DOI:
10.1039/D2RA02673B
(Paper)
RSC Adv., 2022,
12, 16174-16183
Kinetic and mechanistic insights into the degradation of clofibric acid in saline wastewater by Co2+/PMS process: a modeling and theoretical study†
Received
27th April 2022
, Accepted 25th May 2022
First published on 30th May 2022
Abstract
Recently, the degradation of non-chlorinated organic pollutants in saline pharmaceutical wastewater by SO4˙−-based advanced oxidation processes (AOPs) has received widespread attention. However, little is known about the oxidation of chlorinated compounds in SO4˙−-based AOPs. This study chose clofibric acid (CA) as a chlorinated pollutant model; the oxidation kinetics and mechanistic pathway were explored in the Co2+/peroxymonosulfate (PMS) system. Notably, a high removal efficiency (81.0%) but low mineralization rate (9.15%) of CA within 120 min were observed at pH 3.0 during Co2+/PMS treatment. Exogenic Cl− had a dual effect (inhibitory then promoting) on CA degradation. Several undesirable chlorinated by-products were formed in the Co2+/PMS system. This demonstrated endogenic chlorine and exogenic Cl− both reacted with SO4˙− to generate chlorine radicals, which participated in the dechlorination and rechlorination of CA and its by-products. Furthermore, SO4˙− was the dominant species responsible for CA degradation at low Cl− concentrations (≤1 mM), whereas Cl2˙− was the predominant radical at [Cl−]0 > 1 mM. A possible degradation pathway of CA was proposed. Our findings suggested that chlorinated compounds in highly saline pharmaceutical wastewater will be more resistant and deserve more attention.
1 Introduction
It is well known that pharmaceutical industries produce a large amount of toxic and hazardous wastewater, characterized by high biological oxygen demand (BOD), chemical oxygen demand (COD), salinity, and toxicity.1 Pharmaceutical wastewater has severe adverse impacts on aquatic environments and requires pretreatment before discharge.2,3 Clofibric acid (CA), a stable pharmaceutical contaminant with potential ecosystem and human health risks, is persistent and has been commonly detected in aquatic environments.4,5 Due to its complex structure, CA removal was limited to traditional biological degradation.6 Currently, advanced oxidation processes (AOPs) show stronger oxidation abilities to refractory contaminants and have been extensively applied in CA degradation, such as UV/O3,7 UV/TiO2,8 and UV/O3/peroxydisulfate (PDS).9 Among these methods, SO4˙−-based AOPs have garnered substantial scientific interest.9
Typically, SO4˙− is extremely reactive towards a wide range of organic compounds with redox potentials between 2.5–3.1 V. Furthermore, SO4˙− tends to react with organic compounds via one-electron transfer rather than addition or H abstraction, which are less influenced by water matrix components.9 Peroxymonosulfate (PMS) is an efficient precursor of SO4˙− and activated by transition metals,10 UV radiation,11 heat,12 ultrasound,13 and others. Among those methods, Co2+ is the most efficient transition-metal ion for PMS activation, leading to radical-induced contaminant oxidation.14 Co2+/PMS has been universally recognized as a SO4˙−-based system via one-electron oxidation.15 However, recent studies indicated a two-electron transfer pathway involving a non-radical route (i.e., Co4+) occurred at pH = 3.0 in the Co2+/PMS system.16,17 To date, the main reactive oxygen species (ROS), involving radical and non-radical pathways, and their contributions to CA degradation affected by water constituents (i.e., inorganic ions) in the Co2+/PMS system were overlooked.
Chloride ion (Cl−) is ubiquitous in industrial wastewater, whereas the impacts of exogenic Cl− (inorganic form from a water matrix) in oxidations of different chlorinated organic compounds in the Co2+/PMS system are still controversial due to the complicated reaction process. For example, Anipsitakis et al.18 reported a facilitating effect of low dosage Cl− on the degradation of 2,4-dichlorophenol in the Co2+/PMS system. Chan and Chu et al.19 found the presence of Cl− (0–6.8 mM) significantly restrained the removal efficiency and reduced the reaction rate of atrazine. A dual impact (inhibitory then promoting) of Cl− (0–500 mM) was reported in the oxidative degradation of 4-chloro-2-nitrophenol by Co2+/PMS.20 Hence, the concentration of exogenic Cl−, as well as substrate properties might be significant in chlorinated organic compound oxidations in AOPs. Currently, some groups have investigated the influence of exogenic Cl− on the degradation efficiency of CA in AOPs.9,21 Qin et al.9 pointed out that degradation of CA in the UV/O3/PDS system dropped slightly as the Cl− concentration increased from 0 to 10 mM. Tang et al.21 reported that a Cl− concentration increase (0–2 mM) promoted CA degradation in the UV/Cl− system. However, the effect of exogenic Cl− on CA degradation in the Co2+/PMS system was not thoroughly examined, especially under highly saline conditions.
In addition to the effect of exogenic Cl− ions in highly saline wastewater on CA removal, the significance of endogenic Cl− (organic form from the contaminant itself) on the degradation process of CA was also neglected. Endogenic Cl− from chlorinated organic pollutants played an important role in dehalogenation and halogenation processes. Dechlorination was observed during chlorobenzene oxidation with SO4˙−.22,23 The chlorine atoms released from the benzene ring participated in de novo formation of chlorinated by-products by SO4˙−,24 which led to the formation of polychlorinated aromatics during 2,4,6-trichlorophenol (TCP) oxidation.25 However, previous studies have mainly focused on the oxidation efficiency and degradation pathways of CA in AOPs,7–9 and little attention has been paid to the dechlorination and rechlorination of endogenic Cl− during CA oxidation.
This study aims to: (1) evaluate the influence of exogenic Cl− on the degradation kinetics of CA; (2) explore the transformation of endogenic chlorine of CA during Co2+/PMS treatment; (3) investigate the conversion of reactive oxygen species and their contributions to CA degradation; (4) explore the oxidation mechanisms and propose a possible degradation pathway of CA. This work hopes to provide valuable information for studying the role of exogenic Cl− in the oxidation of chlorinated compounds in pharmaceutical wastewater.
2 Experimental
2.1 Materials
Clofibric acid (C10H11ClO3, >99%), acid orange 7 (AO7, C16H11N2O4SNa, >98%), and carbamazepine (CBZ, C15H12N2O, 98%) were obtained from Acros Organics. Oxone® ([2KHSO5·KHSO4·K2SO4] salt, 95%), L-histidine and hexamethyldisilazane, pyridine, and chlorotrimethylsilane were obtained from Sigma-Aldrich. HPLC grade acetonitrile and methanol were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Methyl phenyl sulfoxide (PMSO) and methyl phenyl sulfone (PMSO2) were obtained from Shanghai Rhawn Chemical Technology Co., Ltd. CoSO4·7H2O, NaNO2, NaCl, NaOH, and H2SO4 were of analytical grade, and used without further purification. Milli-Q water (Millipore, France, >18.2 MΩ cm) was used to prepare all reagents and solvents.
2.2 Experimental procedure
All reactions were performed in a 50 mL glass beaker and magnetically stirred (300 rpm) at room temperature (25 ± 2 °C). Reactions were initiated by mixing a specific concentration of PMS into a solution containing CA, CoSO4·7H2O, or other chemicals without pH control. While the effect of pH on the removal efficiency of CA was also studied, NaOH or H2SO4 adjusted the pH of the mixed solution to the desired level. Samples were taken at predetermined times for analysis and quenched with methanol (for HPLC) or NaNO2 (for GC-MS and TOC). All tests were conducted in duplicate to ensure reproducibility and the data are shown as averages with error bars (±5%).
2.3 Analytical methods
Residual CA and CBZ concentrations were measured by a Waters e2695 with 2489 UV/Vis detector using an Atlantis® T3 Waters column (4.6 mm × 250 mm, 5 μm) at 227 and 210 nm, respectively. The mobile phase for CA detection contained 0.5% phosphoric acid/methanol (15/85 (v/v)) at a flow rate of 1.0 mL min−1. The sample injection volume was 20 μL. AO7 concentrations were measured by a Hitachi F-4600 spectrophotometer (Hitachi, Japan) at 485 nm. PMS concentrations were detected spectrophotometrically by iodometric titration at 352 nm.26 A Shimadzu TOC analyzer (TOC-VCPH, Shimadzu, Japan) measured the total organic carbon (TOC). The intermediates of CA were analyzed by GC-MS, (Agilent 8860-GC/5977B-MSD, USA) and the detailed operational parameters are provided in Text S1.†
The oxidation of clofibric acid under various conditions was modeled using pseudo-first-order kinetics eqn (1)
where
C0 and
C are the concentrations of CA at
t = 0 and
t min, respectively,
k is the pseudo-first-order rate constant (min
−1), and
t is the reaction time.
2.4 Kinetic modeling
Kinetic modeling of the Co2+/PMS system was performed by Kintecus 6.51 (https://www.kintecus.com). All radical reactions used as input for kinetic modeling were listed in Table S1.† The formations and transformations of reactive species such as the hydroxyl, sulfate, and chlorine radicals were simulated according to the reactions in Table S1.† Most reaction rate constants were obtained from previous studies,27–29 while some unknown reaction rate constants were calculated by fitting the traces of CA degradation curves by taking these unknown rate constants as adjustable. Due to negligible production at pH 3.0, reactions of chlorite/chlorate were not included in our model.30 Unknown or poorly-defined parameters were optimized to accurately simulate species distributions by using a numerical procedure associated with Kintecus programs.
