, Org. Lett., 2022, 24, 1127–1131, DOI:10.1021/acs.orglett.1c04045.
- H. Huo and Y. Gong, Org. Biomol. Chem., 2022, 20, 3847–3869, 10.1039/D1OB02450G.
- L. Qua, Y. Wu, P. Sun, K. Zhang and Z. Liu, Ploymer, 2017, 114, 36–43, DOI:10.1016/j.polymer.2017.02.071.
- H. L. Ammon, J. Am. Chem. Soc., 1973, 95, 7093–7101, DOI:10.1021/ja00802a033.
- R. Breslow, T. Eicher, A. Krebs, R. A. Peterson and J. Posner, J. Am. Chem. Soc., 1965, 87, 1320–1325, DOI:10.1021/ja01084a029.
- R. Breslow, R. Haynie and J. Mirra, J. Am. Chem. Soc., 1959, 81(1), 247–248 CrossRef CAS.
- O. Körner, R. Gleiter and F. Rominger, Synthesis, 2009, 2009, 3259–3262, DOI:10.1055/s-0029-1216934.
- K. Köhler and W. E. Piers, Can. J. Chem., 1998, 76, 1249–1255, DOI:10.1139/v98-170.
- A. Greenberg, R. P. T. Tomkins, M. Dobrovolny and J. F. Liebman, J. Am. Chem. Soc., 1983, 105, 6855–6858, DOI:10.1021/ja00361a018.
- A. Kascheres, A. C. Joussef and H. C. Duarte, Tetrahedron Lett., 1983, 24, 1837–1840, DOI:10.1016/S0040-4039(00)81785-2.
- P. A. Wender, T. J. Paxton and T. J. Williams, J. Am. Chem. Soc., 2006, 128, 14814–14815, DOI:10.1021/ja065868p.
- T. Matsuda and Y. Sakurai, Eur. J. Org. Chem., 2013, 2013, 4219–4222, DOI:10.1002/ejoc.201300220.
- L. Kong, X. Zhou, Y. Xu and X. Li, Org. Lett., 2017, 19, 3644–3647, DOI:10.1021/acs.orglett.7b01650.
- C. Jenny, D. Obrecht and H. Heimgartner, Tetrahedron Lett., 1982, 23, 3059–3060, DOI:10.1016/S0040-4039(00)87532-2.
- K. T. Potts and J. S. Baum, Chem. Rev., 1974, 74, 189–213, DOI:10.1021/cr60288a003.
- P. C. Venneri and J. Warkentin, Can. J. Chem., 2000, 78, 1194–1203, DOI:10.1139/v00-118.
- R. Noyori, I. Umeda and H. Takaya, Chem. Lett., 1972, 1, 1189–1190, DOI:10.1246/cl.1972.1189.
- A. Rai and L. D. S. Yadav, Eur. J. Org. Chem., 2013, 2013, 1889–1893, DOI:10.1002/ejoc.201300059.
- T. Stach, J. Dräger and P. H. Huy, Org. Lett., 2018, 20, 2980–2983, DOI:10.1021/acs.orglett.8b01023.
- N. K. Urdabayev, A. Poloukhtine and V. V. Popik, Chem. Commun., 2006, 4, 454–456, 10.1039/B513248G.
- A. G. Lvov, N. A. Milevsky, V. Z. Shirinian and M. M. Krayushkin, Chem. Heterocycl. Compd., 2015, 51, 933–935, DOI:10.1007/s10593-015-1800-8.
- C. D. McNitt, H. Cheng, S. Ullrich, V. V. Popik and M. Bjerknes, J. Am. Chem. Soc., 2017, 139, 14029–14032, DOI:10.1021/jacs.7b08472.
- Y. Chiang, A. Kresge and V. Popik, J. Am. Chem. Soc., 1995, 117, 9165–9171, DOI:10.1021/ja00141a007.
- A. Poloukhtine and V. V. Popik, J. Org. Chem., 2003, 68, 7833–7840, DOI:10.1021/jo034869m.
- A. Poloukhtine and V. V. Popik, J. Org. Chem., 2006, 71, 7417–7421, DOI:10.1021/jo061285m.
- K. Mishiro, T. Kimura, T. Furuyama and M. Kunishima, Org. Lett., 2019, 21, 4101–4105, DOI:10.1021/acs.orglett.9b01280.
