
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew dimeric chromanone derivatives from the mutant strains of Penicillium oxalicum and their bioactivities†
Guowei Gu‡
a,
Tao Zhang‡a,
Jianyuan Zhaoa,
Wuli Zhaoa,
Yan Tangab,
Lu Wanga,
Shan Cena,
Liyan Yu*a and
Dewu Zhang *a
*a
aInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, P. R. China. E-mail: yly@cpcc.ac.cn; zhangdewuever@163.com
bSchool of Pharmacy, Yantai University, Yantai 264005, P. R. China
First published on 10th August 2022
Abstract
Three new chromanone dimer derivatives, paecilins F–H (1–3) and ten known compounds (4–13), were obtained from the mutant strains of Penicillium oxalicum 114-2. Their structures were elucidated by extensive analysis of spectroscopic data and comparison with reported data, and the configurations of 1–3 were resolved by quantum chemical calculations of NMR shifts and ECD spectra. Compounds 5 and 11 showed significant anti-influenza A virus activities with IC50 values of 5.6 and 6.9 μM, respectively. Compounds 8 and 9 displayed cytotoxic activities against the MIA-PaCa-2 cell line with IC50 values of 2.6 and 2.1 μM, respectively. Compound 10 exhibited antibacterial activities against Bacillus cereus with a MIC value of 4 μg mL−1.
Introduction
Fungi have been demonstrated to be an important resource which could produce biologically active compounds with diverse chemical structures.1–4 The Penicillium genus composed of over 200 species is one of the largest groups of fungi,5 and a large quantity of secondary metabolites with varied architectural features and potent biological activities are isolated from Penicillium, including polyketides,6–8 terpenoids,9–11 alkaloids,12–14 and macrolides.15–17 The chromanone dimer is a structurally complex polyketide widely distributed in several genera of fungi, exemplified by Aspergillus,18,19 Xylaria,20 Penicillium,21,22 Setophoma,23 and Gonytrichum,24,25 which display cytotoxic,18,19 antimicrobial,23 and innate immune-promoting24,25 activities. In the course of investigating the biosynthesis of oxalicine B, tetrahydroxanthone dimer, secalonic acid D was isolated from the oxalicine B deletion mutant unexpectedly, which is well known for impressive anti-tumor activity. As part of our ongoing research for bioactive tetrahydroxanthone dimer derivatives from the mutant strains of Penicillium oxalicum 114-2, the extracts of the mutant strains of P. oxalicum 114-2 were investigated, which led to the isolation of thirteen polyketides, including three new dihydrochromone dimer derivatives: paecilin F (1), paecilin G (2), and paecilin H (3), along with ten known compounds (4–13) (Fig. 1). The isolated compounds were evaluated for antiviral, cytotoxic, and antibacterial activities. Herein, we describe the isolation, structural elucidation and bioactivities of these secondary metabolites from the mutants of Penicillium oxalicum 114-2.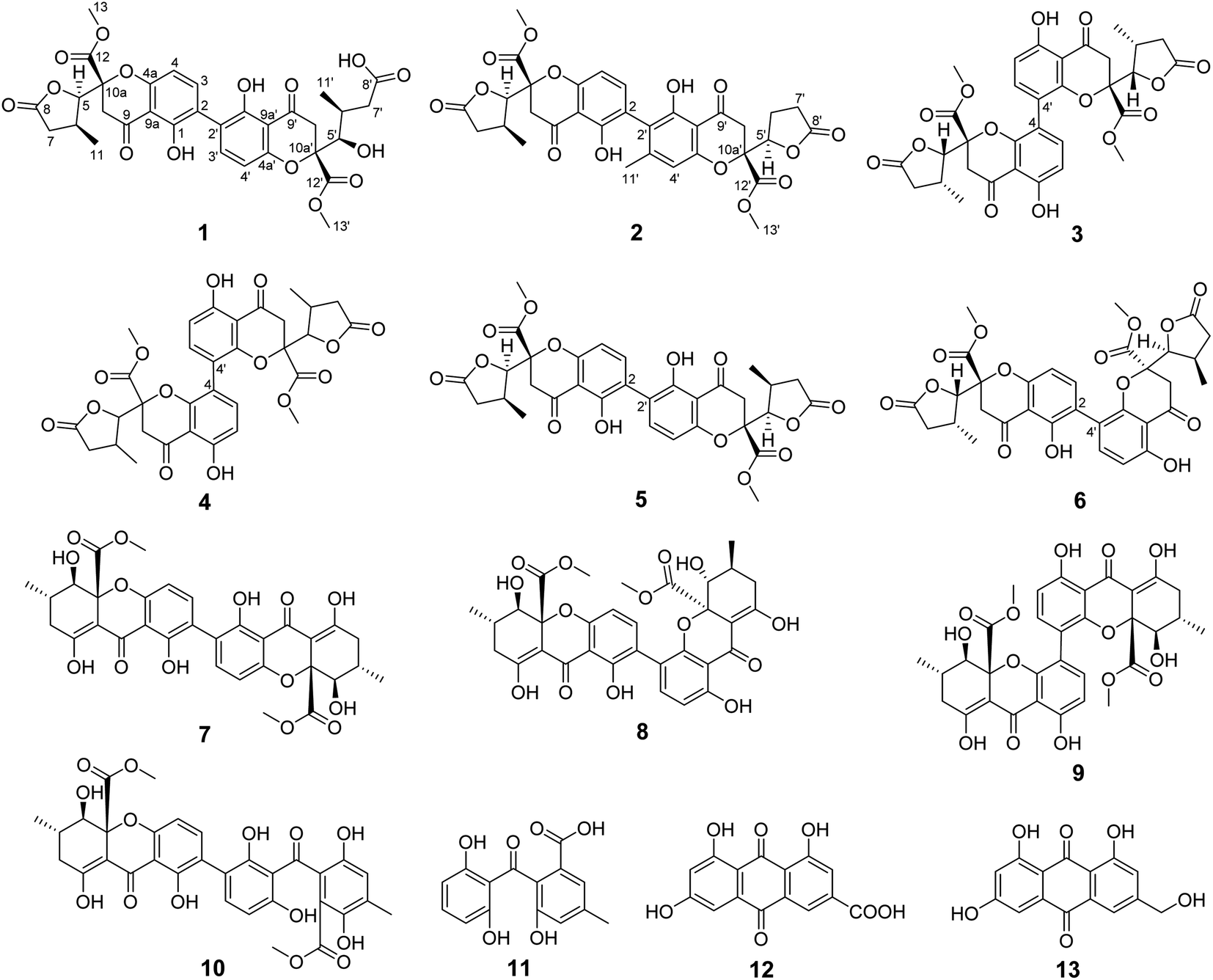 | ||
| Fig. 1 Structures of compounds 1–13. | ||
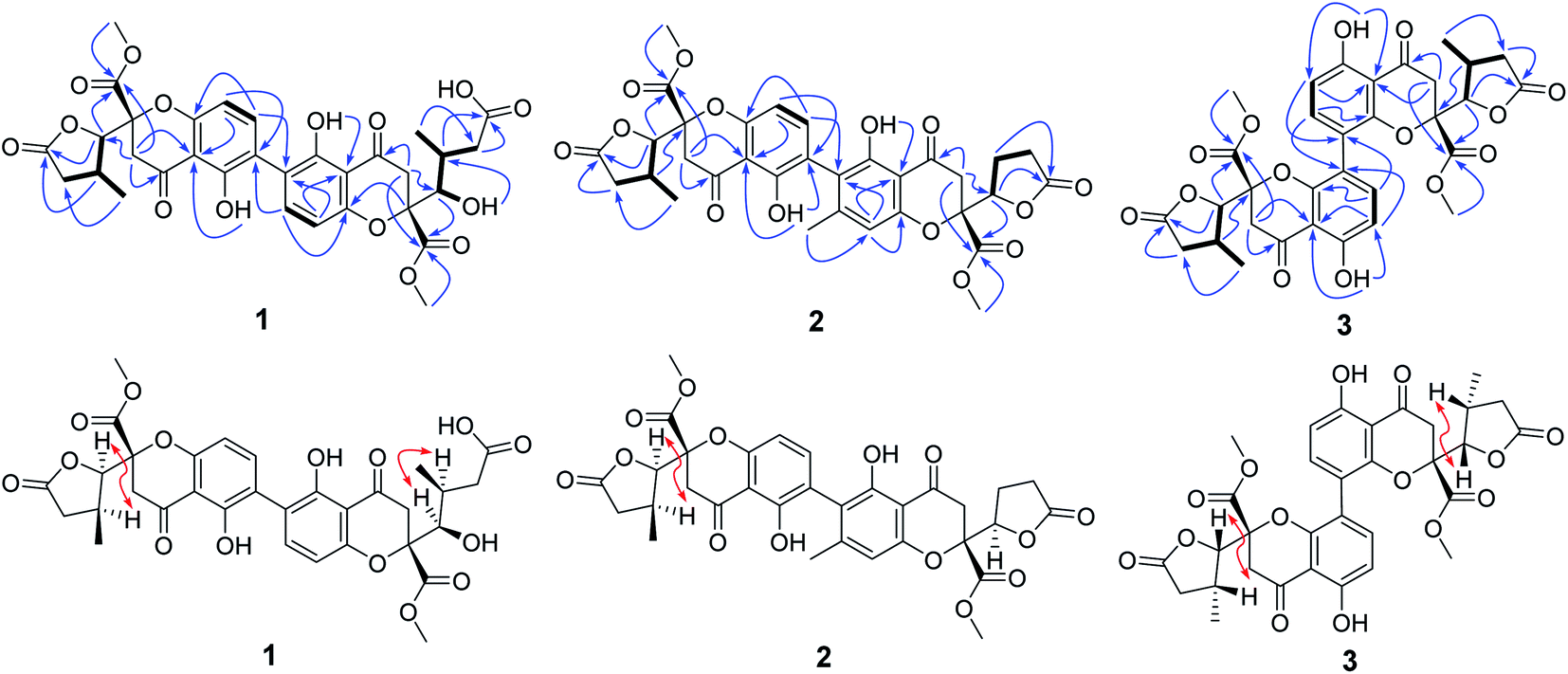 | ||
| Fig. 2 Key COSY, HMBC and NOESY correlations of 1–3. | ||
Results and discussion