3 Results and discussion
3.1 CA degradation
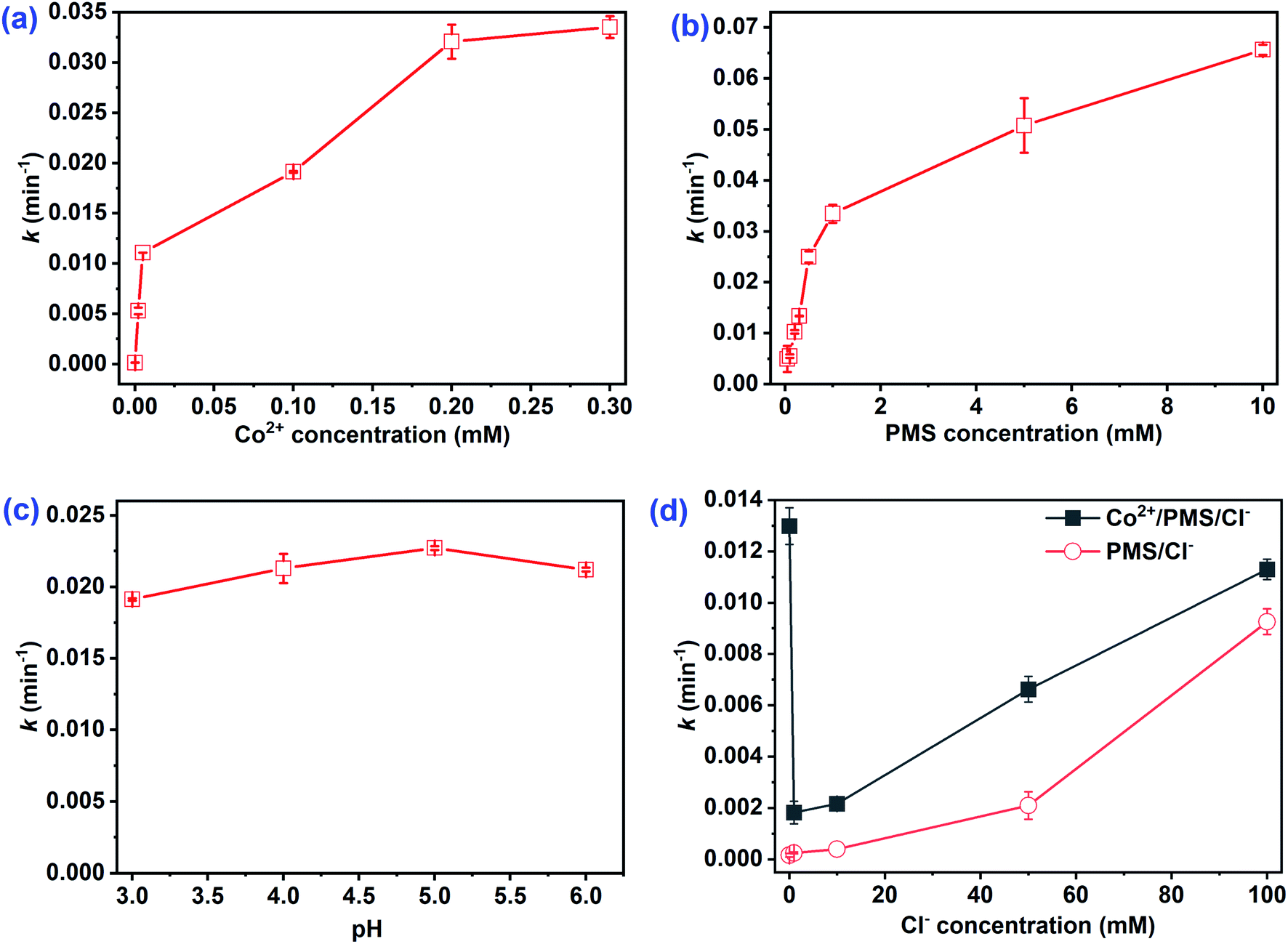
3.1.1 Effect of Co2+ concentration. Co2+ played an important role as a catalyst in the Co2+/PMS system. The impacts of catalyst levels on CA degradation were studied and the degradation kinetics and impacts on the rate constant k are shown in Fig. S1a† and 1a, respectively. No CA oxidation was observed without Co2+ addition (Fig. S1a†), which suggested CA was not oxidized by PMS itself. Furthermore, CA degradation rates increased significantly with Co2+ concentration increased (Fig. 1a). The reaction rate constant k increased from 0 to 3.36 × 10−2 min−1 as Co2+ increased from 0 to 0.3 mM. Dramatic degradation rates and oxidation efficiencies were detected at [Co2+]0 < 0.02 mM, whereas no obvious enhancement was obtained when the Co2+ concentration increased from 0.02 to 0.3 mM. This may be due to Co2+ inducing rapid PMS decomposition with the highest efficiency to generate SO4˙− (eqn (2)). The regeneration of Co2+ with excess PMS (E0(Co3+/Co2+) = 1.45 V) may be a vital step to continue catalysis at low Co2+ levels (≤0.02 mM) (eqn (3)).28 Moreover, the non-radical Co4+ could also exist in the Co2+/PMS system at pH 3.0 (as seen in eqn (4)) and more Co4+ may form at higher Co2+ dosages.16 Co4+ could react with the target pollutant to regenerate Co2+ via eqn (5) and participate in the next oxidation cycle. Notably, the maximum degradation rate appeared when Co2+ concentration increased to 0.3 mM, revealing that 0.5 mM PMS was not completely consumed and continued to produce radicals. Xue et al. also reported a similar phenomenon during thiamphenicol (TAP) and florfenicol (FFC) in the Co2+/PMS system.31 In this study, the optimal dosage of Co2+ was 0.01 mM with an appropriate degradation efficiency (81.0%) and a high rate constant k (3.21 × 10−2 min−1).| | |
HSO5− + Co2+ → Co3+ + OH− + SO4˙−
| (2) |
| | |
HSO5− + Co3+ → Co2+ + H+ + SO5˙−
| (3) |
| | |
HSO5− + Co2+ → CoIVO2+ + H+ + SO42−, k = 2.05 M−1 s−1
| (4) |
| | |
Co4+ + pollutant → Co2+ + products
| (5) |
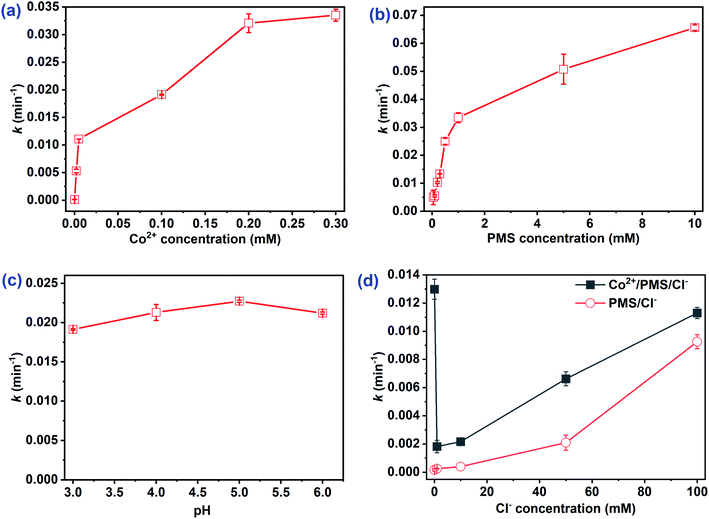 |
| | Fig. 1 Effect of initial (a) Co2+ concentration, (b) PMS concentration, (c) pH, and (d) Cl− concentration on the rate constants k of CA. Experimental conditions: [CA]0 = 0.1 mM, (a, b, d) pH = 3.0; (a, c, d) [PMS]0 = 0.5 mM; (b, c, d) [Co2+]0 = 0.01 mM. | |
3.1.2 Effect of PMS concentration. The impact of initial PMS concentration on CA degradation in the Co2+/PMS system was studied. As illustrated in Fig. 1b, at PMS concentrations between 0–10 mM, the rate constant of CA degradation increased from 0 to 6.56 × 10−2 min−1. Higher levels of PMS accelerated the formation of reactive oxygen species, and the degradation efficiency of CA was proportional to the oxidant dosages under the same catalyst dosage (eqn (2) or eqn (4)). The rate constant increased rapidly when PMS was 0–1 mM, while only a slight improvement was observed at PMS concentrations above 1 mM. Wang et al.10 revealed excessive PMS acted as a radical scavenger, which accelerated the conversion of SO4˙− to less reactive SO5˙− (eqn (6)). Increasing the PMS concentration to 10 mM, the low concentration of Co2+ limited ROS production, which led to the slight degradation rate enhancement. Notably, the degradation efficiency of CA increased gradually when [PMS]0 < 0.5 mM, whereas no evident depletion of CA was obtained at PMS concentrations between 0.5–10 mM (Fig. S1b†). Therefore, 0.5 mM was selected as the optimal PMS concentration.| | |
HSO5˙− + SO4˙− → SO5˙− + HSO4˙−
| (6) |
3.1.3 Effect of pH. The solution pH played a vital role in organic degradation as well as ROS formation in the SO4˙−-based AOP system.31,32 Notably, the CA concentration was not detected by HPLC under neutral or alkaline conditions (Fig. S2†). Due to the strong oxidation capability of PMS under alkaline conditions,28 the impact of initial pH 3.0 to 6.0 was examined. As shown in Fig. 1c, the rate constants at pH levels of 3.0, 4.0, 5.0, and 6.0 were 1.91 × 10−2, 2.13 × 10−2, 2.27 × 10−2, and 2.12 × 10−2 min−1, respectively. Fig. 1c indicated the rate constant increased slightly at pH levels between 3.0–5.0 in the Co2+/PMS system. It is worth noting that HSO5− and Co2+ were the dominant forms of PMS and CoSO4·7H2O at pH < 6.0.10 Additionally, higher pH levels assisted the formation of CoOH+ via Co2+ hydrolysis (eqn (7)) and was the predominant species responsible for PMS decomposition (eqn (8)).33 The rate constant dropped slightly as the pH increased from 5.0 to 6.0, possibly due to HPLC measurement errors as the pH approached neutral or alkaline conditions (Fig. S2†). A similar phenomenon was obtained for the oxidation of TCP25 and FFC.31 Although the rate constant optimized at pH = 5.0, the degradation efficiency (78.0%) decreased slightly after 120 min as compared to pH 3.0 (81.0%). Thus, an initial pH = 3.0 without adjustment was applied in the following experiments.| | |
Co2+ + H2O → CoOH+ + H+
| (7) |
| | |
CoOH+ + HSO5− → CoO + H2O + SO4˙−
| (8) |
