
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural modification of olibergin A, an isoflavonoid, from Dalbergia stipulacea Roxb. and its cytotoxicity†
Supakorn Arthana,
Priyapan Posria,
Sookkawath Walunchaprukb,
Thanaset Senawongb and
Chavi Yenjai *a
*a
aDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Natural Products Research Unit, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: chayen@kku.ac.th; Tel: +66-043-009700 ext. 42174 Tel: +66-043-009700 ext. 42175
bDepartment of Biochemistry, Faculty of Science, Natural Products Research Unit, Khon Kaen University, Khon Kaen 40002, Thailand
First published on 16th June 2022
Abstract
Fifteen derivatives were synthesized from olibergin A, a major isoflavonoid isolated from the stems of Dalbergia stipulacea Roxb. All compounds were evaluated for cytotoxicity against HCT-116, HT-29, MCF-7 and vero cell lines using MTT assay. Cytotoxicity results showed 5-hydroxy-7,2′,4′,5′-tetramethoxyisoflavone (5) was the most active with IC50 values of 19.03 ± 0.70, 10.83 ± 1.65, 12.53 ± 0.70 and 13.53 ± 0.84 μM against HCT-116, HT-29, MCF-7 and vero cell lines, respectively. It should be noted that 5-hydroxy-7,2′,4′,5′-tetramethoxyisoflavone (5) showed two times less toxicity against vero cells than the cisplatin standard (IC50 = 6.55 ± 0.81 μM) while 5 and cisplatin exhibited nearly equal cytotoxicity against the MCF-7 cell line. 5,7,2′,4′,5′-Pentamethoxyisoflavanone (10) showed an IC50 value of 30.34 ± 1.15 μM against the HCT-116 cell line and exhibited weak cytotoxicity against normal cells, the vero cell line. In addition, 5,7,4′-trihydroxy-2′,5′-dimethoxyisoflavan oxime (13) demonstrated cytotoxicity against HT-29 cells with an IC50 value of 31.41 ± 1.38 μM and displayed weak activity toward the vero cell line. The information revealed that these compounds were suitable for development to anticancer agents against HCT-116, HT-29 and MCF-7 cell lines.
1. Introduction
Isoflavonoids are widely distributed in the Fabaceae family, especially in the Dalbergia genus such as D. parviflora, D. olivari, D. verlutina and D. stipulacea. Many isoflavonoids show various biological activities, for example anti-tumor promoting activity on TPA-induced EBV-EA activation,1 cytotoxicity against T-47D and BT20 human breast cancer cell lines,2 human epidermoid carcinoma of the oral cavity (KB), breast adenocarcinoma (MCF-7), human small cell lung cancer (NCI–H187) cell lines3,4 and also estrogenicity.5Dalbergia stipulacea Roxb. belongs to the family Fabaceae. It is a climbing shrub found throughout southern China, eastern India, Myanmar, Laos, Thailand and Vietnam.6 It is believed that this plant has been used as an emmenagogue and for abortion when taken in moderate amounts. It can be used for gonorrhea, syphilis and mouth ulcer.6 Furthermore, the roots of this plant are fish poison.7 Luteolin 4′-rutinoside and luteolin were isolated from the leaves of this plant.8 The chemical constituents from the stems and the roots of this plant are chalcones, isoflavonoids, flavonoids, pterocarpans, diterpenes and coumarins.9–11 R,R-Velucarpin A, a pterocarpan, from the roots of this plant showed cytotoxicity against HeLa cells with an IC50 value of 10.9 ± 0.42 μM (Posri et al., 2021).9 (−)-12α-hydroxyrotenone from the methanol extract of D. stipulacea roots displayed cytotoxicity against HepG2 cell line with an IC50 value of 18.01 ± 1.51 mM.10 It was found that (−)-vestitol from the stems of this plant displayed the strongest antifungal activity against Pythium insidiosum and had higher activity than the amphotericin-B standard.11
Olibergin A, a major isoflavonoid, was isolated from the stems of D. stipulacea.11 In previous studies, this compound has been evaluated for biological activities such as the inhibitory effects on TPA-induced EVB-EA activation and on nitric oxide production in RAW 264.7 cells, antiplasmodial, antifungal activity against Pythium insidiosum and cytotoxicity against KB, HeLa S-3, MCF-7, HepG2, HT-29 and MRC-5 cell lines.1,11–14 It was reported that olibergin A showed cytotoxicity against KB, HeLa and MRC-5 cell lines with IC50 values of 48.06, 59.82 and 25.03 μM, respectively.13,15 In addition, from our screening evaluation on the cytotoxicity of olibergin A against HCT-116, HT-29, MCF-7 and vero cell lines, this compound exhibited moderate to weak activity by showing IC50 values of 79.23 ± 4.78, 32.18 ± 1.73, 72.12 ± 1.12 and 88.83 ± 4.33 μM, respectively. In order to take advantage of this major compound and continuing our research on new anticancer agents with high efficacy and low toxicity from natural products, we planned to modify the chemical structure of olibergin A using simple organic reactions and examine the cytotoxicity of all derivatives against four cell lines including HCT-116, HT-29, MCF-7 and vero cell lines. Fifteen derivatives (2–16) from olibergin A were synthesized including methylation, nitration, acetylation, mesylation, hydrogenation, hydrazone and oxime formation reactions.
2. Results and discussion
Chemistry
The structural modification of isoflavonoid derivatives started from olibergin A (1), a major product from the stems of D. stipulacea.11 In our cytotoxicity screening, this compound showed cytotoxicity against HCT-116, HT-29, MCF-7 and vero cell lines, with IC50 values of 79.23 ± 4.78, 32.18 ± 1.73, 72.12 ± 1.12 and 88.83 ± 4.33 μM, respectively. All derivatives were synthesized into three parts, as here described. For the first part, derivatization of hydroxyl groups at C-5, C-7 and C-4′ were performed to obtain five derivatives (2–6) and also a nitration reaction to provide 7 as shown in Scheme 1. The second part was examined under catalytic hydrogenation leading to reduction at the C-2/C-3 positions and yielded 8–11 as shown in Scheme 2, and the third part was the conversion of the carbonyl group at the C-4 position to hydrazone and oxime derivatives (12–16) as shown in Scheme 3. | ||
| Scheme 1 Derivatives of Olibergin A (1). | ||
 | ||
| Scheme 2 Derivatives of hydrogenation. | ||
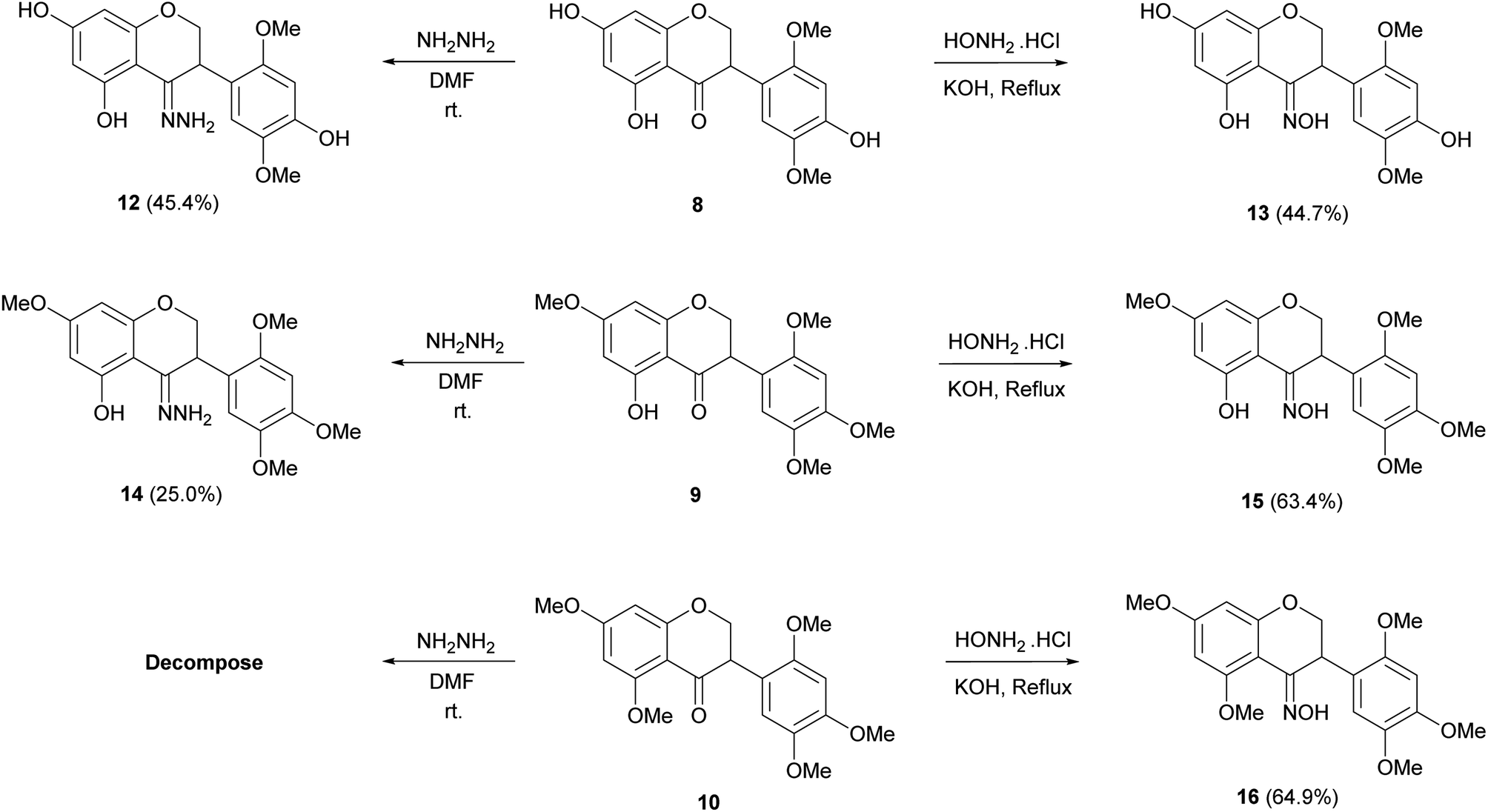 | ||
| Scheme 3 Hydrazone and oxime derivatives. | ||
Treatment of 1 with methanesulfonyl chloride, acetic anhydride and valeroyl chloride in the presence of pyridine provided isoflavone derivatives 2 (13.5%), 3 (15.2%) and 4 (29.4%), respectively (Scheme 1).16 The 1H NMR data of 2 showed a singlet signal of two methyl groups at δH 3.24 corresponding to mesyl groups, while the signals at δH 2.34 of 3 represented two additional acetoxy groups. In addition, the 13C NMR spectrum of 3 and 4 displayed carbonyl signals at δC 180.7 (compound 3) and at δC 181.3 (compound 4). In the case of 4, the proton signals of n-butyl groups showed at δH 2.60, 1.76, 1.47 and 0.98. The methylation of 1 was carried out using CH3I/K2CO3 to obtain the isoflavone derivatives 5 and 6 in 54.3 and 41.0% yield, respectively.17 The 1H NMR spectrum of 5 exhibited four methoxy groups while those of 6 showed five methoxy groups and no intramolecular H-bonding signal around δH 12.8. Compound 1 was treated with a mixture of nitric acid and acetic acid at 0 °C to furnish derivative 7 in 47.8% yield.18 The disappearance of two methoxy protons in the 1H NMR spectral data of the B ring was observed and a singlet proton at δH 6.77 (H-6′) was found. The 13C NMR spectral data showed two signals of α,β-unsaturated ketone groups of a quinone moiety at δC 185.2 (C-2′) and δC 171.3 (C-5′) in the B ring. The HMBC correlations between the proton at δH 6.77 (H-6′) and carbons at δC 116.6 (C-3), δC 171.3 (C-5′) and δC 163.2 (C-4′) confirmed the assignment of compound 7.
Catalytic hydrogenation of 1, 5 and 6 was performed under hydrogen gas, using palladium on charcoal as a catalyst for 8 hours to furnish isoflavanones 8 (93.5%), 9 (66.7%) and 10 (67.9%).19 Isoflavan 11 was obtained from hydrogenation of 6 for a longer time (24 hours) in 70.9% yield (Scheme 2). It should be noted that the reduction reactions of 1 and 5 for a long time gave no isoflavan derivative. This may be due to the chelating effect between the hydroxy group at C-5 and the carbonyl group at C-4 position. The disappearance of the olefinic proton at δH 7.77 (H-2) of 1 and the presence of ABX signals at δH 4.53 (H-2), 4.40 (H-2) and 4.29 (H-3) confirmed the isoflavanone core structure of 8. The isoflavanone 8 was identified as a racemic mixture product, which was confirmed by the ECD spectrum and optical rotation by showing the lack of optical rotation and no obviously observed Cotton effect in the experimental ECD spectrum. This evidence was observed in 9 and 10 as well. In the case of 11, the 1H NMR data showed five alicyclic protons at δH 4.26 and 3.98 (H-2), 3.54 (H-3), 2.87 and 2.69 (H-4) and the absence of a carbonyl carbon signal at the C-4 position.
Hydrazone derivatives 12 (45.4%) and 14 (25.0%) were synthesized by the condensation between compounds 8 and 9 with hydrazine hydrate in DMF as a solvent at room temperature, respectively (Scheme 3).20 In contrast, it was attempted to construct a hydrazone derivative of compound 10 but the product decomposed immediately after purification. The 13C NMR spectra of C-4 in hydrazone derivatives, 12 and 14, showed upfield shift (δC 158.1 and 163.0) while the signals of carbonyl compounds, 8 and 9, showed at δC 197.3 and 197.4. The isoflavanones 8, 9 and 10 were further treated with hydroxylamine hydrochloride in KOH and gave oxime derivatives 13, 15 and 16 in 44.7, 63.4 and 64.9% yield, respectively.20 In the same manner as 12 and 14, oxime carbons (C-4) of these compounds showed upfield shift to δC 160.3–167.3 in the 13C-NMR spectra.
Biological activity
All compounds were evaluated for cytotoxicity against two human colon cancer cell lines, HCT-116 and HT-29, and breast cancer cells (MCF-7) as well as normal cells, vero cells (kidney African Green Monkey cells), using MTT assay. The cytotoxicity results are shown in Table 1.| Compound | HCT-116 | HT-29 | MCF-7 | Vero cell |
|---|---|---|---|---|
| a Inactive at >200 μM. | ||||
| 1 | 79.23 ± 4.78 | 32.18 ± 1.73 | 72.12 ± 1.12 | 88.83 ± 4.33 |
| 2 | Inactive | Inactive | Inactive | Inactive |
| 3 | Inactive | Inactive | Inactive | 111.61 ± 12.26 |
| 4 | Inactive | Inactive | Inactive | Inactive |
| 5 | 19.03 ± 0.70 | 10.83 ± 1.65 | 12.53 ± 0.70 | 13.53 ± 0.84 |
| 6 | 177.62 ± 02.23 | 154.47 ± 12.08 | 98.53 ± 15.36 | >268.55 |
| 7 | 57.18 ± 3.56 | Inactive | 45.91 ± 3.27 | 24.59 ± 3.79 |
| 8 | 92.69 ± 1.84 | 69.92 ± 6.47 | 147.87 ± 1.96 | 93.63 ± 10.23 |
| 9 | 37.07 ± 1.28 | 19.20 ± 1.86 | 30.97 ± 1.89 | 33.52 ± 1.25 |
| 10 | 30.34 ± 1.15 | Inactive | 73.75 ± 0.77 | 116.38 ± 20.19 |
| 11 | 28.94 ± 3.19 | 29.38 ± 0.08 | 21.06 ± 2.61 | 31.13 ± 4.99 |
| 12 | 126.32 ± 0.03 | 97.77 ± 1.73 | 165.59 ± 4.79 | 133.92 ± 1.56 |
| 13 | 82.60 ± 7.89 | 31.41 ± 1.38 | 109.55 ± 2.48 | 103.71 ± 24.96 |
| 14 | Inactive | Inactive | Inactive | Inactive |
| 15 | 26.40 ± 0.05 | 38.55 ± 2.77 | 28.93 ± 3.94 | 29.78 ± 4.85 |
| 16 | 137.80 ± 0.36 | 114.82 ± 2.62 | 110.40 ± 4.52 | 158.70 ± 8.29 |
| Cisplatin | 4.93 ± 0.77 | 5.30 ± 0.53 | 10.42 ± 0.85 | 6.55 ± 0.81 |
Cytotoxicity of isoflavonoid, olibergin A (1), showed weak activity against three cancer cell lines and vero cells as mentioned above (Table 1). Fortunately, some synthesized olibergin A derivatives exhibited stronger activity than the starting material, especially compound 5 which displayed IC50 values of 19.03 ± 0.70, 10.83 ± 1.65 and 12.53 ± 0.70 μM against HCT-116, HT-29 and MCF-7 cancer cell lines, respectively. In the cases of cytotoxicity against HCT-116 cell line, compound 5 showed four times stronger cytotoxicity than 1; it seems that the methoxy groups at C-7 and C-4′ and the hydroxyl group at C-5 in the isoflavone core structure may play an important role for the activity. Similar results were detected for cytotoxicity against MCF-7 cells, which exhibited six time stronger than 1. In addition, this derivative demonstrated three times stronger than the starting material against the HT-29 cell line. It is interesting that 5 showed activity against MCF-7 (IC50 = 12.53 ± 0.70 μM) nearly equal to the cisplatin standard (IC50 = 10.42 ± 0.85 μM), but 5 displayed cytotoxicity against normal cells with an IC50 value of 13.53 ± 0.84 μM, while the cisplatin standard (IC50 = 6.55 ± 0.81 μM) displayed more toxic than 5. This information may be useful for further study on the anticancer agent. In comparison between 5 and 6, compound 6 showed weak activity to all cell lines. The results indicate that the hydroxyl group at the C-5 position of the isoflavone derivative was necessary for the activity. Cytotoxicity results of 2, 3 and 4 exhibited inactivity against all tested cell lines, which means mesyl and ester functional groups at C-7 and C-4′ positions dramatically decrease the activity.
In order to know the effect of the double bond at C-2/C-3, isoflavones were reduced to be isoflavanones 8, 9 and 10 and also isoflavan 11. It was found that 9 and 11 displayed stronger activity against all cell lines than olibergin A (1) by showing IC50 values around 19–37 μM. This information confirmed that presence of a saturated moiety on the C ring may be important for the activity. However, isoflavanone 8 demonstrated weak cytotoxicity (IC50 ≅ 69–147 μM). Comparing the activity between 8 and 9, it seems that methoxy groups at C-7 and C-4′ may be necessary for the activity. It is interesting that 10 showed strong activity against the HCT-116 cell line with an IC50 value of 30.34 ± 1.15 μM, but displayed weak activity against vero cells (IC50 = 116.38 ± 20.19 μM). This information indicates that this compound is suitable for the development of an anticancer agent against HCT-116 cells.
In the cases of oximes 13 and 15, compound 13 displayed cytotoxicity against HT-29 (IC50 = 31.41 ± 1.38 μM) but showed weak cytotoxicity against vero cells (IC50 = 103.71 ± 24.96 μM). There was good evidence for the development of this compound. Compound 15 displayed IC50 around 26–38 μM toward all cell lines. Comparing between 13 and 15, it seems the methoxy groups at C-7 and C-4′ may be important for the cytotoxicity. In the cases of 15 and 16, compound 16 exhibited weaker cytotoxicity against all cell lines. It seems the hydroxy group at the C-5 position is more important for the cytotoxicity than the methoxy group in the same manner as isoflavones 5 and 6.
Hydrazones 12 and 14 showed weak and inactive against four cell lines; these results suggest that this functional group is not suitable for cytotoxicity.
3. Experimental part
General experimental procedures
Melting points were determined on a SANYO Gallenkamp (Leicester, UK) melting point apparatus and are uncorrected. Optical rotations were measured on a JASCO P-1020 digital polarimeter (Japan). UV spectra were measured on an Agilent 8453 UV-Visible spectrophotometer (Waldbronn, Germany). IR spectra were recorded with diamond dust, using an Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR) Bruker TENSOR 27 (Germany). NMR spectra were collected at 400 MHz (1H) and at 100 MHz (13C) using a Bruker AVANCE NEO. The LC-QTOF-MS/MS analysis was performed on an Agilent HPLC 1260 series coupled with a QTOF 6540 UHD accurate mass (Agilent Technologies, Waldbronn, Germany). Circular dichroism spectroscopy is determined using a Jasco J-815 CD Spectrometer. Thin layer chromatography (TLC) was carried out on MERCK silica gel 60 F254 TLC aluminium sheets. Column chromatography separations were carried out on silica gel less than 0.063 mm, 0.063–0.200 mm (Darmstadt, Germany). Preparative thin layer chromatography (PLC) was carried out on glass supported silica gel plates using silica gel 60 PF254 for preparative layer chromatography (Darmstadt, Germany). All solvents were routinely distilled prior to use.Plant material
The stems of D. stipulacea were collected in February 2018 at Ban Na Phaeng village, Wiang Kao district, Khon Kaen province, Thailand (16°41′53.5′′N, 102°20′24.1′′E). Plant material (voucher specimen KKU012018) was identified by Assoc. Prof. Suppachai Tiyaworanant, Faculty of Pharmaceutical Sciences, Khon Kaen University, Thailand.Preparation of isoflavone derivatives
The reaction of 1 with acetic anhydride and valerory chloride at room temperature were examined in the same procedure as described above to construct 3 (15.2%) and 4 (29.4%), respectively.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.25 mL) at 0 °C with stirring for 1 h. The reaction mixture was poured into ice cold water, extracted with EtOAc (3 × 20 mL), washed with water and saturated NaCl, dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was recrystallized to give nitroisoflavone 7 (6.8 mg, 47.8% yield).