Compound 1 was isolated as yellow powder, and HRESI mass spectrum gave a quasi-molecular ion peak at m/z 679.1600 [M + Na]+, corresponding to a molecular formula of C32H32O15 with 17 degrees of unsaturation. The IR spectrum displayed absorption bands at 3422, 1783, 1676, and 1624 cm−1, which indicated the existence of hydroxyl, carbonyl and aromatic moieties. The 1H NMR spectrum (Table 1) of 1 indicated the signals of two phenolic hydroxyl protons at δH 11.91 (1H, s) and 11.82 (1H, s), one carboxyl protons at δH 12.13 (1H, br s), two sets of doublets of ortho-coupled aromatic protons at δH 7.49 (1H, d, J = 8.4 Hz), 6.68 (1H, d, J = 8.4 Hz), 7.47 (1H, d, J = 8.4 Hz), 6.61 (1H, d, J = 8.4 Hz), one hydroxyl protons at δH 5.73 (1H, d, J = 6.6 Hz), two oxymethines at δH 4.94 (1H, d, J = 6.6 Hz) and 3.89 (1H, d, J = 5.4 Hz), two methoxyls at δH 3.70 (3H, s) and 3.64 (3H, s), and two methyls at δH 1.18 (3H, d, J = 7.2 Hz), 0.93 (3H, d, J = 6.0 Hz). The 13C NMR and DEPT spectra (Table 1) exhibited 32 carbon resonances, including sixteen nonprotonated carbons (δC 197.2, 195.3, 175.5, 173.6, 170.4, 169.1, 159.2, 158.3, 158.2, 158.2, 140.8, 140.5, 107.2, 107.2, 87.7, and 83.8, including six carbonyl, four oxygenated aromatic, four aromatic, and two oxygenated), eight methine carbons (δC 140.8, 140.5, 107.2, 107.2, 81.8, 74.8, 32.6, and 30.8, including four aromatic and two oxygenated), four methylene carbons (δC 40.3, 39.8, 39.1, and 36.7), two methoxyl carbons (δC 53.6 and 53.0), and two methyl carbons (δC 14.7 and 13.9). The 1H NMR and 13C NMR spectroscopic data of 1 were similar to those of paecillin D (5) (Tables S10 and S11†).26 The obvious difference was that one γ-butyrolactone group in paecillin D was changed to a linear side chain in 1, which was confirmed by the HMBC correlations (Fig. 2) from H-5′ to C-6′, C-7′, C-10′, C-10a′, C-11′, and C-12′; from H-7′ to C-5′, C-6′, C-8′, and C-11′; from 5′-OH to C-5′, C-6′, and C-10a′, along with the 1H–1H COSY correlations (Fig. 2) of 5′-OH/H-5′/H-6′/H-7′. Furthermore, the HMBC cross-peaks of H-3 with C-2′, H-3′ with C-2 indicated that two monomers had 2–2′ linkage. The large coupling constant of 3JH-5,H-6 (6.6 Hz) and NOESY correlation between H-5 and H-6 established their cis configuration in the γ-butyrolactone group. The small coupling constant of 3JH-5′,H-6′ (1.8 Hz) and NOESY correlation between H-5′ and H-6′ indicated their gauche relationship.19 Because the γ-butyrolactone group and flexible side chain had few effect on the ECD spectrum of 1, a simplified structure of 1A (Fig. S1†), in which two hydroxymethyls replaced γ-butyrolactone group and butanoic acid in 1, was used for the ECD calculations to determine the absolute configurations of C-10a and C-10a′.27 Three stereoisomers, (10aR,10a′R)-1Aa, (10aR,10a′S)-1Ab, (10aS,10a′S)-1Ac existed on the basis of the relative configuration (Fig. S1†). The calculated ECD spectrum of 1Aa was consistent with the experimental one (Fig. 3), suggesting the 10aR,10a′R configuration of 1. Based on above-mentioned relative and absolute configurations analyses, there are four possible configurations for 1: (5S,6R,10aR,5′S,6′R,10a′R)-1a, (5S,6R,10aR,5′R,6′S,10a′R)-1b, (5R,6S,10aR,5′S,6′R,10a′R)-1c, (5R,6S,10aR,5′R,6′S,10a′R)-1d (Fig. S3†). Subsequently, we performed computational predictions of NMR chemical shifts of both the isomers 1a–1d using the gauge-independent atomic orbital (GIAO) method at the mPW1PW91/6-311G(2d,p) level in DMSO (PCM).28,29 Compared of the calculated 1H and 13C NMR data for 1a–1d and the experimental 1H and 13C NMR data for 1 (Tables S1 and S2†) indicated that the possibility of structure 1b can be excluded firstly. In addition, the correlation coefficient (R2) obtained by linear regression analysis, corrected mean absolute deviation (CMAD), corrected largest absolute deviation (CLAD) of 1d were superior to those of 1a and 1c (Table S3†), which indicated that isomer 1d is the most reasonable configuration. Therefore, the structure of 1 was identified as paecilin F.| No. | 1a | 2b | 3a | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a Recorded in DMSO-d6.b Recorded in CDCl3. | ||||||