- A. Schuster-Haberhauer, R. Gleiter, O. Körner, A. Leskovar, D. B. Werz, F. R. Fischer and F. Rominger, Organometallics, 2008, 27, 1361–1366, DOI:10.1021/om701115w.
- T. Okuda, K. Yokose, T. Furumai and H. B. Maruyama, J. Antibiot., 1984, 37, 718–722, DOI:10.7164/antibiotics.37.718.
- F. Bohlmann, J. Jakupovic, L. Müller and A. Schuster, Angew. Chem., Int. Ed., 1981, 20, 292–293, DOI:10.1002/anie.198102921.
- T. Okuda, N. Shimma and T. Furumai, J. Antibiot., 1984, 37, 723–727, DOI:10.7164/antibiotics.37.723.
- H. Kogen, T. Kiho, K. Tago, S. Miyamoto, T. Fujioka, N. Otsuka, K. Suzuki-Konagai and T. Ogita, J. Am. Chem. Soc., 2000, 122, 1842–1843, DOI:10.1021/ja992355s.
- H. Tokuyama, M. Isaka, E. Nakamura, R. Ando and Y. Morinaka, J. Antibiot., 1992, 45, 1148–1154, DOI:10.7164/antibiotics.45.1148.
- R. Thuangtong, S. Varothai, D. Triwongwaranat and C. Rujitharanawong, J. Med. Assoc. Thailand, 2017, 100, 86–92 Search PubMed , PMID: 29911776..
- G. Singh and M. Lavanya, Int. J. Trichol., 2010, 2, 36, DOI:10.4103/0974-7753.66911.
- J. A. Upitis and A. Krol, J. Cutaneous Med. Surg., 2002, 6, 214–217, DOI:10.1007/s10227-001-0050-9.
- K. K. Veverka, J. W. Jakub and C. L. Baum, Dermatol. Surg., 2018, 44, 1501–1508, DOI:10.1097/DSS.0000000000001603.
- C. Robles-Planells, S. A. Michelson, J. Mena, D. Escrig, J. L. Rojas, G. Sanchez-Guerrero, R. Hernández, C. Barrera-Avalos, L. E. Rojo and D. Sauma, Front. Pharmacol., 2019, 10, 1201, DOI:10.3389/fphar.2019.01201.
- R. Breslow and G. Ryan, J. Am. Chem. Soc., 1967, 89, 3073, DOI:10.1021/ja00988a063.
- Z. L. Cheng and Q. Y. Chen, Chin. J. Chem., 2006, 24, 1219–1224, DOI:10.1002/cjoc.200690227.
- M. Nakamura, H. Isobe and F. Nakamura, Chem. Rev., 2003, 103, 1295–1326, DOI:10.1021/cr0100244.
- R. Breslow, L. J. Altman, A. Krebs, E. Mohacsi, I. Murata, R. A. Peterson and J. Posner, J. Am. Chem. Soc., 1965, 87, 1326–1331, DOI:10.1021/ja01084a030.
- D. B. Werz, R. Gleiter and F. Rominger, Eur. J. Org. Chem., 2003, 2003, 151–154, DOI:10.1002/1099-0690(200301)2003:1<151::AID-EJOC151>3.0.CO;2-7.
- J. F. McGarrity, J. Prodolliet and T. Smyth, Org. Magn. Reson., 1981, 17, 59–65, DOI:10.1002/mrc.1270170114.
- P. A. Wender, T. J. Paxton and T. J. Williams, J. Am. Chem. Soc., 2006, 128, 14814–14815, DOI:10.1021/ja065868p.
- Z. X. Zhao, H. Y. Wang, C. Xu and Y. L. Guo, Rapid Commun. Mass Spectrom., 2010, 24, 2665–2672, DOI:10.1002/rcm.4694.
- O. J. Curnow, G. M. Fern and R. J. Pipal, ARKIVOC, 2006,(iii), 43–47 Search PubMed.
- Z. Yoshida, H. Konishi, Y. Tawara, K. Nishikawa and H. Ogoshi, Tetrahedron Lett., 1973, 14, 2619–2622, DOI:10.1016/S0040-4039(00)87412-2.
- K. Elena, K. Tatiana, L. Irina, D. C. Jorge, H. O. Simon, M. I. Daniel and M. G. Marcos, Open Org. Chem. J., 2008, 2, 35–40, DOI:10.2174/1874095200801020035.
- S. W. Tobey and R. West, J. Am. Chem. Soc., 1964, 86, 4215–4216, DOI:10.1021/ja01073a075.
- P. J. Whitman and B. M. Trost, J. Am. Chem. Soc., 1969, 91, 7534–7535, DOI:10.1021/ja01054a067.
- S. S. Nguyen, A. Ferreira, Z. G. Long, T. K. Heiss, R.
S. Dorn, R. D. Row and J. A. Prescher, Org. Lett., 2019, 21, 8695–8699, DOI:10.1021/acs.orglett.9b03298.
- S. Cunha, J. C. Serafim, L. L. B. de Santana, F. Damasceno, J. T. M. Correia, A. O. Santos, M. Oliveira, J. Ribeiro, J. Amparo and S. L. Costa, J. Heterocycl. Chem., 2017, 54, 3700–3710, DOI:10.1002/jhet.2921.
- A. Kascheres, H. C. Schumacher and R. A. Rodrigues, J. Heterocycl. Chem., 1997, 34, 757–759, DOI:10.1002/jhet.5570340309.
- M. A. M. Gomaa, J. Chem. Soc., Perkin Trans. 1, 2002, 3, 341–344, 10.1039/B109711N.
- A. Haito and N. Chatani, Chem. Commun., 2019, 55, 5740–5742, 10.1039/C9CC02397F.
- T. Nanda and P. Ravikumar, Org. Lett., 2020, 22, 1368–1374, DOI:10.1021/acs.orglett.9b04656.
- N. Lozinskaya, S. Sosonyuk, Y. N. Firsova, M. Proskurnina and N. Zefirov, Russ. Chem. Bull., 2011, 60, 1989–1994, DOI:10.1007/s11172-011-0301-x.
- X. Li, C. Han, H. Yao and A. Lin, Org. Lett., 2017, 19, 778–781, DOI:10.1021/acs.orglett.6b03737.
- K. Reitel, K. Kriis, I. Järving and T. Kanger, Chem. Heterocycl. Compd., 2018, 54, 929–933, DOI:10.1007/s10593-018-2372-1.
- T. Matsuda, Y. Tabata and H. Suzuki, New J. Chem., 2018, 42, 19178–19182, 10.1039/C8NJ04579H.
- J. T. Ren, J. X. Wang, H. Tian, J. L. Xu, H. Hu, M. Aslam and M. Sun, Org. Lett., 2018, 20, 6636–6639, DOI:10.1021/acs.orglett.8b02612.
- D. Bai, Y. Yu, H. Guo, J. Chang and X. Li, Angew. Chem., Int. Ed., 2020, 59, 2740–2744, DOI:10.1002/anie.201913130.
- K. Takahashi and S. Tarutani, J. Chem. Soc., Chem. Commun., 1994, 4, 519–520, 10.1039/C39940000519.
- T. Matsuda and Y. Sakurai, J. Org. Chem., 2014, 79, 2739–2745, DOI:10.1021/jo500045n.
- J. Xu, J. Cao, C. Fang, T. Lu and D. Du, Org. Chem. Front., 2017, 4, 560–564, 10.1039/C6QO00734A.
- S. Cunha and Z. N. da Rocha, Quim. Nova, 2008, 31, 788–792, DOI:10.1590/S0100-40422008000400015.
- A. R. Rivero, I. Fernandez, C. R. de Arellano and M. A. Sierra, J. Org. Chem., 2015, 80, 1207–1213, DOI:10.1021/jo502292y.
- J. R. Syu, C. H. Lin, C. W. Kuo and D. Y. Yang, Tetrahedron Lett., 2014, 55, 1207–1211, DOI:10.1016/j.tetlet.2013.12.115.
- Y. Shi, H. Xing, T. Huang, X. Liu, J. Chen, X. Guo, G. B. Li and Y. Wu, Chem. Commun., 2020, 56, 1585–1588, 10.1039/C9CC08926H.
- M. C. Nakhla and J. L. Wood, J. Am. Chem. Soc., 2017, 139, 18504–18507, DOI:10.1021/jacs.7b12570.
- S. Cunha, F. Damasceno and J. Ferrari, Tetrahedron Lett., 2007, 48, 5795–5798, DOI:10.1016/j.tetlet.2007.06.087.
- L. Yao, Q. Hu, L. Bao, W. Zhu and Y. Hu, Org. Lett., 2021, 23, 4971–4975, DOI:10.1021/acs.orglett.1c01304.
- S. J. Zhou, X. Cheng and J. Xuan, Asian J. Org. Chem., 2019, 8, 1376–1379, DOI:10.1002/ajoc.201900272.
- M. Lange, J. C. Tendyck, P. Wegener, A. Hepp, E. U. Würthwein and W. Uhl, Chem.–Eur. J., 2018, 24, 12856–12868, DOI:10.1002/chem.201706089.
- J. Wu, W. X. Gao, X. B. Huang, Y. B. Zhou, M. C. Liu and H. Y. Wu, Org. Lett., 2020, 22, 5555–5560, DOI:10.1021/acs.orglett.0c01914.
- L. H. Li, Y. Jiang, J. Hao, Y. Wei and M. Shi, Adv. Synth. Catal., 2017, 359, 3304–3310, DOI:10.1002/adsc.201700936.
- A. A. Hassan, F. F. Abdel-Latif, A. M. N. El-Din, S. M. Mostafa, M. Nieger and S. Braese, Tetrahedron, 2012, 68, 8487–8492, DOI:10.1016/j.tet.2012.07.063.
- A. A. Aly, A. A. Hassan, M. A. Ameen and A. B. Brown, Tetrahedron Lett., 2008, 49, 4060–4062, DOI:10.1016/j.tetlet.2008.04.066.
- P. Zhou, W. T. Yang, A. U. Rahman, G. Li and B. Jiang, J. Org. Chem., 2019, 85, 360–366, DOI:10.1021/acs.joc.9b02253.
- W. T. Zhao, X. Y. Tang and M. Shi, Eur. J. Org. Chem., 2014, 2014, 2672–2676, DOI:10.1002/ejoc.201400077.
- J. Wallbaum, P. G. Jones and D. B. Werz, J. Org. Chem., 2015, 80, 3730–3734, DOI:10.1021/acs.joc.5b00330.
- L. Liu, J. Li, W. Dai, F. Gao, K. Chen, Y. Zhou and H. Liu, Molecules, 2020, 25, 268, DOI:10.3390/molecules25020268.
- J. Cao, R. Fang, J. Y. Liu, H. Lu, Y. C. Luo and P. F. Xu, Chem.–Eur. J., 2018, 24, 18863–18867, DOI:10.1002/chem.201803861.
- J. Chen, B. Tang, X. Liu, G. Lv, Y. Shi, T. Huang, H. Xing, X. Guo, L. Hai and Y. Wu, Org. Chem. Front., 2020, 7, 2944–2949, 10.1039/D0QO00769B.
- S. W. Liu, C. Yuan, X. F. Jiang, X. X. Wang and H. L. Cui, Asian J. Org. Chem., 2020, 9, 82–85, DOI:10.1002/ajoc.201900630.
- D. H. Wadsworth, C. H. Weidner, S. L. Bender, R. H. Nuttall and H. R. Luss, J. Org. Chem., 1989, 54, 3652–3660, DOI:10.1021/jo00276a028.
- T. Kondo, Y. Kaneko, Y. Taguchi, A. Nakamura, T. Okada, M. Shiotsuki, Y. Ura, K. Wada and T.-a. Mitsudo, J. Am. Chem. Soc., 2002, 124, 6824–6825, DOI:10.1021/ja0260521.
- W.-Q. Zhu, Y. C. Fang, W. Y. Han, F. Li, M. G. Yang and Y.-Z. Chen, Org. Chem. Front., 2021, 8, 3082–3090, 10.1039/D1QO00222H.
- Q. Chen, Y. Teng and F. Xu, Org. Lett., 2021, 23, 4785–4790, DOI:10.1021/acs.orglett.1c01506.
- S. Ahmad, L. Shukla, J. Szawkalo, P. Roszkowski, J. K Maurin and Z. Czarnocki, Catalysis Communications, 2017, 89, 44–47, DOI:10.1016/j.catcom.2016.10.008.
- M. Takahashi, Y. Kadowaki, Y. Uno and Y. Nakano, Heterocycles, 1999, 51(9), 637–639, DOI:10.3987/COM-99-8652.
- A. A. Aly, M. Ramadan, M. A. Al-Aziz, H. M. Fathy, S. Bräse, A. B. Brown and M. Nieger, J. Chem. Res., 2016, 40(10), 637–639, DOI:10.3184/174751916X14743924874916.
- A. A. Aly, A. M. Nour El-Din, M. A.-M. Gomaa, A. B. Brown and A. S. Fahmi, Unusual Reactivity of 2,3-diphenylcyclopropenone towards N-imidoylthioureas; Facile Synthesis of 3-aryl-2,5,6-triphenylpyrimidin-4(3H)-one (PART III), J. Chem. Res., 2007, 2007(8), 439–441, DOI:10.3184/030823407X234563.
- R. D. Mohan and A. Jose, Asian J. Chem., 2018, 30, 1075–1077 CrossRef CAS.
- B. Niu, B. Jiang, L. Z. Yu and M. Shi, Org. Chem. Front., 2018, 5, 1267–1271, 10.1039/C8QO00091C.
- Y. F. Yang, X. B. Huang, W. X. Gao, Y. B. Zhou, M. C. Liu and H. Y. Wu, Org. Biomol. Chem., 2020, 18, 5822–5825, 10.1039/D0OB00601G.
- L. Sizhan, C. Mingyue, W. Bowen, H. Chunmei, Z. Yingying, L. Jing, X. Xuetao, W. Zhen and W. Shaohua, Chin. J. Org. Chem., 2021, 41, 1622–1630, DOI:10.6023/cjoc202010041.
- T. Matsuda, K. Yamanaka, Y. Tabata and T. Shiomi, Tetrahedron Lett., 2018, 59, 1458–1460, DOI:10.1016/j.tetlet.2018.02.084.
- A. A. Hassan, N. K. Mohamed, A. A. Aly, H. N. Tawfeek, H. Hopf, S. Bräse and M. Nieger, Mol. Diversity, 2019, 23, 821–828, DOI:10.1007/s11030-018-09912-5.
- E. M. El-Sheref, J. Sulfur Chem., 2017, 38, 625–634, DOI:10.1080/17415993.2017.1337121.
- Y. Shi, T. Huang, T. Wang, J. Chen, X. Liu, Z. Wu, X. Huang, Y. Zheng, Z. Yang and Y. Wu, Chem.–Eur. J., 2021, 27, 13346–13351, DOI:10.1002/chem.202101839.
- S. Ahmad, L. Shukla, J. Szawkało, P. Roszkowski, J. K. Maurin and Z. Czarnocki, Catal. Commun., 2017, 89, 44–47, DOI:10.1016/j.catcom.2016.10.008.
- O. B. Østby, L. L. Gundersen, F. Rise, Ø. Antonsen, K. Fosnes, V. Larsen, A. Bast, I. Custers and G. R. Haenen, Arch. Pharm., 2001, 334, 21–24, DOI:10.1002/1521-4184(200101)334:1<21::AID-ARDP21>3.0.CO;2-W.
- A. Molchanov, D. Sipkin, Y. B. Koptelov and R. Kostikov, Russ. J. Org. Chem., 2005, 41, 567–574, DOI:10.1007/s11178-005-0205-z.
- A. A. Aly, A. A. Hassan, M. A.-M. Gomaa and E. M. El-Sheref, ARKIVOC, 2007, xiv, 1–11, DOI:10.3998/ark.5550190.0008.e01.
- K. Matsumoto, A. Okada, T. Girek, Y. Ikemi, J. C. Kim, N. Hayashi, H. Yoshida and A. Kakehi, Heterocycl. Commun., 2002, 8, 325–328, DOI:10.1515/HC.2002.8.4.325.
- W. T. Zhao, F. Gao and D. Zhao, Angew. Chem., 2018, 130, 6437–6440, DOI:10.1002/ange.201803156.
- K. Hemming, P. A. O'Gorman and M. I. Page, Tetrahedron Lett., 2006, 47, 425–428, DOI:10.1016/j.tetlet.2005.11.081.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Alaa A. Hassana,
Sara M. Mostafa
*a,
Alaa A. Hassana,
Sara M. Mostafa