
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed stereo- and regioselective hydrophosphorylation of terminal alkynes: scope and mechanistic study†
Junchen Li,
Zhenhua Gao,
Yongbiao Guo ,
Haibo Liu,
Peichao Zhao,
Xiaojing Bi*,
Enxue Shi* and
Junhua Xiao*
,
Haibo Liu,
Peichao Zhao,
Xiaojing Bi*,
Enxue Shi* and
Junhua Xiao*
State Key Laboratory of NBC Protection for Civilian, Beijing, P. R. China. E-mail: xiao.junhua@pku.edu.cn; exshi@sina.com; xiaojingbimail@yeah.net
First published on 29th June 2022
Abstract
Herein, a protocol for copper-catalyzed highly stereo- and regioselective hydrophosphorylation of terminal alkynes to E-alkenylphosphorus compounds was well developed. It represents a general and practical hydrophosphorylation method, of which diarylphosphine oxide, dialkylphosphine oxide and dialkyl phosphite all had effective P(O)H parts to react with different types of terminal alkynes. Contrary to previous air-sensitive reports, all the reactions proceeded well under air. This methodology is quite attractive owing to the high stereo- and regioselectivity, good functional group tolerance, scalability and facile late-stage derivatization of some natural product derivatives and commercially available herbicides. What's more, investigations on the reaction mechanism with deuterium-labeling experiments and DFT studies firstly disclosed the deprotonation–protonation equilibrium of terminal alkynes and P(O)H part during the catalytic hydrophosphorylation process.
Introduction
The importance of the ubiquitous organophosphorus motifs is underscored by their abundance in the applied sciences, especially with respect to catalysis, pharmaceuticals, agrochemicals, and flame retardants.1 The phosphorus-based reagents enable both the effective diversification of simple materials and late-stage modification of complex molecular architectures.2 In particular, alkenylphosphorus compounds have served as a robust type of synthon for the preparation of versatile functional compounds.3 Despite the importance of alkenylphosphorus compounds, general and efficient methods for their preparation are rather limited.Transition metal-catalyzed protocols based on the catalysts of palladium,4 nickel,5 rhodium6 and copper7 were proved to be effective for synthesis of alkenylphosphorus compounds. Since Tanaka and Han's pioneering work,4g catalytic hydrophosphorylation of carbon–carbon triple bonds with different types of P(O)H compounds has become one of the most straightforward and efficient methods. In addition to the selectivity of Markovnikov and anti-Markovnikov addition for hydrophosphorylation, the precise regulation of E/Z-selectivity of the multisubstituted alkenylphosphorus products is still a hot research topic (Scheme 1a). Recently, Han et al., successively realized Pd4f,g-, Ni5a-, and Rh6a-catalyzed hydrophosphorylation of alkynes with different types of P(O)H compounds (Scheme 1b). Remarkably, the absystem that Pd-catalyzed phosphorylation of alkynes adjusted to a very wide spectrum of P(O)H compounds, such as H-phosphonates, H-phosphinates, secondary phosphine oxides, and hypophosphinic acid while using expensive and toxic palladium catalyst.4f Zhao et al., disclosed that the catalyst system of CuI/ethylenediamine was quite effective for catalyzing anti-Markovnikov hydrophosphorylation of terminal alkynes for synthesis of E-alkenylphosphine oxides7b,d (Scheme 1c). Their systematic mechanistic investigation and DFT studies indicated that oxygen-free phosphorylation is favorable either kinetically or thermodynamically under their conditions.7d,e Beletskaya et al., reported a catalytic system of Cu(acac)2 without any base for the hydrophosphoryhlation of alkynes under argon atmosphere.7c Though having achieved these gratifying research results, these protocols also suffer from relatively narrow substrate scope and lack of stereoselectivity for some designated kinds of substrates, thus limiting their further applications.
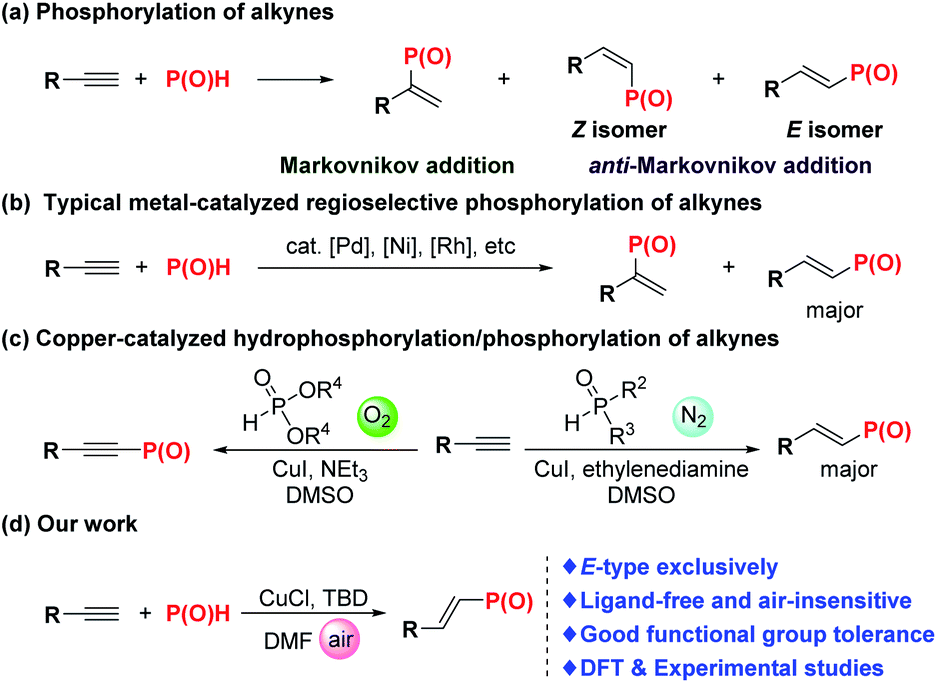 | ||
| Scheme 1 Typical hydrophosphorylation of terminal alkynes and our work. | ||
Herein, we report one general, efficient and highly stereo- and regioselective methodology for hydrophosphorylation of terminal alkynes to E-alkenylphosphorus products catalyzed by a copper salt under ligand-free conditions. Unlike the previous catalytic system, oxygen shows no obvious side effect on this reaction and N2 protection is unnecessary, which makes this protocol more convenient to be handled. Moreover, diarylphosphine oxide, dialkylphosphine oxide and dialkyl phosphonate were all verified to be effective for this hydrophosphorylation reaction. Note that practicality of this reaction got well demonstrated via its scalability and facile late-stage derivatization of some natural product derivatives and one commercially available herbicide. Most importantly, the catalytic mechanism was well investigated through DFT studies along with several mechanistic experiments.
Results and discussion
As our continuous research interests for developing efficient and convenient methodology on constructing organophosphorus compounds, the catalytic hydrophosphorylation of terminal alkynes to E-alkenylphosphorus compounds was explored. Firstly, methyl propiolate and diethyl phosphite were selected as the model substrates to carry out the solvent evaluation under the catalysis of CuI with N,N,N′,N′-tetramethylethylenediamine (TMEDA) as the additive (for details of solvent evaluation, please see ESI†). The results indicated that DMF was the best solvent of choice, and this system was regio- and stereospecific, affording only the E-alkenylphosphorus product 3aa in 13% yield without detection of any other isomers or byproducts (Table 1, entry 1, for details, please see ESI†). Then different kinds of organic bases were screened as the reaction additive, among which DBN, MTBD, DBU, DABCO and Et3N all led to the failure of detecting the desired product (Table 1, entries 2–4 and 6–7). It's very different from previous work7d,e that neither alkenylphosphonate nor alkynylphosphonate was produced under previous catalytic system of CuI/Et3N in DMF (entry 7). When TMG was utilized as the additive, product 3aa could be detected in 11% yield (entry 5). Gratifyingly, 29% yield was obtained when stronger organic base TBD was employed in the hydrophosphorylation reaction (entry 8). After that, a slightly better yield was obtained with increase of the reaction temperature to 80 °C (entry 9). Surprisingly, further augment of the temperature to 100 °C, the yield was sharply reached to 84% (entry 10). While only 67% yield was observed when the reaction was conducted at 120 °C (entry 11). Therefore, we set down 100 °C as the optimized reaction temperature. Frustratingly, further lowering the catalyst loading from 30 mol% to 15 mol% resulted in a dramatic loss of yield (entry 12). Finally, a series of copper salts were tested to catalyze the title reaction (entries 13–18). Among them, CuCl gave the best catalytic performance with a shorter reaction time, providing the title product in 88% yield (entry 14). However, Cu(II)Cl2 showed no catalytic activity at all as compared with its Cu(I) counterpart (entries 14 &15). Interestingly, almost the same yield was gained when carrying out the reaction under N2 atmosphere (entries 14 & 19). Note that the reaction did not work at all in the absence of TBD (entry 20). Therefore, based on the above investigation of reaction conditions, it was disclosed that a system consisting of CuCl (30 mol%), TBD (30 mol%) in DMF at 100 °C was optimal and the reaction time is about 5 h.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Temp. (oC) | Yield (%)b |
| a Conditions: 1a (0.3 mmol), diethyl phosphite 2a (0.36 mmol), copper salt (30 mol%), additive (30 mol%), DMF (2 mL), air, 14 h.b Yields determined by GC-MS.c 15 mol% of CuI instead.d 5 h.e 5 h with in N2 atmosphere. DBN: 1,5-Diazabicyclo[4.3.0]non-5-ene; MTBD: 1,3,4,6,7,8-Hexahydro-1-methyl-2H-pyrimidol[1,2-a]pyrimidine; TBD: 1,3,4,6,7,8-Hexahydro-2H-pyrimidol[1,2-a]pyrimidine; DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene; DABCO: 1,4-Diazabicyclo[2.2.2]octane. | ||||
| 1 | CuI | TMEDA | r.t | 13 |
| 2 | CuI | DBN | r.t | 0 |
| 3 | CuI | MTBD | r.t | 0 |
| 4 | CuI | DBU | r.t | 0 |
| 5 | CuI | TMG | r.t | 11 |
| 6 | CuI | DABCO | r.t | 0 |
| 7 | CuI | Et3N | r.t | 0 |
| 8 | CuI | TBD | r.t | 29 |
| 9 | CuI | TBD | 80 | 32 |
| 10 | CuI | TBD | 100 | 84 |
| 11 | CuI | TBD | 120 | 67 |
| 12c | CuI | TBD | 100 | 32 |
| 13 | CuBr | TBD | 100 | 83 |
| 14d | CuCl | TBD | 100 | 88 |
| 15 | CuCl2 | TBD | 100 | 0 |
| 16 | CuCN | TBD | 100 | 12 |
| 17 | CuOTf | TBD | 100 | 6 |
| 18 | Cu(MeCN)4PF6 | TBD | 100 | 48 |
| 19e | CuCl | TBD | 100 | 87 |
| 20 | CuCl | — | 100 | 0 |

Using the optimal conditions, we carried out reactions of methyl propiolate with different types of P(O)H compounds to explore the generality of this transformation (Scheme 2). At first, some H-phosphonates were subjected to the optimal condition, which all readily reacted with methyl propiolate (1a) to afford a series of the corresponding E-hydrophosphorylation products. Among them, diethyl, diisopropyl, dibutyl, diisobutyl, diphenyl and dibenzyl phosphonates and 4,4,5,5-tetramethyl-1,3,2-dioxaphospholane 2-oxide were smoothly converted to the desired products with good to excellent yields (3aa, 3ac–3ah). Dimethyl phosphonate showed relatively lower reactivity and afforded the product with only 49% isolated yield (3ab). After that, two of H-phosphinates were also tested under the optimal conditions, giving the corresponding products in 81% and 91% yields, respectively (3ai and 3aj). Then, a series of different substituted diarylphosphine oxides were found to be suitable for the title hydrophosphorylation with 1a. Diphenylphosphine oxide was proved to be quite active, thus offering the product 3ak in excellent yield (92% yield). The ortho-substituents on the phenyl ring showed a little negative effect on this reaction with formation of products 3al and 3am in 63% and 86% yields respectively. The more sterically hindered mesityl group led to a further decrease of the reaction yield (3an, 55% yield). After that, substituents at para position of the aryl group in diarylphosphine oxides were also investigated. No matter electron-donating methyl and tert-butyl group or electron-withdrawing CF3 group all had nearly no effect on the reaction efficiency, smoothly providing the desired E-alkenylphosphine oxides in good to excellent yields (3ao–3aq). Whereas, di(p-PhC6H4)phosphine oxide only gave 35% yield of compound 3ar under the optimal condition. 3,5-Disubstituted diphenylphosphine oxides were proved to be proper substrates for this hydrophosphorylation reaction yielding the corresponding products in 64% and 84% yields (3as and 3at). Note that α- and β-naphthyl substituted phosphine oxides showed the similar reactivity with the desired products both in 74% yield (3au and 3av).