| 1 | 158.2, s | 159.3, s | 160.2, s | |||
| 2 | 117.0, s | 116.6, s | 109.5, d | 6.49, d (8.4) | ||
| 3 | 140.8, d | 7.49, d (8.4) | 141.7, d | 7.37, d (8.4) | 141.0, d | 7.75, d (8.4) |
| 4 | 107.2, d | 6.68, d (8.4) | 107.9, d | 6.67, d (8.4) | 114.0, s | |
| 4a | 158.3, s | 159.3, s | 155.4, s | |||
| 5 | 81.8, d | 4.94, d (6.6) | 82.7, d | 4.80, d (6.6) | 81.6, d | 4.85, d (6.6) |
| 6 | 32.6, d | 2.98, m | 33.7, d | 3.00, m | 32.8, d | 2.88, m |
| 7 | 36.7, t | 2.83, dd (17.4, 8.4), 2.33, dd (17.4, 6.0) | 36.8, t | 2.69, dd (17.4, 8.4), 2.50, dd (17.4, 8.4) | 35.8, t | 2.45, dd (16.8, 8.4), 1.72, dd (16.8, 9.0) |
| 8 | 175.5, s | 175.0, s | 174.9, s | |||
| 9 | 195.3, s | 194.2, s | 194.6, s | |||
| 9a | 107.1, s | 107.7, s | 107.3, s | |||
| 10 | 39.1, t | 3.61, d (17.4), 3.06, d (17.4) | 40.0, t | 3.27, d (17.4), 3.20, d (17.4) | 39.2, t | 3.56, d (18.0), 3.17, d (18.0) |
| 10a | 83.8, s | 84.7, s | 85.3, s | |||
| 11 | 14.7, q | 1.18, d (7.2) | 15.0, q | 1.36, d (7.2) | 14.3, q | 1.00, d (7.2) |
| 12 | 169.1, s | 169.1, s | 169.1, s | |||
| 13 | 53.6, q | 3.70, s | 53.7, q | 3.77, s | 53.6, q | 3.75, s |
| 1′ | 158.2, s | 159.6, s | 160.2, s | |||
| 2′ | 115.5, s | 117.6, s | 109.5, d | 6.49, d (8.4) | ||
| 3′ | 140.5, d | 7.47, d (8.4) | 151.5, s | 141.0, d | 7.75, d (8.4) | |
| 4′ | 107.2, d | 6.61, d (8.4) | 109.1, d | 6.57, s | 114.0, s | |
| 4a′ | 159.2, s | 158.9, s | 155.4, s | |||
| 5′ | 74.8, d | 3.89, d (5.4) | 81.1, d | 4.88, d (8.4, 6.0) | 81.6, d | 4.85, d (6.6) |
| 6′ | 30.8, d | 2.22, m | 22.2, t | 2.45, m | 32.8, d | 2.88, m |
| 7 | 39.8, t | 2.41, m, 2.19, m | 27.8, t | 2.72, m, 2.60, m | 35.8, t | 2.45, dd (16.8, 8.4), 1.72, dd (16.8, 9.0) |
| 8′ | 173.6, s | 175.7, s | 174.9, s | |||
| 9′ | 197.2, s | 193.2, s | 194.6, s | |||
| 9a′ | 106.9, s | 105.8, s | 107.3, s | |||
| 10′ | 40.3, t | 3.50, d (17.4), 3.01, d (17.4) | 39.6, t | 3.11, d (16.8), 2.97, d (16.8) | 39.2, t | 3.56, d (18.0), 3.17, d (18.0) |
| 10a′ | 87.7, s | 84.2, s | 85.3, s | |||
| 11′ | 13.9, q | 0.93, d (6.0) | 21.5, q | 2.12, s | 14.3, q | 1.00, d (7.2) |
| 12′ | 170.4, s | 169.1, s | 169.1, s | |||
| 13′ | 53.0, q | 3.64, s | 53.9, q | 3.79, s | 53.6, q | 3.75, s |
| 1-OH | 11.82, s | 11.74, s | 11.57, s | |||
| 1′-OH | 11.91, s | 11.73, s | 11.57, s | |||
| 5′-OH | 5.73, d, (6.6) | |||||
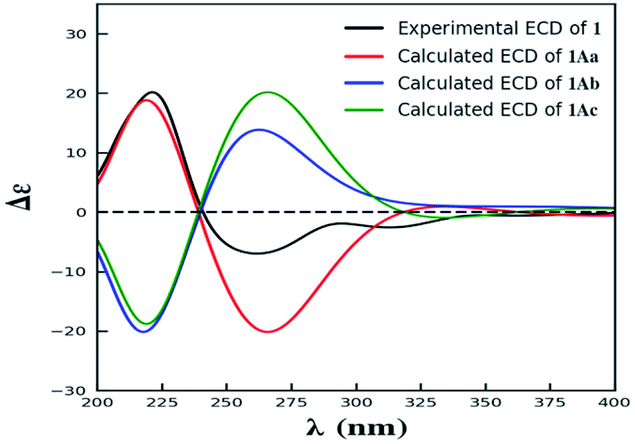 | ||
| Fig. 3 The experimental ECD spectrum of 1, and the calculated ECD spectra of 1Aa–1Ac. | ||
Compound 2 was isolated as yellow powder, and its molecular formula was established as C32H30O14 based on HR-ESI peak ion m/z 677.1267 [M + K]+. Its 1H and 13C NMR data were similar to those of paecilin D (5) (Tables S10 and S11†). The obvious differences include the presence of an aromatic proton at δH 6.57 (1H, s) and one aryl methyl moiety at δH 2.12 (3H, s) in 2 instead of two ortho-coupled aromatic protons at δH 7.54 (1H, d, J = 9.0 Hz), 6.63 (1H, d, J = 9.0 Hz) and a methyl moiety at δH 1.14 (1H, d, J = 7.2 Hz) in 5. The HMBC correlations from H-4′ to C-2′, C-4a′, and C-9a′; from H-11′ to C-2′, C-3′, and C-4′ assigned the location of the aryl methyl group. Meanwhile, the typical methyl in one γ-butyrolactone group in 2 disappeared compared with 5, suggesting the existence of the γ-butyrolactone group without methyl moiety in 2, which was further determined by the 1H–1H COSY correlations of H-5′/H2-6′/H2-7′ and the HMBC correlations from H-5′ to C-6′, C-7′, C-8′, and C-12′. The large coupling constant of 3JH-5,H-6 (6.6 Hz) and