DOI:
10.1039/D2RA01919A
(Paper)
RSC Adv., 2022,
12, 17821-17836
Structure-based design and synthesis of a novel long-chain 4′′-alkyl ether derivative of EGCG as potent EGFR inhibitor: in vitro and in silico studies†
Received
24th March 2022
, Accepted 7th June 2022
First published on 16th June 2022
Abstract
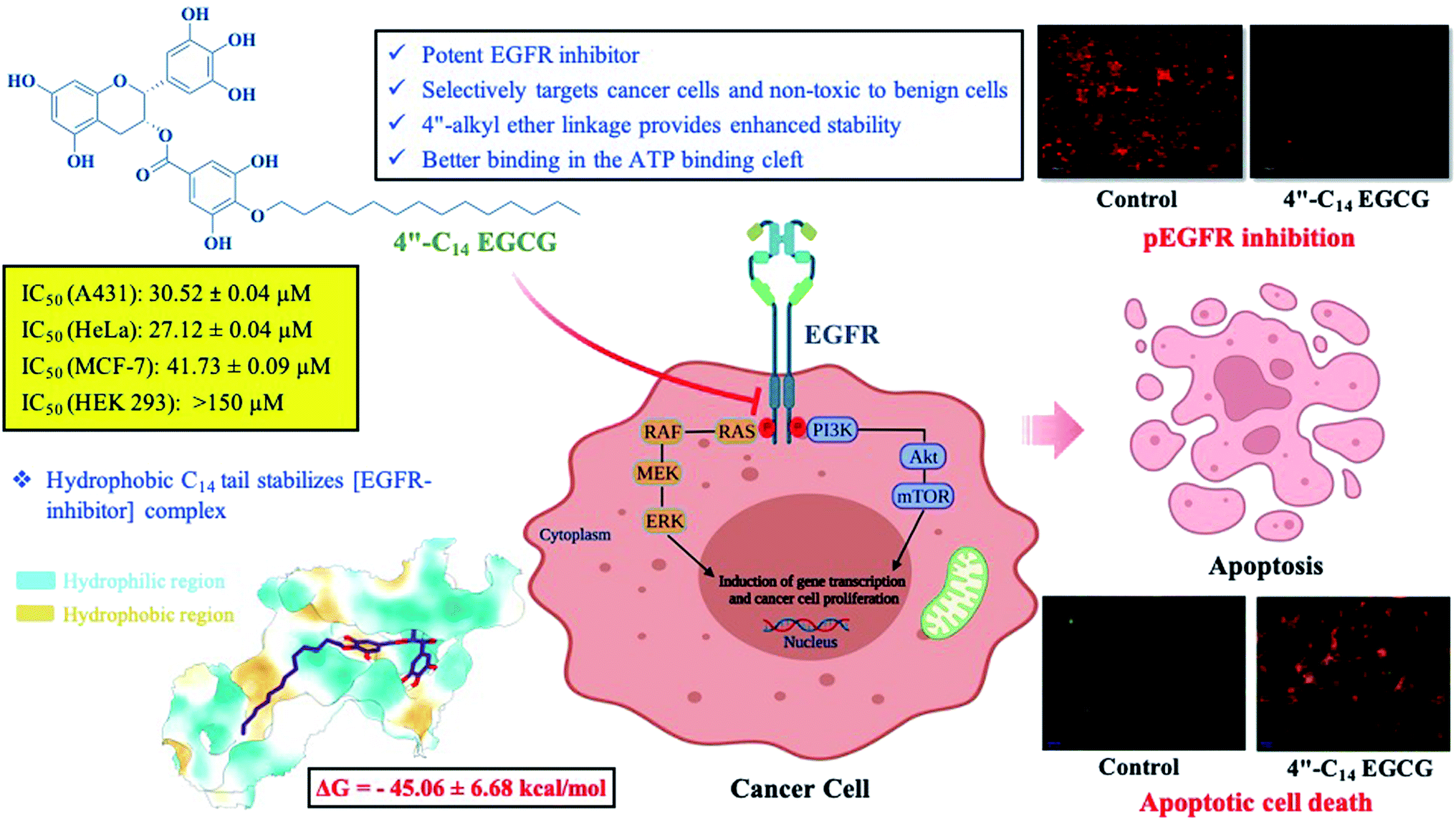
Herein, we report the discovery of a novel long-chain ether derivative of (−)-epigallocatechin-3-gallate (EGCG), a major green tea polyphenol as a potent EGFR inhibitor. A series of 4′′-alkyl EGCG derivatives have been synthesized via regio-selectively alkylating the 4′′ hydroxyl group in the D-ring of EGCG and tested for their antiproliferative activities against high (A431), moderate (HeLa), and low (MCF-7) EGFR-expressing cancer cell lines. The most potent compound, 4′′-C14 EGCG showed the lowest IC50 values across all the tested cell lines. 4′′-C14 EGCG was also found to be significantly more stable than EGCG under physiological conditions (PBS at pH 7.4). Further western blot analysis and imaging data revealed that 4′′-C14 EGCG induced cell death in A431 cells with shrunken nuclei, nuclear fragmentation, membrane blebbing, and increased population of apoptotic cells where BAX upregulation and BCLXL downregulation were observed. In addition, autophosphorylation of EGFR and its downstream signalling proteins Akt and ERK were markedly inhibited by 4′′-C14 EGCG. MD simulation and the MM/PBSA analysis disclosed the binding mode of 4′′-C14 EGCG in the ATP-binding site of EGFR kinase domain. Taken together, our findings demonstrate that 4′′-C14 EGCG can act as a promising potent EGFR inhibitor with enhanced stability.
1. Introduction
The epidermal growth factor receptor (EGFR) protein is a member of the ErbB family of tyrosine kinases that regulates several biological processes such as cell proliferation, migration, apoptosis, survival, differentiation, etc.1 Frequent mutation or overexpression of EGFR has been shown to be involved in the progression of various types of cancer including non-small cell lung cancer, prostate, skin, and breast cancer.2 Thus, targeting EGFR via EGFR-specific tyrosine kinase inhibitors (TKIs) and monoclonal antibodies (mAbs) has become an attractive therapeutic approach for cancer treatment over the years. Several EGFR TKIs such as gefitinib,3 erlotinib,4 afatinib,5 rociletinib,6 and osimertinib7 have been approved by the United States Food and Drug Administration (FDA) for the treatment of patients harbouring activating mutations such as L858R, T790M or exon 19 deletions.8 EGFR developed resistance against the first-generation TKIs due to their reversible nature and T790M gatekeeper mutation. The second-generation TKIs were able to effectively inhibit T790M, L858R, and exon 19 deletion mutant forms of EGFR.9 However, EGFR also acquired resistance against the second-generation TKIs and the later exhibited off-target affinity towards the wild-type EGFR. Further emergence of C797S mutation resulted the resistance in osimertinib (a third generation TKIs) therapy as the mutation averts the covalent bond formation between the inhibitor and C797.9 Despite showing great efficacy against EGFR, significant side effects have also been observed for these TKIs.10 In addition, drug resistance to several FDA-approved TKIs are also reported.11,12 These warrant further research works on the development of effective natural or synthetic inhibitors that will specifically target the EGFR with minimal to zero side effects.
Structure–activity relationship (SAR) approach have been proven to be instrumental in targeted drug discovery applications.13–15 Several potent EGFR inhibitors were discovered based on the comprehensive SAR studies on the medicinally important scaffolds.16–19 Various drug delivery systems have also been explored to reduce the doses and in that context, metal–phenolic networks have recently been identified as promising agent for the development of cancer nanomedicine due to its less toxicity and pH based drug release.20 On the other hand, targeted theranostic nano vehicle with immunostimulatory activities showed promising results to avert the drug resistance and remove cancer causing stem cells.21 In the quest of searching novel TKIs with less toxicity profile, compounds from natural origin might prove to be promising. Despite significant advances in the cancer treatments using potent small molecule synthetic drugs, the resistance to chemotherapy and off-target toxicity makes the use of synthetic molecules difficult and sometimes unsuitable. On the other hand, several natural products have recently been used in cancer treatment and have shown very promising efficacy as well as selectivity.22–24 Small molecules of natural origin display a wide range of structural diversity and promising biological activities with fewer side effects and less toxicity towards benign cells. Over 60% of the anticancer drugs presently in the clinical application with proven efficacy against several types of cancer are of natural origin such as plants, microorganisms, marine organisms, etc.25 In addition, semisynthetic molecules bearing minor chemical modifications on the parent natural products have also been shown to exhibit increased biological efficacy, pharmacokinetics with fewer side effects.25–27 Various novel strategies have been introduced to discover the molecular targets of natural products without chemically modifying them, such as drug affinity responsive target stability, stability of proteins from rates of oxidation, cellular thermal shift assay, thermal proteome profiling, and bioinformatics-based analysis of connectivity. These analytical methods play critical roles in identifying the mechanisms of action of drugs of natural origin.28
Plant-based polyphenols are one such class of natural products comprised of a large structurally diverse molecules with multiple bioactivities exhibiting a plethora of health beneficial effects. One of the promising biologically active polyphenols is (−)-epigallocatechin-3-gallate (EGCG), a major ingredient in green tea. The FDA and European Food Safety Authority (EFSA) have classified EGCG as “Generally Recognized as Safe” (GRAS) for its health beneficial effects.29 Green tea polyphenols mainly EGCG, exhibits various health-beneficial and disease-preventive activities including immunomodulatory,30 anti-oxidant,31 anti-cancer,32 and anti-bacterial.31 EGCG has shown anticancer activities such as inhibition of cancer cell proliferation,33 activation of apoptotic pathways,34 inhibition of angiogenesis, invasion, and metastasis in various types of cancer.35 Nevertheless, despite EGCG possessing great therapeutic and chemopreventive properties, its use in clinical trials has been rather limited because of poor bioavailability,36,37 chemical instability,38,39 low membrane permeability,40 and rapid metabolism.41 To overcome the aforementioned shortcomings, various structural modifications have been done with EGCG to achieve enhanced biological properties, increased stability, and improved bioavailability.42–47
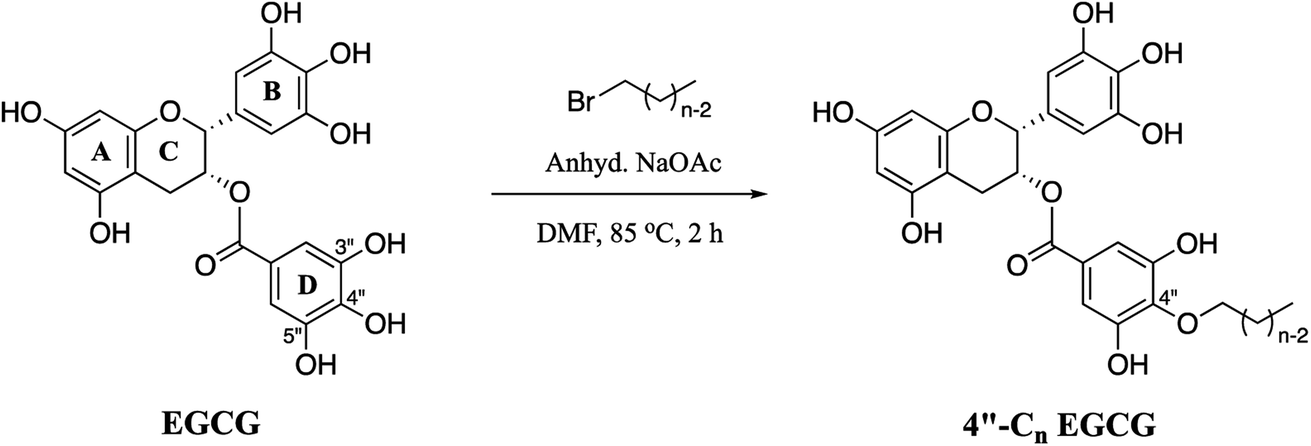
Herein, we decided to regio-selectively introduce alkyl chains of varied length at 4′′ position of EGCG through an ether linkage (Scheme 1), due to its resistance to hydrolysis, and test them for their potential to inhibit EGFR. Seven novel 4′′-alkyl EGCG derivatives were synthesized and characterized by 1D and 2D-NMR. Cytotoxicity profiles were evaluated on high, moderate, and low endogenous EGFR-expressing cell lines A431, HeLa, and MCF-7, respectively. The compounds were also tested in a non-cancerous cell line (HEK-293) to ensure their tumor-targeting ability. Finally, the most promising compound, 4′′-C14 EGCG, was evaluated for its ability to induce apoptosis. The inhibitory activity of 4′′-C14 EGCG was examined against pEGFR and its downstream signalling proteins pAKT and pERK. Further, the stability of 4′′-C14 EGCG was evaluated and compared with EGCG under physiological conditions (PBS, pH 7.4). Molecular docking and molecular dynamic (MD) simulation studies were also carried out against the ATP binding pocket of the EGFR kinase domain to elucidate its binding mode with 4′′-C14 EGCG. The results obtained from the above analysis suggest that 4′′-C14 EGCG acts as a potent EGFR inhibitor (Fig. 1).