3.1.4 Effect of Cl− concentration. Pharmaceutical wastewater usually contains many salts, which may influence the oxidation efficiency of pharmaceutical contaminants. Thus, the impact of chloride ions on CA degradation was investigated. As illustrated in Fig. 1d, Cl− had a dual effect (inhibitory then promoting) on the CA degradation. Low Cl− concentrations (≤1 mM) had a negative impact on CA oxidation, but facilitated oxidation when [Cl−]0 > 1 mM (Fig. S3a†). The chloride ion was supposed to scavenge SO4˙− to generate less reactive chlorine radicals (Cl˙/Cl2˙−), and lead to the inhibitory effect of Cl− on CA degradation.34 Further addition of chloride and formation of chlorine species (HOCl/Cl2) shown in eqn (9) and (10) may facilitate rapid CA degradation.35 For comparison, removal of AO7 and CBZ under the same conditions was also studied. As shown in Fig. S4,† a similar phenomenon (inhibitory then promoting) was observed and critical points also appeared at low Cl− concentrations. Moreover, the same dual effect of Cl− was observed for 4-chloro-2-nitrophenol removal in saline wastewater by Co2+/PMS,20 whereas an opposite dual effect (promoting then inhibitory) was obtained on chlorinated azo dye oxidation in the UV/PS system.36 However, CA degradation efficiency gradually increased with added Cl− in the PMS/Cl− system (Fig. S3b†). The removal rate of CA was 75.1% after 120 min in the presence of 100 mM Cl− by PMS/Cl− but reached 85.4% when Co2+ and Cl− coexisted. According to previous studies, PMS can oxidize Cl− directly to generate reactive chlorine species (HClO/Cl2) to degrade organic compounds.35 However, this dual effect of Cl− was not present in H2O2-based AOPs, such as Fenton and photo-Fenton systems,37 because Cl− cannot activate H2O2 to oxidize the contaminants.| | |
Cl− + HSO5− → SO42− + HOCl
| (9) |
| | |
2Cl− + HSO5− + H+ → SO42− + Cl2 + H2O
| (10) |
3.2 Mineralization
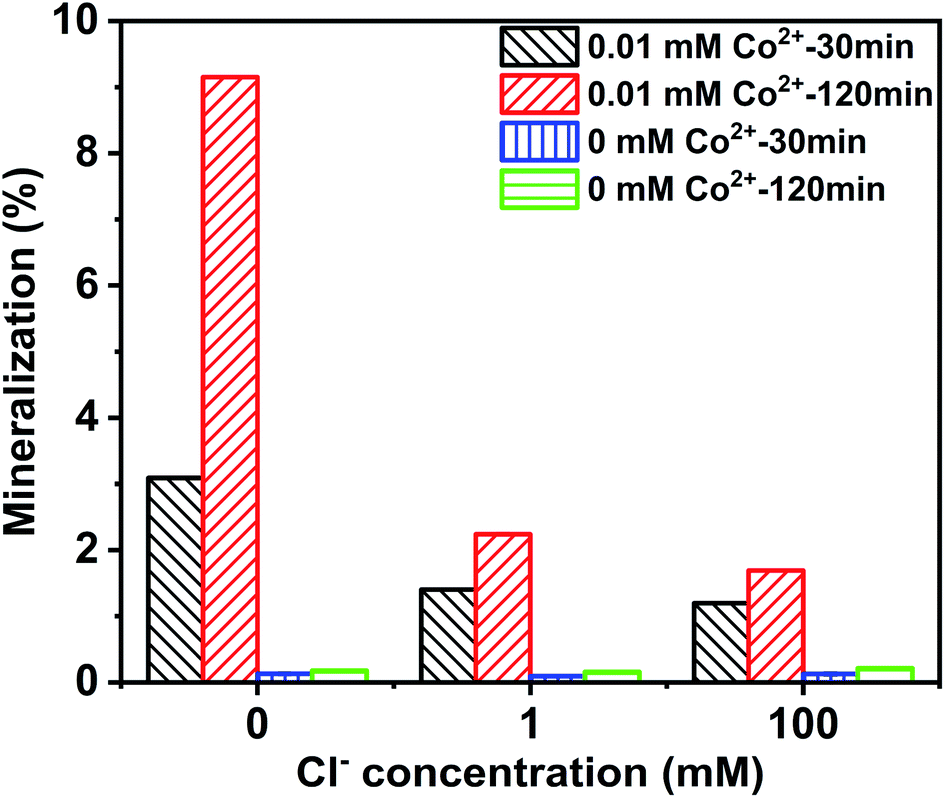
The mineralization of CA was evaluated by measuring TOC removal efficiency. As depicted in Fig. 2, the mineralization rate increased with prolonged reaction times, while TOC removal in the Co2+/PMS system was much higher than in the absence of Co2+. TOC removal was 3.09% and 9.15% with 0 mM Cl− in the Co2+/PMS system after 30 and 120 min, respectively. However, the mineralization degree of CA was 2.24%, and 1.69% after 120 min at 1 mM, and 100 mM Cl− in the Co2+/PMS system, respectively. TOC removal decreased with additional Cl− in the Co2+/PMS system. Interestingly, the degradation efficiency of CA improved at high Cl− (300 mM) in the PMS/Cl− system (Fig. S3b†) but the TOC removal rate was lower than 0.2%. In general, the mineralization degree of CA was quite low in the Co2+/PMS system. The high degradation efficiency but low mineralization degree of CA suggested the formation of refractory intermediates in a saline environment, especially chlorinated by-products.38
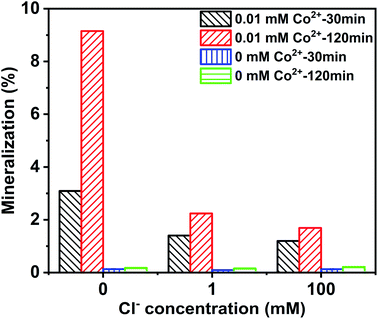 |
| | Fig. 2 Mineralization of CA degradation with different Cl− concentrations at different reaction times in the Co2+/PMS/Cl− and PMS/Cl− system, respectively. Experimental conditions: [CA]0 = 0.1 mM, [Co2+]0 = 0.01 mM, [PMS]0 = 0.5 mM, pH 3.0. | |
3.3 Free radical identification
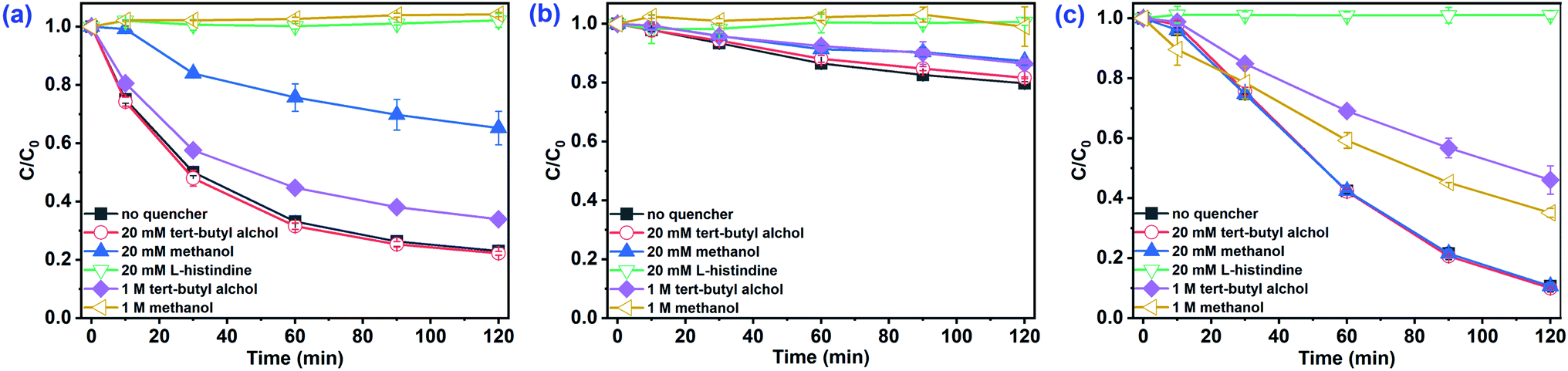
SO4˙− and ˙OH are regarded as the primary radicals yielded in PMS-based systems.34,39 Hence, quenching experiments were conducted to detect the major ROS during the oxidation of organic pollutants in AOPs. Both ˙OH and SO4˙− were scavenged by methanol (MeOH) with the rate constants of 9.7 × 108 M−1 s−1 and 2.5 × 107 M−1 s−1, respectively. tert-Butyl alcohol (TBA) reacts with ˙OH (kTBA, ˙OH = (3.8–7.6) × 108 M−1 s−1), but not SO4˙− 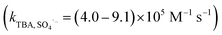 .40,41 In the absence of Cl−, as shown in Fig. 3a, 20 mM TBA had almost no influence on CA degradation, while the degradation rate of CA decreased from 81.0% to 66.0% by adding 1 M TBA. In contrast, MeOH inhibited CA degradation. Adding 20 mM MeOH reduced the degradation rate of CA from 81.0% to 34.8%, and CA removal stopped completely when 1 M MeOH was added. This result indicated that SO4˙− was the primary oxidant in the Co2+/PMS system without Cl− addition. In the presence of Cl−, as shown in Fig. 3b and c, there was no obvious inhibitory effect on CA degradation in the Co2+/PMS system with 20 mM TBA, while CA degradation decreased slightly as TBA increased to 1 M. This indicated ˙OH was not the primary radical because it is difficult to form in an acidic solution (eqn (11)).34 Besides, partial inhibitory on CA removal by adding 20 mM MeOH and complete inhibitory with 1 M MeOH were observed (see Fig. 3b), which suggested SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− concentration (1 mM). However, in the presence of 100 mM Cl− (shown in Fig. 3c), only partially restrained on CA removal was detected by adding 1 M MeOH. That revealed SO4˙− was not the dominant oxidant in the presence of 100 mM Cl−. Therefore, SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− levels (≤1 mM), whereas other reactive oxygen species might be predominant at high Cl− concentrations.
.40,41 In the absence of Cl−, as shown in Fig. 3a, 20 mM TBA had almost no influence on CA degradation, while the degradation rate of CA decreased from 81.0% to 66.0% by adding 1 M TBA. In contrast, MeOH inhibited CA degradation. Adding 20 mM MeOH reduced the degradation rate of CA from 81.0% to 34.8%, and CA removal stopped completely when 1 M MeOH was added. This result indicated that SO4˙− was the primary oxidant in the Co2+/PMS system without Cl− addition. In the presence of Cl−, as shown in Fig. 3b and c, there was no obvious inhibitory effect on CA degradation in the Co2+/PMS system with 20 mM TBA, while CA degradation decreased slightly as TBA increased to 1 M. This indicated ˙OH was not the primary radical because it is difficult to form in an acidic solution (eqn (11)).34 Besides, partial inhibitory on CA removal by adding 20 mM MeOH and complete inhibitory with 1 M MeOH were observed (see Fig. 3b), which suggested SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− concentration (1 mM). However, in the presence of 100 mM Cl− (shown in Fig. 3c), only partially restrained on CA removal was detected by adding 1 M MeOH. That revealed SO4˙− was not the dominant oxidant in the presence of 100 mM Cl−. Therefore, SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− levels (≤1 mM), whereas other reactive oxygen species might be predominant at high Cl− concentrations.| | |
SO4˙− + HO− → ˙OH + SO42−, k = (6.5 ± 1 × 107) M−1 s−1
| (11) |
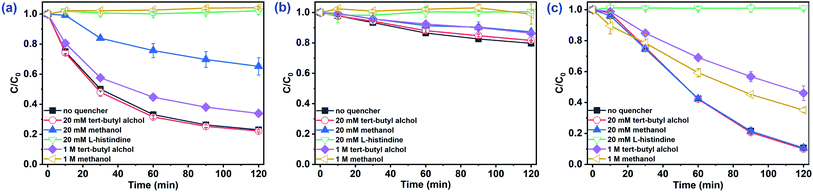 |
| | Fig. 3 Effect of radical scavengers on CA degradation in the Co2+/PMS system. Experimental conditions: [CA]0 = 0.1 mM, [Co2+]0 = 0.01 mM, [PMS]0 = 0.5 mM, pH 3.0, (a) 0 mM Cl−; (b) 1 mM Cl−; (c) 100 mM Cl−. | |
3.4 Non-radical identification
The low solubility of Co3+ (ksp = 1.6 × 10−44) limits its presence in solutions at pH 3.0.17 Hence, although Co3+ could exist in the Co2+/PMS system, it contributes very little to CA oxidation.
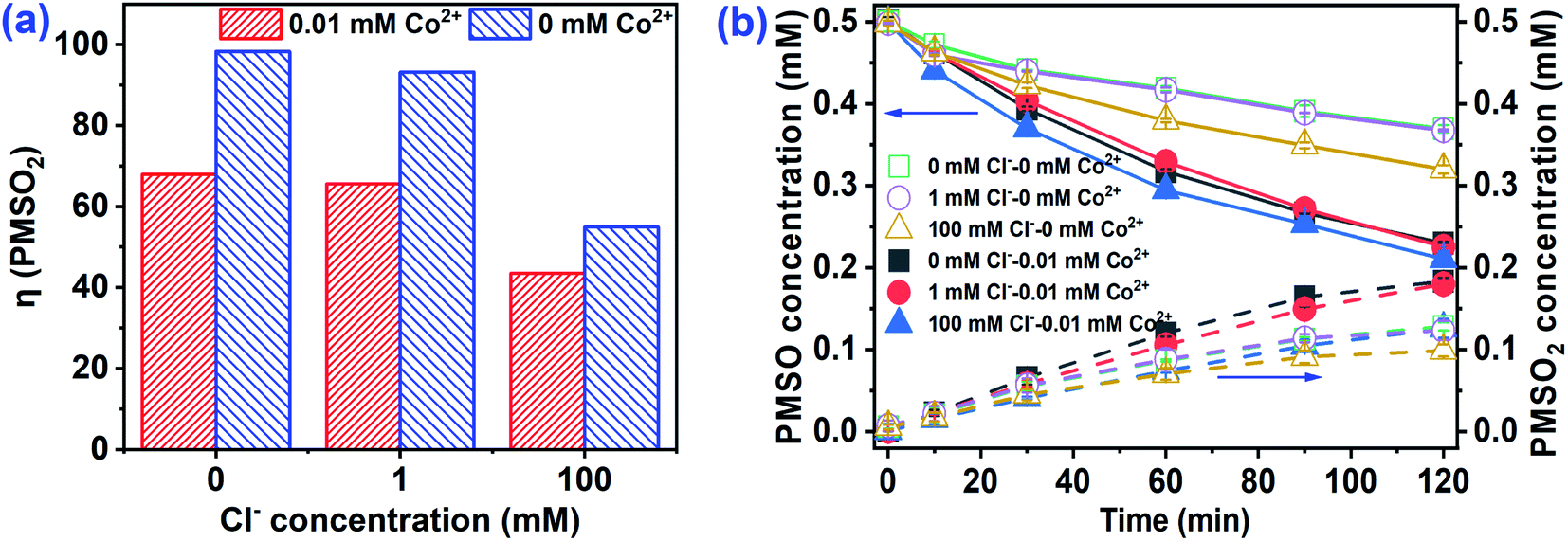
Recent studies confirmed that high-valent cobalt-oxo [Co4+] species played a significant role in the oxidation of organic pollutants by Co2+/PMS.16,17 PMSO reacts readily with Co4+ to produce PMSO2, though SO4˙− and ˙OH cannot oxidize PMSO to PMSO2.17 Hence, the consumption of PMSO and formation of PMSO2 were conducted to identify the presence of Co4+. As shown in Fig. 4a, PMSO directly oxidizes to PMSO2 with η-(PMSO2) (represents the conversion ratio of PMSO to PMSO2) approaching 100% by PMS alone. For the PMS/Cl− system, PMSO oxidation increased as the Cl− increased from 1 to 100 mM, but formation of PMSO2 decreased in the co-presence of PMS and Cl−. For example, the η-(PMSO2) was 93.2% at 1 mM Cl−, however, it was only 54.9% at [Cl−]0 = 100 mM. Some reactive chlorine may have formed when Cl− was 100 mM and oxidized PMSO to produce non-PMSO2 products that lowered the PMSO2 formation rate. However, the loss rate of PMSO accelerated in the Co2+/PMS/Cl− system as compared to the PMS/Cl− system (Fig. 4b). Additional consumption of PMSO after Co2+ addition suggested that other oxidants (e.g., Co4+, SO4˙− or ˙OH) might transform PMSO. At the same time, the formation of PMSO2 was higher in the presence of Co2+ (Fig. 4b), which indicated formation of Co4+ and rapid oxidation of PMSO to PMSO2 (rate constant, 2.0 × 106 M−1 s−1). Additional consumption of PMSO was much higher than PMSO2 formation when Co2+ was involved (Fig. S5†), which confirmed that Co4+ was not the only reactive oxygen species responsible for PMSO oxidation. Moreover, Liu et al.17 reported Co4+, SO4˙−, and ˙OH existed in solution when the PMS/Co2+ ratio >10. The PMS/Co2+ ratio in this study was 10 and our results agreed with those of Liu et al.17
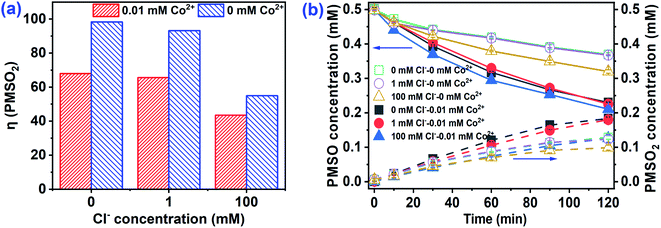 |
| | Fig. 4 (a) η(PMSO2), (b) PMSO loss and PMSO2 corresponding production in Co2+/PMS at different Cl− concentrations. Experimental conditions: [PMS]0 = 0.5 mM, [PMSO]0 = 0.5 mM, pH 3.0. | |
As depicted in Fig. S6,† the effect of PMSO on CA degradation was investigated. CA degradation dropped significantly in the Co2+/PMS system in the presence of both PMSO and Cl−. This confirmed that PMSO may act as a scavenging agent for high-valent cobalt, PMS, and free radicals, which led to the inhibitory degradation of CA in the Co2+/PMS system. For the PMS/Cl− system, the degradation efficiency of CA reached 75.1% in 100 mM Cl−, while complete inhibition of CA degradation with added PMSO. Chlorine species (HOCl/Cl2) generated by the reaction of Cl− and PMS could preferentially oxidize PMSO rather than CA.
3.5 Kinetic modeling
These results revealed that multiple ROS, including SO4˙−, ˙OH, and Co4+, were produced in the Co2+/PMS system. Kinetic modeling helped understand the various reactive species and their contributions to the overall degradation (see equations Table S1†). Fig. S7† shows a typical kinetic plot with a fitting degree above 98.0%.
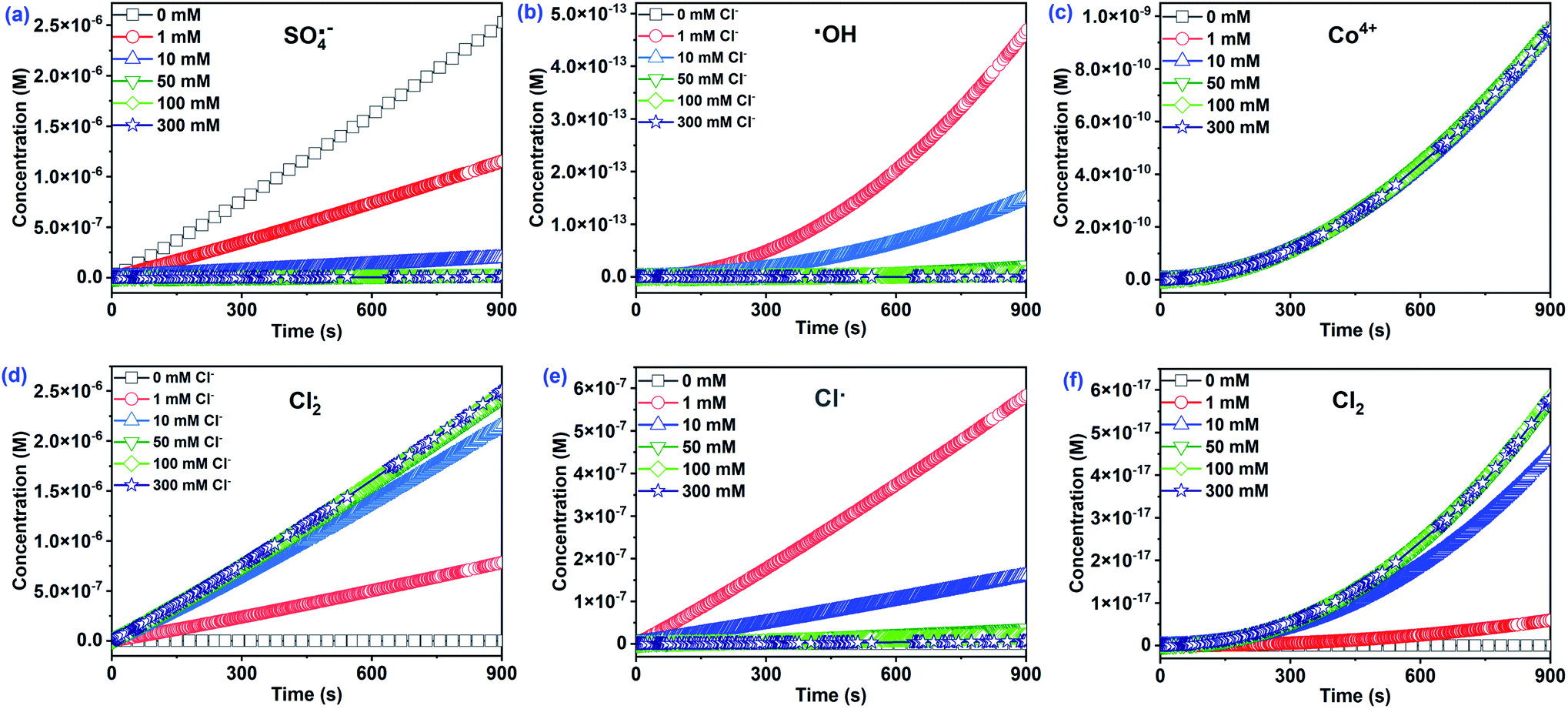
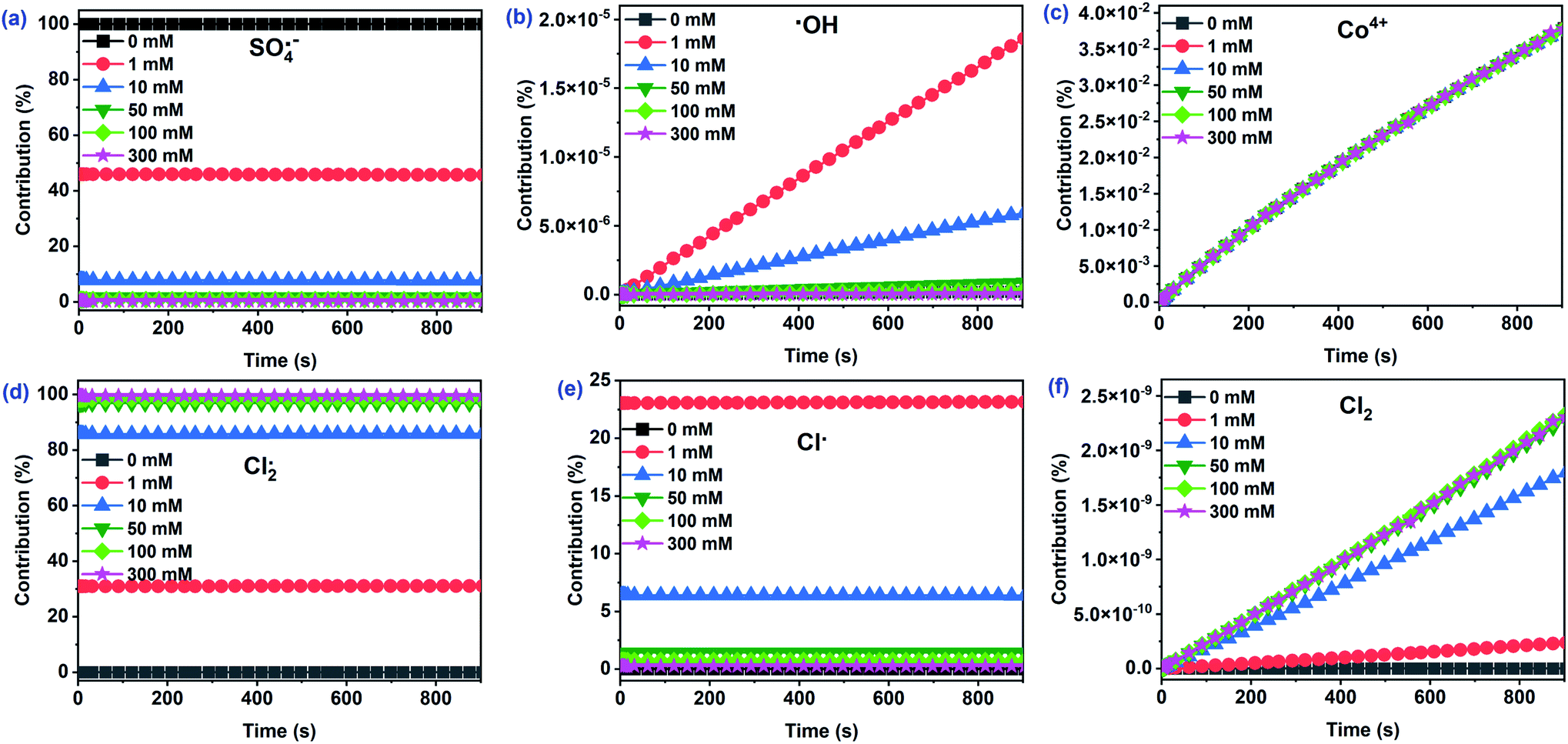
As depicted in Fig. 5a–c, three main reactive species (SO4˙−, ˙OH, and Co4+) may oxidize CA during Co2+/PMS in the absence of Cl−. The concentration of SO4˙− (∼10−6 M) was approximately nine and four times higher than ˙OH, and Co4+, respectively. Thus, the contribution of SO4˙− to CA oxidation reached 99.96%, while the contribution of Co4+ (∼0.03%) and ˙OH (∼0.01%) were negligible (Fig. 6). This agreed with quenching experiments and further revealed that SO4˙− plays a primary role in the Co2+/PMS system without Cl− addition.
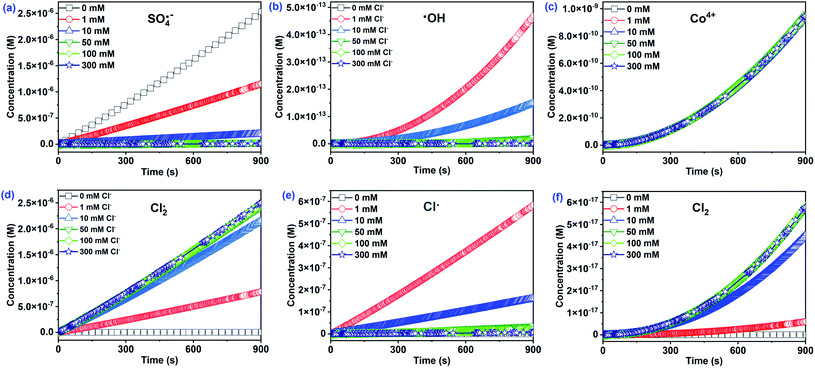 |
| | Fig. 5 The estimated concentration of primary reactive species at different Cl− concentrations in the Co2+/PMS/Cl− system. Experimental conditions: [CA]0 = 0.1 mM, [Co2+]0 = 0.01 mM, [PMS]0 = 0.5 mM, pH 3.0. | |
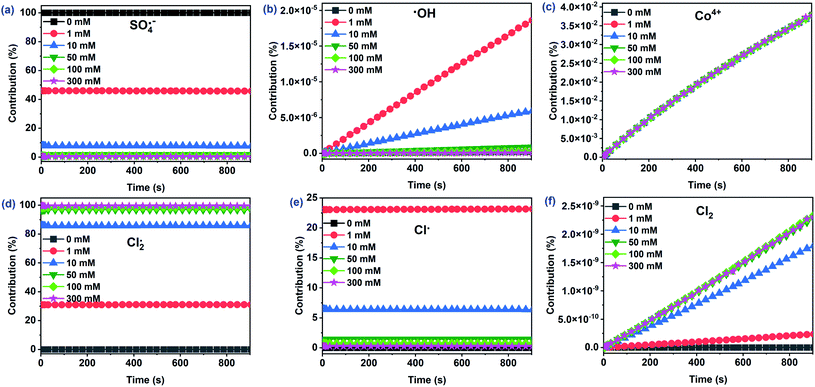 |
| | Fig. 6 The contribution of primary reactive species to CA degradation at different Cl− concentrations in the Co2+/PMS/Cl− system. Experimental conditions: [CA]0 = 0.1 mM, [Co2+]0 = 0.01 mM, [PMS]0 = 0.5 mM, pH 3.0. | |
A rapid adverse impact of Cl− on the yield of SO4˙− should be related to the quenching reaction of SO4˙− by Cl− with a high rate constant of 2.7 × 108 M−1 s−1 (eqn (12)). As a result, the contribution of SO4˙− dropped significantly from 99.96% to 0.28% as Cl− levels increased from 0 to 300 mM. Notably, Cl− facilitated the formation of ˙OH (∼10−13 M) at low Cl− concentrations (≤1 mM), while the ˙OH levels decreased with additional Cl− (Fig. 5). Overall, the contribution of ˙OH was small regardless of the amount of Cl− present and agreed with quenching test results. It has been reported that ˙OH level was relatively low under acidic conditions.35,42 However, the Co4+ concentration was relatively stable (∼10−10 M) as the initial Cl− level increased from 0 to 300 mM. Consequently, the contribution of Co4+ was less than 0.04%. This was likely due to its low reactivity with CA (k = 2.82 × 10−2 M−1 s−1) and the transformation of Co4+ to SO4˙−/˙OH when the molar ratio of PMS/Co2+ exceeded 10.
| | |
SO4˙− + Cl− → SO42− + Cl˙, kf = 2.7 × 108 M−1 s−1
| (12) |
| | |
Cl˙ + Cl− → Cl2˙−, k = 6.5 × 109 M−1 s−1
| (13) |
| | |
Cl2˙− + Cl2˙− → Cl2 + 2Cl−
| (14) |
Interestingly, the Cl2˙− concentration was overwhelming (∼10−5 M) among all chlorine reactive species and significantly increased with Cl−. As a result, the contribution of Cl2˙− to CA degradation reached 99.44% with 300 mM Cl−. The absolute predominance of Cl2˙− was due to two things—the high rate constant of the reaction between CA and Cl2˙− (k = 1.41 × 108 M−1 s−1) played a significant role in CA oxidation. Secondly, the transformation of Cl˙ to Cl2˙− with a high rate constant of 6.5 × 109 M−1 s−1 (eqn (13)) relates to the high concentration of Cl2˙−. Furthermore, generation of Cl˙ increased (0–1 mM) and then decreased (1–300 mM) with added Cl−. In the presence of 1 mM Cl−, Cl˙ accounted for 23.16% of CA oxidation due to its high CA reactivity (k = 5.5 × 109 M−1 s−1) and high production (∼10−7 M). Cl2 concentration had a similar increase as Cl2˙−, due to the conversion of the Cl2˙− to Cl2 as shown in eqn (14). The other three reactive species, (HOCl, ClO˙, and ClOH˙−) at relatively low concentrations, initially increased, then decreased with added Cl− (Fig. S8†). Consequently, their contributions to CA were relatively low and were neglected. Cl2˙− should be the dominant reactive species for CA oxidation at [Cl−]0 > 1 mM, while SO4˙− played a critical role at low Cl− concentrations (≤1 mM) in the Co2+/PMS system.
3.6 Mechanism and pathways
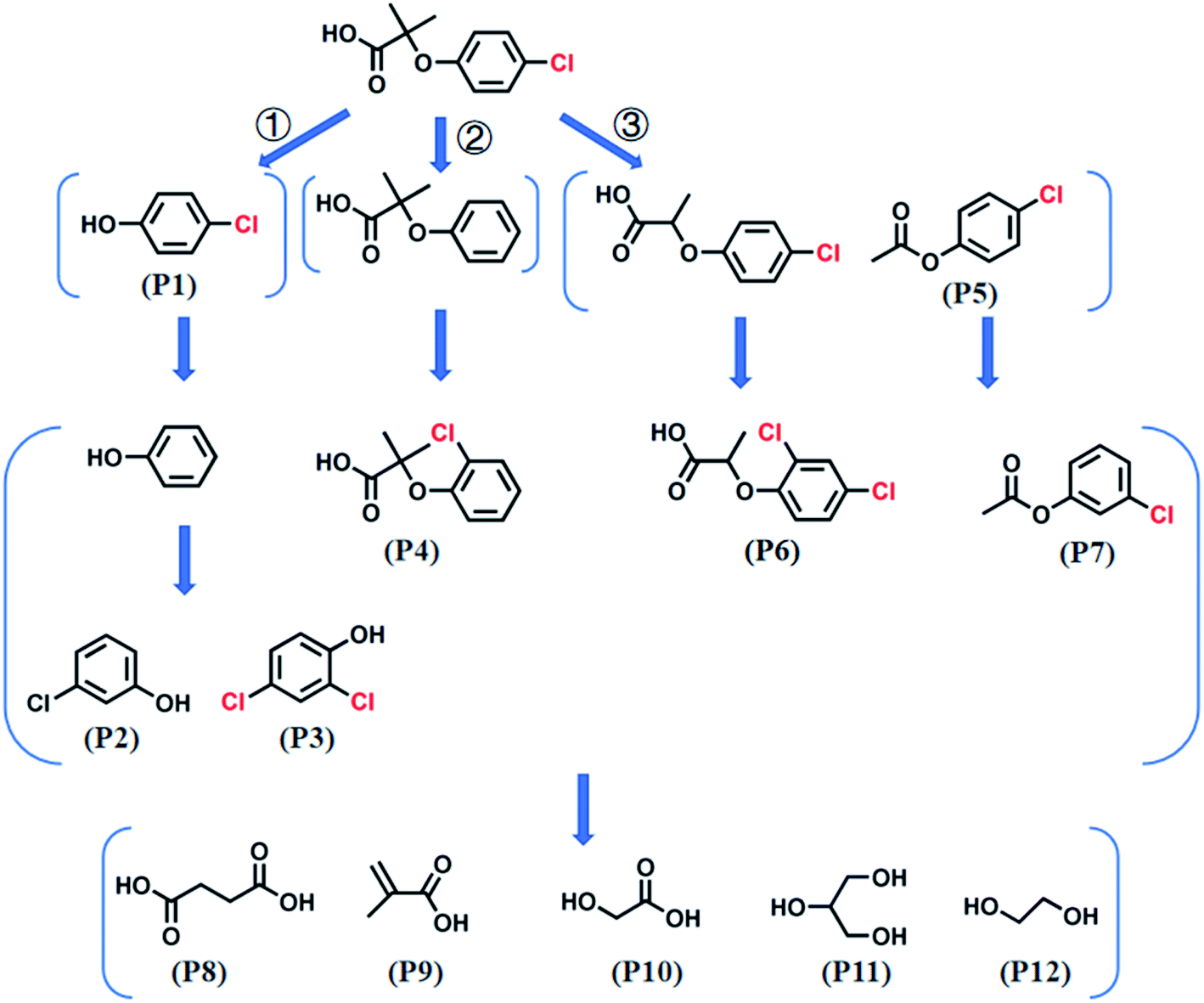
GC-MS analyzed the CA intermediates. The primary chlorinated by-products at different Cl− concentrations (i.e., 0, 1, and 100 mM) at 30 and 120 min are shown in Table S2.† The chlorinated by-products increased with added Cl− or reaction times. For example, 11 kinds of chlorinated by-products were detected in the Co2+/PMS system without Cl− addition, whereas 22 and 23 refractory chlorinated by-products at 1 mM and 100 mM Cl− were detected, respectively (Table S2†). Based on the identified products and previous studies,9,43 possible mechanisms and a CA degradation pathway were proposed (Fig. 7).
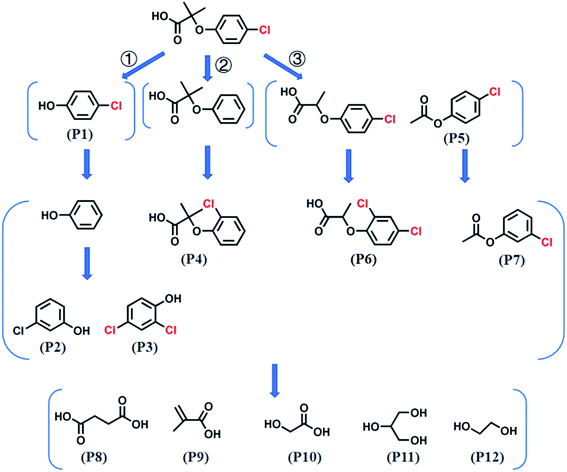 |
| | Fig. 7 A possible CA oxidation pathway in the Co2+/PMS system. Experimental conditions: [CA]0 = 0.1 mM, [Co2+]0 = 0.01 mM, [PMS]0 = 0.5 mM, pH 3.0. | |
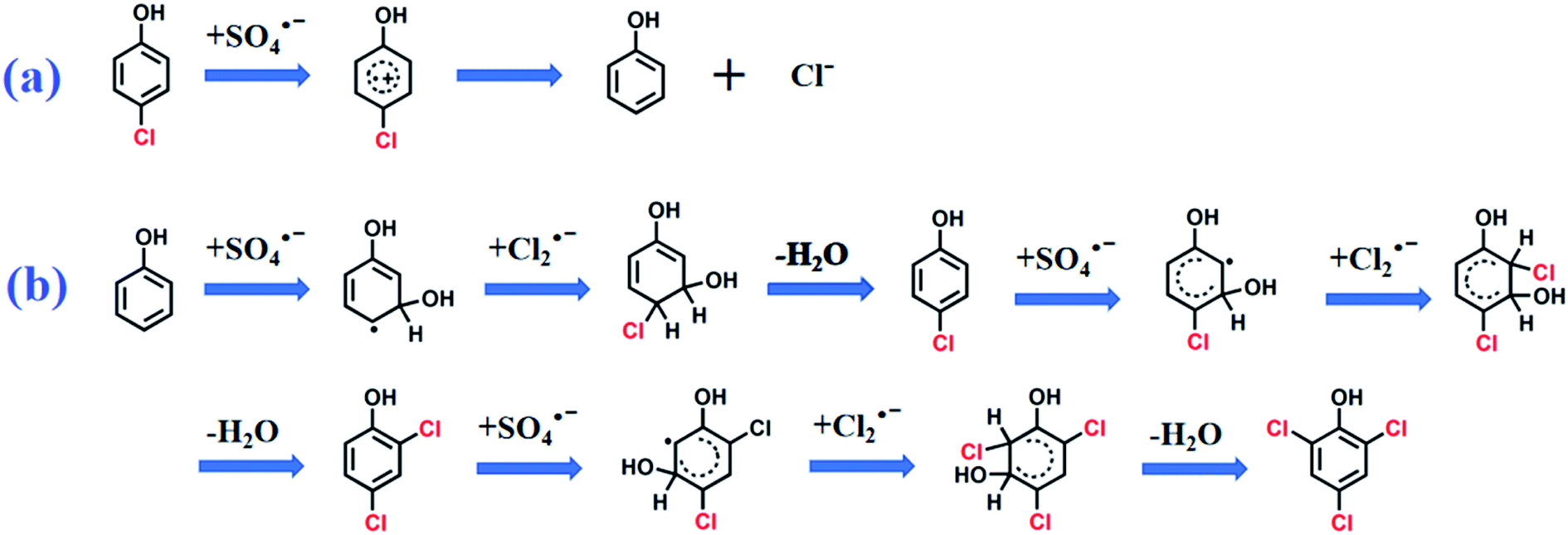
In the absence of Cl−, SO4˙− was the main reactive species in CA degradation. Thus, CA oxidation could proceed by three different pathways, including C1–O bond breaking, dechlorination, and attack on the C7 position of CA. Firstly, cleavage of the C1–O bond formed 4-chlorophenol (P1). Dechlorination of 4-chlorophenol attacked by SO4˙− may generate phenol (not detected, which implied in the present study) and the release of chloride ions (shown in Scheme 1a).44 Interestingly, chloride ions undergo oxidization by SO4˙− to produce chlorine radicals that form chlorinated aromatic by-products (P2 and P3) by nucleophilic addition. Peng et al.36 also indicated chlorine atoms of chlorinated azo dyes might be partially released as inorganic ions, which undergo subsequent rechlorination. Alternatively, SO4˙− initially reacts with CA, which led to CA dechlorination, followed by oxidization of the intermediate by SO4˙− to generate chlorinated by-products (P4). In the last way, the C7 position of CA was attacked by SO4˙− followed by demethylation and decarboxylation intermediates, such as 2-(4-chloro-phenoxy)-propionic acid (not detected) and acetic acid-4-chlorophenyl ester (P5), which was further transformed to chlorinated by-products (P6 and P7) through chlorination, respectively. Furthermore, the aromatic ring was gradually cleaved to produce organic acid compounds such as butanedioic acid (P8), methacrylic acid (P9), and glycolic acid (P10). Finally, short-chain alcohol compounds (e.g., glycerol (P11) and ethylene glycol (P12)) formed and eventually oxidized into CO2 and H2O. The identification of polychlorinated by-products (P2, P3, and P6) during CA removal without exogenous Cl− addition provided direct evidence for dechlorination and rechlorination of endogenic Cl−. Furthermore, the formation of various chlorinated by-products also explains the high degradation efficiency (81.0%) but low mineralization rate (9.15%) within 120 min during CA removal in the absence of Cl−.
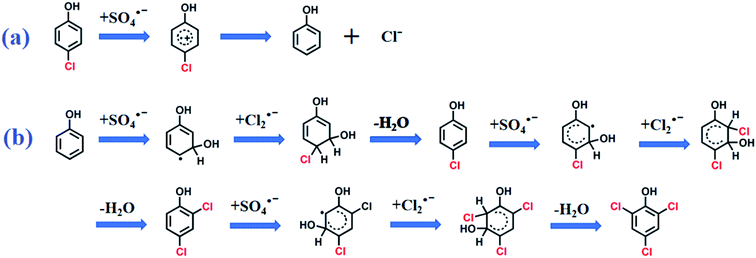 |
| | Scheme 1 The possible pathway of (a) dechlorination and (b) chlorination on CA degradation intermediates. | |
In the presence of Cl−, SO4˙− was scavenged by Cl− to produce less reactive chlorine radicals (Cl˙/Cl2˙−), which resulted in the reduction of CA oxidation. The inhibition of Cl− is vital at low chloride levels (≤1 mM). Additional Cl− dramatically enhanced CA oxidation, whereas the overall degradation rate was still lower than in the absence of Cl−. Hence, Cl2˙− was the dominant chlorine species contributing to CA degradation under chloride-rich conditions. More refractory polychlorinated by-products (chlorine atom number ≥ 2) were produced with additional Cl−, such as 3,4-dichlorophenol, 2,3-dichlorophenol, and 2,5-dichlorophenol. Thus, the mineralization rate dropped as the Cl− concentration increased. The oxidation pathways of CA involving Cl− might include C1–O bond cleavage, chlorination, dechlorination, and rechloridation process. Notably, 2,4,6-trichlorophenol was generated as the chlorinated by-product of CA in the presence of exogenous Cl−. It was assumed that chlorination phenol was initiated by Cl2˙− attack of the degradation product radicals, yielding 2,4,6-trichlorophenol (Scheme 1b). This result further proved that a higher exogenous Cl− concentration produced additional chlorine radicals, and then oxidized CA to produce more chlorinated by-products, consistent with previous reports.25,31
4 Conclusions
The degradation kinetics and mechanism of CA were studied in the Co2+/PMS system. The reaction rates of CA increased with added Co2+ and PMS though varied slightly with initial pH. Furthermore, Cl− had a dual effect (inhibitory then promoting) on CA degradation. Intermediate identification by GC-MS revealed that more refractory polychlorinated by-products (chlorine atom number ≥ 2) were produced in the Co2+/PMS/Cl− system, which resulted in a decreased CA mineralization rate. According to quenching experiments and kinetic modeling, SO4˙− (45.7%) played the main role in CA degradation, followed by Cl2˙− (31.1%) and Cl˙ (23.2%) at low Cl− levels (≤1 mM), whereas Cl2˙− was the predominant radical when [Cl−]0 ˃ 1 mM. However, the contribution of Co4+ to CA degradation was relatively low regardless of added Cl−. A possible CA degradation pathway in saline wastewater was proposed, including cleavage of the C1–O bond, chlorination, dechlorination, and rechloridation process. Our findings indicated the introduction of exogenic Cl− increased the difficulty in oxidative degradation of CA. Hence, it is imperative to explore other effective technologies for CA treatment in saline wastewater.
Conflicts of interest
There are no conflicts to declare.
Author contributions
Jiale Wang: Conceptualization, Visualization, Writing-original draft, Data curation. Siyi Fan: Data curation, Resources, Data curation. Zhirui Xu: Resources, Validation. Jiaqi Gao: Investigation, Resources. Ying Huang: Conceptualization, Funding acquisition, Software, Writing-review & editing, Supervision. Xubiao Yu: Methodology, Funding acquisition, Supervision. Huihui Gan: Visualization, Investigation, Funding acquisition.
Acknowledgements
This work was supported by Natural Science Foundation of Ningbo (No. 202003N4135), the General Research Project of Zhejiang Provincial Department of Education (No. Y202043966), National Natural Science Foundation of China (No. 41977152, No. 41676104 and No. 52070103), Zhejiang Provincial Natural Science Foundation (No. LGF19E090006), the Commonweal Project of Ningbo Science and Technology Bureau (No. 2019C10024), the fund by Laboratory of Sustainable Urban Drainage of Ningbo University, and the K. C. Wong Magna Fund in Ningbo University.
Notes and references
- D. Sreekanth, D. Sivaramakrishna, V. Himabindu and Y. Anjaneyulu, Bioresour. Technol., 2009, 100, 2534–2539 CrossRef CAS PubMed.
- K. K. Ng, X. Shi, S. L. Ong, C.-F. Lin and H. Y. Ng, Chem. Eng. J., 2016, 295, 317–325 CrossRef CAS.
- X. Shi, O. Lefebvre, K. K. Ng and H. Y. Ng, Bioresour. Technol., 2014, 153, 79–86 CrossRef CAS PubMed.
- M. Isidori, A. Nardelli, L. Pascarella, M. Rubino and A. Parrella, Environ. Int., 2007, 33, 635–641 CrossRef CAS PubMed.
- T. J. Runnalls, D. N. Hala and J. P. Sumpter, Aquat. Toxicol., 2007, 84, 111–118 CrossRef CAS PubMed.
- H. Lin, X. Tang, J. Wang, Q. Zeng, H. Chen, W. Ren, J. Sun and H. Zhang, J. Hazard. Mater., 2021, 405, 124204 CrossRef CAS PubMed.
- Y. Wang, H. Y. Li, P. Yi and H. Zhang, J. Hazard. Mater., 2019, 379, 120771 CrossRef CAS PubMed.