NOESY correlation between H-5 and H-6 indicated their cis configuration in the γ-butyrolactone group. The similar positive Cotton effect around 214 nm and negative Cotton effect around 245 nm (Fig. S31†) compared to those of 1 suggested the 10aR,10a′R absolute configuration of 2. To assign the configurations of C-5, C-6, and C-5′, quantum chemical calculations of the NMR data of four diastereoisomers: (5S,6R,10aR,5′S,10a′R)-2a, (5S,6R,10aR,5′R,10a′R)-2b, (5R,6S,10aR,5′S,10a′R)-2c, (5R,6S,10aR,5′R,10a′R)-2d (Fig. S4†), were performed using GIAO method at the mPW1PW91/6-311G(2d,p) level in chloroform (PCM).28,29 The calculated chemical shifts of 2a–2d (Tables S4 and S5†), especially the calculated NMR data of 2d, were in good agreement with the experimental values. Moreover, the R2, CMAD, and CLAD values of 2d were superior to those of 2a–2c (Tables S6†). The deduction was further supported by DP4+ probability analysis (Fig. S8†), which showed that isomer 2d was the most reasonable configuration with a probability of 99.84% for the combination of 1H and 13C NMR data. These results indicated that the configuration of 2d was the most plausible. Accordingly, the structure of 2 was elucidated to be paecilin G.
Compound 3 was obtained as yellow powder. The molecular formula was determined to be C32H30O14 by HR-ESI mass spectrum, which showed a quasi-molecular ion peak at m/z 639.1677 [M + H]+. Its 1H and 13C NMR spectroscopic data were great similar to those of paecilin A (4).30 Interpretation of its 1D and 2D NMR data of 3 established the same planar structure as 4. A literature search revealed that a total of five stereoisomers for 3 have been reported: paecilin A (4), paecilin C (3A),21 dimethyl (2S,2′S)-5,5′-dihydroxy-2,2′-bis[(2S,3S)-3-methyl-5-oxotetrahydrofuran-2-yl]4,4′-dioxo-[8,8′-bichroman]-2,2′-dicarboxylate (3B),31 dimethyl (2S,2′S)-5,5′-dihydroxy-2,2′-bis([2R,3R]-3-methyl-5-oxotetrahydrofuran-2-yl)-4,4′-dioxo-[8,8′-bichromane]-2,2′-dicarboxylate (3C),32 dimethyl (2S,2′S)-5,5′-dihydroxy-2-([2S,3S]-3-methyl-5-oxotetrahydrofuran-2-yl)-2'-([2R,3R]-3-methyl-5-oxotetrahydrofuran-2-yl)-4,4′-dioxo-[8,8′-bichromane]-2,2′-dicarboxylate (3D) (Fig. S9†).32 Detailed analysis of NMR data of these stereoisomers indicated that 3 was new chromanone dimer (Tables 1, S12, and S13†). The large coupling constant of 3JH-5,H-6 (6.6 Hz) and 3JH-5′,H-6′ (6.6 Hz), as well as NOESY correlation between H-5 and H-6 indicated their cis configuration in the γ-butyrolactone group. The absolute configuration of C-10a and C-10a′ in 3 was determined as described for 1 by comparison of its experimental and calculated ECD spectra. Similarly, a simplified structure 3A was subjected to ECD calculations.27 Only two possible stereoisomers for 3: (10aS,10a′S)-3Aa and (10aR,10a′R)-3Ab, remained on the basis of its NMR data (Fig. S5†). The calculated ECD spectrum of 3Ab was consistent with the experimental one (Fig. 4), establishing the 10aR,10a′R absolute configuration of 3. After the relative configuration analysis, there are two diastereoisomers: (5R,6S,10aR,5′R,6′S,10a′R)-3a, (5S,6R,10aR,5′S,6′R,10a′R)-3b (Fig. S7†). Chemical shifts of isomers 3a and 3b were predicted using the GIAO method at the mPW1PW91/6-311G(2d,p) level in DMSO.28,29 The calculated 1H and 13C NMR data of 3b matched well with the experimental values (Tables S7 and S8†), and the R2, CMAD, and CLAD values of calculated 1H NMR data for 3b were also superior to those of 3a (Tables S9†), indicating the configuration of 3b was more reasonable than 3a. Thus, the structure of 3 was determined to be paecilin H.