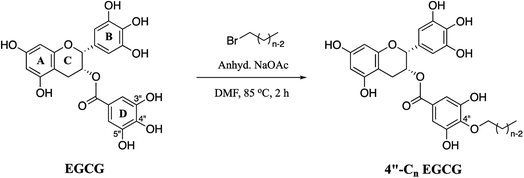 |
| | Scheme 1 Synthetic route for 4′′-alkyl EGCG derivatives (4′′-Cn EGCG), where n = 6, 8, 10, 12, 14, 16, and 18. | |
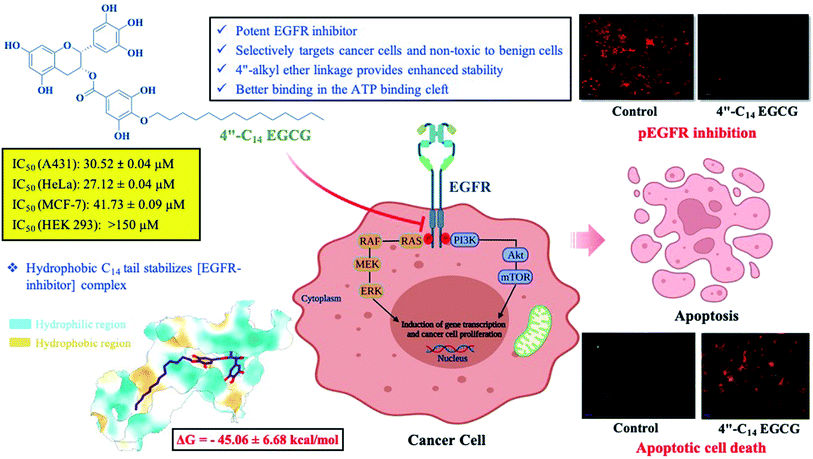 |
| | Fig. 1 Schematic representation of 4′′-C14 EGCG mediated pEGFR inhibition and induction of apoptosis. | |
2. Materials and methods
2.1. Chemistry
EGCG was purchased from Carbosynth Ltd, UK. 1-Bromoalkanes of different chain lengths (C6, C8, C10, C12, C14, C16, and C18) were bought from Sisco Research Laboratories (SRL) Pvt. Ltd, India. Silica gel (230–400 mesh) for column chromatography was purchased from Spectrochem Pvt. Ltd, India. All the solvents used were of reagent grade. LC-MS was recorded in ESI mode on LC-MS-IT-TOF (Shimadzu Pvt. Ltd) instrument. 1H, 13C, and HMBC NMR were recorded in Bruker AVANCE III 500 FT NMR spectrometer. FTIR spectrum was recorded using PerkinElmer Spectrum 100 spectrometer based on a universal attenuated total reflectance (ATR) sensor.
2.1.1. General procedure for the synthesis of 4′′-alkyl EGCG (4′′-Cn EGCG) derivatives. To a stirred mixture of (−)-epigallocatechin-3-gallate (EGCG) (0.6545 mmol, 1 eq.) and anhydrous sodium acetate (1.9373 mmol, 3 eq.) in 1 mL dried N,N-dimethyl formamide (DMF), 1-bromoalkane (5.2360 mmol, 8 eq.) was added dropwise. The resulting mixture was allowed to stir at 85 °C for 2 h. The progress of the reaction was monitored by thin-layer chromatography. The reaction was quenched by adding water (2 mL) and extracted with ethyl acetate (2 mL × 3). The organic layer was then washed with water (2 mL × 3) to remove DMF and dried over anhydrous sodium sulphate. The solvent was evaporated in rotavapor and the obtained crude product was purified by silica gel column chromatography (Silica: 230–400 mesh, eluent: acetone–dichloromethane) to afford the 4′′-alkyl EGCG compounds as brownish solid. The synthesized compounds were confirmed by LC-MS, FT-IR, 1H NMR and 13C NMR.
2.2. Biological evaluation
2.2.1. Cell culture, reagents, and antibodies. A431, MCF-7, HeLa, and HEK-293 cell lines were obtained from National Centre for Cell Science (NCCS), Pune, India. The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco, Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco Thermo Fisher Scientific, USA). Cells were grown in humidified 5% CO2 incubator at 37 °C. EGFR inhibitor, gefitinib, and DMSO were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-BCLXL, anti-BAX, anti-p-EGFR (Tyr1068), anti-p-Akt (Ser473), anti-p-Erk1/2, anti-Erk1/2, anti-Akt antibodies were purchased from Cell Signaling Technology (Beverly, MA, United States). Anti-EGFR, anti-β-actin, anti-Gapdh antibodies were purchased from Santa Cruz (Dallas, TX, United States).
2.2.2. Antiproliferative assay. The synthesized 4′′-alkyl EGCG derivatives were tested for anticancer activity against three human cancer cell lines (A431, HeLa, and MCF-7) and HEK293 cells using the standard Cell Counting Kit-8 (CCK8) methods (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). The CCK8 test utilizes WST-8 dye[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] which further gets reduced by dehydrogenases present in cells into an orange-coloured water-soluble formazan dye. Briefly, 1 × 104 cells were seeded in each well of 96-well plates and cultured in the CO2 incubator for 24 h. The 4′′-alkyl EGCG derivatives were dissolved in DMSO at various concentrations. Afterward, the culture medium was aspirated from the adherent cells, and the compounds at different concentrations prepared above were added into the wells (final concentration of DMSO was 0.1%). After 24 h of incubation with the drugs, 10 μL of CCK8 detection solution was added to each well followed by culturing them in a CO2 incubator for another 2 h. In this experiment, EGCG was taken as reference compound, and gefitinib as a positive control. The absorbance of the formazan dye was measured at 450 nm using a microplate reader (Synergy H1 microplate reader, Biotek).
2.2.3. AO/EtBr and DAPI staining. Apoptotic morphological changes of A431 cells were assessed by AO/EtBr dual staining method. Briefly, 1 × 105 cells were seeded on glass coverslips in 24-well plates for 24 h, and then treated with 4′′-C14 EGCG and EGCG at different concentrations (25 μM, 50 μM and 75 μM) for 24 h. The cells were washed twice with cold 1X phosphate buffer saline (PBS), followed by fixation with 4% paraformaldehyde and stained with AO (10 μg mL−1) and EtBr (10 μg mL−1) or DAPI (10 μg mL−1) for 20 min. The stained cells were washed twice with PBS, and coverslips were mounted on glass slides. The cells were then observed under the fluorescence microscope (Olympus).
2.2.4. Wound healing assay. The A431 cells were seeded in 12-well plates at a density of 2 × 105 cells per well and cultured for 24 h. To create a wound, the cell monolayers in 12-well plates were scratched by using a sterile 200 μL pipette tip. After scratch, cells were washed with PBS to remove detached cells and were further incubated with mediums containing 25 μM, 50 μM, and 75 μM of 4′′-C14 EGCG or reference compound EGCG. Gefitinib at 5 μM was taken as positive control, while 0.1% DMSO-treated cells were taken as control. Nikon inverted microscope was used to take the images of the scratch at 0 h and 24 h after creating the wound. The gap width area was measured with the Image J software (NIH).
2.2.5. Western blot analysis. A431 cells were seeded in a 6-well plate at a density of 4 × 105 cells per well for 24 h. Cells were treated with 25 μM, 50 μM and 75 μM of 4′′-C14 EGCG or reference compound EGCG and gefitinib at 5 μM concentration for next 24 h followed by EGF stimulation (50 ng mL−1) given for 15 minutes. After harvesting the cells, cell lysates were prepared in RIPA lysis buffer (1 M Tris pH 7.4, 2 M NaCl, 0.1 M EDTA, 1 M PMSF, protease inhibitor cocktail, 100 mM DTT and 1 mM sodium orthovanadate, Triton-X-100, and Glycerol) and stored at −80 °C. Proteins were separated in 8–15% SDS-PAGE and transferred to nitrocellulose membrane (BioRad) for 2 h at 100 volts. Membrane was blocked with 5% BSA or skimmed milk in 1X PBST (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl and 0.1% Tween 20) for 1 h. Followed by membrane were incubated with primary rabbit/mouse IgG antibodies (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) overnight at 4 °C and then washed using 1X PBST. Next, membranes were incubated with HRP-conjugated anti-rabbit/anti-mouse IgG secondary antibodies in 1X PBST (1:2000) for 2 h at room temperature. The membrane was washed using 1X PBST, developed using standard chemiluminescent substrate (BioRad) and images were captured on a Fusion Solo S chemidoc system (Vilber). β-Actin and Gapdh were used as loading controls. Protein band intensities were quantified by using Image J software (NIH) with respect to their corresponding loading controls.
:1000) overnight at 4 °C and then washed using 1X PBST. Next, membranes were incubated with HRP-conjugated anti-rabbit/anti-mouse IgG secondary antibodies in 1X PBST (1:2000) for 2 h at room temperature. The membrane was washed using 1X PBST, developed using standard chemiluminescent substrate (BioRad) and images were captured on a Fusion Solo S chemidoc system (Vilber). β-Actin and Gapdh were used as loading controls. Protein band intensities were quantified by using Image J software (NIH) with respect to their corresponding loading controls.
2.2.6. Immunocytochemistry analysis. A431 cells were seeded at 1 × 105 cells per well on round coverslips in 24-well tissue culture plates. Next, cells were incubated with the above-mentioned concentrations of compounds for 24 h. Followed by that, cells were fixed with 4% of PFA for 20 min. The cells were permeabilized using 0.5% saponin for 10 min and blocked with 5% BSA for an h at 37 °C. After blocking, cells were incubated with primary antibody (1:200) overnight at 4 °C. Then, the cells were washed thrice with 1X cold PBS and incubated with Alexa Fluor conjugated secondary antibody (1:500) in the dark at room temperature for 2 h. Upon completion of the incubation, the coverslips were mounted on glass slides using Antifade Gold DAPI (Invitrogen) and sealed with nail paint. Slides were imaged by using a confocal laser scanning microscope (FV1200MPE, IX83 Model, Olympus).
2.2.7. Stability analysis. A stock solution of EGCG and 4′′-C14 EGCG of concentration 1 mg mL−1 was made with 100 mM PBS buffer of pH 7.4. From this, 60 μL was made up to 3 mL with the buffer and the absorbance was measured at λmax (213 nm) using a UV-visible spectrophotometer at the time of mixing (0 h), after 5 h, and 24 h. The stability was calculated based in the decrease in the absorbance at λmax over the incubation time. The experiment was repeated twice.
2.2.8. DPPH assay. Different stock solution of EGCG and 4′′-C14 EGCG (0.4, 2, 4, 8, 16, 24, 32, 40, 80, 200 μM) were prepared in DMSO. From the stock solutions, 50 μL was mixed with 150 μL of 0.2 mM DPPH (2,2-diphenyl-1-picryl-hydrazyl-hydrate) (prepared in 95% methanol) and incubated for 1 h in dark at room temperature. After that, the absorbance was recorded at 517 nm using the microplate reader.
2.3. Theoretical section
2.3.1. Computational docking. Molecular docking was performed for EGCG and 4′′-C14 EGCG against the wild-type EGFR crystal structure (PDB ID 4I23), which is available in a complex with dacomitinib.48 We performed molecular docking using the GLIDE module of the Schrödinger suite.49–51 The EGFR structure was treated with the help of Protein Preparation Wizard of the Schrödinger suite to optimize the hydrogen bonds and conducted minimization up to 0.3 Å RMSD to converging heavy atoms. During docking, the protein structure was kept rigid. The docking grid was generated by taking the native ligand of the crystal structure in the center. EGCG and its modified molecule were prepared in a neutral ionization state using the LigPrep module of Schrödinger. The ligands were minimized using the OPLS3 force field after the addition of hydrogen atoms.52 The docking run was conducted under the GLIDE module's extra precision (XP) protocol to achieve high accuracy. The best binding pose was selected by estimating GScore, which includes ligand–protein interactions, hydrophobic interactions, hydrogen bonds, stacking interactions, desolvation, etc. The best binding pose for both the ligands was used for the conventional molecular dynamics (cMD) study.
2.3.2. Molecular dynamics simulation of active EGFR in complex with 4′′-C14 EGCG and EGCG. Molecular dynamics simulations were performed using the pmemd.cuda module of AMBER18.53 The antechamber module of the AMBER18 suite was used to derive the parameters for both ligands.54,55 The AMBER FF14SB and Generalized Amber Force Field (GAFF2) were used for the preparation of protein and ligands, respectively.56 For both systems, a truncated octahedron box was used for solvation using TIP3P water molecules,57 and a 10 Å buffer distance was kept from all sides of the systems. The SHAKE algorithm was used to restrain all bonds, including hydrogen atoms.58 The temperature (300 K) and pressure (1 bar) were controlled by the Langevin thermostat and Berendsen barostat, respectively.59,60 The long-range electrostatic interactions with a non-bonded cut-off 10 Å were considered using the Particle mesh Ewald (PME) method.61 Two-step minimizations, followed by heating and equilibrium steps, were performed before production runs. A detailed simulation protocol was discussed in our previous studies.62–64 Finally, a 200 ns production run was conducted for both systems with a time-step of 2 fs in the NPT ensemble. A total of 20000 configurations was generated for both systems, which were further used for the trajectory analysis using the Cpptraj module of AmberTools.62,65The protein–ligand binding free energy was calculated using the Molecular Mechanics Poisson Boltzmann Surface Area (MMPBSA).66–68 The binding free energy was estimated using the following equation.69
| | |
ΔGbind = ΔH − TΔS ≈ ΔEinternal + ΔGsolv − TΔS
| (1) |
The total binding free energy (ΔGbind) comprises internal energy (ΔEinternal), desolvation free energy (ΔGsolv), and configurational entropy (TΔS). For the high computational cost, the entropic contribution was not considered in our study. The binding free energy was estimated using 10000 configurations obtained from the last 100 ns. Further, we decomposed the total binding free energy at the per-residue level using the molecular mechanics generalized Born surface area (MMGBSA) scheme.70
3. Results and discussion
3.1. Chemistry
EGCG has three aromatic rings (A, B, and D, Scheme 1) and they are connected by a pyran ring (C, Scheme 1). The antioxidant property of EGCG results from the transfer of hydrogen atoms or single-electron transfer reactions involving multiple phenolic hydroxyl groups. Among all the hydroxy groups present in EGCG, the 4′′-OH is uniquely posed and most acidic because of the presence of the ester group at its para position. We wanted to take this advantage of the 4′′-OH position to regio-selectively incorporate the alkyl chains. Of note, previous reports71,72 suggest that the loss of 4′′-OH group from EGCG does not significantly alter its biological activities as compared to parent analog, EGCG.
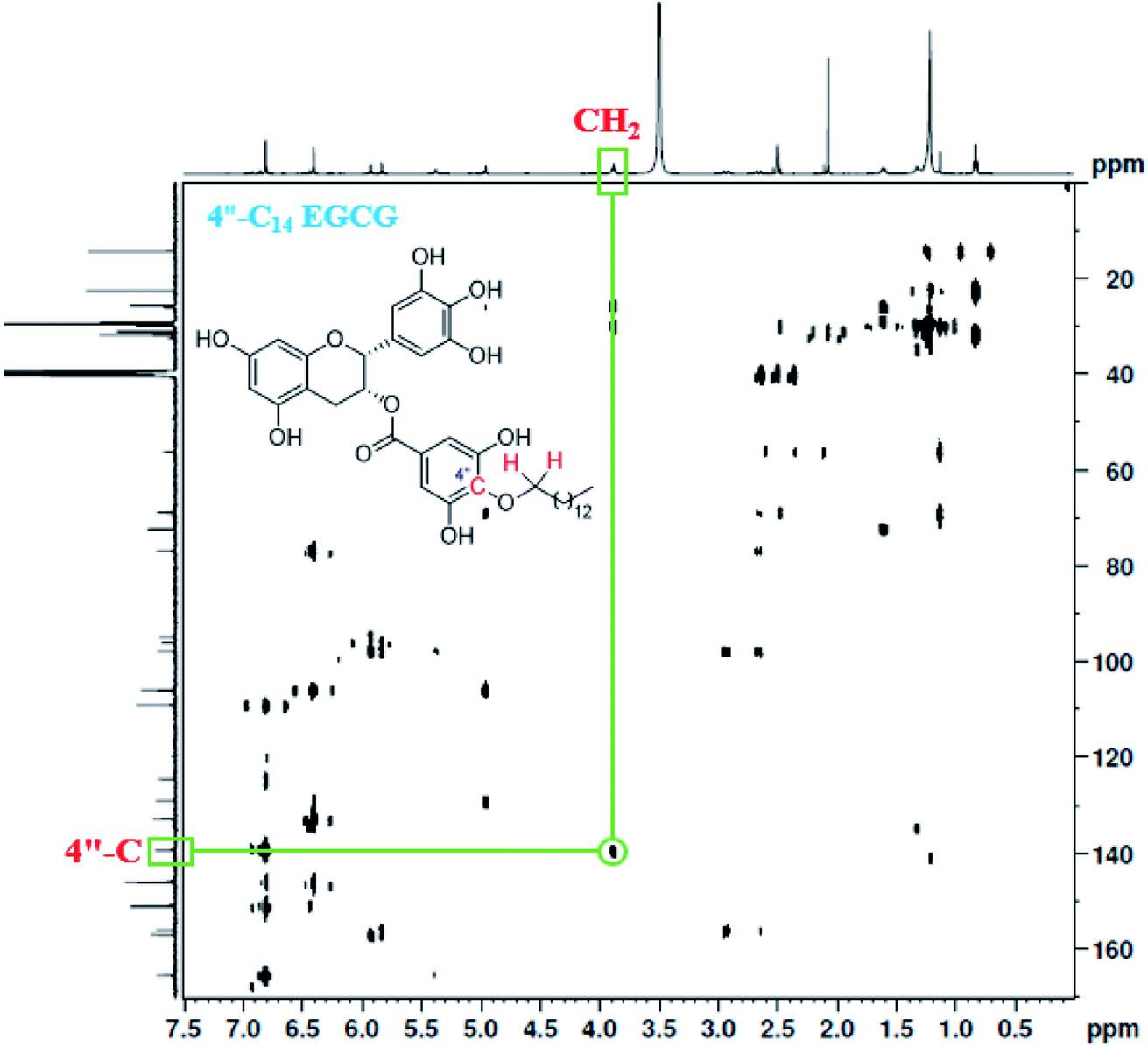
A total of seven 4′′-alkyl EGCG derivatives with varied hydrophobicity were synthesised as described in Scheme 1. Purity (>98%) of the compounds were confirmed using high-performance liquid chromatography (HPLC), and their structures were characterized using ESI-LC-MS, FT-IR, 1H NMR, and 13C NMR. The positioning of the alkyl chain in 4′′-alkyl EGCG derivatives (taking an example of 4′′-C14 EGCG) was further confirmed by the Heteronuclear Multiple-Bond Correlation (HMBC) 2D-NMR experiment (Fig. 2). The 4′′-C assignment was made by comparing the 13C-NMR of EGCG and 4′′-alkyl derivatives of EGCG. As expected, significant differences in the chemical shift values were observed for C-1′′, C-3′′, C-4′′, and C-5′′, which implies that the alkylation happened in the D-ring of EGCG. Further, the assignment is in well agreement with the reported literature.73,74
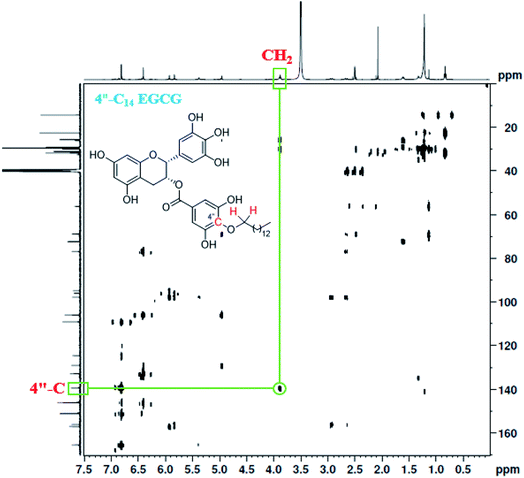 |
| | Fig. 2 HMBC NMR of 4′′-C14 EGCG. The highlighted cross peak indicates that the carbon at 4′′ position is directly connected to –OCH2– (as shown in the inset). | |
3.2. Biological evaluation
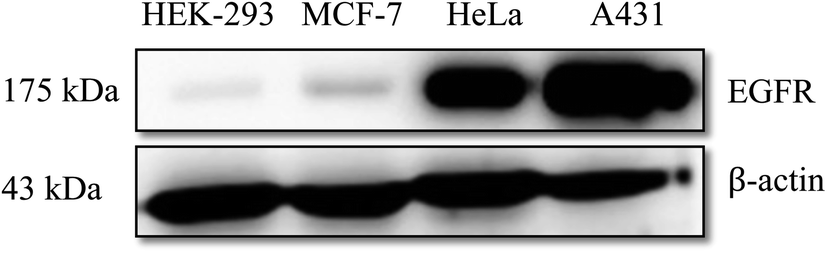
3.2.1. 4′′-Alkyl EGCG derivatives suppresses the proliferation of cancer cells. Cytotoxicity of 4′′-alkyl EGCG derivatives was assessed using standard CCK8 assay on A431, HeLa, and MCF-7 cell lines exhibiting high, moderate, and low endogenous EGFR expression levels, respectively.75 EGCG was used as a reference compound in this experiment. The 4′′-alkyl derivatives displayed variable levels of cytotoxic activity against all the tested human cancer cell lines. We validated the EGFR expression profile across the four cell lines which were used in this study and as shown in Fig. 3, A431 had the highest EGFR expression and HeLa and MCF-7 had medium and low EGFR expression, respectively. HEK-293 cells showed very low EGFR expression.
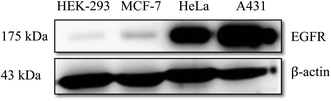 |
| | Fig. 3 Endogenous EGFR expression profile in HEK-293, MCF-7, HeLa and A431 cell lines. | |
For A431 cells, 4′′-C6 EGCG and 4′′-C8 EGCG showed the highest IC50 value (105.09 ± 0.03 μM and 109 ± 0.02), while a successive addition of two methylene groups, i.e. –(CH2)2– in the alkyl chain such as 4′′-C10 EGCG, 4′′-C12 EGCG and 4′′-C14 EGCG decreased the IC50 values to 100.04 ± 0.02 μM, 50.53 ± 0.06 μM and 30.52 ± 0.04 μM, respectively. While adding more hydrophobicity to the alkyl chain as can be seen in 4′′-C16 EGCG and 4′′-C18 EGCG, the IC50 values further increased (84.94 ± 0.02 μM, 94.59 ± 0.01 μM respectively) as compared to 4′′-C14 EGCG. This suggested that among the seven derivatives, 4′′-C14 EGCG exhibited the most potent antiproliferative activity against A431 cells. Next, we studied the cytotoxicity of the 4′′-alkyl derivatives on HeLa cells. Here also, we found 4′′-C14 EGCG displaying the lowest IC50 value (27.12 ± 0.04 μM) among the compounds tested. In the case of MCF-7 cells, 4′′-C8 EGCG exhibited a high IC50 value of 156.5 ± 0.58 μM. More hydrophobic derivatives showed lower IC50 values, thus correlating well with the hydrophobicity of the alkyl chain as can be seen in 4′′-C10 EGCG (99.04 ± 0.42 μM), 4′′-C12 EGCG (96.92 ± 0.19 μM), and 4′′-C14 EGCG (41.73 ± 0.09 μM). Further, increase in hydrophobicity resulted in increased IC50 values (compared to 4′′-C14 EGCG) as can be seen for 4′′-C16 EGCG (51.25 ± 0.08 μM) and 4′′-C18 EGCG (133.97 ± 0.02 μM). Taken together, the results obtained from all the three tested human cancer cell lines (A431, HeLa, and MCF-7) suggested that the introduction of a fourteen-carbon long aliphatic hydrocarbon at 4′′ position of EGCG (4′′-C14 EGCG) exhibited the most potent antiproliferative activity (see Table 1) among the tested compounds. Therefore, we selected 4′′-C14 EGCG for further studies. In addition, to ensure that the 4′′-alkyl EGCG derivatives are not toxic to the non-cancerous cells, their antiproliferative activities were carried out against the HEK-293 cell line. After incubating the HEK-293 cells with 4′′-alkyl EGCG derivatives, none of them displayed any cytotoxicity till 150 μM (See Table 1). This indicates that the synthesized 4′′-alkyl EGCG derivatives are selective towards the cancer cells and not toxic to the non-cancerous cells. We also checked the anti-proliferative activity of gefitinib, an FDA-approved first generation EGFR inhibitor in all the cell lines tested. It showed an IC50 of 6.68 ± 0.06, 5.53 ± 0.09 and 9.16 ± 0.07 μM, against A431, HeLa and MCF-7 cell lines, respectively. Although, 4′′-C14 EGCG displayed lower IC50 value in HeLa cells as compared to A431, the endogenous expression of EGFR in A431 is much higher than that in HeLa and this prompted us to select A431 over HeLa for further studies. Of note, the over-expression of EGFR has been recognized as one of the major cancer-driving mechanisms for the development and progression of various types of cancer including pancreatic cancer, skin cancer, lung cancer, breast cancer, etc.18
Table 1 Cytotoxicity profiles of 4′′-alkyl EGCG derivatives against high (A431), moderate (HeLa), low (MCF-7) EGFR expressing cancer cell lines as well as a non-cancerous cell line (HEK-293). The IC50 values are expressed as (mean ± SEM). Experiments were performed in triplicates and one representative set of data is shown from two biological replicates
| Compounds |
IC50 (μM) |
| A431 |
HeLa |
MCF-7 |
HEK-293 |
| 4′′-C6 EGCG |
105.9 ± 0.03 |
114.6 ± 0.23 |
50.51 ± 0.23 |
>150 |
| 4′′-C8 EGCG |
109 ± 0.02 |
88.17 ± 0.04 |
156.5 ± 0.58 |
>150 |
| 4′′-C10 EGCG |
100.04 ± 0.02 |
112.3 ± 0.02 |
99.04 ± 0.42 |
>150 |
| 4′′-C12 EGCG |
50.53 ± 0.06 |
81.32 ± 0.06 |
96.92 ± 0.19 |
>150 |
| 4′′-C14 EGCG |
30.52 ± 0.04 |
27.12 ± 0.04 |
41.73 ± 0.09 |
>150 |
| 4′′-C16 EGCG |
84.94 ± 0.02 |
104.6 ± 0.05 |
51.25 ± 0.08 |
>150 |
| 4′′-C18 EGCG |
94.59 ± 0.01 |
60.59 ± 0.10 |
133.97 ± 0.02 |
>150 |
| EGCG |
53.78 ± 0.04 |
61.63 ± 0.03 |
72.83 ± 0.04 |
>150 |
| Gefitinib |
6.68 ± 0.06 |
5.53 ± 0.09 |
9.16 ± 0.07 |
24.48 ± 0.04 |
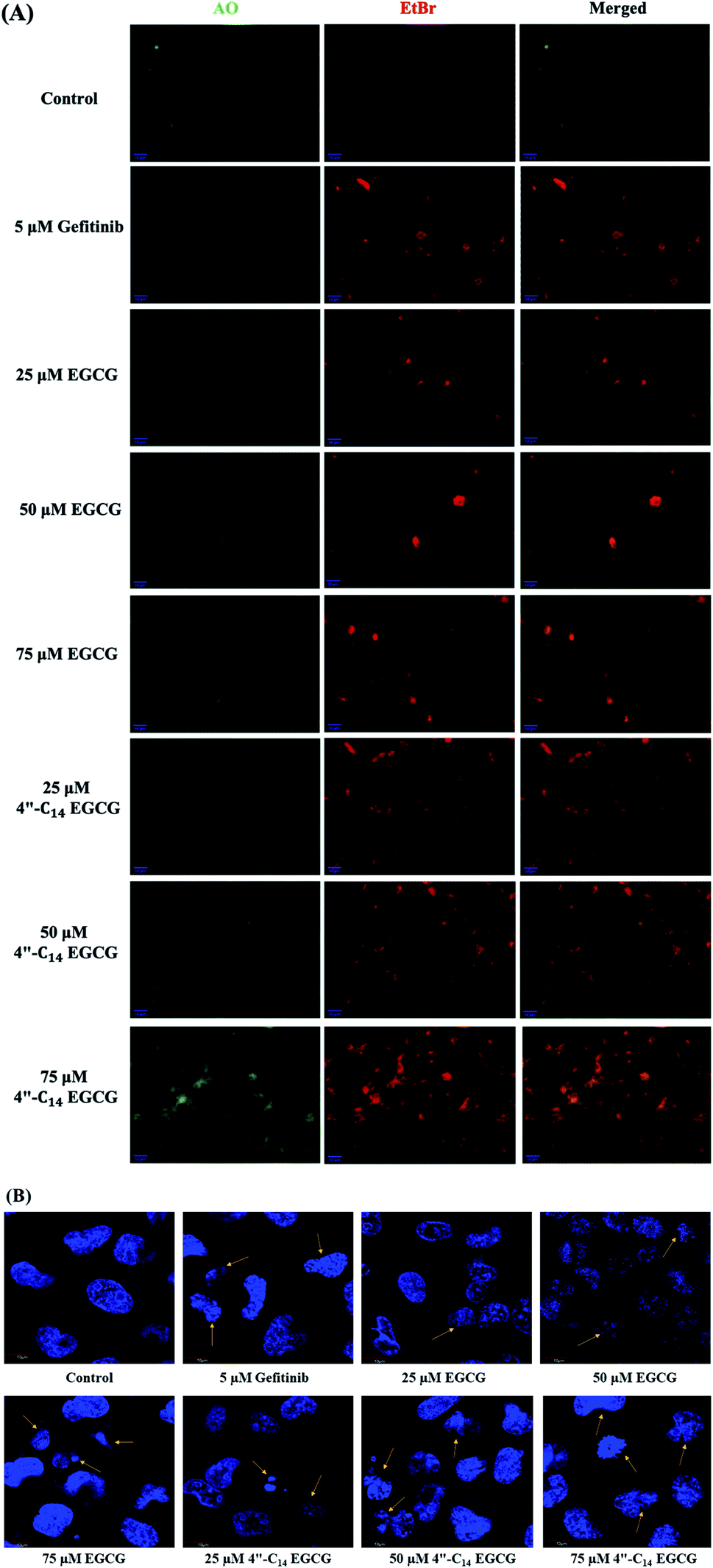
3.2.2. 4′′-C14 EGCG induces apoptosis. Apoptosis is the preferred pathway for anticancer agent-induced cell death. The effect of 4′′-C14 EGCG on the induction of apoptosis was examined using the acridine orange and ethidium bromide (AO/EtBr) dual staining method.76 Membrane permeable cationic dye, AO stains both live and dead cells and emits green fluorescence whereas EtBr only stains (in red) the cells that have lost their membrane integrity or undergone cell death. AO/EtBr staining showed more green fluorescent with no orange or red fluorescence in control cells, indicating the live and healthy condition of the cells (Fig. 4A, panel ‘control’). As shown in Fig. 4, treatment of A431 cells with 4′′-C14 EGCG for 24 h was able to induce apoptosis in a concentration-dependent manner. First-generation TKI, gefitinib was taken as a positive control, which induced cell death at 5 μM in A431 cells.
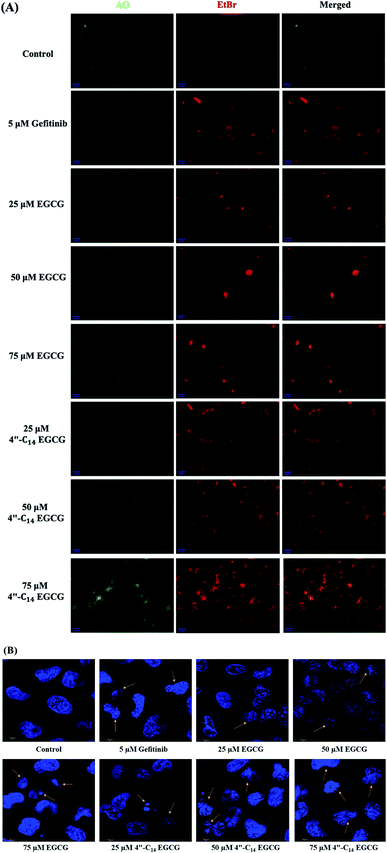 |
| | Fig. 4 (A) Morphological changes and cell death were observed by AO/EtBr staining of A431 cells treated with gefitinib, EGCG, and 4′′-C14 EGCG. Apoptotic characteristics such as chromatin condensation, membrane blebbing, and apoptotic body formations were assessed in treated cells with increasing concentrations. (B) Detection of altered nuclear morphology by DAPI staining of gefitinib, EGCG, and 4′′-C14 EGCG-treated (24 h) A431 cells showed nuclear apoptotic features such as nuclear fragmentation and shrunken nuclei. Two independent experiments were performed, and the data from a representative experiment are shown (scale bar = 10 μm). | |
We also carried out 4′,6-diamidino-2-phenylindole (DAPI) staining to distinguish the normal and apoptotic cells by observing their nuclear morphological changes upon treatment of A431 cells with 4′′-C14 EGCG, EGCG, and gefitinib treatment for 24 h (Fig. 4B). Obtained results suggested that the nuclear structure was intact in the control cells, whereas 4′′-C14 EGCG, EGCG, and gefitinib treated cells exerted the enhanced nuclear damage in a concentration-dependent manner.
3.2.3. The anti-migration activity of 4′′-C14 EGCG. Cell migration is a critical process and known to be involved in several physiological processes such as angiogenesis, tumor metastasis, wound healing and thus plays a crucial role in the cancer progression.77 Herein, we investigated the anti-migration activity of gefitinib, EGCG, and 4′′-C14 EGCG on A431 cells by using wound healing assay in vitro. As shown in Fig. 5, cells exposed to different concentrations of gefitinib, EGCG, and 4′′-C14 EGCG showed considerable inhibition of cell migration compared to the untreated cells. Of note, 4′′-C14 EGCG (at 50 μM and 75 μM) showed a greater level of inhibition of cell migration in A431 when compared to EGCG (Fig. 5).
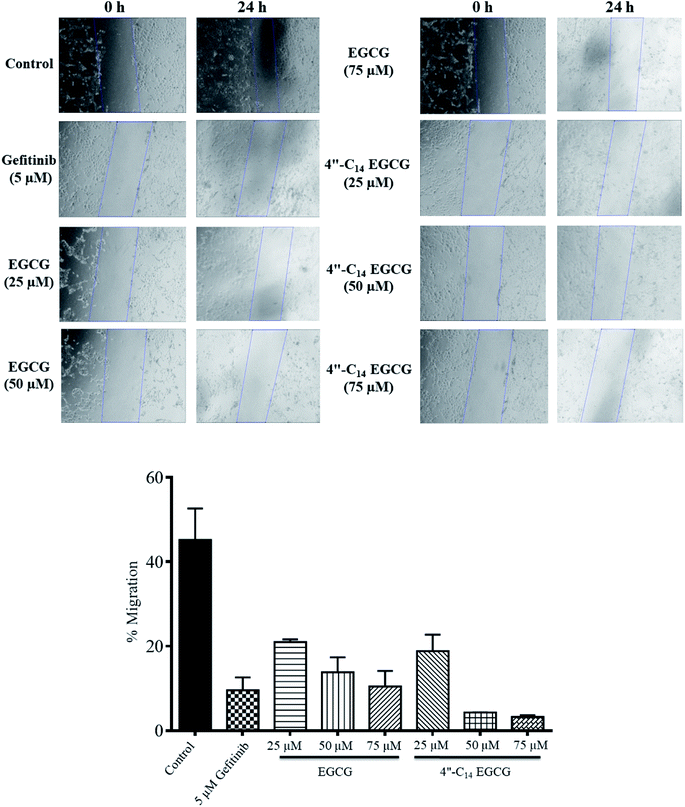 |
| | Fig. 5 Effect of gefitinib, EGCG, and 4′′-C14 EGCG on migration of A431 cells. Wounds were created with a sterile 200 μL pipette tip, and cells were treated with different concentrations (25 μM, 50 μM, 75 μM) of EGCG and 4′′-C14 EGCG. Images were captured using phase-contrast microscopy 0 h and 24 h post-treatment. Two independent experiments were performed, and the data from a representative experiment are shown. Data are represented as (mean ± SEM). | |
3.2.4. 4′′-C14 EGCG induces apoptotic cell death. The BCL-2 family of proteins are known to be involved in regulation of apoptotic cell death and includes both pro-apoptotic (e.g., BAX) as well as anti-apoptotic (e.g., BCLXL) proteins. To further investigate whether 4′′-C14 EGCG triggers apoptosis, cells were incubated with varying concentrations of the inhibitors for 24 h. Western blot analysis showed a concentration-dependent reduction in the expression of BCLXL upon EGCG and 4′′-C14 EGCG treatment (Fig. 6). A431 cells treated with 4′′-C14 EGCG at 25 μM concentration resulted in significant downregulation of the BCLXL expression compared to the control cells. On the other hand, EGCG at 25 μM concentration was unable to inhibit the BCLXL expression level, and its expression was inhibited only at 75 μM. In comparison to untreated and DMSO treated cells an enhanced expression of BAX was observed after treatment with 4′′-C14 EGCG (above 50 μM) for 24 h (Fig. 6). Taken together, these results suggest that 4′′-C14 EGCG induces cell death via apoptosis.
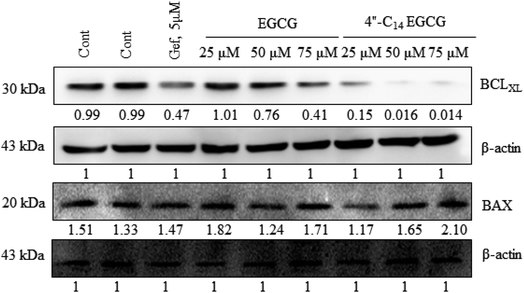 |
| | Fig. 6 Effect of gefitinib, EGCG, and 4′′-C14 EGCG on the expression of apoptotic proteins. Western blot analysis was carried out by using BCLXL and BAX-specific antibodies. A431 cells treated with 4′′-C14 EGCG were compared with control (untreated), DMSO-treated, positive control (gefitinib), and reference compound (EGCG). β-Actin was taken as an internal loading control. Two independent experiments were performed, and the data from a representative experiment are shown. Densitometry analysis have been carried out by using Image J software and obtained values are given underneath each band. | |
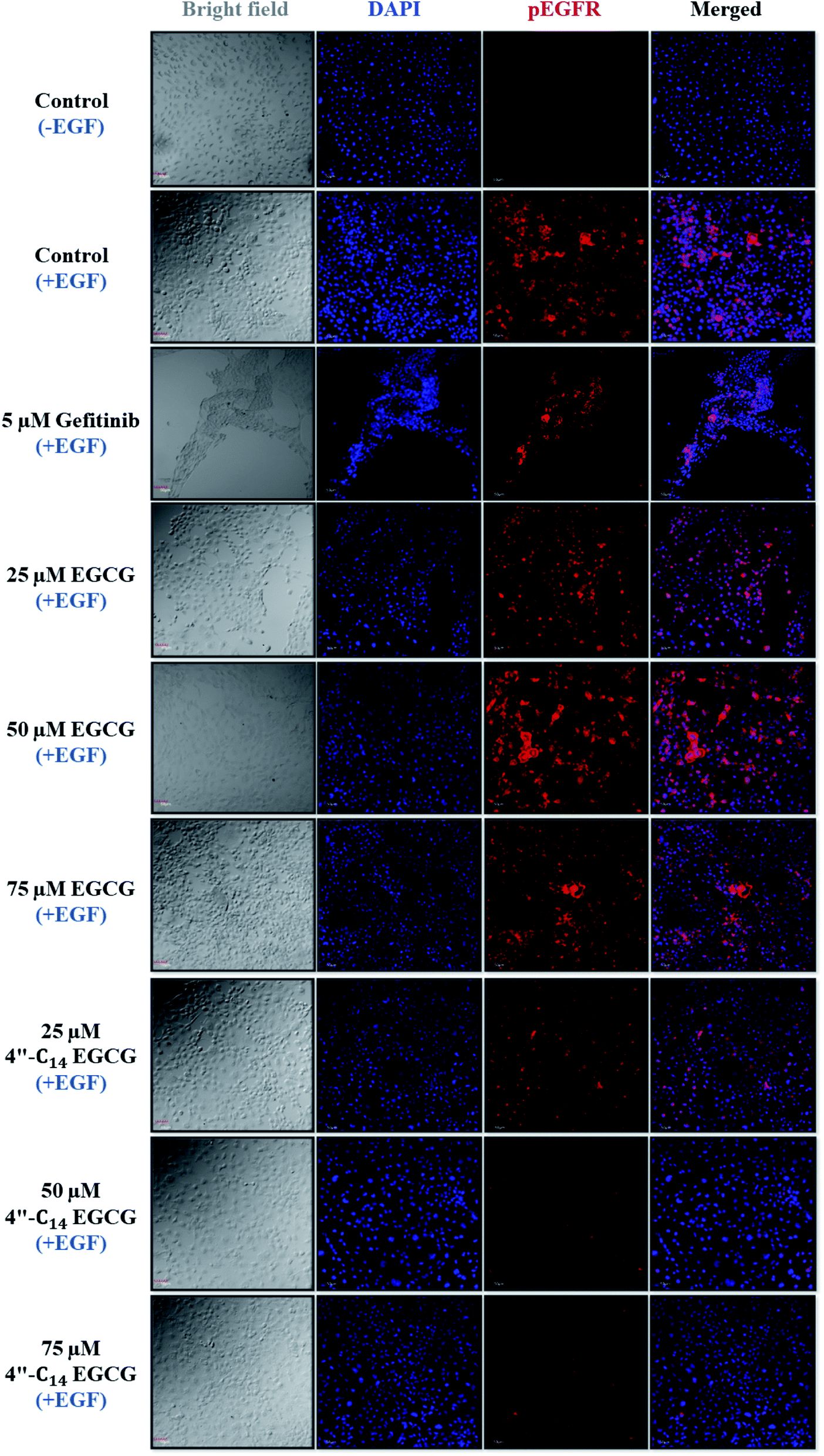
3.2.5. Effect of 4′′-C14 EGCG on phosphorylation of EGFR and its downstream signalling pathways. The binding of the epidermal growth factor (EGF) to the extracellular binding domain of EGFR leads to the activation of the receptor, which further causes either homodimerization with another EGFR or heterodimerization with other members of ErBb family.78 Homo or heterodimerization subsequently stimulates the autophosphorylation of the EGFR, which then causes activation of downstream proteins that are important for cell proliferation and differentiation.79 Both the overexpression and activation of EGFR are associated with the progression of several types of cancer.80–82 Therefore, we wanted to see the effect of 4′′-C14 EGCG on EGF-induced phosphorylation of EGFR. Our results showed that the exposure of A431 cells to various concentrations of gefitinib, EGCG, and 4′′-C14 EGCG for 24 h reduced the level of phosphorylated EGFR (pEGFR). As shown in Fig. 7, treatment with 50 μM of 4′′-C14 EGCG significantly inhibited the phosphorylation of EGFR. Of note, 75 μM 4′′-C14 EGCG completely inhibited the phosphorylation of EGFR. Furthermore, we also investigated the phosphorylation level of ERK and Akt, which are the downstream signalling proteins of EGFR. Notably, 4′′-C14 EGCG showed more than 90% inhibition in the phosphorylation of ERK at 25 μM, wherein EGCG displayed a similar level of inhibition of ERK phosphorylation at a much higher concentration (75 μM). Furthermore, 4′′-C14 EGCG was also able to inhibit the phosphorylation of Akt at 25 μM concentration while no such inhibition was observed even after 75 μM EGCG treatment. Altogether, these data suggest that 4′′-C14 EGCG exhibits anticancer activity through inhibition of EGFR activation. The ability of 4′′-C14 EGCG to inhibit the kinases downstream of EGFR, such as ERK or Akt, further enhances its potential as a promising EGFR inhibitor.
 |
| | Fig. 7 Effect of 4′′-C14 EGCG on EGFR, ERK, and Akt phosphorylation. A431 cells were treated with various concentrations (25, 50, 75 μM) of 4′′-C14 EGCG, EGCG, and positive control gefitinib (5 μM) for 24 h followed by 50 ng mL−1 EGF stimulation for 15 min. The inhibitory profiles were assessed by the western blotting using β-actin and/or Gapdh as the loading control. 4′′-C14 EGCG effectively blocked the phosphorylation of EGFR, ERK, and Akt. Two independent experiments were performed, and the data from a representative experiment are shown. Densitometry analysis have been carried out by using Image J software and obtained values are given underneath each band. | |
3.2.6. Immunocytochemistry analysis of 4′′-C14 EGCG-mediated pEGFR inhibition. Next, we performed immunocytochemistry analysis to study the expression of pEGFR after treatment with gefitinib, EGCG, and 4′′-C14 EGCG for 24 h using a pEGFR-specific antibody. Treatment of A431 with 4′′-C14 EGCG at 50 μM for 24 h resulted in a significant reduction in the pEGFR expression level (red) (Fig. 8) as compared to the EGCG treatment. Consistent with our western blot results as shown in Fig. 7, immunocytochemistry analysis also confirmed that 4′′-C14 EGCG strongly inhibited the autophosphorylation of EGFR.
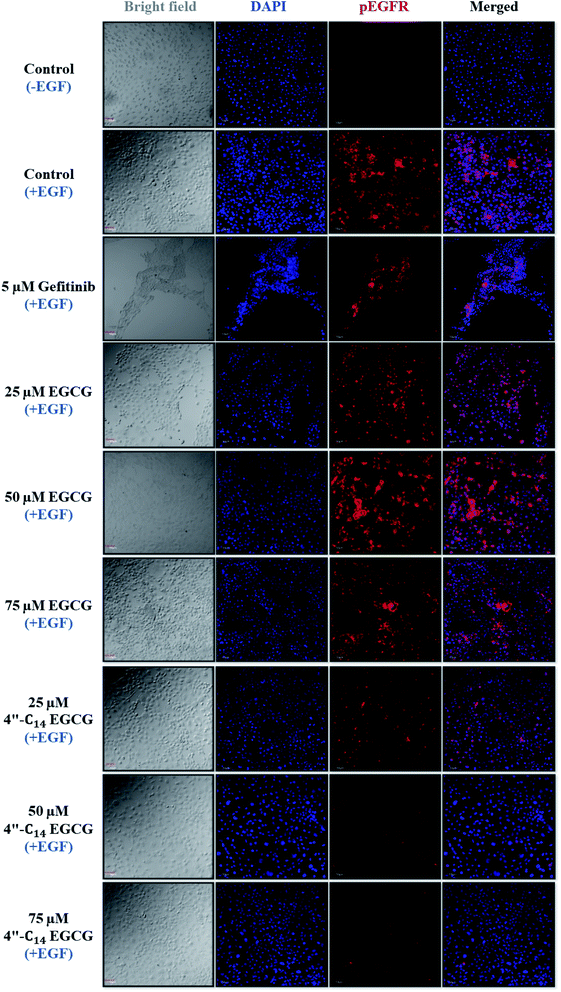 |
| | Fig. 8 Effect of 4′′-C14 EGCG treatment on EGF-stimulated EGFR phosphorylation. A431 cells were pre-treated with 25, 50, 75 μM of 4′′-C14 EGCG, EGCG, and 5 μM of gefitinib for 24 h followed by 50 ng mL−1 EGF stimulation for 15 min, and immune-stained for phospho-EGFR (red) and the nucleus were stained with DAPI (blue). 4′′-C14 EGCG blocked the phosphorylation of EGFR at 50 and 75 μM concentration. Two independent experiments were performed and the data from a representative experiment are shown (scale bar = 50 μm). | |
3.2.7. Improved stability of 4′′-C14 EGCG. It is well-documented that EGCG suffers from poor stability and is prone to rapid degradation in alkaline environment.83 EGCG undergoes auto-oxidation at physiological conditions (pH 7.4, 37 °C), and convert into ortho-quinone via non-enzymatic dehydrogenation of phenolic hydroxyl groups.84 Herein, we wanted to compare the stability of EGCG and 4′′-C14 EGCG. The stability of EGCG and 4′′-C14 EGCG was evaluated by spectrophotometric (UV-Vis) analysis (Fig. S36†). Obtained results showed that under physiological conditions (PBS at pH 7.4), EGCG degraded 36.52 ± 3.06% and 50.72 ± 2.27% after 5 h and 24 h, respectively. On the other hand, 4′′-C14 EGCG showed only 20.51 ± 0.93% and 27.80 ± 1.43% degradation after 5 h and 24 h, respectively suggesting an enhanced stability of 4′′-C14 EGCG than EGCG (Table 2).
Table 2 Stability assay of EGCG and 4′′-C14 EGCG in PBS buffer at pH 7.4. UV-Visible absorption experiments were performed in duplicates and one representative set of data is shown. The data are expressed as (mean ± SEM)
| Time |
Degradation (%) in PBS at pH 7.4 |
| EGCG |
4′′-C14 EGCG |
| 5 h |
36.52 ± 3.06 |
20.51 ± 0.93 |
| 24 h |
50.72 ± 2.27 |
27.80 ± 1.43 |
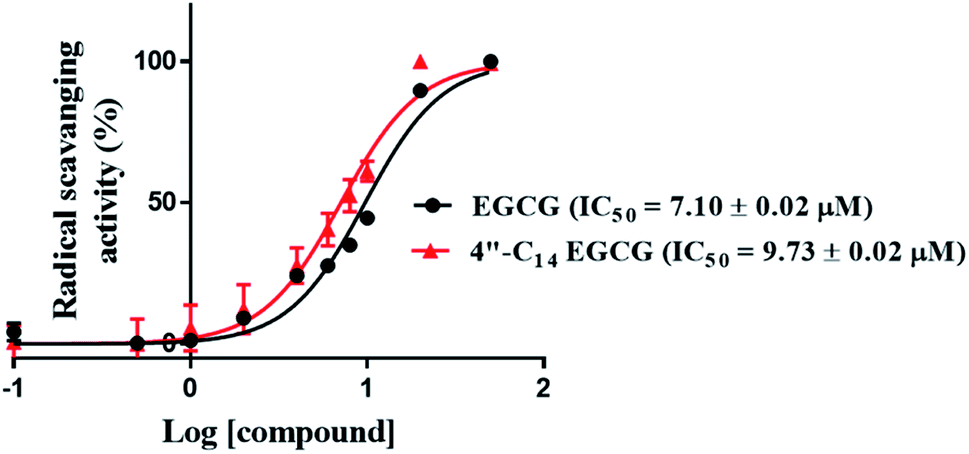
3.2.8. 4′′-C14 EGCG and EGCG showed the equivalent antioxidant property. EGCG is very well known for its antioxidant activity.85 Antioxidant activity of EGCG prevents cells from ROS-mediated DNA damage, thus protecting the occurrence of cancer.85–87 Several natural or synthetic molecules have showed chemo-preventive properties via regulating the cellular redox level. Thus, we explored the in vitro antioxidant capability of 4′′-C14 EGCG and EGCG by using DPPH assay. The antioxidant activity is measured as the reducing ability of the antioxidant compounds towards DPPH radical (Fig. 9). The IC50 of EGCG and 4′′-C14 EGCG were found to be 7.10 ± 0.02 μM and 9.73 ± 0.02 μM respectively. Obtained results indicate that the antioxidant property of 4′′-C14 EGCG is not significantly different from the EGCG.
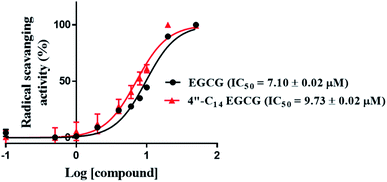 |
| | Fig. 9 Radical scavenging activities of EGCG and 4′′-C14 EGCG. Two independent experiments were performed and data are expressed as (mean ± SEM). | |
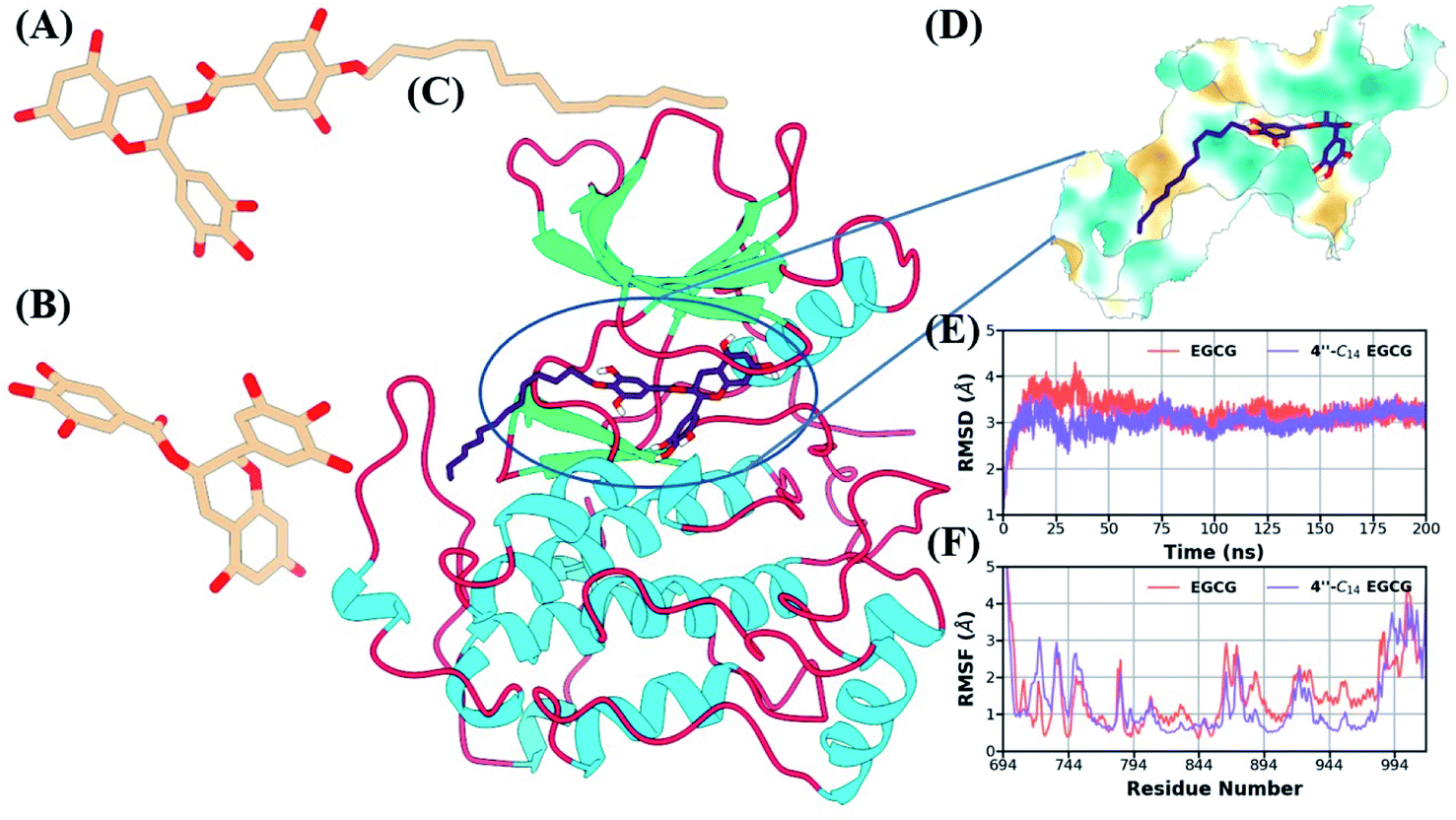
3.2.9. Elucidating the binding mode of 4′′-C14 EGCG against EGFR. To elucidate the binding mechanism of 4′′-C14 EGCG, we performed molecular docking analysis along with MD simulation on the active EGFR kinase (PDB ID 4I23) crystalized with a tyrosine kinase inhibitor dacomitinib.88 The structures of 4′′-C14 EGCG and EGCG are shown in Fig. 10A and 10B, along with the crystal structure of the EGFR kinase domain (Fig. 10C). Using the XP protocol of the GLIDE module (Schrödinger suite), the docking study revealed a strong binding score of −11.29 and −10.79 for 4′′-C14 EGCG and EGCG, respectively. We also estimated the lipophilicity of the binding cavity region using the MLP (molecular lipophilicity potential) program embedded in ChimeraX89 and displayed in the Fig. 10D. As is evident from Fig. 10D, the outer area of the binding pocket is hydrophilic, whereas the inside is hydrophobic, that helps to bind the ligand strongly. To get a deeper insight into the binding mechanism of 4′′-C14 EGCG and explore the stability of the complex, the docked complex was subjected to molecular dynamics (MD) simulation for 200 ns. The root-mean-square deviation (RMSD) of the backbone atoms of the EGFR kinase for both complexes were calculated with respect to the corresponding initial docked structure and shown in the Fig. 10E. As shown in Fig. 10E, both systems got stabilized after 50 ns and remained stable throughout the remaining simulation time length. The average RMSD over the last 100 ns is 3.15 Å and 3.08 Å for EGCG and 4′′-C14 EGCG, respectively. It indicates the stability of both complexes. The flexibility of each residue was studied by estimating the root-mean-square fluctuation (RMSF) of Cα atoms and shown in Fig. 10F. The RMSF value of most of the amino acids are below 2 Å indicating the stable protein structure. The significant lowering in RMSF value for 4′′-C14 EGCG implies more stability compared to the EGFR/EGCG complex. The terminal region, especially the flexible C terminal and functional regions like P-loop, α-loop, αC helix and A-loop showed higher RMSF value compared to other regions.
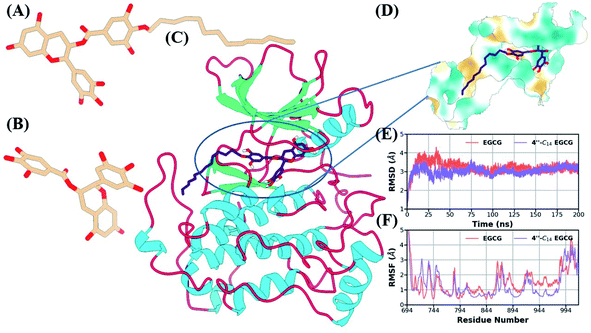 |
| | Fig. 10 3D depiction of the ligands used in our study (A) 4′′-C14 EGCG, (B) EGCG. (C) Cartoon representation of the EGFR kinase domain along with the initial binding pose of the ligand (4′′-C14 EGCG). Secondary structure is shown using the following color scheme; loop region: red, helix: cyan, and sheet: green. (D) Zoomed surface representation of the lipophilic potential around the binding cavity (dark cyan: hydrophilic, goldenrod: hydrophobic). (E) The time evolution of root-mean-square deviation (RMSD) of backbone atoms for EGFR/EGCG and EGFR/4′′-C14 EGCG. (F) The root-mean-square fluctuation (RMSF) of Cα atoms of EGFR/EGCG and EGFR/4′′-C14 EGCG. | |
We also estimated the solvent-accessible surface area (SASA) and radius of gyration (RoG) to estimate the degree of solvent exposure and structural compactness of the kinase domain (Fig. S37A and B in the ESI†). Both the parameters also showed stability after the initial 50 ns. The RMSD value for EGCG was relatively lower compared to 4′′-C14 EGCG because of the missing hydrophobic tail region (Fig. S37C in the ESI†). Initially, the tail region of the EGCG derivative showed flexibility which got stabilized after 25 ns. We also estimated the RMSD distribution of the binding pocket (5 Å surrounding the molecule) for both complexes (Fig. S37D in the ESI†). The binding pocket for 4′′-C14 EGCG had increased flexibility, which got stabilized after 100 ns to reach equilibrium. On the other hand, in the case of EGFR/EGCG, the binding pocket remained stable throughout the simulation. The flexibility of the binding of EGFR/4′′-C14 EGCG increased mainly because of fourteen carbon alkyl chain attached to the 4′′ position in D ring of EGCG.
3.2.10. Binding free energy analysis. The binding free energy estimation was conducted for the last 100 ns of the trajectories using the MM/PBSA scheme. Prior to the binding free energy estimation, the center of mass distance was calculated between ligand and protein as well as the number of hydrogen bonds which also showed similar stability (Fig. S38A and B in the ESI†). Fig. S38† illustrates different components of the binding free energy graphically, and individual values are listed in the Table 3. 4′′-C14 EGCG (−45.06 kcal mol−1) showed almost 10 kcal mol−1 more negative binding free energy than EGCG (−35.52 kcal mol−1) suggesting that the binding of 4′′-C14 EGCG is more favourable than its parent natural compound. In both cases, the intermolecular electrostatic and van der Waals interactions and nonpolar solvation free energy favour the complexation. In the case of EGFR/EGCG, ΔEelec (−81.59 kcal mol−1) is more favourable than EGFR/4′′-C14 EGCG (−60.56 kcal mol−1) by an amount of −21.03 kcal mol−1. However, in the case of EGFR/4′′-C14 EGCG (−57.11 kcal mol−1), ΔEvdW is more favourable compared to EGFR/EGCG (−31.94 kcal mol−1) by an amount of −25.17 kcal mol−1. Further, ΔGnp contributes more favourably to the complexation of EGFR/4′′-C14 EGCG than EGFR/EGCG. This suggests that 4′′-C14 EGCG displays a stronger affinity than EGCG due to the increased favourable contribution of the net nonpolar (ΔEvdW + ΔGnp).
Table 3 The calculated binding free energy (kcal mol−1) and its components for EGCG and 4′′-C14 EGCGc
| Systems |
ΔEvdW |
ΔEelec |
ΔGpol |
ΔGnp |
ΔEMMa |
ΔGsolvb |
ΔG |
| ΔEvdW+ ΔEelec. ΔGpol + ΔGnp. The standard deviation (SD) is provided in the parenthesis. |
| EGCG |
−31.94 (4.07) |
−81.59 (11.08) |
82.36 (6.95) |
−4.34 (0.11) |
−113.53 (9.90) |
78.02 (6.92) |
−35.52 (4.90) |
| 4′′-C14 EGCG |
−57.11 (5.77) |
−60.56 (12.38) |
79.40 (9.75) |
−6.79 (0.38) |
−117.67 (12.72) |
72.61 (9.64) |
−45.06 (6.68) |
Next, we identified the critical residues for both systems, which contribute significantly to the binding (see Table S1 in the ESI†). The contribution of each residue to the total binding free energy was displayed in Fig. 11A and B. It is evident from Table S1† and Fig. 11A and B that D855 and D800 are the highest contributing residues for EGCG and 4′′-C14 EGCG, respectively. Several other residues, such as V726, L718, G796, contribute significantly to the binding for complexes. The location of all these key residues is shown in the ball and stick model for both cases (Fig. 11C and D).
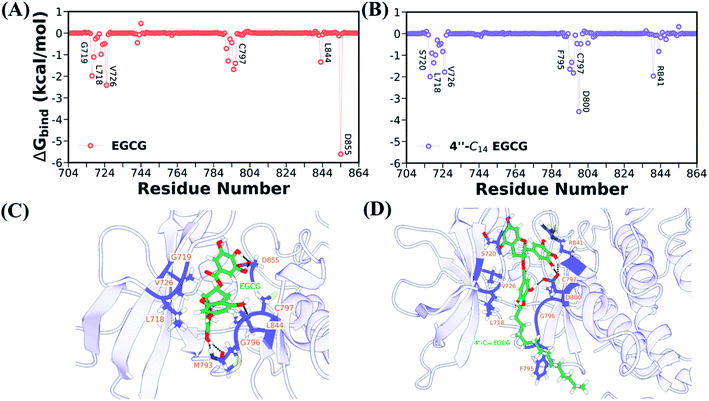 |
| | Fig. 11 Decomposition of binding free energy in residual level for (A) EGCG, (B) 4′′-C14 EGCG. Receptor ligand 3D interaction profile for (C) EGCG, (D) 4′′-C14 EGCG. Black dashed lines are used to depict hydrogen bonds between protein and ligands. | |
Further, we also conducted the hydrogen bond analysis to complement the binding free energy, and the time evolution of protein-ligand hydrogen bonds is shown in Fig. S38B in the ESI.† The average number of hydrogen bonds is higher in EGCG, which is also evident from the electrostatic contribution to the total binding. However, both the ligands show a steady number of hydrogen bonds throughout the simulation length, indicating a stable and strong interaction. We also listed the important hydrogen bonds along with their occupancy present in both the systems (Table S2 in the ESI†). In case of EGCG, we found five critical hydrogen bonds (more than 35% occupancy) involving M793 and different oxygen atoms of D855, whereas only two hydrogen bonds were observed in case of 4′′-C14 EGCG. Regardless of those steady hydrogen bonds, a greater number of hydrogen bonds with lesser occupancy was observed in the case of 4′′-C14 EGCG (see Table S2 in the ESI†), which helps in stable binding and conferring stability of its tail region. We further support our finding with the help of Ligplot analysis which shows possible hydrophobic contacts and hydrogen bonds (Fig. S40 in the ESI†).
4. Conclusion
In this work, we designed and synthesized a series of 4′′-alkyl derivatives of EGCG and among them 4′′-C14 EGCG displayed the most promising inhibitory activity against EGFR tyrosine kinase. 4′′-C14 EGCG exhibited enhanced stability compared to EGCG in PBS buffer of pH 7.4. A comprehensive study on their cytotoxicity in selected cancer cell lines was performed by CCK8 assay, AO/EtBr staining, DAPI staining, western blotting, and immunocytochemistry analysis. Importantly, 4′′-alkyl EGCG derivatives were very selective in targeting the cancer cells and did not show any toxicity in the non-cancerous cell line. 4′′-C14 EGCG showed the lowest IC50 on A431, HeLa, and MCF-7 cells and it was even better than the parent molecule, EGCG. The event of apoptotic cell death was evident from the AO/EtBr and DAPI staining results. The wound-healing assay demonstrated that the migration of A431 cells were significantly reduced after the treatment of 4′′-C14 EGCG at 50 μM concentration. Western blot analysis revealed that A431 cells were undergoing apoptosis through downregulation of anti-apoptotic protein, BCLXL, and upregulation of pro-apoptotic protein, BAX. Further, our results showed that 4′′-C14 EGCG, unlike EGCG, was able to completely inhibit the phosphorylation of EGFR at 75 μM. The results from the immunocytochemistry experiments were also in agreement with the western blot analysis, further confirming the inhibition of EGFR autophosphorylation by 4′′-C14 EGCG. Our results also showed that 4′′-C14 EGCG was able to effectively inhibit the phosphorylation of the downstream signalling proteins of EGFR, namely ERK and Akt. Antioxidant activity of 4′′-C14 EGCG was not found to be significantly different from the parent molecule, EGCG. In addition, the binding mode of 4′′-C14 EGCG with EGFR kinase domain was studied and compared with EGCG by using molecular docking and MD simulation studies. It was evident from the MD simulation study that the addition of a fourteen-carbon long aliphatic hydrocarbon chain at 4′′ position (4′′-C14 EGCG) enhanced the structural framework of EGCG, resulting in an increased binding affinity with EGFR compared to the parent molecule, EGCG. Thus, the obtained results from in silico studies support our experimental findings. Taken together, we have identified a novel lipophilic derivative of EGCG, namely 4′′-C14 EGCG, as potent EGFR inhibitor and anticancer agent with improved stability and biological activity.
Conflicts of interest
The authors declare that there is no conflict of interest.
Acknowledgements
The authors gratefully acknowledge the financial support from the Indian Institute of Technology Indore and Indian Institute of Technology Palakkad for the provision of conducting research and the scholarship provided to the students by Govt. of India. This work was also supported by the Department of Science and Technology-Science & Engineering Research Board (DST-SERB), Govt. of India (ECR/2017/002082) and Council of Scientific & Industrial Research (CSIR), Govt. of India (02(0434)/21/EMR-II).
References
- M. Wieduwilt and M. Moasser, Cell. Mol. Life Sci., 2008, 65, 1566–1584 CrossRef CAS PubMed.
- Y. Yarden and G. Pines, Nat. Rev. Cancer, 2012, 12, 553–563 CrossRef CAS PubMed.
- M. H. Cohen, G. A. Williams, R. Sridhara, G. Chen and R. Pazdur, Oncologist, 2003, 8, 303–306 CrossRef CAS PubMed.
- J. R. Johnson, M. Cohen, R. Sridhara, Y.-F. Chen, G. M. Williams, J. Duan, J. Gobburu, B. Booth, K. Benson and J. Leighton, Clin. Cancer Res., 2005, 11, 6414–6421 CrossRef CAS PubMed.
- R. T. Dungo and G. M. Keating, Drugs, 2013, 73, 1503–1515 CrossRef CAS PubMed.
- J. C. Chuang, A. A. Salahudeen and H. A. Wakelee, Expert Opin. Pharmacother., 2016, 17, 989–993 CrossRef CAS PubMed.
- A. L. Koch, P. J. Vellanki, N. Drezner, X. Li, P. S. Mishra-Kalyani, Y. L. Shen, H. Xia, Y. Li, J. Liu and J. F. Zirkelbach, Clin. Cancer Res., 2021, 27, 6638–6643 CrossRef CAS PubMed.
- S. Wang, S. Cang and D. Liu, J. Hematol. Oncol., 2016, 9, 1–7 CrossRef PubMed.
- K. S. Thress, C. P. Paweletz, E. Felip, B. C. Cho, D. Stetson, B. Dougherty, Z. Lai, A. Markovets, A. Vivancos and Y. Kuang, Nat. Med., 2015, 21, 560–562 CrossRef CAS PubMed.
- W.-H. Hsu, J.-H. Yang, T. Mok and H. Loong, Ann. Oncol., 2018, 29, i3–i9 CrossRef PubMed.
- L. Huang and L. Fu, Acta Pharm. Sin. B, 2015, 5, 390–401 CrossRef PubMed.
- Q. Liu, S. Yu, W. Zhao, S. Qin, Q. Chu and K. Wu, Mol. Cancer, 2018, 17, 1–9 Search PubMed.
- T. Wang, M.-B. Wu, J.-P. Lin and L.-R. Yang, Expert Opin. Drug Discovery, 2015, 10, 1283–1300 CrossRef CAS PubMed.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- S. K. Liew, S. Malagobadan, N. M. Arshad and N. H. Nagoor, Biomolecules, 2020, 10, 138 CrossRef CAS PubMed.
- P. F. Lamie, A. M. El-Kalaawy, N. S. A. Latif, L. A. Rashed and J. N. Philoppes, Eur. J. Med. Chem., 2021, 214, 113222 CrossRef CAS PubMed.
- S. Goyal, S. Jamal, A. Shanker and A. Grover, BMC Genomics, 2015, 16, 1–9 CrossRef PubMed.
- P. Bhatia, V. Sharma, O. Alam, A. Manaithiya, P. Alam, M. T. Alam and M. Imran, Eur. J. Med. Chem., 2020, 204, 112640 CrossRef CAS PubMed.
- N. Jiang, Y. Bu, Y. Wang, M. Nie, D. Zhang and X. Zhai, Molecules, 2016, 21, 1572 CrossRef PubMed.
- Z. Zhang, L. Xie, Y. Ju and Y. Dai, Small, 2021, 17, 2100314 CrossRef CAS PubMed.
- M. M. Joseph, A. N. Ramya, V. M. Vijayan, J. B. Nair, B. T. Bastian, R. K. Pillai, S. T. Therakathinal and K. K. Maiti, Small, 2020, 16, 2003309 CrossRef CAS PubMed.
- S. F. Nabavi, A. G. Atanasov, H. Khan, D. Barreca, D. Trombetta, L. Testai, A. Sureda, S. Tejada, R. A. Vacca and V. Pittalà, Cancer Lett., 2018, 434, 101–113 CrossRef CAS PubMed.
- M. Huang, J.-J. Lu and J. Ding, Nat. Prod. Bioprospect., 2021, 1–9 Search PubMed.
- Â. Bisol, P. S. de Campos and M. L. Lamers, Phytother. Res., 2020, 34, 568–582 CrossRef PubMed.
- M. Gordaliza, Clin. Transl. Oncol., 2007, 9, 767–776 CrossRef CAS PubMed.
- M. Fridlender, Y. Kapulnik and H. Koltai, Front. Plant Sci., 2015, 6, 799 Search PubMed.
- J. S. Arya, M. M. Joseph, D. R. Sherin, J. B. Nair, T. K. Manojkumar and K. K. Maiti, J. Med. Chem., 2019, 62, 8311–8329 CrossRef CAS PubMed.
- J. Chang, Y. Kim and H. Kwon, Nat. Prod. Rep., 2016, 33, 719–730 RSC.
- E. Cione, C. La Torre, R. Cannataro, M. C. Caroleo, P. Plastina and L. Gallelli, Molecules, 2020, 25, 63 CrossRef CAS PubMed.
- S. Wang, Z. Li, Y. Ma, Y. Liu, C.-C. Lin, S. Li, J. Zhan and C.-T. Ho, Molecules, 2021, 26, 3755 CrossRef CAS PubMed.
- M. Nikoo, J. M. Regenstein and H. Ahmadi Gavlighi, Compr. Rev. Food Sci. Food Saf., 2018, 17, 732–753 CrossRef CAS PubMed.
- K.-j. Min and T. K. Kwon, Integr. Med. Res., 2014, 3, 16–24 CrossRef PubMed.
- K.-W. Luo, W.-Y. Lung, X.-L. L. Chun-Xie and W.-R. Huang, Oncotarget, 2018, 9, 12261 CrossRef PubMed.
- J. Gu, K. Qiao, P. Sun, P. Chen and Q. Li, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 4557–4563 Search PubMed.
- S. Shankar, S. Ganapathy, S. R. Hingorani and R. K. Srivastava, Front. Biosci., 2008, 13, 440–452 CrossRef PubMed.
- Z.-Y. Cai, X.-M. Li, J.-P. Liang, L.-P. Xiang, K.-R. Wang, Y.-L. Shi, R. Yang, M. Shi, J.-H. Ye and J.-L. Lu, Molecules, 2018, 23, 2346 CrossRef PubMed.
- W. Dai, C. Ruan, Y. Zhang, J. Wang, J. Han, Z. Shao, Y. Sun and J. Liang, J. Funct. Foods, 2020, 65, 103732 CrossRef CAS.
- Q. Y. Zhu, A. Zhang, D. Tsang, Y. Huang and Z.-Y. Chen, J. Agric. Food Chem., 1997, 45, 4624–4628 CrossRef CAS.
- N. Li, L. S. Taylor, M. G. Ferruzzi and L. J. Mauer, Food Res. Int., 2013, 53, 909–921 CrossRef CAS.
- A. Faralli, E. Shekarforoush, A. C. Mendes and I. S. Chronakis, Pharmaceutics, 2019, 11, 155 CrossRef CAS PubMed.
- T. Miyazawa, Biofactors, 2000, 13, 55–59 CrossRef CAS PubMed.
- X. Chen, B. Liu, R. Tong, S. Ding, J. Wu, Q. Lei and W. Fang, Langmuir, 2021, 37, 969–977 CrossRef CAS PubMed.
- X. Zhang, J. Wang, J.-M. Hu, Y.-W. Huang, X.-Y. Wu, C.-T. Zi, X.-J. Wang and J. Sheng, Molecules, 2016, 21, 620 CrossRef PubMed.
- J. L. Gonzalez-Alfonso, P. Peñalver, A. O. Ballesteros, J. C. Morales and F. J. Plou, Front. Nutr., 2019, 6, 30 CrossRef PubMed.
- K. Osanai, K. R. Landis-Piwowar, Q. P. Dou and T. H. Chan, Bioorg. Med. Chem., 2007, 15, 5076–5082 CrossRef CAS PubMed.
- Y. Oritani, Y. Setoguchi, R. Ito, H. Maruki-Uchida, T. Ichiyanagi and T. Ito, Biol. Pharm. Bull., 2013, 36, 1577–1582 CrossRef CAS PubMed.
- C. Ping, T. Yao, S. Dong and Z. Xiao-ming, J. Zhejiang Univ., Sci., A, 2003, 4, 714–718 CrossRef PubMed.
- S. Lindert, N. Alexander, N. Wötzel, M. Karakaş, P. L. Stewart and J. Meiler, Structure, 2012, 20, 464–478 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- R. Friesner, R. JLB, T. Murphy, J. Halgren and D. Klicic, J. Med. Chem., 2004, 47, 1739–1750 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren and J. L. Knight, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, 2018, University of California, San Francisco Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- B. Wang and K. M. Merz, J. Chem. Theory Comput., 2006, 2, 209–215 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- D. J. Price and C. L. Brooks III, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed.
- V. Kräutler, W. F. Van Gunsteren and P. H. Hünenberger, J. Comput. Chem., 2001, 22, 501–508 CrossRef.
- R. W. Pastor, B. R. Brooks and A. Szabo, Mol. Phys., 1988, 65, 1409–1419 CrossRef.
- H. J. Berendsen, J. v. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- P. mesh Ewald, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef.
- R. Roy, B. Ghosh and P. Kar, ACS Omega, 2020, 5, 3932–3942 CrossRef CAS PubMed.
- R. Roy, A. Mishra, S. Poddar, D. Nayak and P. Kar, J. Biomol. Struct. Dyn., 2020, 40, 2302–2315 CrossRef PubMed.
- S. Singh, M. F. Sk, A. Sonawane, P. Kar and S. Sadhukhan, J. Biomol. Struct. Dyn., 2020, 39, 6249–6264 CrossRef PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- R. Roy, M. F. Sk, N. A. Jonniya, S. Poddar and P. Kar, J. Biomol. Struct. Dyn., 2021, 1–13 Search PubMed.
- L. Thurakkal, S. Singh, R. Roy, P. Kar, S. Sadhukhan and M. Porel, Chem. Phys. Lett., 2021, 763, 138193 CrossRef CAS PubMed.
- M. F. Sk, R. Roy and P. Kar, J. Biomol. Struct. Dyn., 2020, 39, 3649–3661 CrossRef PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan and W. Wang, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- H. Gohlke, C. Kiel and D. A. Case, J. Mol. Biol., 2003, 330, 891–913 CrossRef CAS PubMed.
- M. Maeda-Yamamoto, N. Inagaki, J. Kitaura, T. Chikumoto, H. Kawahara, Y. Kawakami, M. Sano, T. Miyase, H. Tachibana and H. Nagai, J. Immunol., 2004, 172, 4486–4492 CrossRef CAS PubMed.
- M. Suzuki, K. Yoshino, M. Maeda-Yamamoto, T. Miyase and M. Sano, J. Agric. Food Chem., 2000, 48, 5649–5653 CrossRef CAS PubMed.
- C. Minnelli, R. Galeazzi, E. Laudadio, A. Amici, D. Rusciano, T. Armeni, M. Cantarini, P. Stipa and G. Mobbili, Antioxidants, 2020, 9, 208 CrossRef CAS PubMed.
- K. Kida, M. Suzuki, N. Matsumoto, F. Nanjo and Y. Hara, J. Agric. Food Chem., 2000, 48, 4151–4155 CrossRef CAS PubMed.
- Z. Weihua, R. Tsan, W.-C. Huang, Q. Wu, C.-H. Chiu, I. J. Fidler and M.-C. Hung, Cancer Cell, 2008, 13, 385–393 CrossRef PubMed.
- B. S. Cummings, L. P. Wills and R. G. Schnellmann, Curr. Protoc. Pharmacol., 2012, 56, 12.18.11–12.18.24 Search PubMed.
- P. Friedl, E. Sahai, S. Weiss and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2012, 13, 743–747 CrossRef CAS PubMed.
- P. Wee and Z. Wang, Cancers, 2017, 9, 52 CrossRef PubMed.
- D. Singh, B. K. Attri, R. K. Gill and J. Bariwal, Mini-Rev. Med. Chem., 2016, 16, 1134–1166 CrossRef CAS PubMed.
- G. da Cunha Santos, F. A. Shepherd and M. S. Tsao, Annu. Rev. Pathol.: Mech. Dis., 2011, 6, 49–69 CrossRef PubMed.
- R. Ali and M. K. Wendt, Signal Transduction Targeted Ther., 2017, 2, 1–7 Search PubMed.
- T. E Taylor, F. B Furnari and W. K Cavenee, Curr. Cancer Drug Targets, 2012, 12, 197–209 CrossRef PubMed.
- S. Sang, M.-J. Lee, Z. Hou, C.-T. Ho and C. S. Yang, J. Agric. Food Chem., 2005, 53, 9478–9484 CrossRef CAS PubMed.
- M. Akagawa, T. Shigemitsu and K. Suyama, Biosci., Biotechnol., Biochem., 2003, 67, 2632–2640 CrossRef CAS PubMed.
- J. D. Lambert and R. J. Elias, Arch. Biochem. Biophys., 2010, 501, 65–72 CrossRef CAS PubMed.
- R. Sahadevan, S. Singh, A. Binoy and S. Sadhukhan, Crit. Rev. Food Sci. Nutr., 2022, 1–30, DOI:10.1080/10408398.2022.2068500.
- M. Wang, H. Zhang, L. Yi, P. Högger, R. Arroo, V. K. Bajpai, M.-A. Prieto, J. Simal-Gandara, S. Wang and H. Cao, Food Chem., 2022, 366, 130521 CrossRef CAS PubMed.
- K. S. Gajiwala, J. Feng, R. Ferre, K. Ryan, O. Brodsky, S. Weinrich, J. C. Kath and A. Stewart, Structure, 2013, 21, 209–219 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Revathy Sahadevan
a,
Revathy Sahadevan