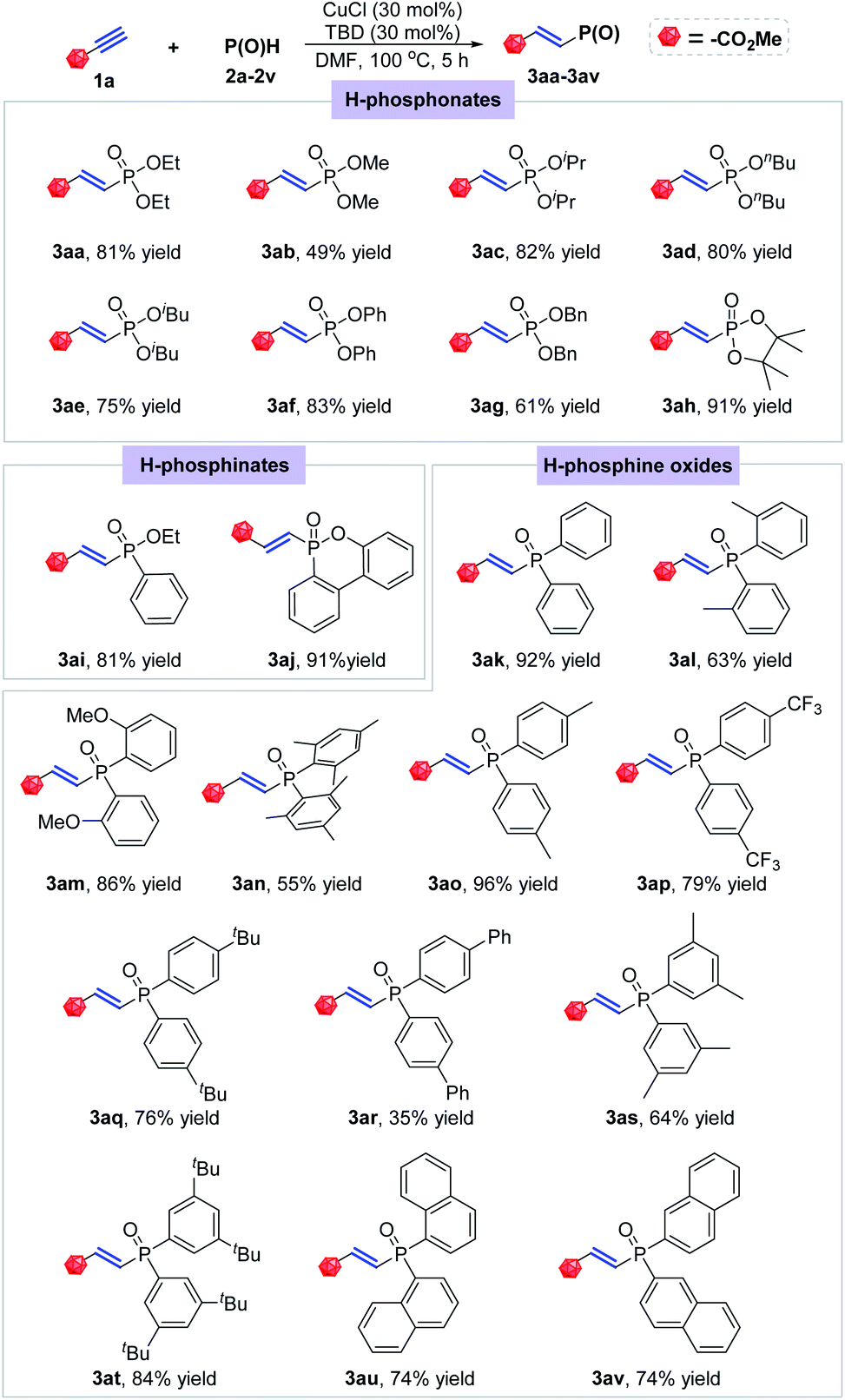 | ||
| Scheme 2 Substrate scope for P(O)H. Conditions: 1a (0.3 mmol), P(O)H (0.36 mmol), CuCl (30 mol%), TBD (30 mol%), DMF (2 mL), 100 °C, 5 h. | ||
In order to further explore the generality of this catalytic system, different kinds of terminal alkynes were tested to react with diphenylphosphine oxide (2k) in the presence of CuCl and organic base TBD (Scheme 3). Firstly, a series of propargyl esters were selected to react with 2k under the standard conditions, among which ethyl, isopropyl, isobutyl, tert-butyl and pentyl propiolates (1b–1f) were all smoothly converted to the desired E-alkenylphosphine oxides in good to excellent yields (3bk–3fk). Note that amide group in the propargyl ester substrate kept untouched under such conditions, providing the desired product 3gk and 3hk in 81% and 92% yields, respectively. After that, aliphatic terminal alkynes were also introduced to the catalytic conditions, offering the E-alkenylphosphine oxides smoothly with medium to excellent yields (3jk–3pk). Notably, the hydroxyl group had nearly no effect on the title reaction (3lk and 3mk). Finally, another kind of terminal alkynes bearing P(O) groups were also verified to be effective substrates for the title hydrophosphorylation (3qk–3vk), from which the products possibly acted as significant intermediates for bidentate phosphine ligand synthesis.
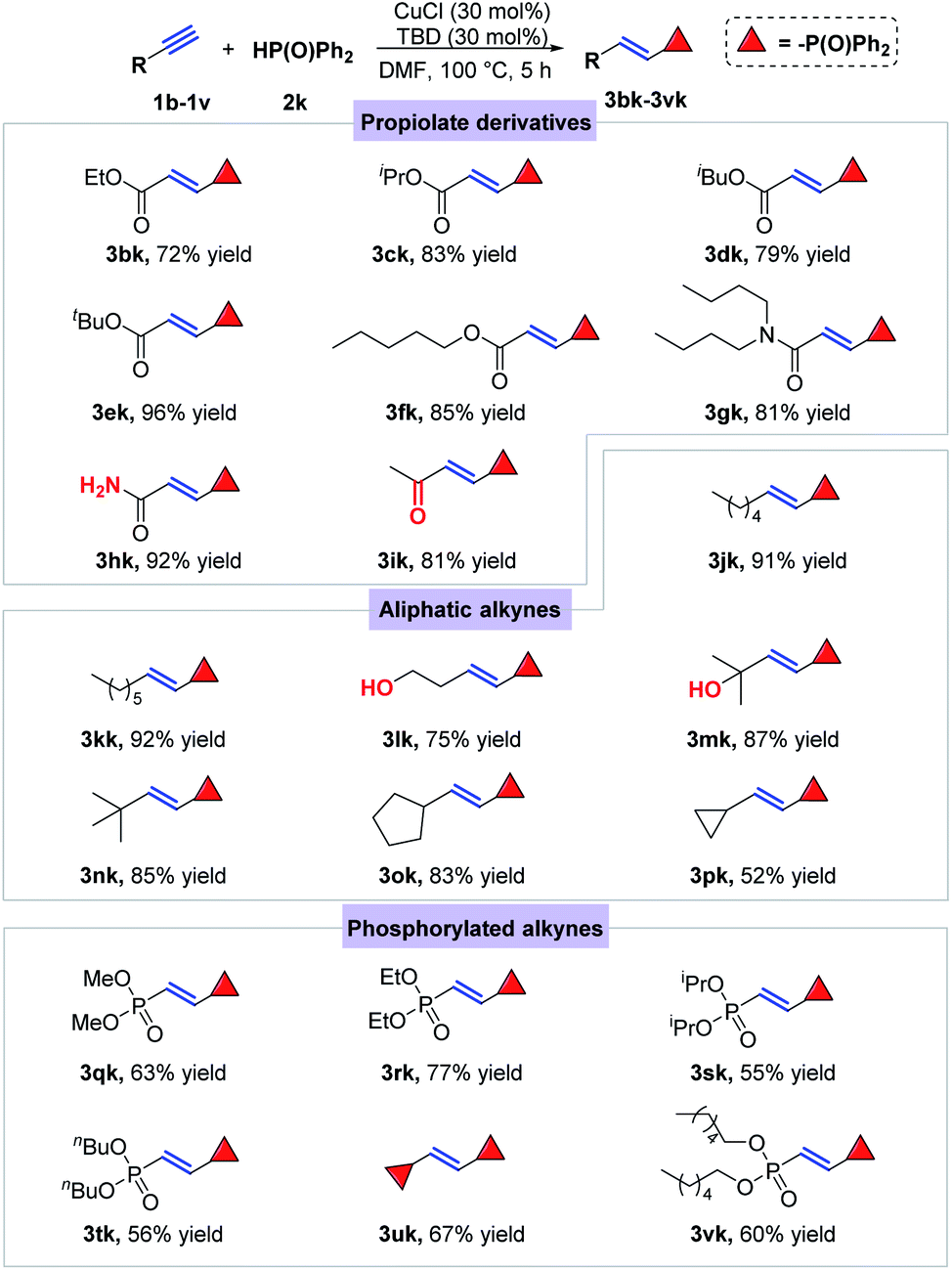 | ||
| Scheme 3 Substrate scope for terminal alkynes. Conditions: alkyne (0.3 mmol), 2k (0.36 mmol), CuCl (30 mol%), TBD (30 mol%), DMF (2 mL), 100 °C, 5 h. | ||
Next, a series of different substituted arylalkynes were tested to react with diphenylphosphine oxide under the standard conditions (Scheme 4). Hydrophosphorylation of phenylacetylene readily took place to afford the desired product 5ak in 92% isolated yield. Then, para substituents with different electronic property on the phenyl ring were explored. Firstly, substrates bearing electron-donating groups like OMe, NEt2 and Et were all successfully converted to the title compounds in good to excellent yields (5bk, 5ck and 5ek).