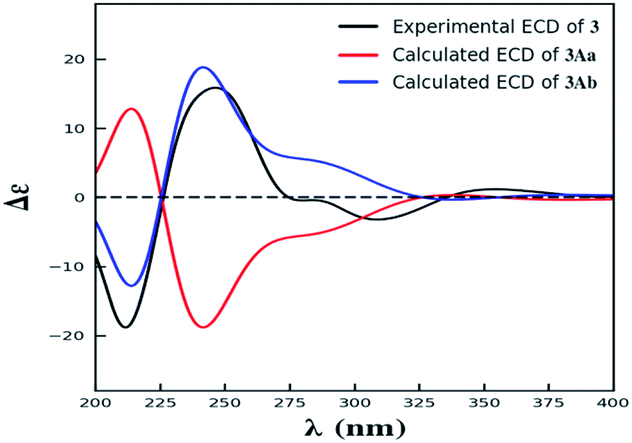 | ||
| Fig. 4 The experimental ECD spectrum of 3, and the calculated ECD spectra of 3Aa and 3Ab. | ||
Additionally, we analyzed the coupling constant of 3JH-5,H-6 for 1–6 and those of its stereoisomers reported in the literature (Table 1, S11 and S12†),21,26,30–33 indicating that the large coupling constant of 3JH-5,H-6 (J > 6 Hz) assigned the cis configuration of γ-butyrolactone group, and small coupling constant of 3JH-5,H-6 (J < 5 Hz) established the trans configuration of γ-butyrolactone group, which can provide guidance for determining the relative configuration of C-5 and C-6 for chromanone dimer and monomer derivatives.
The known compounds were identified as paecilin A (4),30 paecilin D (5),26 paecilin E (6),33 secalonic acid D (7),34 2,4′-secalonic acid D (8),35 4,4′-secalonic acid D (9),36 secalonic acid H (10),22 monodictyphenone (11),37 emodic acid (12),38 and citreorosein (13)39 by comparison of their NMR and MS data with those reported in the literatures.
Compounds 1–13 were evaluated for anti-influenza A virus (IAV), cytotoxic, and antibacterial activities. Compounds 5 and 11 exhibited significant anti-IAV (A/WSN/33, H1N1) activities with the IC50 values of 5.6 and 6.9 μM, respectively (Table 2). Compounds 8 and 9 displayed cytotoxic activities against MIA-PaCa-2 cell line with the IC50 values of 2.6 and 2.1 μM, respectively (Table S14†). Compounds 1–13 were tested for antibacterial activities against Escherichia coli ATCC 25922, Enterococcus faecalis ATCC 29212, Pseudomonas syringae CPCC 101099, Bacillus cereus CPCC 101254, Acinetobacter baumannii 12-8, Pseudomonas aeruginosa 13-17, Klebsiella pneumoniae ATCC BAA-2146, Enterococcs faecium ATCC 700221 and Staphylococcus aureus ATCC 33591. Compounds 8 and 10 exhibited moderate antibacterial activities against Bacillus cereus CPCC 101254 with the MIC values of 32 μg mL−1 and 4 μg mL−1, respectively, and others showed no antibacterial activities (Table S15†).
| Compounds | Inhibition rate (%) in 10 μM | IC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|---|
| 1 | 23.5 ± 1.0 | — | — | |
| 2 | 27.1 ± 3.1 | — | — | |
| 3 | 18.7 ± 6.1 | — | — | |
| 4 | 27.1 ± 4.6 | — | — | |
| 5 | 90.8 ± 2.1 | 5.6 ± 0.7 | 46.2 ± 2.0 | 8.3 |
| 6 | 16.4 ± 1.5 | — | — | |
| 7 | 74.7 ± 7.1 | — | 12.2 ± 1.2 | |
| 8 | 65.5 ± 3.1 | — | 10.5 ± 3.4 | |
| 9 | 49.6 ± 5.4 | — | 13.2 ± 2.5 | |
| 10 | 40.2 ± 7.4 | — | — | |
| 11 | 97.2 ± 1.8 | 6.9 ± 1.1 | >100 | >14.5 |
| 12 | 15.0 ± 4.8 | — | — | |
| 13 | 49.9 ± 1.0 | — | — | |
| Ribavirin | 90.3 ± 5.2 (50 μM) | 28.2 ± 4.4 | >100 | >3.5 |
Conclusions
In conclusion, thirteen secondary metabolites including three new chromanone dimers, paecilins F–H (1–3) were isolated from the mutants of Penicillium oxalicum 114-2. Their structures were elucidated by detailed interpretation of the spectroscopic data, and the stereochemistry of 1–3 were established by computational predictions of NMR chemical shifts and calculated ECD spectra. Compounds 5 and 11 displayed significant anti-IAV activities with IC50 values of 5.6 and 6.9 μM, respectively, which were stronger than the positive control, ribavirin (IC50 = 28.2 μM), indicating that 5 and 11 might be promising natural agents for antiviral drug discovery.Experimental
General experimental procedures
Optical rotations were recorded on an Autopol IV automatic polarimeter with a 10 cm glass microcell at 25 °C (Rudolph Research Analytical, NJ, USA). The CD spectra were measured on a JASCO J-815 spectropolarimeter using CH3OH as the solvent at room temperature (Jasco Corporation, Tokyo, Japan). UV spectra were obtained in CH3OH on a Persee TU-1901 UV-vis spectrometer (Beijing Purkinje General Instrument Co., Ltd, Beijing, China). IR spectra were acquired on a PerkinElmer FT-IR/NIR spectrometer (PerkinElmer, Waltham, MA, USA). 1D and 2D NMR spectra were performed at 600 MHz for 1H NMR and 150 MHz for 13C NMR on Bruker ARX-600 spectrometer using solvent signals as the internal standard (Bruker, Switzerland). Chemical shifts (δ) are given in ppm, and coupling constants (J) are given in hertz (Hz). ESIMS data were recorded on a Thermo LTQ mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). HRESIMS data were measured using a Thermo LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Column chromatography (CC) were carried out with silica gel (200–300 mesh, Qingdao Marine Chemical Inc. Qingdao, PR China). Analytical TLC was carried out on pre-coated silica gel GF254 plates (Qingdao Marine Chemical Industry, Qingdao, China), and spots were visualized under UV light or by spraying with 10% H2SO4 in 90% EtOH followed by heating at 120 °C.Fungal material