- T. E. Doll and F. H. Frimmelm, Water Res., 2004, 38(4), 955–964 CrossRef CAS PubMed.
- W. Qin, Z. Lin, H. Dong, X. Yuan, Z. Qiang, S. Liu and D. Xia, Water Res., 2020, 186, 116336 CrossRef CAS PubMed.
- X. Wang, Z. Wang, Y. Tang, D. Xiao, D. Zhang, Y. Huang, Y. Guo and J. Liu, Chem. Eng. J., 2019, 368, 999–1012 CrossRef CAS.
- G. Wen, X. Xu, H. Zhu, T. Huang and J. Ma, Chem. Eng. J., 2017, 328, 619–628 CrossRef CAS.
- Y.-Y. Ahn, J. Choi, M. Kim, M. S. Kim, D. Lee, W. H. Bang, E.-T. Yun, H. Lee, J.-H. Lee and C. Lee, Environ. Sci. Technol., 2021, 55, 5382–5392 CrossRef CAS PubMed.
- N. Li, S. Tang, Y. Rao, J. Qi, Q. Zhang and D. Yuan, Electrochim. Acta, 2019, 298, 59–69 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- Z. Zhang and J. O. Edwards, Inorg. Chem., 1992, 31, 3514–3517 CrossRef CAS.
- Y. Zong, X. Guan, J. Xu, Y. Feng, Y. Mao, L. Xu, H. Chu and D. Wu, Environ. Sci. Technol., 2020, 54, 16231–16239 CrossRef CAS PubMed.
- B. Liu, W. Guo, H. Wang, S. Zheng, Q. Si, Q. Zhao, H. Luo and N. Ren, J. Hazard. Mater., 2021, 416, 125679 CrossRef CAS PubMed.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 CrossRef CAS PubMed.
- K. H. Chan and W. Chu, Water Res., 2009, 43, 2513–2521 CrossRef CAS PubMed.
- J. Zhou, J. Xiao, D. Xiao, Y. Guo, C. Fang, X. Lou, Z. Wang and J. Liu, Chemosphere, 2015, 134, 446–451 CrossRef CAS PubMed.
- Y. Tang, X. Shi, Y. Liu, L. Feng and L. Zhang, R. Soc. Open Sci., 2018, 5, 171372 CrossRef PubMed.
- Y. Sun, J. Zhao, B.-T. Zhang, J. Li, Y. Shi and Y. Zhang, Chem. Eng. J., 2019, 368, 553–563 CrossRef CAS.
- L. Zhou, Y. Zhang, R. Ying, G. Wang, T. Long, J. Li and Y. Lin, Environ. Sci. Pollut. Res., 2017, 24, 11549–11558 CrossRef CAS PubMed.
- B. Sheng, F. Yang, Y. Huang, Z. Wang, R. Yuan, Y. Guo, X. Lou and J. Liu, Chem. Eng. J., 2020, 381, 122634 CrossRef CAS.
- L. Xu, R. Yuan, Y. Guo, D. Xiao, Y. Cao, Z. Wang and J. Liu, Chem. Eng. J., 2013, 217, 169–173 CrossRef CAS.
- C. Liang, C.-F. Huang, N. Mohanty and R. M. Kurakalva, Chemosphere, 2008, 73, 1540–1543 CrossRef CAS PubMed.
- J. E. Grebel, J. J. Pignatello and W. A. Mitch, Environ. Sci. Technol., 2010, 44, 6822–6828 CrossRef CAS PubMed.
- Y.-H. Guan, J. Ma, X.-C. Li, J.-Y. Fang and L.-W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- Y. Lei, S. Cheng, N. Luo, X. Yang and T. An, Environ. Sci. Technol., 2019, 53, 11170–11182 CrossRef CAS PubMed.
- M. Jaksić, B. Nikolić, I. Csonka and A. Djordjevic, J. Electrochem. Soc., 1969, 116, 684 CrossRef.
- Y. Xue, Z. Wang, R. Bush, F. Yang, R. Yuan, J. Liu, N. Smith, M. Huang, R. Dharmarajan and P. Annamalai, Chem. Eng. J., 2021, 415, 129041 CrossRef CAS.
- Y. Huang, M. Jiang, S. Gao, W. Wang, Z. Liu and R. Yuan, Sep. Purif. Technol., 2022, 120921 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- Z. Wang, R. Yuan, Y. Guo, L. Xu and J. Liu, J. Hazard. Mater., 2011, 190, 1083–1087 CrossRef CAS PubMed.
- R. Yuan, S. N. Ramjaun, Z. Wang and J. Liu, J. Hazard. Mater., 2011, 196, 173–179 CrossRef CAS PubMed.
- W. Peng, Y. Fu, L. Wang, Y. Wang, Y. Dong, Y. Huang and Z. Wang, Chin. Chem. Lett., 2021, 32, 2544–2550 CrossRef CAS.
- Z. Wang, M. Feng, C. Fang, Y. Huang, L. Ai, F. Yang, Y. Xue, W. Liu and J. Liu, RSC Adv., 2017, 7, 12318–12321 RSC.
- C. Fang, X. Zhang, X. Lou, Y. Shi, Y. Tang, D. Huang, J. Liu and Y. Guo, Int. J. Environ. Anal. Chem., 2022, 1–15 CrossRef CAS.
- F. Yang, Y. Huang, C. Fang, Y. Xue, L. Ai, J. Liu and Z. Wang, Chemosphere, 2018, 199, 84–88 CrossRef CAS PubMed.
- C. Qi, X. Liu, Y. Li, C. Lin, J. Ma, X. Li and H. Zhang, J. Hazard. Mater., 2017, 328, 98–107 CrossRef CAS PubMed.
- J. Miao, J. Sunarso, C. Su, W. Zhou, S. Wang and Z. Shao, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- C. Liang, Z.-S. Wang and C. J. Bruell, Chemosphere, 2007, 66, 106–113 CrossRef CAS PubMed.
- K. Zhu, X. Wang, M. Geng, D. Chen, H. Lin and H. Zhang, Chem. Eng. J., 2019, 374, 1253–1263 CrossRef CAS.
- C. Cai, X. Duan, X. Xie, S. Kang, C. Liao, J. Dong, Y. Liu, S. Xiang and D. D. Dionysiou, J. Hazard. Mater., 2021, 410, 124604 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Xubiao Yu* and
Huihui Gan
*,
Xubiao Yu* and
Huihui Gan
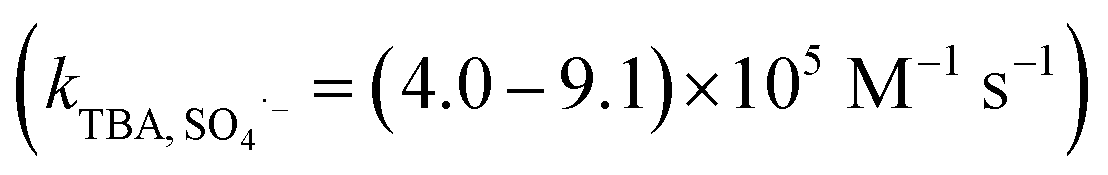 .40,41 In the absence of Cl−, as shown in Fig. 3a, 20 mM TBA had almost no influence on CA degradation, while the degradation rate of CA decreased from 81.0% to 66.0% by adding 1 M TBA. In contrast, MeOH inhibited CA degradation. Adding 20 mM MeOH reduced the degradation rate of CA from 81.0% to 34.8%, and CA removal stopped completely when 1 M MeOH was added. This result indicated that SO4˙− was the primary oxidant in the Co2+/PMS system without Cl− addition. In the presence of Cl−, as shown in Fig. 3b and c, there was no obvious inhibitory effect on CA degradation in the Co2+/PMS system with 20 mM TBA, while CA degradation decreased slightly as TBA increased to 1 M. This indicated ˙OH was not the primary radical because it is difficult to form in an acidic solution (eqn (11)).34 Besides, partial inhibitory on CA removal by adding 20 mM MeOH and complete inhibitory with 1 M MeOH were observed (see Fig. 3b), which suggested SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− concentration (1 mM). However, in the presence of 100 mM Cl− (shown in Fig. 3c), only partially restrained on CA removal was detected by adding 1 M MeOH. That revealed SO4˙− was not the dominant oxidant in the presence of 100 mM Cl−. Therefore, SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− levels (≤1 mM), whereas other reactive oxygen species might be predominant at high Cl− concentrations.
.40,41 In the absence of Cl−, as shown in Fig. 3a, 20 mM TBA had almost no influence on CA degradation, while the degradation rate of CA decreased from 81.0% to 66.0% by adding 1 M TBA. In contrast, MeOH inhibited CA degradation. Adding 20 mM MeOH reduced the degradation rate of CA from 81.0% to 34.8%, and CA removal stopped completely when 1 M MeOH was added. This result indicated that SO4˙− was the primary oxidant in the Co2+/PMS system without Cl− addition. In the presence of Cl−, as shown in Fig. 3b and c, there was no obvious inhibitory effect on CA degradation in the Co2+/PMS system with 20 mM TBA, while CA degradation decreased slightly as TBA increased to 1 M. This indicated ˙OH was not the primary radical because it is difficult to form in an acidic solution (eqn (11)).34 Besides, partial inhibitory on CA removal by adding 20 mM MeOH and complete inhibitory with 1 M MeOH were observed (see Fig. 3b), which suggested SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− concentration (1 mM). However, in the presence of 100 mM Cl− (shown in Fig. 3c), only partially restrained on CA removal was detected by adding 1 M MeOH. That revealed SO4˙− was not the dominant oxidant in the presence of 100 mM Cl−. Therefore, SO4˙− was the primary oxidant in the Co2+/PMS system at low Cl− levels (≤1 mM), whereas other reactive oxygen species might be predominant at high Cl− concentrations.