:1, 0.25 mL) at 0 °C with stirring for 1 h. The reaction mixture was poured into ice cold water, extracted with EtOAc (3 × 20 mL), washed with water and saturated NaCl, dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was recrystallized to give nitroisoflavone 7 (6.8 mg, 47.8% yield).The hydrogenations of 5 and 6 were examined in the same procedure as described above to furnish isoflavanones 9 (62.2 mg, 66.7%) and 10 (32.7 mg, 67.9%), respectively. Moreover, the hydrogenation of 6 was done in the same procedure as described above but took 24 hours to give isoflavan 11 (55.7 mg, 88.6%).
The reactions of 9 (0.0916 mmol) and 10 (0.1349 mmol) with NH2OH·HCl (2.76 and 0.67 mmol) were examined in the same procedure as described above and then 15 (21.8 mg, 63.4%) and 16 (34.1 mg, 64.9%), respectively, were obtained.
The preparation of hydrazone derivatives from 9 (0.1249 mmol) and 10 (0.1130 mmol) with NH2NH2·H2O was examined in the same procedure as described above and then 14 (21.8 mg, 63.4%) was obtained. Unfortunately, the hydrazone derivative from 10 decomposed.
Spectroscopic data
Olibergin A (1) yellow solid; mp 240–242 °C; FT-IR (neat) νmax cm−1: 3254, 2963, 2940, 1655, 1577, 1620, 1455, 1309, 1177, 1037, 861; 1H NMR (400 MHz, CDCl3): δ 12.87 (1H, s, OH-5), 7.77 (1H, s, H-2), 6.78 (1H, s, H-6′), 6.56 (1H, s, H-3′), 6.30 (1H, d, J = 2.1 Hz, H-8), 6.21 (1H, d, J = 2.1 Hz, H-6), 3.78 (3H, s, OMe-5′), 3.66 (3H, s, OMe-2′) ppm; 13C NMR (100 MHz, CDCl3); 180.7 (C-4), 163.8 (C-7), 161.9 (C-5), 158.1 (C-8a), 154.7 (C-2), 152.2 (C-2′), 147 (C-4′), 140.7 (C-5′), 120.2 (C-3), 114.9 (C-1′), 109.8 (C-6′), 105.4 (C-4a), 100.3 (C-6), 99.1 (C-3′), 94.1 (C-8), 56.6 (OMe-5′), 56.2 (OMe-2′) ppm; HRESI-MS m/z 331.0818 [M + H]+ (calcd for C22H23O7, 331.0815).5-Hydroxy-7,4′-dimesyl-2′,5′-dimethoxyisoflavone (2) white powder; mp 218–220 °C; FT-IR (neat) νmax cm−1: 2945, 2843, 1644, 1590, 1508, 1461, 1354, 1273, 1212, 1172, 1110, 967, 891; 1H NMR (400 MHz, CDCl3): δ 12.82 (1H, s, OH-5), 8.04 (1H, s, H-2), 6.75 (1H, d, J = 2.2 Hz, H-6), 6.96 (1H, d, J = 2.2 Hz, H-8), 7.00 (1H, s, H-3′), 7.07 (1H, s, H-6′), 3.78 (3H, s, OMe-2′), 3.88 (3H, s, OMe-5′), 3.24 (6H, s, MeSO2O-7,4′) ppm; 13C NMR (100 MHz, CDCl3); 181.1 (C-4), 163.2 (C-5), 157.3 (C-7), 156.7 (C-8a), 154.2 (C-2), 151.8 (C-2′), 145.5 (C-4′), 139.2 (C-5′), 120.7 (C-3), 118.4 (C-6′), 117 (C-1′), 110.8 (C-4a), 109.5 (C-3′), 105.8 (C-6), 101.7 (C-8), 57.2 (OMe-2′), 57.0 (OMe-5), 38.6, 38.9 (MeSO2O-7,4′) ppm; HRESI-MS m/z 487.0366 [M + H]+ (calcd for C19H19O11S2, 487.0369).
7,4′-Diacetoxy-5-hydroxy-2′,5′-dimethoxyisoflavone (3) white solid; mp 242–244 °C; FT-IR (neat) νmax cm−1: 2968, 2935, 1772, 1752, 1649, 1509, 1453, 1193, 1168, 1129, 1031, 882; 1H NMR (400 MHz, CDCl3): δ 12.78 (1H, s, OH-5), 7.97 (1H, s, H-2), 6.59 (1H, d, J = 2.0 Hz, H-6), 6.77 (1H, d, J = 2.0 Hz, H-8), 7.75 (1H, s, H-3′), 7.00 (1H, s, H-6′), 3.74 (3H, s, OMe-2′), 3.82 (3H, s, OMe-5′), 2.34 (3H, s, M![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) COO-7), 2.34 (3H, s, MCOO-4′) ppm; 13C NMR (100 MHz, CDCl3); 180.6 (C-4), 168.8 (Me
COO-7), 2.34 (3H, s, MCOO-4′) ppm; 13C NMR (100 MHz, CDCl3); 180.6 (C-4), 168.8 (Me![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) OO-7), 168 (MeOO-4′), 162.1 (C-5), 156.5 (C-7), 155.6 (C-8a), 155.5 (C-2), 151.2 (C-2′), 144.1 (C-4′), 140.3 (C-5′), 120.4 (C-3), 116.6 (C-6′), 116 (C-1′), 109.3 (C-4a), 107.3 (C-3′), 105.3 (C-6), 100.7 (C-8), 56.4 (OMe-2′), 56.3 (OMe-5′), 21 (MCOO-7), 20.5 (MeCOO-4′) ppm; HRESI-MS m/z 415.1033 [M + H]+ (calcd for C21H19O9, 415.1029).
OO-7), 168 (MeOO-4′), 162.1 (C-5), 156.5 (C-7), 155.6 (C-8a), 155.5 (C-2), 151.2 (C-2′), 144.1 (C-4′), 140.3 (C-5′), 120.4 (C-3), 116.6 (C-6′), 116 (C-1′), 109.3 (C-4a), 107.3 (C-3′), 105.3 (C-6), 100.7 (C-8), 56.4 (OMe-2′), 56.3 (OMe-5′), 21 (MCOO-7), 20.5 (MeCOO-4′) ppm; HRESI-MS m/z 415.1033 [M + H]+ (calcd for C21H19O9, 415.1029).
7,4′-Dibutoxy-5-hydroxy-2′,5′-dimethoxyisoflavone (4) yellow powder; mp 135–137 °C; FT-IR (neat) νmax cm−1: 2951, 2868, 1765, 1646, 1589, 1511, 1462, 1277, 1218, 1039; 1H NMR (400 MHz, CDCl3): δ 12.78 (1H, s, OH-5), 7.96 (1H, s, H-2), 6.57 (1H, d, J = 2.1 Hz, H-6), 6.75 (1H, d, J = 2.1 Hz, H-8), 6.73 (1H, s, H-3′), 6.99 (1H, s, H-6′), 3.74 (3H, s, OMe-2′), 3.80 (3H, s, OMe-5′), 2.60 (2H, q, J = 7.4 Hz,  ), 1.76 (2H, dq, J = 14.9, 7.3 Hz,
), 1.76 (2H, dq, J = 14.9, 7.3 Hz,  ), 1.47 (2H, dq, J = 14.5, 7.3 Hz,
), 1.47 (2H, dq, J = 14.5, 7.3 Hz,  ), 0.98 (3H, t, J = 7.4 Hz,
), 0.98 (3H, t, J = 7.4 Hz, 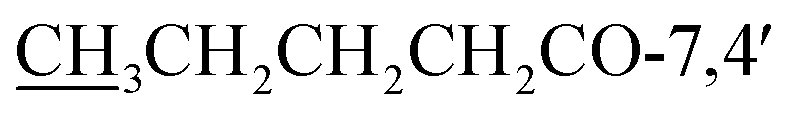 ) ppm; 13C NMR (100 MHz, CDCl3); 181.3 (C-4), 171.7/172.2 (CH3CH2CH2CH2O-7,4′), 162.7 (C-5), 157.3 (C-7), 156.4 (C-8a), 156.1 (C-2), 151.9 (C-2′), 145.4 (C-5′), 141.1 (C-4′), 121.1 (C-3), 117.1 (C-1′), 116.6 (C-3′), 109.9 (C-6′), 107.9 (C-4a), 105.9 (C-6), 101.4 (C-8), 57.1 (OMe-2′), 56.9 (OMe-5′), 34.2/34.5 (
) ppm; 13C NMR (100 MHz, CDCl3); 181.3 (C-4), 171.7/172.2 (CH3CH2CH2CH2O-7,4′), 162.7 (C-5), 157.3 (C-7), 156.4 (C-8a), 156.1 (C-2), 151.9 (C-2′), 145.4 (C-5′), 141.1 (C-4′), 121.1 (C-3), 117.1 (C-1′), 116.6 (C-3′), 109.9 (C-6′), 107.9 (C-4a), 105.9 (C-6), 101.4 (C-8), 57.1 (OMe-2′), 56.9 (OMe-5′), 34.2/34.5 ( ), 27.2/27.5 (
), 27.2/27.5 (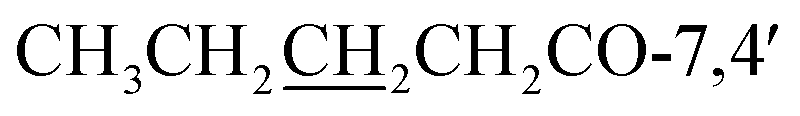 ), 22.6 (
), 22.6 (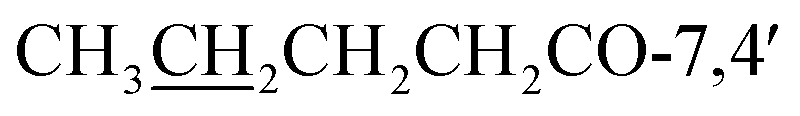 ), 13.8 (
), 13.8 (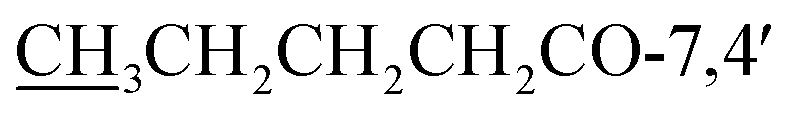 ) ppm; HRESI-MS m/z 499.1971 [M + H]+ (calcd for C27H31O9, 499.1968).
) ppm; HRESI-MS m/z 499.1971 [M + H]+ (calcd for C27H31O9, 499.1968).
5-Hydroxy-7,2′,4′,5′-tetramethoxyisoflavone (5) Yellow powder; mp 164–165 °C; FT-IR (neat) νmax cm−1: 2942, 2839, 1656, 1610, 1514, 1499, 1294, 1207, 1155, 1034, 819; 1H NMR (400 MHz, CDCl3): δ 7.97 (1H, s, H-2), 12.86 (1H, s, OH-5), 6.35 (1H, d, J = 2.3 Hz, H-6), 6.96 (1H, d, J = 2.2 Hz, H-8), 6.61 (1H, s, H-3′), 6.88 (1H, s, H-6′), 3.77 (3H, s, OMe-7), 3.91 (3H, s, OMe-2′), 3.84 (3H, s, OMe-4′), 3.85 (3H, s, OMe-5′) ppm; 13C NMR (100 MHz, CDCl3); 180.8 (C-4), 165.4 (C-7), 162.6 (C-5), 157.9 (C-8a), 154.9 (C-2), 152 (C-2′), 150.1 (C-4′), 143.1 (C-5′), 120.6 (C-3), 115.1 (C-1′), 110.7 (C-6′), 106.3 (C-4a), 98.1 (C-6), 98.1(C-3′), 92.4 (C-8), 56.8 (OMe-2′), 56.4 (OMe-4), 56.2 (OMe-5), 55.8 (OMe-7) ppm; HRESI-MS m/z 359.1129 [M + H]+ (calcd for C19H19O7, 359.1131).
5,7,2′,4′,5′-Pentamethoxyisoflavone (6) black needles; mp 188–189 °C; FT-IR (neat) νmax cm−1: 2942, 2845, 1615, 1457, 1282, 1210, 1150; 1H NMR (400 MHz, CDCl3): δ 7.97 (1H, s, H-2), 6.35 (1H, d, J = 2.3 Hz, H-6), 6.96 (1H, d, J = 2.2 Hz, H-8), 6.61 (1H, s, H-3′), 6.88 (1H, s, H-6′), 3.77 (3H, s, OMe-7), 3.91 (3H, s, OMe-2′), 3.84 (3H, s, OMe-4′), 3.85 (3H, s, OMe-5′), 3.85 (3H, s, OMe-5′) ppm; 13C NMR (100 MHz, CDCl3); 175.1 (C-4), 163.5 (C-7), 161.1 (C-5), 159.7 (C-8a), 152.1 (C-2), 151.6 (C-2′), 149.3 (C-4′), 142.6 (C-5′), 122.2 (C-3), 115.5 (C-1′), 112.2 (C-6′), 109.8 (C-4a), 97.9 (C-3′), 95.9 (C-6), 92.4 (C-8), 56.7 (OMe-2′), 56.3 (OMe-4′), 56.1 (OMe-5′), 55.9 (OMe-5), 55.5 (OMe-7) ppm; HRESI-MS m/z 373.1284 [M + H]+ (calcd for C20H21O7, 373.1287).
1-(5,7-dihydroxy-4-oxo-4H-1-benzopyran-3-yl)-4-hydroxy-3-nitro-2,5-quinone (7) brown solid; mp 190–191 °C; FT-IR (neat) νmax cm−1: 3377, 2924, 2855, 1620, 1495, 1325, 1285, 1233, 1161, 1047; 1H NMR (400 MHz, DMSO-d6): δ 12.57 (1H, s, OH-5), 8.30 (1H, s, H-2), 6.21 (1H, s, H-6), 6.37 (1H, s, H-8), 6.75 (1H, s, H-6′) ppm; 13C NMR (100 MHz, CDCl3); 185.4 (C-2′), 179.1 (C-4), 171.5 (C-5′), 166.1 (C-7), 163.4 (C-4′), 161.9 (C-5), 157.4 (C-8a), 157.2 (C-2), 140.7 (C-1′), 134.4 (C-3′), 131.5 (C-6′), 116.8 (C-3), 103.8 (C-4a), 99.9 (C-6), 94.4 (C-8) ppm; HRESI-MS m/z 346.0193 [M + H]+ (calcd for C15H8NO9, 346.0194).
5,7,4′-Trihydroxy-2′,5′-dimethoxyisoflavanone (8) brown solid; mp 176–177 °C; FT-IR (neat) νmax cm−1: 3343, 2939, 2843, 1629, 1512, 1455, 1380, 1267, 1196, 1165, 1094, 1035; 1H NMR (400 MHz, CDCl3 + CD3OD): δ 12.27 (1H, s, OH-5), 6.60 (1H, s, H-3′), 6.63 (1H, s, H-6′), 5.99 (1H, d, J = 2.2 Hz, H-8), 5.94 (1H, d, J = 2.2 Hz, H-6), 4.53 (1H, t, J = 11.4 Hz, H-2), 4.40 (1H, dd, J = 11.0, 5.6 Hz, H-2), 4.29 (1H, dd, J = 11.9, 5.6 Hz, H-3), 3.82 (3H, s, OMe-2′), 3.73 (3H, s, OMe-5′) ppm; 13C NMR (100 MHz, CDCl3); 197.3 (C-4), 166.3 (C-7), 164.1 (C-5), 163.4 (C-8a), 152.2 (C-2′), 146.4 (C-4′), 141.0 (C-5′), 113.8 (C-1′), 113.1 (C-6′), 102.7 (C-4a), 100.4 (C-3′), 96.4 (C-6), 95.3 (C-8), 70.3 (C-2), 56.7 (OMe-2), 56.0 (OMe-5′), 46.9 (C-3) ppm; HRESI-MS m/z 334.1034 [M + 2H]+ (calcd for C17H18O7, 334.1042).
5-Hydroxy-7,2′,4′,5′-tetramethoxyisoflavanone (9) white powder; mp 97–99 °C; FT-IR (neat) νmax cm−1: 2995, 2938, 2836, 1640, 1575, 1511, 1437, 1316, 1271, 1205, 1156, 1033; 1H NMR (400 MHz, CDCl3): δ 12.24 (1H, s, OH-5), 6.66 (1H, s, H-6′), 6.57 (1H, s, H-3′), 6.07 (1H, d, J = 2.2 Hz, H-8), 6.00 (1H, d, J = 2.2 Hz, H-6), 4.53 (1H, t, J = 12.0 Hz, H-2), 4.43 (1H, dd, J = 10.9, 5.7 Hz, H-2), 4.28 (1H, dd, J = 11.9, 5.6 Hz, H-3), 3.88 (3H, s, OMe-2′), 3.82 (3H, s, OMe-5′), 3.80 (3H, s, OMe-4′), 3.77 (3H, s, OMe-7) ppm; 13C NMR (100 MHz, CDCl3); 197.3 (C-4), 167.7 (C-7), 164.6 (C-5), 163.3 (C-8a), 151.9 (C-2′), 149.7 (C-4′), 143.3 (C-5′), 114.3 (C-1′), 113.9 (C-6′), 103.4 (C-4a), 98.2 (C-3′), 95 (C-8), 94 (C-6), 70.4 (C-2), 55.7 (OMe-7), 56.7 (OMe-2′), 56.5 (OMe-4′), 56.2 (OMe-5′), 47.3 (C-3) ppm; HRESI-MS m/z 361.1288 [M + H]+ (calcd for C19H21O7, 361.1287).
5,7,2′,4′,5′-Pentamethoxyisoflavanone (10) white powder; mp 121–123 °C; FT-IR (neat) νmax cm−1: 2940, 2843, 1675, 1606, 1514, 1460, 1213, 1160, 1118, 1032; 1H NMR (400 MHz, CDCl3): δ 6.65 (1H, s, H-6′), 6.54 (1H, s, H-3′), 6.09 (1H, s, H-6), 6.09 (1H, s, H-8), 4.52 (1H, t, J = 12.0 Hz, H-2), 4.44 (1H, dd, J = 10.8, 5.3 Hz, H-2), 4.24 (1H, dd, J = 11.2, 5.3 Hz, H-3), 3.87 (3H, s, OMe-5), 3.77 (3H, s, OMe-7), 3.87 (3H, s, OMe-2′), 3.77 (3H, s, OMe-4′), 3.83 (3H, s, OMe-5′) ppm; 13C NMR (100 MHz, CDCl3); 190.1 (C-4), 165.7 (C-7), 165.3 (C-5), 162.7 (C-8a), 152.1 (C-2′), 149.2 (C-4′), 143.1 (C-5′), 115.6 (C-1′), 114.3 (C-6′), 106.8 (C-4a), 98.1 (C-3′), 93.4 (C-8), 93.1 (C-6), 70.8 (C-2), 56.8 (OMe-2′), 56.7 (OMe-4′), 56.2 (OMe-5), 55.7 (OMe-7), 53.2 (OMe-5′), 48.0 (C-3) ppm; HRESI-MS m/z 375.1444 [M + H]+ (calcd for C20H23O7, 375.1444).
5,7,2′,4′,5′-Pentamethoxyisoflavan (11) brown liquid; FT-IR (neat) νmax cm−1: 2937, 2843, 1603, 1509, 1457, 1316, 1206, 1117, 1037; 1H NMR (400 MHz, CDCl3): δ 6.71 (1H, s, H-6′), 6.55 (1H, s, H-3′), 6.08 (1H, s, H-6), 6.08 (1H, s, H-8), 4.26 (1H, ddd, J = 10.2, 3.4, 2.0 Hz, H-2), 3.98 (1H, t, J = 10.0 Hz, H-2), 3.89 (3H, s, OMe-5), 3.81 (3H, s, OMe-2′), 3.80 (3H, s, OMe-5′), 3.79 (3H, s, OMe-4′), 3.72 (3H, s, OMe-7), 3.54 (1H, m, Hz, H-3), 2.87 (1H, dd, J = 5.5, 1.8 Hz, H-4), 2.69 (1H, dd, J = 16.4, 10.8 Hz, H-4) ppm; 13C NMR (100 MHz, CDCl3); δ 159.4 (C-7), 158.7 (C-5), 155.8 (C-8a), 151.8 (C-2′), 148.4 (C-4′), 143.2 (C-5′), 121.3 (C-1′), 112.1 (C-6′), 103.9 (C-4a), 97.9 (C-3′), 93.3 (C-8), 91.3 (C-6), 70 (C-2), 56.9 (OMe-2′), 56.5 (OMe-4′), 56.3 (OMe-5), 55.5 (OMe-7), 55.4 (OMe-5′), 31.5 (C-3), 25.2 (C-4) ppm; HRESI-MS m/z 361.1650 [M + Na]+ (calcd for C20H24O6Na, 361.1651).
5,7,4′-Trihydroxy-2′,5′-dimethoxyisoflavan hydrazone (12) light brown solid; mp 245–246 °C, FT-IR (neat) νmax cm−1: 3363, 2940, 2841, 1630, 1600, 1513, 1460, 1236, 1194, 1038; 1H NMR (400 MHz, CDCl3 + CD3OD): δ 4.13 (2H, br s, H-2), 4.39 (1H, s, H-3), 5.85 (1H, d, J = 2.2 Hz, H-6), 5.96 (1H, d, J = 2.2 Hz, H-8), 6.39 (1H, s, H-3′), 6.46 (1H, s, H-6′), 3.71 (3H, s, OMe-2′), 3.48 (3H, s, OMe-5′) ppm; 13C NMR (100 MHz, CDCl3); δ 160.6 (C-4), 159.9 (C-7), 152.3 (C-8a), 158.1 (C-5), 151.3 (C-2′), 146.7 (C-4′), 141.2 (C-5′), 113.7 (C-1′), 112.2 (C-6′), 99.8 (C-4a), 99.2 (C-3′), 97.0 (C-6), 95.4 (C-8), 69.6 (C-2), 56.6 (OMe-2′), 55.9 (OMe-5′), 32.2 (C-3) ppm; HRESI-MS m/z 347.1235 [M + H]+ (calcd for C17H19N2O6, 347.1243).
5,7,4′-Trihydroxy-2′,5′-dimethoxyisoflavan oxime (13) brown solid; mp 172–174 °C; FT-IR (neat) νmax cm−1: 3353, 2939, 2843, 1629, 1512, 1455, 11380, 1288, 1165, 1158, 1093, 1035, 826; 1H NMR (400 MHz, CDCl3 + CD3OD): δ 6.50 (1H, s, H-6′), 6.32 (1H, s, H-3′), 6.02 (1H, s, H-8), 4.81 (1H, br s, H-3), 4.25 (1H, d, J = 11.0, Hz, H-2), 4.13 (1H, dd, J = 11.0, 4.0 Hz, H-2), 5.89 (1H, d, s, H-6), 3.74 (3H, s, OMe-2′), 3.50 (3H, s, OMe-5′) ppm; 13C NMR (100 MHz, CDCl3); δ 160.2 (C-4), 159.1 (C-7), 158.2 (C-5), 155.1 (C-8a), 151.5 (C-2′), 145.7 (C-4′), 140.5 (C-5′), 116.4 (C-1′), 112.3 (C-6′), 100.0 (C-4a), 98.3 (C-3′), 96.8 (C-6), 95.4 (C-8), 69.5 (C-2), 56.7 (OMe-2′), 56.0 (OMe-5′), 32.1 (C-3) ppm; HRESI-MS m/z 349.1142 [M + 2H]+ (calcd for C17H19NO7, 349.1151).
5-Hydroxy-7,2′,4′,5′-tetramethoxyisoflavanone hydrazone (14) dark brown solid; mp 103–105 °C; FT-IR (neat) νmax cm−1: 2994, 2937, 2842, 1632, 1588, 1512, 1461, 1314, 1204, 1154, 1034; 1H NMR (400 MHz, CDCl3): δ 6.57 (1H, s, H-6′), 6.56 (1H, s, H-3′), 6.26 (1H, d, J = 2.2 Hz, H-8), 6.03 (1H, d, J = 2.4 Hz, H-6), 4.56 (1H, br s, H-3), 4.29 (2H, br s, H-2), 3.89 (3H, s, OMe-2′), 3.88 (3H, s, OMe-5′) 3.78 (3H, s, OMe-4′), 3.61 (3H, s, OMe-7) ppm; 13C NMR (100 MHz, CDCl3); δ 163.1 (C-4), 160.8 (C-7), 158.2 (C-5), 152.3 (C-8a), 151.1 (C-2′), 149.8 (C-4′), 143.5 (C-5′), 114.7 (C-1′), 112.6 (C-6′), 100.1 (C-4a), 97.4 (C-3′), 95.9 (C-8), 94.3 (C-6), 69.8 (C-2), 56.8 (OMe-2′), 56.4 (OMe-4′), 56.3 (OMe-5′), 55.5 (OMe-7), 32.6 (C-3) ppm; HRESI-MS m/z 375.1555 [M + H]+ (calcd for C19H23N2O6, 375.1556).
5-Hydroxy-7,2′,4′,5′-tetramethoxyisoflavanone oxime (15) light yellow solid; mp 213–214 °C; FT-IR (neat) νmax cm−1: 3408, 2943, 2847, 2353, 1632, 1576, 1515, 1456, 1203, 1155, 1089, 1034; 1H NMR (400 MHz, CDCl3): δ 6.57 (1H, s, H-6′), 6.42 (1H, s, H-3′), 6.29 (1H, d, J = 2.4 Hz, H-8), 6.05 (1H, d, J = 2.4 Hz, H-6), 4.92 (1H, m, H-3), 4.43 (1H, dd, J = 11.2, 1.4 Hz, H-2), 4.22 (1H, dd, J = 11.2, 3.4 Hz, H-2), 3.88 (3H, s, OMe-2′), 3.87 (3H, s, OMe-5′), 3.79 (3H, s, OMe-4′), 3.59 (3H, s, OMe-7) ppm; 13C NMR (100 MHz, CDCl3); δ 163.4 (C-4), 159.7 (C-7), 158.6 (C-5), 156.5 (C-8a), 151.0 (C-2′), 149.1 (C-4′), 142.8 (C-5′), 116.7 (C-1′), 112.5 (C-6′), 98.3 (C-4a), 97.7 (C-3′), 95.7 (C-8), 94.4 (C-6), 69.5 (C-2), 56.7 (OMe-2′), 56.4 (OMe-4′), 56.1 (OMe-5′), 55.4 (OMe-7), 32.2 (C-3) ppm. HRESI-MS m/z 376.1396 [M + H]+ (calcd for C19H22NO7, 376.1396).
5,7,2′,4′,5′-Pentamethoxyisoflavan oxime (16) dark brown solid; mp 194–196 °C; FT-IR (neat) νmax cm−1: 3180, 2939, 2844, 1609, 1511, 1460, 1310, 1210, 1157, 1114, 1031, 931; 1H NMR (400 MHz, CDCl3): δ 6.55 (1H, s, H-3′), 6.55 (1H, s, H-6′), 6.22 (1H, d, J = 2.2 Hz, H-8), 6.20 (1H, d, J = 2.1 Hz, H-6), 4.94 (1H, m, H-3), 4.41 (1H, dd, J = 11.4, 1.4 Hz, H-2), 4.31 (1H, dd, J = 11.4, 3.7 Hz, H-2), 4.15 (3H, s, OMe-5′), 3.87 (3H, s, OMe-5), 3.86 (3H, s, OMe-4′), 3.85 (3H, s, OMe-7), 3.59 (3H, s, OMe-2′), ppm; 13C NMR (100 MHz, CDCl3); δ 167.4 (C-4), 162.1 (C-7), 161.1 (C-5), 153.0 (C-8a), 151.6 (C-2′), 150.3 (C-4′), 143.0 (C-5′), 113.5 (C-1′), 112.9 (C-6′), 98.0 (C-4a), 95.9 (C-3′), 95.3 (C-8), 93.8 (C-6), 69.5 (C-2), 57.5 (OMe-2), 57.4 (OMe-4′), 56.67 (OMe-5′), 56.2 (OMe-7), 56.2 (OMe-5), 33.1 (C-3) ppm; HRESI-MS m/z 390.1554 [M + H]+ (calcd for C20H24NO7, 390.1553).
Cytotoxicity assay
:1 DMSO/EtOH). After incubation, the medium was replaced with 110 μL of fresh medium containing MTT (0.5 mg mL−1 in PBS) (Sigma Chemical Co., St Louis, MO, USA) and incubated for 2 h. The formazan formed after conversion of MTT was dissolved in DMSO. The absorbance of formazan was measured with a microplate reader (Bio-Rad Laboratories, USA) at the wavelength of 550 nm using 655 nm as a reference wavelength. Each assay was replicated four times. The percentage of viable cells which corresponds to the production of formazan was calculated as previously described.25| Cell viability (%) = [sample(A550 − A655)/control(A550 − A655)] × 100 |
4. Conclusions
The major constituent from the stems of D. stipulacea was found as a isoflavonoid, olibergin A. Due to the cytotoxicity of olibergin A showed weak activity against HCT-116, HT-29, MCF-7 and vero cell lines, then this compound was used as the starting material for structural modification and led to sixteen derivatives. All derivatives were evaluated for the activity using MTT assay and the results displayed isoflavone 5 was the most active compound (IC50 ≅ 10–19 μM). It should be note that isoflavanone 10 and oxime 13 exhibited strong cytotoxicity against HCT-116 and HT-29 cell lines, respectively, and showed weak activity toward normal cells. In addition, oxime 15 displayed cytotoxicity against all cell lines with IC50 values around 26–38 μM. These information may be useful to develop the structure and cytotoxicity of isoflavonoid derivatives.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation (Implementation Unit, Khon Kaen), Thailand is acknowledged.References
- C. Ito, M. Itoigawa, T. Kanematsu, N. Ruangrungsi, T. Mukainaka, H. Tokuda, H. Nishino and H. Furukawa, Phytochemistry, 2003, 64, 1265–1268, DOI:10.1016/j.phytochem.2003.08.010.
- K. Umehara, K. Nemoto, K. Kimijima, A. Matsushita, E. Terada, O. Monthakantirat, W. De-Eknamkul, T. Miyase, T. Warashina, M. Degawa and H. Noguchi, Phytochemistry, 2008, 69, 546–552, DOI:10.1016/j.phytochem.2007.07.011.
- U. Songsiang, S. Wanich, S. Pitchuanchom, S. Netsopa, K. Uanporn and C. Yenjai, Fitoterapia, 2009, 80, 427–431, DOI:10.1016/j.fitote.2009.06.002.
- U. Songsiang, C. Hahnvajanawong and C. Yenjai, Fitoterapia, 2011, 82, 1169–1174, DOI:10.1016/j.fitote.2011.07.015.
- K. Umehara, K. Nemoto, A. Matsushita, E. Terada, O. Monthakantirat, W. De-Eknamkul, T. Miyase, T. Warashina, M. Degawa and H. Noguchi, J. Nat. Prod., 2009, 72, 2163–2168, DOI:10.1021/np900676y.
- R. Nongmaithem, S. D. Yumkham, N. P. Devi, S. Salam, A. K. Das and P. K. Singh, Indian J. Tradit. Knowl., 2018, 17(4), 754–762 Search PubMed . https://nopr.niscair.res.in/handle/123456789/45057.
- P. Bhatt and R. Dayal, Phytochemistry, 1992, 31(2), 719–721, DOI:10.1016/0031-9422(92)90074-Z.
- P. Borai and R. Dayal, Phytochemistry, 1993, 33(3), 731–732, DOI:10.1016/0031-9422(93)85488-D.
- P. Posri, T. Sribuhom, S. Walunchapruk, T. Senawong, S. Tontapha, V. Amornkitbamrung and C. Yenjai, RSC Adv., 2021, 11, 37643–37648, 10.1039/D1RA07041J.
- T. Sribuhom, P. Posri, W. Khankeaw, C. Pornchoo, A. Prawan, S. Tontapha, V. Amornkitbamrung and C. Yenjai, Nat. Pro. Res., 2022, 1–9, DOI:10.1080/14786419.2022.2053852.
- P. Posri, J. Suthiwong, Y. Thongsrim and C. Yenjai, Nat. Pro. Res., 2021, 35, 2823–2830, DOI:10.1080/14786419.2019.1672068.
- C. Lee, J. W. Lee, Q. Jin, D. S. Jang, S. J. Lee, D. Lee, J. T. Hong, Y. Kim, M. K. Lee and B. Y. Hwang, Bioorg. Med. Chem. Lett., 2013, 23, 4263–4266, DOI:10.1016/j.bmcl.2013.04.032.
- S. Kaennakam, P. Siripong and S. Tip-pyang, Nat. Pro. Res., 2016, 30, 1493–1498, DOI:10.1080/14786419.2015.1114936.
- S. Kaennakam, E. R. Sukandar, K. Rassamee, P. Siripong and S. Tip-pyang, Phytochem. Lett., 2019, 31, 187–191, DOI:10.1016/j.phytol.2019.04.005.
- E. Tuenter, Y. Zarev, A. Matheeussen, E. Elgorashi, L. Pieters and K. Foubert, Phytochem. Lett., 2019, 30, 169–172, DOI:10.1016/j.phytol.2019.02.001.
- J. Suthiwong, J. Wandee, S. Pitchuanchom, P. Sojikul, V. Kukongviriyapan and C. Yenjai, Med. Chem. Res., 2018, 27(8), 2042–2049, DOI:10.1007/s00044-018-2214-9.
- N. Mitsuru, O. Susuma and M. Takanao, Bull. Chem. Soc. Jpn., 1980, 53, 831–832, DOI:10.1246/bcsj.53.831.
- R. Larget, B. Lockhart, P. Renard and M. Largeron, Bioorg. Med. Chem. Lett., 2000, 10, 835–838, DOI:10.1016/S0960-894X(00)00110-4.
- C. Yenjai and S. Wanich, Bioorg. Med. Chem. Lett., 2010, 20, 2821–2823, DOI:10.1016/j.bmcl.2010.03.054.
- C. Yenjai, S. Wanich, S. Pitchuanchom and B. Sripanidkulchai, Arch. Pharmacal Res., 2009, 32(9), 1179–1184, DOI:10.1007/s12272-009-1900-z.
- S. S. Chibber and R. P. Sharma, Phytochemistry, 1979, 18, 1082, DOI:10.1016/S0031-9422(00)91494-8.
- N. Mitsuru, O. Susumu and M. Takanao, Bull. Chem. Soc. Jpn., 1980, 53, 831–832, DOI:10.1246/bcsj.53.831.
- O. Pancharoen, A. Athipornchai, A. Panthong and W. C. Taylor, Chem. Pharm. Bull., 2008, 56(6), 835–838, DOI:10.1248/cpb.56.835.
- C. Tantapakul, V. Suthiphasilp, A. Payaka, B. Chaiyosang, D. J. Harding, W. Phuphong, S. Tontapha and S. Laphookhieo, Phytochemistry, 2022, 198, 113168, DOI:10.1016/j.phytochem.2022.113168.
- P. Kumnerdkhonkaen, S. Saenglee, M. Asgar, G. Senawong, K. Khongsukwiwat and T. Senawong, BMC Complementary Altern. Med., 2018, 18, 130, DOI:10.1186/s12906-018-2185-x.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02865d |
| This journal is © The Royal Society of Chemistry 2022 |