The wild type strain P. oxalicum 114-2 was kindly provided by Prof Qu Yinbo from National Glycoengineering Research Center, Shandong University.40 The oxalicine B deletion mutants derived from parental strain 114-2 including ΔoxaB, ΔoxaL, and ΔoxaK were obtained during our research on biosynthesis of oxalicine B. All the strains used in this study are deposited in the China Pharmaceutical Culture Collection (CPCC), Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College.Fermentation, extraction and isolation
The fungal strain was spread onto slants of PDA medium (potato starch 0.4%, dextrose 2.0%, agar 1.5%; the media were autoclaved at 121 °C for 15 min), and incubated at 28 °C for 5–7 days to generate spores. The spores were inoculated in PDA plate medium at 28 °C for 5 days to prepare spore suspension (seed culture). Fermentation was carried out on plate, each plate contained about 50 ml medium (2% malt extract broth, 2% agar and 0.2% soybean meal, autoclaving at 121 °C for 30 min). After cooling to solid medium, each plate was added into 200 μL of the spore suspension, coating evenly, and incubated at 28 °C for 7 days.The fermented material was extracted repeatedly with EtOAc (3 × 3 L), and the organic solvent was evaporated to dryness under vacuum to yield the crude extract, which was initially subjected to silica gel (Qingdao Marine Chemical Inc. Qingdao, China) column chromatography (CC) eluting with dichloromethane–acetone gradient (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:100, v/v) to produce six fractions (Fr.1–Fr.6) based on the HPLC analysis. Fr.2–Fr.4 and Fr.6 were subjected to reversed-phase silica gel column chromatography eluting with CH3CN–H2O gradient (10:90–100:0, v/v), respectively. Fr.2.2 (30 mg) was subjected to Sephadex LH-20 CC to give compound 11 (3 mg). Fr.2.4 (16 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) (45:55) at 4 ml min−1 to yield 13 (2.0 mg, tR = 8.5 min) and 12 (3.0 mg, tR = 18 min). Fr.3.2 (7 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) (40:60) at 4 ml min−1 to yield 1 (1.2 mg, tR = 22.6 min). Fr.3.4 (40 mg) was subjected to Sephadex LH-20 CC to produce four fractions (Fr.3.4.1–Fr.3.4.4). Fr.3.4.2 (9 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) (40:60, v/v) at 4 ml min−1 to obtain 2 (1.4 mg, tR = 33.4 min). Fr.3.4.3 (22 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (40:60–100:0, v/v) at 4 ml min−1 to obtain 3 (4 mg, tR = 20.9 min). Fr.4.2 (46 mg) was subjected to Sephadex LH-20 CC to produce four fractions (Fr.4.2.1–Fr.4.2.4). Fr.4.2.1 (15 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (50:50, v/v) at 4 ml min−1 to obtain 5 (8 mg, tR = 17.0 min). Fr.4.2.3 (10 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (45:55–100:0, v/v) at 4 ml min−1 to obtain 10 (3.5 mg, tR = 18.3 min). Fr.4.2.4 (11 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (45:55–100:0, v/v) at 4 ml min−1 to obtain 4 (1.5 mg, tR = 17.0 min). Fr.4.3 (14 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (45:55, v/v) at 4 ml min−1 to obtain 6 (3.3 mg, tR = 32.4 min). Fr.5 subjected to recrystallization to obtain 7 (1.1 g). Fr.6.2 (380 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (50:50–100:0 for, v/v) at 4 ml min−1 to obtain 9 (15 mg, tR = 17.9 min) and 8 (26 mg, tR = 18.0 min).
:0–0:100, v/v) to produce six fractions (Fr.1–Fr.6) based on the HPLC analysis. Fr.2–Fr.4 and Fr.6 were subjected to reversed-phase silica gel column chromatography eluting with CH3CN–H2O gradient (10:90–100:0, v/v), respectively. Fr.2.2 (30 mg) was subjected to Sephadex LH-20 CC to give compound 11 (3 mg). Fr.2.4 (16 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) (45:55) at 4 ml min−1 to yield 13 (2.0 mg, tR = 8.5 min) and 12 (3.0 mg, tR = 18 min). Fr.3.2 (7 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) (40:60) at 4 ml min−1 to yield 1 (1.2 mg, tR = 22.6 min). Fr.3.4 (40 mg) was subjected to Sephadex LH-20 CC to produce four fractions (Fr.3.4.1–Fr.3.4.4). Fr.3.4.2 (9 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) (40:60, v/v) at 4 ml min−1 to obtain 2 (1.4 mg, tR = 33.4 min). Fr.3.4.3 (22 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (40:60–100:0, v/v) at 4 ml min−1 to obtain 3 (4 mg, tR = 20.9 min). Fr.4.2 (46 mg) was subjected to Sephadex LH-20 CC to produce four fractions (Fr.4.2.1–Fr.4.2.4). Fr.4.2.1 (15 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (50:50, v/v) at 4 ml min−1 to obtain 5 (8 mg, tR = 17.0 min). Fr.4.2.3 (10 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (45:55–100:0, v/v) at 4 ml min−1 to obtain 10 (3.5 mg, tR = 18.3 min). Fr.4.2.4 (11 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (45:55–100:0, v/v) at 4 ml min−1 to obtain 4 (1.5 mg, tR = 17.0 min). Fr.4.3 (14 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (45:55, v/v) at 4 ml min−1 to obtain 6 (3.3 mg, tR = 32.4 min). Fr.5 subjected to recrystallization to obtain 7 (1.1 g). Fr.6.2 (380 mg) was purified by reversed-phase semi-preparative HPLC eluting with CH3CN–H2O (0.1% TFA) gradient (50:50–100:0 for, v/v) at 4 ml min−1 to obtain 9 (15 mg, tR = 17.9 min) and 8 (26 mg, tR = 18.0 min).