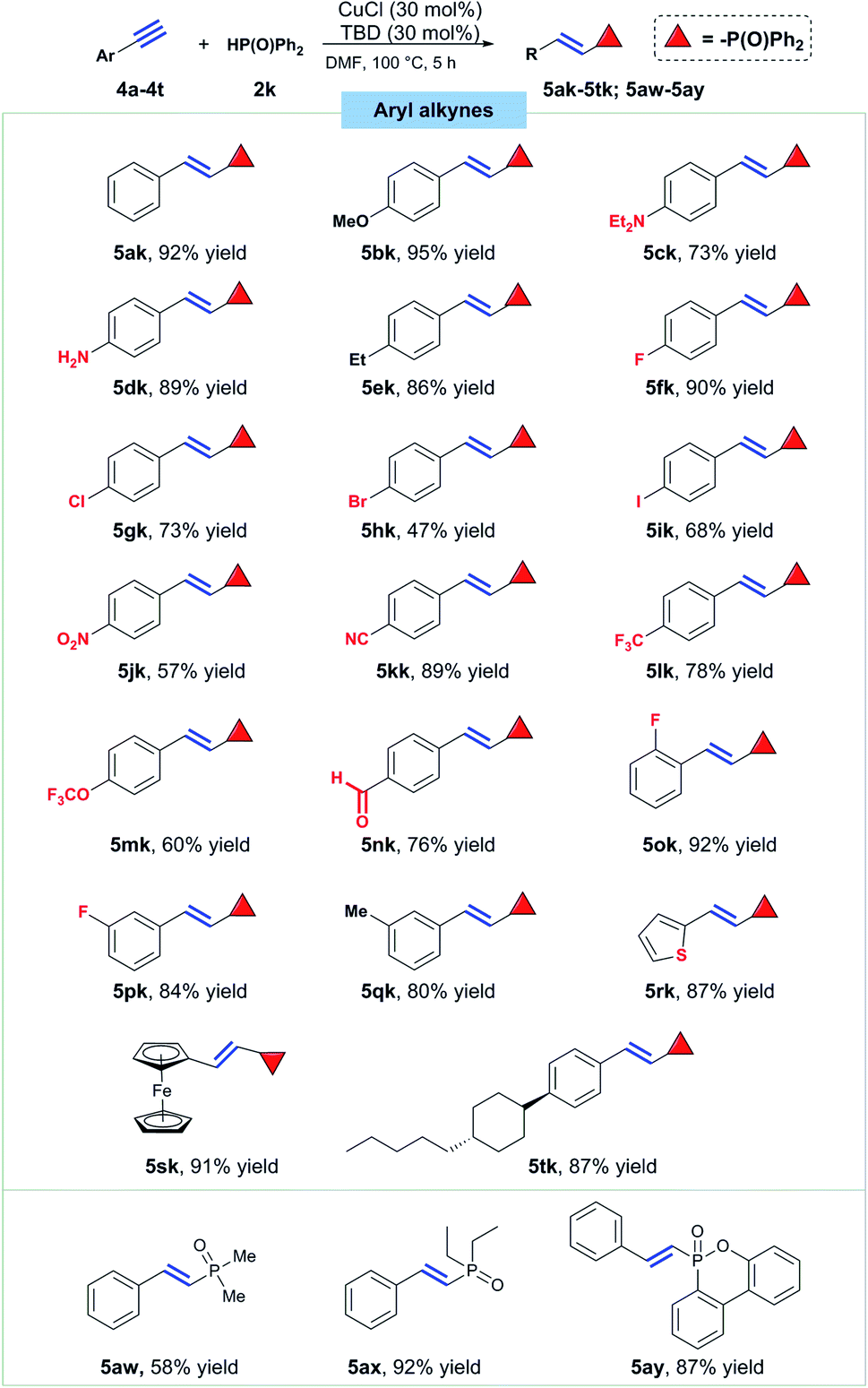 | ||
| Scheme 4 Substrate scope for aryl alkynes. Conditions: alkyne (0.3 mmol), 2k (0.36 mmol), CuCl (30 mol%), TBD (30 mol%), DMF (2 mL), 100 °C, 5 h. | ||
What's noteworthy, free amine group was kept intact under such copper catalysis conditions with the formation of E-alkenylphosphine oxide 5dk in 89% yield. After that, the effect of halogen groups at the para position on this reaction was also studied and the results disclosed that fluoro, chloro, bromo and iodo groups all had nearly no effect on the hydrophosphorylation (5fk–5ik). Then electron-withdrawing groups like nitro, nitrile, trifluoromethyl, trifluoromethoxyl and formyl groups, were all well tolerated, affording the corresponding E-alkenylphosphine oxides in medium to good yields (5jk–5nk). Groups at ortho and meta position did not impede the title reaction to happen, yielding product 5ok, 5pk and 5qk in 92%, 82% and 80% yields, respectively. Thiophene and ferrocene-based terminal alkynes both presented good reactivity towards the titled hydrophosphorylation with 87% and 91% yield of the products respectively (5rk and 5sk). A key intermediate for synthesis of liquid crystal material also proved suitable to furnish the titled transformation effectively (5tk, 87% yield). Finally, dimethylphosphine oxide and diethylphsophine oxide were tried to react with phenylacetylene and the corresponding hydrophosphorylation smoothly took place with formation of the product 5aw and 5ax in 58% and 92% yield, respectively. What's more, a flame retardant dibenzo[c,e][1,2]oxaphosphinine 6-oxide (DOPO) was confirmed to be also reactive and product 5ay was isolated in 87% yield.
Encouraged by our success in hydrophosphorylation of simple terminal alkynes, more complex substrates were then investigated. To our delight, the hydrophosphorylation of one estradiol derivative smoothly proceeded under the standard conditions to yield the corresponding product 5uk in 75% yield (Scheme 5a). The phenolic and alcoholic hydroxyl group both kept untouched. Ethisteronean, an orally active steroidal contraceptive agent, afforded the corresponding hydrophosphorylated product 5vk in 81% yield. Furthermore, other molecules derived from complex natural products were successfully hydrophosphorylated with our method. For instance, the transformation of a derivative of β-D-glucopyranoside gave the desired product 5wk in 57% yield. A derivative of 2′-deoxyuridine provided the corresponding E-alkenylphosphine oxide product 5xk in 62% yield. The hydrophosphorylation selectively happened at the terminal alkyne moiety with the primary and secondary alcohol being untouched. What's more, hydrophosphorylation of a commonly used herbicide clodinafop-propargyl yielded the corresponding product in 61% yield (5yk).
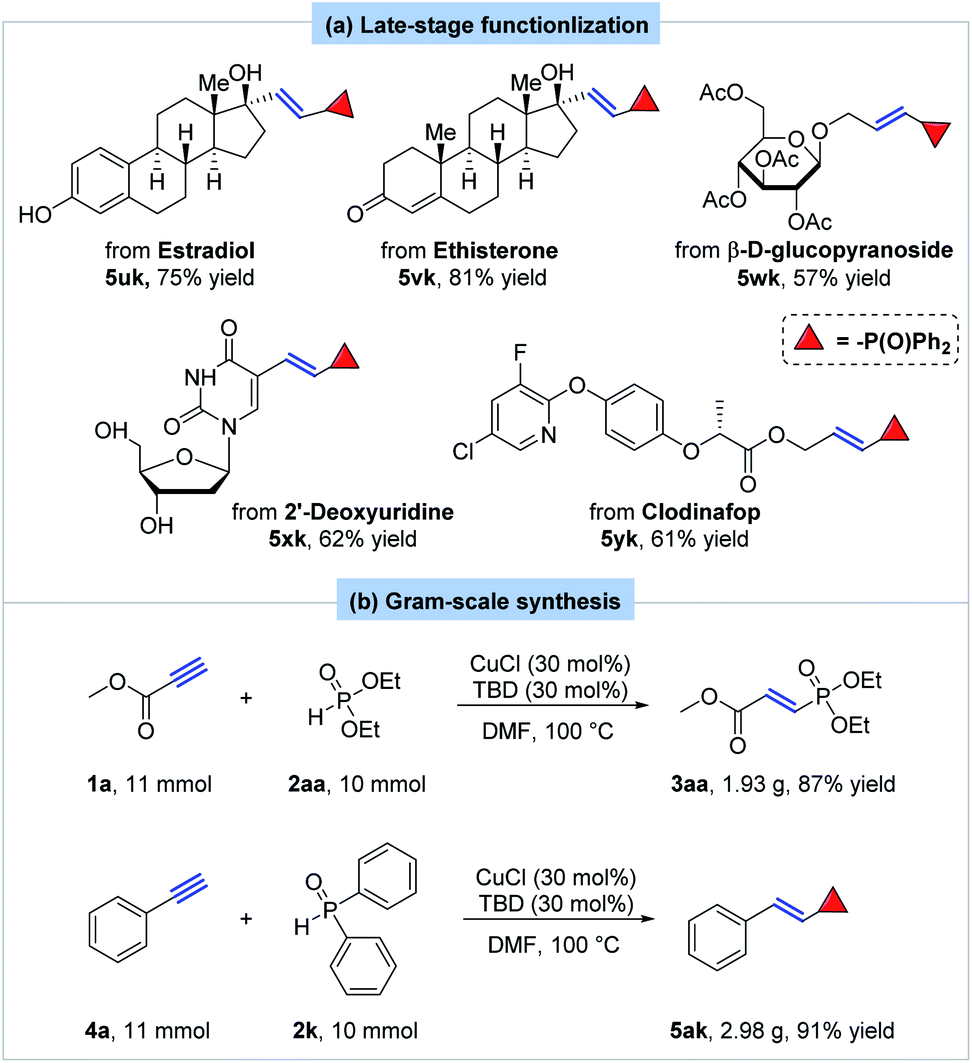 | ||
| Scheme 5 Late-stage functionalization of natural product derivatives and gram-scale synthesis. | ||
To demonstrate the practicality and scalability of our methodology, the gram-scale hydrophosphorylation of methyl propiolate with diethyl phosphite was carried out on 10 mmol to provide 3aa in 87% yield (1.93 g) (Scheme 5b). Moreover, the reaction of phenylacetylene with diphenylphosphine oxide was also performed at 10 mmol with formation of product 5ak in 91% yield (2.98 g).
After that, several competitive reactions were designed and conducted to figure out the relative reaction trends of different P(O)H compounds and terminal alkynes, which might provide clues for the reaction mechanism (Scheme 6). When equal amount of diethyl phosphite (2a) and diphenylphosphine oxide (2k) competitively reacted with methyl propiolate in one pot under the standard conditions, product 3bk was much preferably formed in 98% yield, albeit with 2% yield of 3aa (eq. 1). Also, phenylacetylene was tested to react with diethyl phosphite and diphenylphosphine oxide and only product 5ak was observed (85% yield) (eq. 2). These results indicated that diphenylphosphine oxide showed much higher reactivity than that of diethyl phosphite under such hydrophosphorylation condition. After that, the reaction of equal amount of 1-ethynyl-4-methoxybenzene and 1-ethynyl-4-(trifluoromethyl)benzene with diphenylphosphine oxide was performed with preferential formation of product 5lk and the result disclosed that electron-deficient alkynes possess a higher reactivity than electron-rich alkynes (eq. 3).
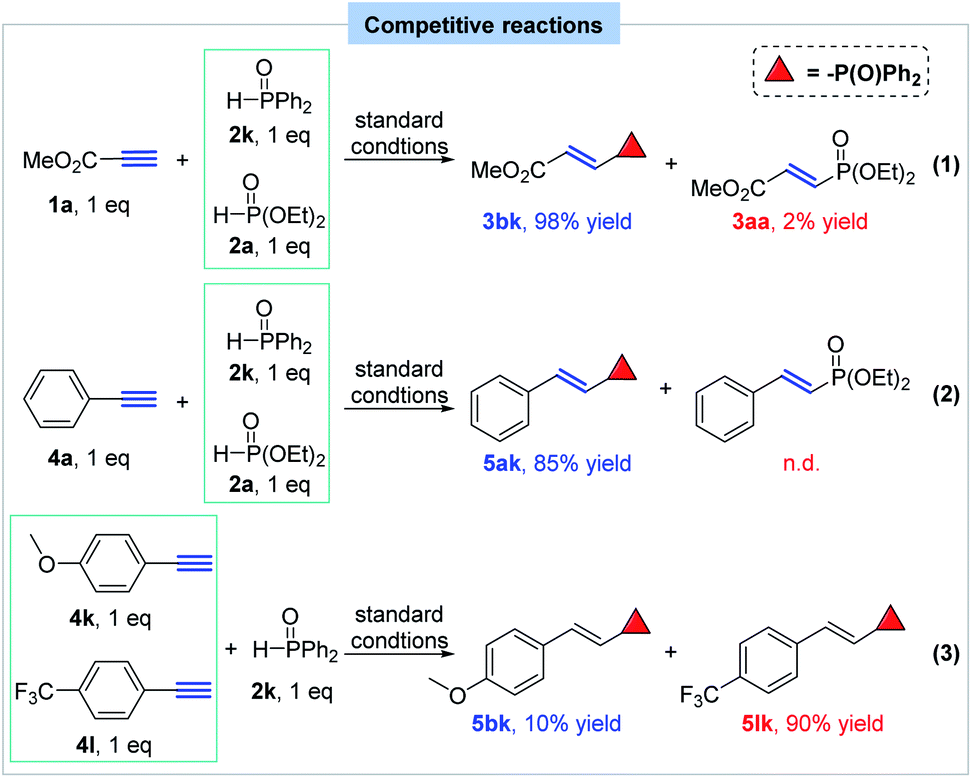 | ||
| Scheme 6 Competitive reactions. | ||
To gain insight in the reaction mechanism, several control and deuterium-labeling experiments were designed and carried out (Scheme 7). Firstly, treatment of (phenylethynyl)copper with 1 equivalent of diphenylsphospine oxide in DMF did not yield the desired E-alkenyldiphenylphosphine oxide at all whether in the presence of TBD or not, which indicated that (phenylethynyl)copper was not the active species in the hydrophosphorylation reaction (Scheme 7a, eq. 4 and eq. 5). Then deuterium labeling experiments were also performed to make sure the possible hydrogen source of the hydrophosphorylation process. When the reaction proceeded in d7-DMF, no deuterium-labeled product was observed, indicating no transfer of hydrogen from solvent to the product (Scheme 7b, eq. 6). Note that when deuterium-labeled diphenylphosphine oxide [D]-2k (90% deuterium content) and phenylacetylene 4a was subjected to the standard condition, α- and β-deuterated product 5ak was obtained in 94% yield with 68% deuterium content (Scheme 7b, eq. 7). Interestingly, when [D]-4a (90% deuterium content) reacted with 2k, [D]-5ak was also observed with 30% deuterium content at α position and 62% deuterium content at β position (Scheme 7b, eq. 8). These results revealed that hydrogen of the alkenyl moiety in 5ak partially came from both substrate 2k and 4a.
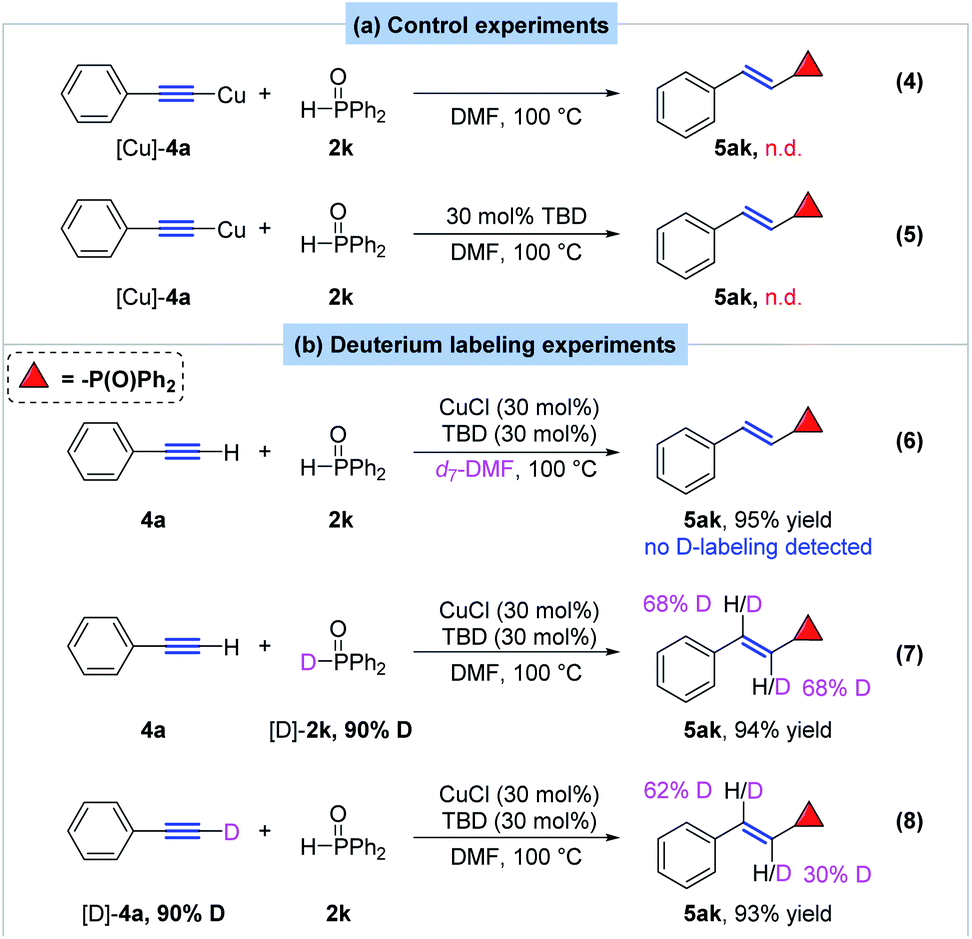 | ||
| Scheme 7 Control and deuterium-labeling experiments. | ||
DFT studies were also conducted to better understand the catalytic pathway and the origin of regio- and stereoselectivity for this hydrophosphorylation reaction and the possible intermolecular hydrogen exchange process (Fig. 1). Hydrophosphorylation of phenylacetylene with diphenylphosphine oxide catalyzed by Copper(I) chloride was considered in the DFT calculations, among which the van der Waals complex A of Y-shape Cu(I) complex with the base TBD was calculated as the more stable species and set as the starting point. TBD deprotonates the coordinated hydroxydiphenylphosphane and gives an ion pair B in a barrierless way. From the intermediate B, the diphenylphosphoryl group can insert into the carbon–carbon triple bond of ethynylbenzene. It is interesting to find that the diphenylphosphoryl nucleophilicly attack the hydrogen atom-bonded carbon gives the favorable transition state (TSB-C) to form the anti-Markovnikov product 5ak (after protonation of the alkenyl ligand) with an energy barrier of 29.3 kcal mol−1 respect to complex B. The final step is the ligand exchange to replace the product 5ak with the reactants phenylacetylene and diphenylphosphine oxide to complete the cycle.
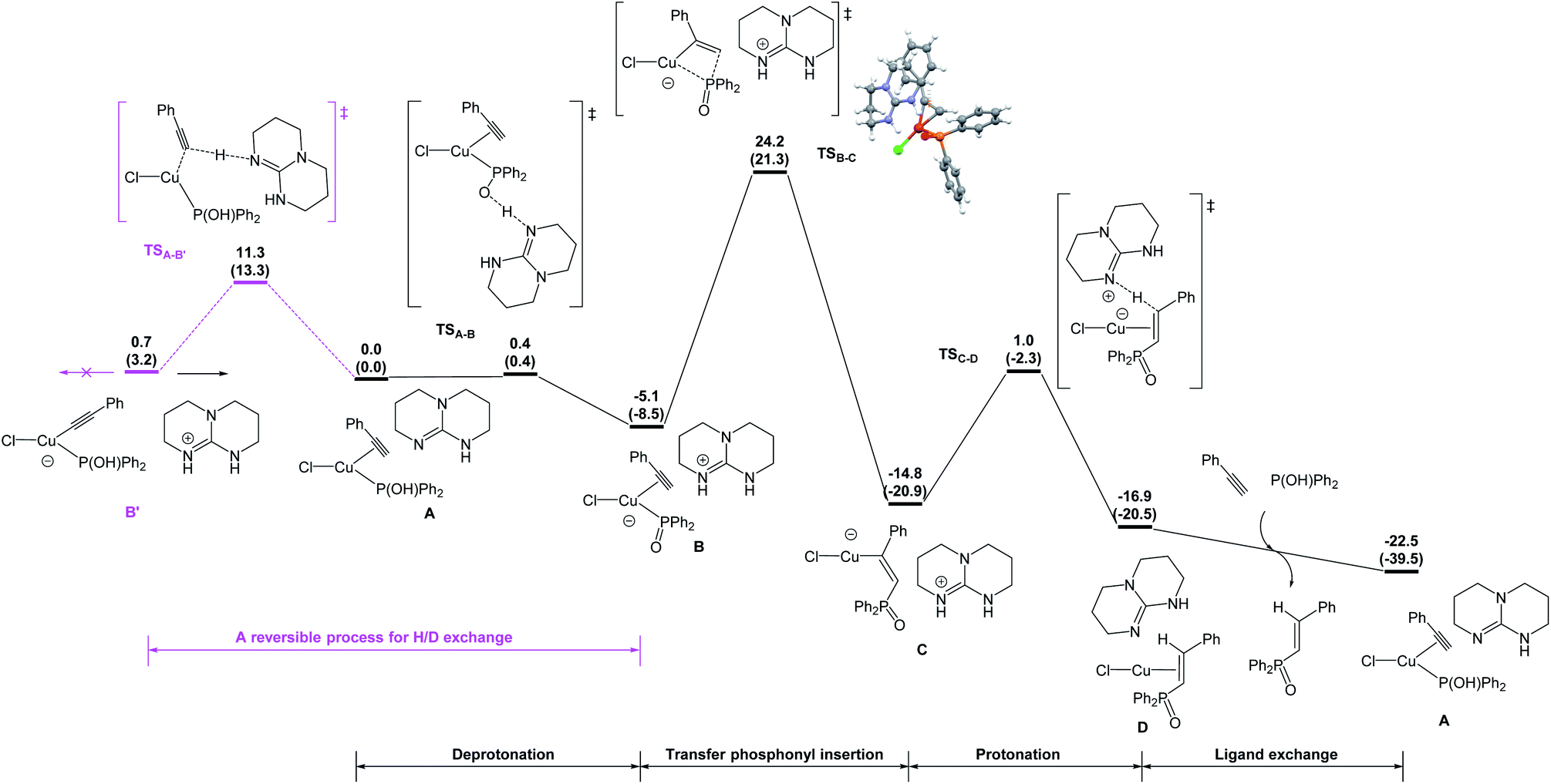 | ||
| Fig. 1 Energy profile calculated for the copper-catalyzed hydrophosphorylation of phenylacetylene with diphenylphosphine oxide. Relative free energies and electronic energies (in parentheses) are given in kcal mol−1. | ||
On the other hand, the barrier of the unfavorable transition state (TS′B-C′) is 3.0 kcal mol−1 higher to the Markovnikov product than that of TSB-C (Fig. 2). Electronically, the phenyl as a π-withdrawing group makes the hydrogen atom-bonded carbon more electrophilic to be attacked. Furthermore, in the TS′B-C′ the phenyl group of ethynylbenzene induce a steric repulsion with the diphenylphosphoryl group. Here, the electronic and steric effects consistently favor the formation of the anti-Markovnikov product. The syn hydrophosphorylation process in our proposed pathway can account for the formation of the E-diphenyl(styryl)phosphine oxide.
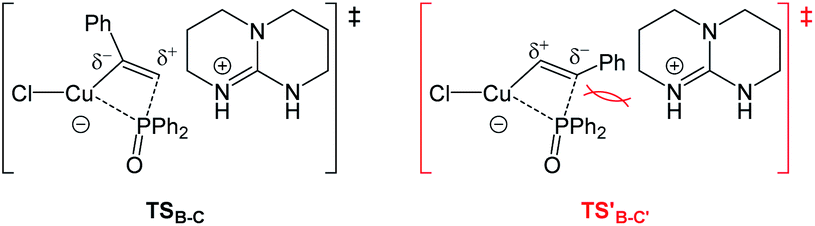 | ||
| Fig. 2 Structures of transition states for explaining the regioselectivity. | ||
We also postulated a possible pathway for above H/D exchange. At the starting point (van der Waals complex A), the base TBD could not only deprotonate the hydroxydiphenylphosphane, but also deprotonate the coordinated ethynylbenzene to produce the phenylethynyl Cu(I) complex B′ (Fig. 1). However, further reaction from B′ encounters much higher barriers (for details, please see SI). The inactivity of the phenylethynyl Cu(I) complex was also proved experimentally (Scheme 7a). The possible reversible pathway between A, B and B′ could explain the H/D exchange in above transformations. When deuterium-labeled diphenylphosphine oxide [D]-2k was employed as the substrate, the β-deuterated product 5ak was obtained without H/D exchange from [D]-A1. At the same time, D+ transfer from the coordinated [D]-2K to the phenylethynyl ligand led to the formation of [D]-B, which was subsequently converted to the α-deuterated product (Fig. 3a). Similarly, using deuterium-labeled phenylacetylene [D]-4a as the substrate, the α-deuterated product 5ak was obtained without H/D exchange from [D]-A2. However, the TBD could deprotonate the D+ and re-protonate the phenylethynyl ligand with the unlabeled H+ and gave the β-deuterated product 5ak (Fig. 3b).
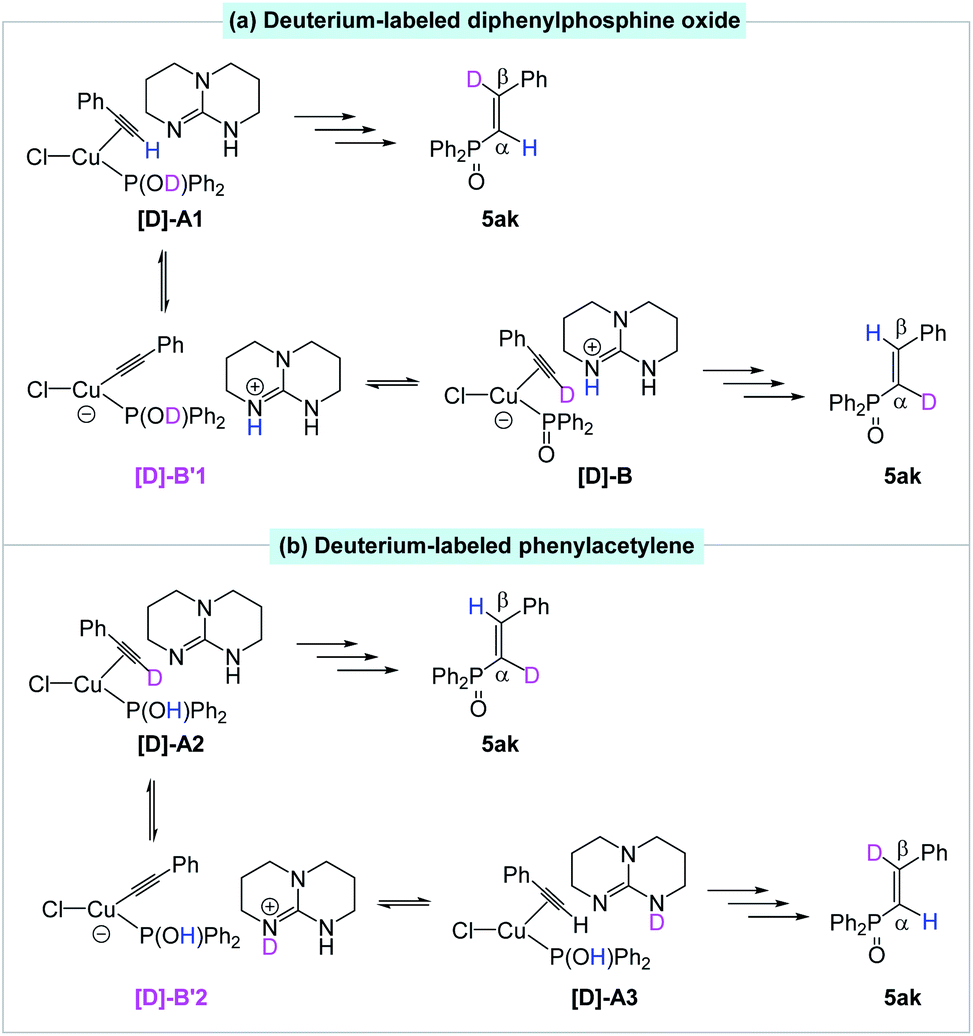 | ||
| Fig. 3 Proposed pathways for H/D exchange. | ||
Based on the above results of experimental and DFT studies, a plausible reaction pathway was depicted in Scheme 8. Firstly, the catalyst copper(I) chloride coordinates with one molecule of diphenylphosphine oxide and phenylacetylene to form the complex A. Then, in the presence of a base, deprotonation occurred at the hydroxyl group of phosphine part to provide intermediate B, followed by transfer insertion of diphenylphosphine oxide part to the triple bond to give copper complex C. Protonation of C produces D, which further coordinates with another diphenylphosphine oxide and phenylacetylene to release the desired product and regenerate the complex A.
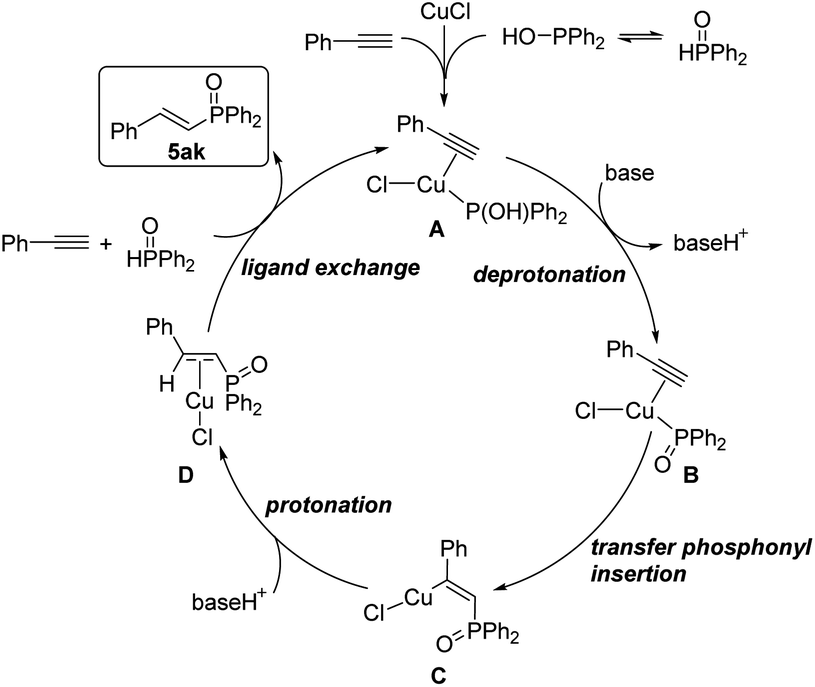 | ||
| Scheme 8 Proposed catalytic pathway. | ||
Conclusions
In conclusion, we developed a highly general and efficient method for hydrophosphorylation of different types of terminal alkynes catalyzed by copper(I) chloride under ligand free and open-air conditions, which was more convenient to be handled. Moreover, diarylphosphine oxide, dialkylphosphine oxide and dialkyl phosphite were all effective for this hydrophosphorylation reaction. Practicality of this reaction got well demonstrated via gram-scale synthesis and late-stage functionalization of several natural product derivatives and one commercially available herbicide. Mechanistic experiments and DFT studies preliminarily demonstrated the deprotonation–protonation equilibrium of terminal alkynes and P(O)H compounds during the catalytic hydrophosphorylation process.Conflicts of interest
The authors declare no conflict of interest.Notes and references
-
(a) Handbook of Organophosphorus Chemistry, ed R. Engel, Marcel Dekker, Inc., New York, 1992 Search PubMed
; (b) The Chemistry of Organophosphorus Compounds Vol. 4, ed F. R. Hartley, John Wiley & Sons, Chichester, 1996 Search PubMed
-
(a) P. J. Murphy, Organophosphorus Reagents: a Practical Approach in Chemistry, Oxford University Press, Oxford, UK, 2004 Search PubMed
- For selected examples, please see:
(a) K. V. Sajna, V. Srinivas and K. C. Kumara Swamy, Efficient Palladium-Catalyzed Double Arylation of Phosphonoalkynes and Diarylalkynes in Water: Use of a Dinuclear Palladium(I) Catalyst, Adv. Synth. Catal., 2010, 352, 3069–3081 CrossRef CAS
-
(a) Y. Xu, J. Xia and H. Guo, Palladium-Catalysed Synthesis of Alkenyldiphenyl- and Alkenylbenzylphenylphosphine Oxides, Synthesis, 1986, 1986, 691–692 CrossRef
-
(a) L.-B. Han, C. Zhang, H. Yazawa and S. Shimada, Efficient and Selective Nickel-Catalyzed Addition of H−P(O) and H−S Bonds to Alkynes, J. Am. Chem. Soc., 2004, 126, 5080–5081 CrossRef CAS PubMed
-
(a) L.-B. Han, C.-Q. Zhao and M. Tanaka, Rhodium-Catalyzed Regio- and Stereoselective Addition of Diphenylphosphine Oxide to Alkynes, J. Org. Chem., 2001, 66, 5929–5932 CrossRef CAS PubMed
-
(a) M. Niu, H. Fu, Y. Jiang and Y. Zhao, Copper-catalyzed Addition of H-phosphine Oxides to Alkynes Forming Alkenylphosphine Oxides, Chem. Commun., 2007, 3, 272–274 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02908a |
| This journal is © The Royal Society of Chemistry 2022 |