ε): 206 (2.03), 257 (1.46) and 364 (0.49) nm; IR vmax: 3422, 2925, 1783, 1734, 1676, 1624, 1433, 1357, 1202, 1135, 800 and 722 cm−1; CD (MeOH) λmax (Δε): 221.5 (+20.17), 261.5 (−6.98), 313.5 (−2.59) nm; ESIMS m/z: 657.46 [M + H]+; HR-ESIMS m/z: 679.16003 [M + Na]+ (calcd for C32H32NaO15, 679.16334); 1H and 13C NMR data, see Table 1.ε): 206 (1.65), 276 (0.83) and 360 (0.36) nm; IR vmax: 3421, 2923, 1787, 1677, 1646, 1456, 1435, 1203, 1136, 1027, 802 and 724 cm−1; CD (MeOH) λmax (Δε): 214.0 (+12.62), 245.0 (−5.00), 281.5 (−2.48), 382.0 (+0.52) nm; ESIMS m/z: 639.54 [M + H]+; HR-ESIMS m/z: 677.12360 [M + K]+ (calcd for C32H30KO14, 677.12671). 1H and 13C NMR data, see Table 1.ε): 208 (2.26), 254 (1.84) and 360 (0.52) nm; IR vmax: 3420, 2980, 1789, 1741, 1650, 1587, 1463, 1345, 1278, 1228, 1202, 1158, 1025, 998 and 736 cm−1; CD (MeOH) λmax (Δε) 211.5 (−18.83), 246.5 (+15.85), 309.0 (−3.21), 354.5 (+1.17) nm; ESIMS m/z: 639.43 [M + H]+; HR-ESIMS m/z: 639.16766 [M + H]+ (calcd for C32H31O14, 639.17083). 1H and 13CNMR data, see Table 1.ECD calculations
The simplified structures 1A and 3A (Fig. S1 and S5†) were used as the model compounds of 1 and 3, respectively. Conformational analyses of 1A and 3A were performed by using the MMFF94 molecular mechanics force field. According to the relative energy within 6 kcal mol−1, the molecules of 1Aa, 1Ab, and 3Aa showed 9 and 7 conformers with distributions higher than 1%, respectively (Fig. S2 and S6†). The resultant conformers were further optimized and checked as the true minima of potential energy surface by the density functional theory method at the B3LYP/6-31G(d) level, and 30 lowest electronic transitions were calculated. ECD spectra of different conformers were simulated using a Gaussian function with a half-bandwidth of 0.4 eV. The overall theoretical ECD spectra were given on the basis of the Boltzmann weighting of each conformers. In the 200–400 nm region, the theoretically calculated ECD and UV spectra of 1A and 3A compared with the experimental ECD data of 1 and 3, which led the assignment of the absolute configurations of C-10a and C-10a′ for 1 and 3.27Quantum chemical calculations
All theoretical calculations were carried out using the Gaussian 09 program package. Conformation search by MMFF94 molecular force fields with 6 kcal mol−1 energy window limit was performed for all possible isomers, providing corresponding stable conformers with distributions higher than 1%. The conformers were optimized using the semiempirical PM6 method available in Gaussian 09 software package. The resulting geometries were further optimized at the HF/6-31G(d) and B3LYP/6-31G9d levels of theory in the gas phase. The predominant conformers were subjected to theoretical NMR calculations at the mPW1PW91/6-311G(2d,p) level of theory. The Boltzmann distributions were performed using the Gibbs free energies as weighting factors. The linear regression approach was used to convert NMR shielding tensors into NMR chemical shifts, and the NMR chemical shifts were considered as the average values of the same atoms in the different conformers. The theoretical NMR data were statistically analyzed with experimental chemical shifts, using DP4+ probability.28,29Anti-IAV activity assays
All the metabolites were evaluated for their anti-IAV activities as previously described methods.41Cytotoxic activity assays
All the metabolites were tested for their cytotoxic activities as previously reported methods.42Antibacterial activity assays
All the metabolites were tested for their antibacterial activities as previously described methods.41Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (82073744, 31872617), CAMS Innovation Fund for Medical Sciences (2021-I2M-1-055, 2019-I2M-1-005), the National Mega-project for Innovative Drugs (2019ZX09721001-004-006), and the National Microbial Resource Center (NMRC-2021-3). We thank the CAMS Collection Center of Pathogenic Microorganisms (CAMS-CCPM-A) for providing valuable reagents. We are grateful to Prof. Qu Yinbo (National Glycoengineering Research Center, Shandong University, Jinan, China) for sharing the strain P. oxalicium 114-2.Notes and references
- S. Chen, R. Cai, Z. Liu, H. Cui and Z. She, Nat. Prod. Rep., 2022, 39, 560–595 RSC.
- T. El-Elimat, H. A. Raja, M. Figueroa, A. H. Al Sharie, R. L. Bunch and N. H. Oberlies, J. Nat. Prod., 2021, 84, 898–916 CrossRef CAS.
- A. Schueffler and T. Anke, Nat. Prod. Rep., 2014, 31, 1425–1448 RSC.
- X. Yang, J. Liu, J. Mei, R. Jiang, S. Tu, H. Deng, J. Liu, S. Yang and J. Li, Mini-Rev. Med. Chem., 2021, 21, 2000–2019 CrossRef CAS.
- R. M. K. Toghueo and F. F. Boyom, 3 Biotech., 2020, 10, 107 CrossRef.
- J. Xue, H. Li, P. Wu, L. Xu, Y. Yuan and X. Wei, J. Nat. Prod., 2020, 83, 1480–1487 CrossRef CAS.
- Z. F. Wang, Z. C. Sun, L. Xiao, Y. M. Zhou and F. Y. Du, J. Agric. Food Chem., 2019, 67, 14102–14109 CrossRef CAS PubMed.
- T. Asai, D. Luo, K. Yamashita and Y. Oshima, Org. Lett., 2013, 15, 1020–1023 CrossRef CAS.
- N. P. Ariantari, E. Ancheeva, C. Wang, A. Mándi, T. O. Knedel, T. Kurtán, C. Chaidir, W. E. G. Müller, M. U. Kassack, C. Janiak, G. Daletos and P. Proksch, J. Nat. Prod., 2019, 82, 1412–1423 CrossRef CAS.
- J. W. Tang, L. M. Kong, W. Y. Zu, K. Hu, X. N. Li, B. C. Yan, W. G. Wang, H. D. Sun, Y. Li and P. T. Puno, Org. Lett., 2019, 21, 771–775 CrossRef CAS.
- H. L. Li, R. Xu, X. M. Li, S. Q. Yang, L. H. Meng and B. G. Wang, Org. Lett., 2018, 20, 1465–1468 CrossRef CAS.
- D. Zhang, L. Zhao, L. Wang, X. Fang, J. Zhao, X. Wang, L. Li, H. Liu, Y. Wei, X. You, S. Cen and L. Yu, J. Nat. Prod., 2017, 80, 371–376 CrossRef CAS PubMed.
- L. H. Meng, C. Y. Wang, A. Mándi, X. M. Li, X. Y. Hu, M. U. Kassack, T. Kurtán and B. G. Wang, Org. Lett., 2016, 18, 5304–5307 CrossRef CAS.
- T. Song, M. Chen, Z. W. Ge, W. Chai, X. C. Li, Z. Zhang and X. Y. Lian, J. Org. Chem., 2018, 83, 13395–13401 CrossRef CAS.
- Y. Zhang, J. Bai, L. Zhang, C. Zhang, B. Liu and Y. Hu, Angew. Chem., Int. Ed. Engl., 2021, 60, 6639–6645 CrossRef CAS.
- L. H. Meng, X. M. Li, C. T. Lv, C. S. Li, G. M. Xu, C. G. Huang and B. G. Wang, J. Nat. Prod., 2013, 76, 2145–2149 CrossRef CAS.
- M. Okabe, T. Sugita, K. Kinoshita and K. Koyama, J. Nat. Prod., 2016, 79, 1208–1212 CrossRef CAS.
- G. Wu, X. Qi, X. Mo, G. Yu, Q. Wang, T. Zhu, Q. Gu, M. Liu, J. Li and D. Li, Eur. J. Med. Chem., 2018, 148, 268–278 CrossRef CAS PubMed.
- G. Wu, G. Yu, T. Kurtán, A. Mándi, J. Peng, X. Mo, M. Liu, H. Li, X. Sun, J. Li, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2015, 78, 2691–2698 CrossRef CAS PubMed.
- A. Maha, V. Rukachaisirikul, S. Phongpaichit, W. Poonsuwan and J. Sakayaroj, Tetrahedron, 2016, 72, 2874–2879 CrossRef CAS.
- J. Bao, Y. L. Sun, X. Y. Zhang, Z. Han, H. C. Gao, F. He, P. Y. Qian and S. H. Qi, J. Antibiot., 2013, 66, 219–223 CrossRef CAS.
- L. Chen, Y. X. Bi, Y. P. Li, X. X. Li, Q. Y. Liu, M. G. Ying, Q. H. Zheng, L. Du and Q. Q. Zhang, Heterocycles, 2017, 94, 1766–1774 CrossRef CAS.
- T. El-Elimat, M. Figueroa, H. A. Raja, T. N. Graf, S. M. Swanson, J. O. Falkinham 3rd, M. C. Wani, C. J. Pearce and N. H. Oberlies, Eur. J. Org. Chem., 2015, 2015, 109–121 CrossRef CAS PubMed.
- H. Kikuchi, M. Isobe, M. Sekiya, Y. Abe, T. Hoshikawa, K. Ueda, S. Kurata, Y. Katou and Y. Oshima, Org. Lett., 2011, 13, 4624–4627 CrossRef CAS PubMed.
- H. Kikuchi, M. Isobe, S. Kurata, Y. Katou and Y. Oshima, Tetrahedron, 2012, 68, 6218–6223 CrossRef CAS.
- P. H. F. da Silva, M. P. de Souza, E. A. Bianco, S. R. S. da Silva, L. N. Soares, E. V. Costa, F. M. A. da Silva, A. Barison, M. R. Forim, Q. B. Cass, A. D. L. de Souza, H. H. F. Koolen and A. Q. L. de Souza, J. Braz. Chem. Soc., 2017, 29, 622–630 Search PubMed.
- D. Zhang, X. Tao, R. Chen, J. Liu, L. Li, X. Fang, L. Yu and J. Dai, Org. Lett., 2015, 17, 4304–4307 CrossRef CAS PubMed.
- S. G. Smith and J. M. Goodman, J. Org. Chem., 2009, 74, 4597–4607 CrossRef CAS PubMed.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS.
- Z. Guo, Z. She, C. Shao, L. Wen, F. Liu, Z. Zheng and Y. Lin, Magn. Reson. Chem., 2007, 45, 777–780 CrossRef CAS.
- L. F. Tietze, L. Ma, S. Jackenkroll, J. R. Reiner, J. Hierold, B. Gnanaprakasam and S. Heidemann, Heterocycles, 2014, 88, 1101–1119 CrossRef CAS.
- G. Valdomir, S. Senthilkumar, D. Ganapathy, Y. Zhang and L. F. Tietze, Chem.–Asian J., 2018, 13, 1888–1891 CrossRef CAS PubMed.
- D. Kumla, T. S. Aung, S. Buttachon, T. Dethoup, L. Gales, J. A. Pereira, Â. Inácio, P. M. Costa, M. Lee, N. Sekeroglu, A. M. S. Silva, M. M. M. Pinto and A. Kijjoa, Mar. Drugs, 2017, 15, 375 CrossRef PubMed.
- H. Ren, L. Tian, Q. Gu and W. Zhu, Arch. Pharmacal Res., 2006, 29, 59–63 CrossRef CAS.
- T. Qin, T. Iwata, T. T. Ransom, J. A. Beutler and J. A. Porco Jr, J. Am. Chem. Soc., 2015, 137, 15225–15233 CrossRef CAS.
- L. Chen, Y. P. Li, X. X. Li, Z. H. Lu, Q. H. Zheng and Q. Y. Liu, J. Antibiot., 2019, 72, 34–44 CrossRef CAS PubMed.
- B. Liu, H. F. Wang, L. H. Zhang, F. Liu, F. J. He, J. Bai, H. M. Hua, G. Chen and Y. H. Pei, Chem. Nat. Compd., 2016, 52, 821–823 CrossRef CAS.
- K. A. Alvi, B. Nair, C. Gallo and D. Baker, J. Antibiot., 1997, 50, 264–266 CrossRef CAS.
- J. L. Liang, H. C. Cha, S. H. Lee, J. K. Son, H. W. Chang, J. E. Eom, Y. Kwon and Y. Jahng, Arch. Pharmacal Res., 2012, 35, 447–454 CrossRef CAS.
- G. Yao, R. Wu, Q. Kan, L. Gao, M. Liu, P. Yang, J. Du, Z. Li and Y. Qu, Biotechnol. Biofuels, 2016, 9, 78 CrossRef.
- D. Zhang, G. Gu, B. Zhang, Y. Wang, J. Bai, Y. Fang, T. Zhang, S. Dai, S. Cen and L. Yu, RSC Adv., 2021, 11, 22489–22494 RSC.
- D. Zhang, X. Tao, G. Gu, Y. Wang, W. Zhao, W. Zhao, Y. Ren, S. Dai and L. Yu, Front. Microbiol., 2021, 12, 662321 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESIMS, IR, UV, ECD, 1D and 2D NMR for 1–3; cytotoxic and antibacterial activities of 1–13. See https://doi.org/10.1039/d2ra02639b |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |