
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrazolone-type compounds: synthesis and in silico assessment of antiviral potential against key viral proteins of SARS-CoV-2†
Jovica Brankovića,
Vesna M. Milovanović b,
Dušica Simijonovićc,
Slađana Novakovićd,
Zorica D. Petrovića,
Snežana S. Trifunoviće,
Goran A. Bogdanovićd and
Vladimir P. Petrović*a
b,
Dušica Simijonovićc,
Slađana Novakovićd,
Zorica D. Petrovića,
Snežana S. Trifunoviće,
Goran A. Bogdanovićd and
Vladimir P. Petrović*a
aUniversity of Kragujevac, Faculty of Science, Department of Chemistry, R. Domanovića 12, 34000 Kragujevac, Serbia. E-mail: vladimir.petrovic@pmf.kg.ac.rs
bUniversity of Kragujevac, Faculty of Agronomy, Department of Chemistry and Chemical Engineering, Cara Dušana 34, 32000 Čačak, Serbia
cUniversity of Kragujevac, Institute for Information Technologies Kragujevac, Department of Science, Jovana Cvijića bb, 34000 Kragujevac, Serbia
dUniversity of Belgrade, “VINCA” Institute of Nuclear Sciences-National Institute of the Republic of Serbia, Department of Theoretical Physics and Condensed Matter Physics, 11001 Belgrade, Serbia
eUniversity of Belgrade, Faculty of Chemistry, Studentski trg 12–16, 11000 Belgrade, Serbia
First published on 27th May 2022
Abstract
Coronavirus outbreak is still a major public health concern. The high mutation ability of SARS-CoV-2 periodically delivers more transmissible and dangerous variants. Hence, the necessity for an efficient and inexpensive antiviral agent is urgent. In this work, pyrazolone-type compounds were synthesised, characterised using spectroscopic methods and theoretical tools, and evaluated in silico against proteins of SARS-CoV-2 responsible for host cell entry and reproduction processes, i.e., spike protein (S), Mpro, and PLpro. Five of twenty compounds are newly synthesised. In addition, the crystal structure of a pyrazolone derivative bearing a vanillin moiety is determined. The obtained in silico results indicate a more favourable binding affinity of pyrazolone analogues towards Mpro, and PLpro in comparison to drugs lopinavir, remdesivir, chloroquine, and favipiravir, while in the case of S protein only lopinavir exerted higher binding affinity. Also, the investigations were performed on ACE2 and the spike RBD-ACE2 complex. The obtained results for these proteins suggest that selected compounds could express antiviral properties by blocking the binding to the host cell and viral spreading, also. Moreover, several derivatives expressed multitarget antiviral action, blocking both binding and reproduction processes. Additionally, in silico ADME/T calculations predicted favourable features of the synthesised compounds, i.e., drug-likeness, oral bioavailability, as well as good pharmacokinetic parameters related to absorption, metabolism, and toxicity. The obtained results imply the great potential of synthesised pyrazolones as multitarget agents against SARS-CoV-2 and represent a valuable background for further in vitro investigations.
Introduction
The viral strain SARS-CoV-2 causing severe acute respiratory disease remains a major public health concern. According to the statistical data acquired from World Health Organization (WHO), up to this point, a total of 520 million cases of COVID-19 have been confirmed, including approximately 6.3 million deaths reported worldwide. New viral forms are periodically emerging, which are feared to be more contagious, transmissible, and dangerous than previous strains. The WHO and U.S. Centers for Disease Control and Prevention (CDC) declared the Delta (B.1.617.2) and Omicron (B.1.1.529) strains as “variants of concern”. The risk for hospitalisation was doubled for the patients infected with delta variant, in comparison with those infected with alpha.1 Furthermore, the omicron variant possesses the ability to evade immunity from prior infection.2,3 Recent findings indicate that omicron can dodge the neutralizing antibodies in vaccinated individuals.4–7 Enormous efforts were made in vaccine development to repress the coronavirus outbreak. Even with more than 11.6 billion vaccine doses administered globally, the COVID-19 situation is still complicated. As an aggravating circumstance, the SARS-CoV-2 high mutation ability could influence vaccine effectiveness.8 Regarding this, the potent antiviral compound may overcome this challenge and contribute to the fight against coronavirus.8 Different strategies were employed to face the COVID-19 emergency, including the prevention of synthesis of viral RNA.9 The SARS-CoV-2 comprises two overlapping polyproteins (pp1a and pp1ab) encoded with ∼30 kb RNA genome, which cleavage is essential for replication and transcription processes.10–13 These cleavage processes are regulated by non-structural viral proteins, such as the main protease Mpro (also known as 3-chymotrypsin-like protease 3CLpro) and papain-like protease PLpro.8–11 The Mpro protein is a homodimer structure, where the substrate-binding site consists of five sub-pockets responsible for the proteolytic activity through a multi-step mechanism, involving an uncommon Cys145–His41 catalytical dyad with the help of a water molecule.8,9 Compound interacting with catalytic amino acid residues of these subpockets can inhibit the proteolytic action of SARS-CoV-2 main protease.12 Furthermore, the Mpro divides the polypeptide chain after Gln residue, unlike all known human proteases.14 Hence, these atypical features and the engagement in the viral lifecycle designated Mpro as an attractive antiviral target.10–12,15 Similarly, the cysteine protease (PLpro) is engaged in multiple processes linked with viral maturation and spread, as well as in mechanisms of evasion host antiviral immune response.16,17 On the other hand, inhibition of the receptor-binding processes and blocking the entry into the host cell are also part of antiviral strategies. SARS-CoV-2 invades human cells via interaction of the homotrimeric transmembrane spike-shaped (S) glycoprotein located on the virion surface with extracellular domains of the host angiotensin-converting enzyme 2 (ACE2) receptor.18 ACE2 is linked with a variety of physiological functions and is extensively expressed in the lungs, cardiovascular system, gut, kidneys, central nervous system, and adipose tissue.19 Two functional subunits of spike protein, S1 and S2, empower the entry of the viral cell. The receptor-binding domain (RBD) is located in the S1 subunit, whereas the S2 is involved in the membrane fusion processes.20 Furthermore, SARS-CoV-2 possesses the ability to facilitate its cell entry by exploiting host cell proteases, such as cathepsin, elastase, furin, and transmembrane protease serine 2.19 Such unusual behaviour of the SARS-CoV-2 virus triggered the various approaches in the search for potential antiviral candidates, i.e. computational screening of existing drugs, drug repurposing, and in silico design of new potential inhibitors of viral key proteins.21–24 Molecular docking is also identified as a cost-effective and less time-consuming method for the search of promising antiviral candidates, particularly against SARS-CoV-2.8,9,12,23,25–27 Such molecular docking analysis identified various potential compounds that can interact with Mpro and S proteins of virus SARS-CoV-2, including pyrazolone-type compounds.28,29 Moreover, pyrazolone-based compounds were investigated on the SARS-CoV and MERS-CoV proteases and designated as a good base for the development of antiviral agents.30,31 Pyrazolone structural motif is frequently utilised for the development of novel hybrid molecules with various biological activities,32 including antiviral,33 antioxidant,34 antimicrobial,35 analgesic,36 anti-inflammatory,37 cytotoxic,38 and many other activities. Furthermore, these compounds express inhibitory activity on many enzymes, such as cyclooxygenase,39 phosphodiesterase,40 carboxylesterase,41 and α-glucosidase,42 which also illustrate the versatility of pyrazolone pharmacophore. Their diverse biological activities encouraged us to synthesise a series of pyrazolone analogues with potential anti-SARS-CoV-2 activity. Molecular docking analyses were performed with viral S, Mpro, and PLpro proteins to get insight into the binding affinity with pyrazolone derivatives. On the other hand, the binding of some compound to the RBD-ACE2 complex could block the virus spread.43 Additionally, some pyrazolones were identified as potent inhibitors of angiotensin-converting enzyme (ACE), which has a number of pathological roles.44 This prompted us to investigate the binding of selected pyrazolones to human ACE2 cell receptors, as well as to the spike RBD-ACE2 complex, with the idea that in such case, the SARS-CoV-2 cell entry and spreading could also be prevented.Results and discussion
Synthesis of pyrazolone derivatives
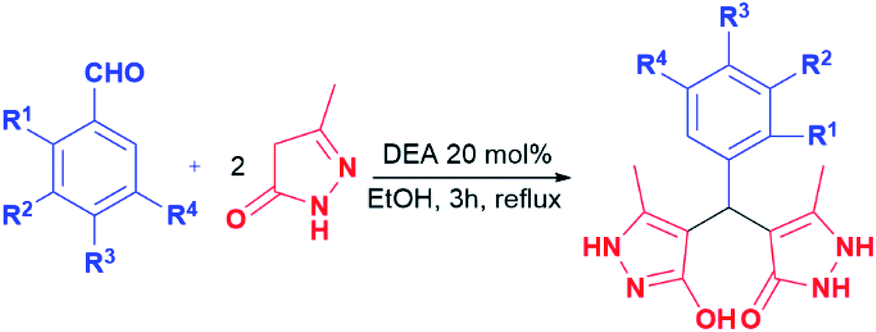
Generally, the literature on these particular types of compounds is relatively scarce, and the research is mainly focused on the development of novel eco-friendly synthetic methods employing various catalysts, mostly ionic liquids and nanoparticles.45–52 Following such a trend, the synthesis of pyrazolone analogues a–t was accomplished in the reactions of 5-methyl-2,4-dihydro-3H-pyrazol-3-one with different aromatic aldehydes in the presence of diethanolamine as a catalyst, and in ethanol and under reflux. To optimise reaction conditions, the model reaction between benzaldehyde and 5-methyl-2,4-dihydro-3H-pyrazol-3-one was investigated in the presence of various catalysts and solvents (Table 1).| Model reaction 1 | |||
|---|---|---|---|
|
|||
| Reaction conditions and product yields | |||
| H2O | EtOH | H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (1:1) :EtOH (1:1) |
|
| a Reaction conditions: aromatic aldehyde (1 mmol), pyrazolone (2 mmol), catalyst 20 mol%, solvent 3.0 mL, reflux 3 h. | |||
| No catalyst | 43% | 67% | 68% |
| [HDEA][OAc] | 54% | 77% | 68% |
| DEA | 73% | 82% | 65% |
| [HTEA][OAc] | 70% | 73% | 70% |
| TEA | 77% | 77% | 77% |
| Model reaction 2 | ||||
|---|---|---|---|---|
|
||||
| Reaction conditions and product yields | ||||
| R3 | [HDEA][OAc] | DEA | TEA | |
| OH | EtOH | 69% | 72% | 53% |
| Cl | 58% | 76% | 76% | |
In this model reaction, diethanolamine (DEA), triethanolamine (TEA), and their corresponding acetate salts ([HDEA][OAc] and [HTEA][OAc]) were tested as catalysts, while as solvents water, ethanol, and a mixture of water and ethanol were used. The selection of the corresponding ethanolamine catalysts was based on their known environmental behaviour, i.e., ready biodegradation and low bioaccumulation potential, cost-effectiveness, as well as on their extensive application in industry and common consumer products.53–55 The reaction progress was monitored using thin-layer chromatography. The optimal time for conversion of starting material in all cases was 3 h. It is worth pointing out, that formation of the product was observed in all tested reactions. However, the results presented in Table 1 pointed out DEA as the most efficient among all tested catalysts, where the best yields were achieved in ethanol as solvent. Furthermore, the presence of groups with electron-donating and the electron-withdrawal effect was considered in the catalyst selection. For that purpose, aromatic aldehydes bearing the –OH and –Cl groups were used, Table 1. The obtained results revealed that the best yields were achieved in the presence of DEA, as well. Based on the obtained results, the optimal conditions for performing the reactions were 20 mol% of DEA as a catalyst, ethanol as solvent, and reflux for 3 h. These reaction conditions were used for the synthesis of pyrazolone derivatives a–t. It is important to emphasise that five of twenty analogues (m, o, q, r, and t) are newly synthesised compounds. To our knowledge, these new compounds were not obtained by previously reported methods, while the other compounds are known.50,56–60 The products were obtained from good to excellent yield, except derivatives m, q, and r which were isolated in 31–45% yields (Table 2).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Yield (%) |
| a | H | H | H | H | 82 |
| b | OH | H | H | H | 64 |
| c | H | OH | H | H | 72 |
| d | H | H | OH | H | 72 |
| e | H | Cl | H | H | 72 |
| f | H | H | Cl | H | 76 |
| g | H | H | F | H | 70 |
| h | NO2 | H | H | H | 72 |
| i | H | NO2 | H | H | 78 |
| j | H | H | NO2 | H | 85 |
| k | CH3 | H | H | H | 91 |
| l | H | CH3 | H | H | 70 |
| m | OH | OH | H | H | 45 |
| n | H | OH | OH | H | 71 |
| o | OH | OCH3 | H | H | 61 |
| p | H | OCH3 | OH | H | 85 |
| q | H | OCH3 | OH | OCH3 | 31 |
| r | H | OH | OCH3 | OCH3 | 31 |
| s | H | OCH3 | OCH3 | OCH3 | 87 |
| t | OH | Cl | H | Cl | 94 |
Characterisation of compounds a–t
All synthesised pyrazolone derivatives were characterised using 1H NMR, 13C NMR, UV-Vis, and FT-IR spectra. Additionally, simulations of IR and UV-Vis spectra were performed to confirm the assignation of experimental bands. All spectra are provided in the ESI.†In the 1H NMR spectra, for all compounds, the signals originating from methyl groups protons were observed around 2.0 ppm as sharp singlets (Fig. S1–S20†). For the compounds containing methoxy group(s), the peaks that correspond to these protons were noted at around 3.6 ppm. Furthermore, sharp singlets in the 5.1–4.6 ppm region correspond to the proton from the methine group that joints aldehyde and pyrazolone moieties. The signals in the 8.1–6.3 ppm area were assigned to the aromatic protons from the aldehyde moiety. Depending on the structure of the compound, these protons resonated mostly as sharp singlets, doublets, doublets of doublets, or multiplets. On the other hand, the peaks originating from the phenolic –OH proton were mainly noticed in the 9.0–8.0 ppm region, resonating as sharp or broad singlets. Finally, in the 11.4–11.0 ppm region, broad singlets originating from –OH and –NH groups of the pyrazolone rings were observed. The 13C NMR spectra consist of multiple peaks originating from methyl, methoxy, methinic, and aromatic carbons, as well as those from the pyrazolone unit. Generally, the signals at the highest chemical shifts (above 160.0 ppm) correspond to the carbonyl carbon. In the 160.0–103.0 ppm region multiple signals were found, where those at higher chemical shifts correspond to more substituted carbon atoms, from both aldehyde and pyrazolone moieties (i.e., those bearing –OH, –OCH3, etc.). On the other hand, peaks at the lower ppm values in the aforementioned region were assigned to less substituted carbons. For compounds bearing –OCH3 groups, the signals related to methoxy carbon were in the 60.0–55.0 ppm area. Finally, around 10.0 ppm, the signals from the carbon of the methyl group were observed, whereas those from the methine group were noted near 32.0 ppm.
Generally, all IR spectra share a great resemblance, which is indicating the similar structure of all compounds. Moreover, a high agreement between experimental and simulated spectra was achieved (Fig. S24–S28†). Here, the bands located in the 3600–3200 cm−1 region were assigned to the O–H and N–H stretching vibrations. Furthermore, aromatic C–H stretching vibrations were identified somewhat above 3000 cm−1, while the bands originating from aliphatic C–H were observed in the region from 3000 cm−1 to 2800 cm−1. The bands located around 1600 cm−1 originate from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, whereas those near 1530 cm−1 are assigned to the stretching vibration of the CN bond. Also, the bands around 1500 cm−1 and 1450 cm−1 were identified because of bending HCC and CC stretching vibrations, while those in the 1400–1350 cm−1 region were assigned to the bending HNN and HOC. In the 1330–1000 cm−1 multiple bands were identified, such as stretching C–O, CC, and N–N, whereas below 1000 cm−1 the bands originating from torsion HOCC, HNCC, and HCCC vibrations were mainly observed. Also, for analogues bearing –NO2, –F, and –Cl groups, the bands originating from stretching NO, C–F, and C–Cl were identified.
O stretching vibration, whereas those near 1530 cm−1 are assigned to the stretching vibration of the CN bond. Also, the bands around 1500 cm−1 and 1450 cm−1 were identified because of bending HCC and CC stretching vibrations, while those in the 1400–1350 cm−1 region were assigned to the bending HNN and HOC. In the 1330–1000 cm−1 multiple bands were identified, such as stretching C–O, CC, and N–N, whereas below 1000 cm−1 the bands originating from torsion HOCC, HNCC, and HCCC vibrations were mainly observed. Also, for analogues bearing –NO2, –F, and –Cl groups, the bands originating from stretching NO, C–F, and C–Cl were identified.
The experimental UV-Vis spectra were in excellent agreement with simulated ones (Fig. S29–S33†). For the newly synthesised compounds, Kohn–Sham orbitals were constructed to identify the electronic transitions responsible for the appearance of experimental bands (Fig. S34–S38†). Generally, the experimental UV-Vis spectra revealed absorption bands around 202, 230, 250, and 280 nm. The electronic transitions for the newly synthesised compound o are presented in Table 3, while transitions for the other new compounds (m, q, r, and t) are provided in Table S1.† The electronic transitions were arranged in descending order of contribution. For compound o, the absorption band at 282 nm is a consequence of HOMO to LUMO electronic transition. In all cases, the bands around 250 nm mainly originate from HOMO to LUMO and HOMO−1 to LUMO transitions. For compounds m, o, and r, the bands around 230 nm mostly originate from HOMO−3 to LUMO transition. In the case of t, the band at 232 nm was assigned to HOMO−1 to LUMO−1, whereas for q the band at 230 nm mainly corresponds to HOMO−4 to LUMO transition. For compounds m, o, and q, the bands around 202 nm primarily originate from transitions to high-level LUMOs, such as LUMO+4, LUMO+5, LUMO+6, and LUMO+7. On the other hand, for compounds r and t, these bands appeared as a consequence of the electronic transitions from low-level HOMOs (HOMO−4, HOMO−5, and HOMO−6) to LUMO, LUMO+1, and LUMO+2.
| λ (nm) | Transition | |
|---|---|---|
| 282 | HOMO | LUMO |
| 249 | HOMO | LUMO |
| HOMO−1 | LUMO | |
| HOMO | LUMO+1 | |
| 227 | HOMO−3 | LUMO |
| HOMO−1 | LUMO+1 | |
| HOMO−2 | LUMO | |
| HOMO−1 | LUMO+3 | |
| HOMO | LUMO+3 | |
| HOMO−2 | LUMO+1 | |
| 202 | HOMO−2 | LUMO+5 |
| HOMO−2 | LUMO+6 | |
| HOMO−4 | LUMO+1 | |
| HOMO−6 | LUMO | |
| HOMO−1 | LUMO+6 | |
Crystal structure analysis
The crystal structure of compound p is given in Fig. 1, while selected bond lengths and valence angles are listed in Table 4, which are here reported for the first time. To our knowledge, crystal data on these particular compounds are insufficient. Moreover, all articles related to these compounds presented the OH-tautomeric forms of both pyrazolone rings,45–52 including the crystal structure of compound a.57 Single crystal X-ray analysis shows that the compound consists of a substituted phenyl ring and two 3-methyl-5-pyrazolone rings connected through the benzyl C1 atom. The two pyrazolone fragments are present in keto–enol tautomeric forms. The carbonyl acceptor and hydroxy donor resulting from tautomerisation suitably orient to form a strong intramolecular O2–H⋯O1 hydrogen bond which stabilises this crystal structure (Fig. 1). The phenyl ring is nearly symmetrically positioned with respect to the pyrazolone rings. Thus, the dihedral angle between the phenyl and N–H, and the phenyl and O–H pyrazolone ring is 77.26(7) and 70.85(7)°, respectively. The dihedral angle between the pyrazole rings is 47.44(10)°. The bond distances and angles in p are in good agreement with the values of previously reported crystal structures, comprising similar set of rings.61–64 The different position of proton in two pyrazolones is primarily reflected in the lengths of C3–O1 and C7–O2 bonds (Table 4), also the bonds vicinal to protonated N1 atom are slightly longer in comparison to those involving deprotonated N3 atom. Valence angles at benzyl C1 atom notably deviate from the regular tetrahedral geometry (Table 4). The strong intramolecular O2–H⋯O1 hydrogen bond formed between hydroxy and carbonyl residues is a specific feature of this crystal structure (Fig. 1).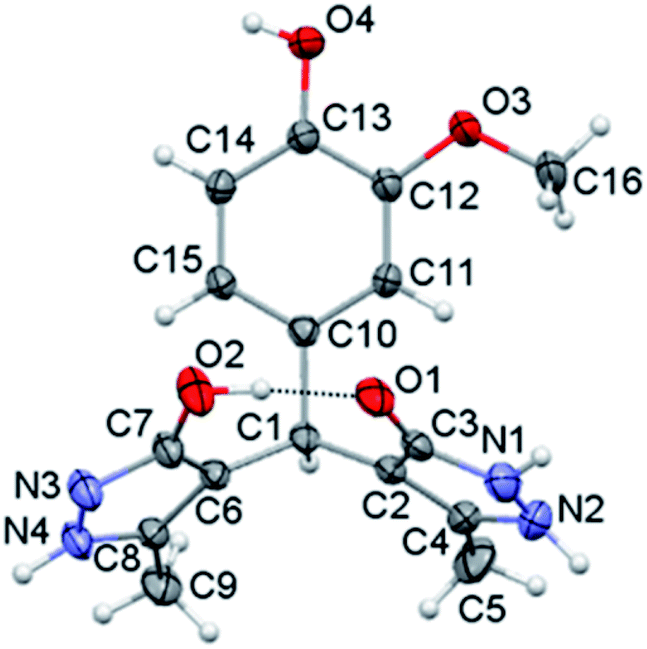 | ||
| Fig. 1 Crystal structure of molecule p. Thermal ellipsoids are shown at the 30% probability level. | ||
| Bond | Angle | ||
|---|---|---|---|
| C3–O1 | 1.267(3) | N1–N2–C4 | 107.6(2) |
| C7–O2 | 1.346(3) | N3–N4–C8 | 112.5(2) |
| N1–C3 | 1.355(3) | C3–N1–N2 | 109.4(2) |
| N2–C4 | 1.358(3) | C7–N3–N4 | 112.5(2) |
| N3–C7 | 1.327(3) | C2–C3–N1 | 106.3(2) |
| N4–C8 | 1.340(3) | C6–C7–N3 | 111.9(2) |
| N1–N2 | 1.371(4) | C1–C2–C3 | 125.9(2) |
| N3–N4 | 1.354(3) | C1–C6–C7 | 129.4(2) |
| C2–C3 | 1.424(3) | C2–C1–C6 | 114.6(2) |
| C2–C4 | 1.364(3) | C10–C1–C6 | 112.6(2) |
| C6–C7 | 1.412(3) | C10–C1–C2 | 111.5(2) |
| C6–C8 | 1.375(3) | C11–C10–C1 | 120.8(2) |
The eight-membered S(8) ring motif65 formed in this way partly locks the orientation of the pyrazolone rings with an almost linear distribution of the donor and acceptor sites. In the crystal packing the molecules arrange by strong intermolecular O–H⋯N and N–H⋯O hydrogen bonds (Table 5).
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) | Symmetry codes |
|---|---|---|---|---|---|
| a Cg1 = C10/C15; Cg2 = N3/C7. | |||||
| O2–H1⋯O1 | 0.94(5) | 1.64(5) | 2.579(3) | 174(4) | x, y, z |
| N4–H1n4⋯O1 | 0.93(3) | 1.93(4) | 2.842(3) | 168(3) | x − 1/2, y, −z + 1/2 |
| N1–H1n1⋯O3 | 0.85(4) | 2.58(3) | 3.164(3) | 127(3) | −x + 1/2, y − 1/2, z − 1/2 |
| N1–H1n1⋯O4 | 0.85(4) | 1.95(4) | 2.777(3) | 164(3) | −x + 1/2, y − 1/2, z − 1/2 |
| O4–H1o4⋯N3 | 0.82(4) | 1.83(4) | 2.648(3) | 169(4) | −x, y + 1/2, −z + 1 |
| C9–H9a⋯Cg1 | 0.96 | 3.05 | 3.769(3) | 132 | x + 1/2, y, −z + 1/2 |
| C16–H16a⋯Cg2 | 0.96 | 3.20 | 3.919(3) | 133 | −x, y + 1/2, −z |
The packing can be described in terms of R33(9) hydrogen bonded trimers which mutually connect to form a complex three-dimensional network (Fig. 2). The three strong hydrogen bonds joined in the trimer, individually define chains extending in a particular direction. Thus, the strongest interaction in the system O4–H⋯N3 arranges the molecules into a zig-zag chain extending along the b crystallographic axis, while the N4–H⋯O1 interaction formed between the pyrazolone rings builds the chain along a crystallographic axis. Finally, the pyrazolone donor N1–H1 interacts with the O⋯O donor system formed by phenyl ring substituents66 and connects the molecules in [011] direction (Fig. 2).
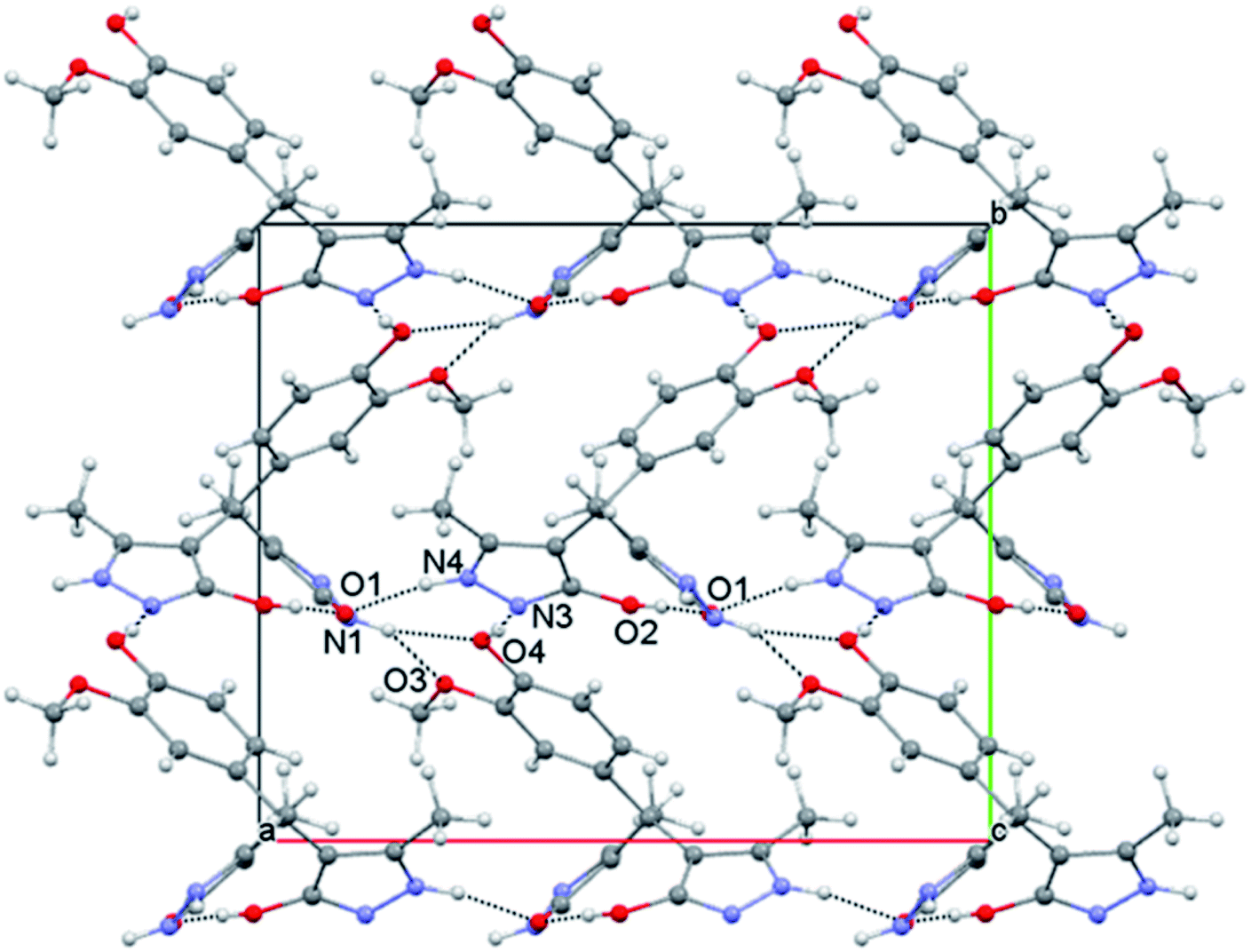 | ||
| Fig. 2 View of the molecular packing with the strongest hydrogen bonds. | ||
In silico inhibitory activity towards SARS-CoV-2 proteins
All pyrazolones a–t were subjected to in silico investigation of potential antiviral activity against SARS-CoV-2. Molecular docking was performed on the protein responsible for attachment of the virus to the host cells (glycoprotein spike protein (S)), as well as on proteins involved in viral reproduction processes (main protease (Mpro), and papain-like protease (PLpro)). Also, docking was conducted on human cell receptor angiotensin-converting enzyme-related carboxypeptidase (ACE2), and on spike receptor-binding domain complexed with its receptor ACE2 (Spike RBD-ACE2). Blind docking was performed for all proteins and all compounds. Such an approach was chosen over site-specific docking to get the full insight. Furthermore, CastP server67 and CHARMM-GUI68 were used for the prediction of active site pockets and respective amino acid residues (Fig. 3). Good agreement between these two predictions was achieved. It is important to emphasise that all compounds were positioned within the predicted pockets of investigated proteins.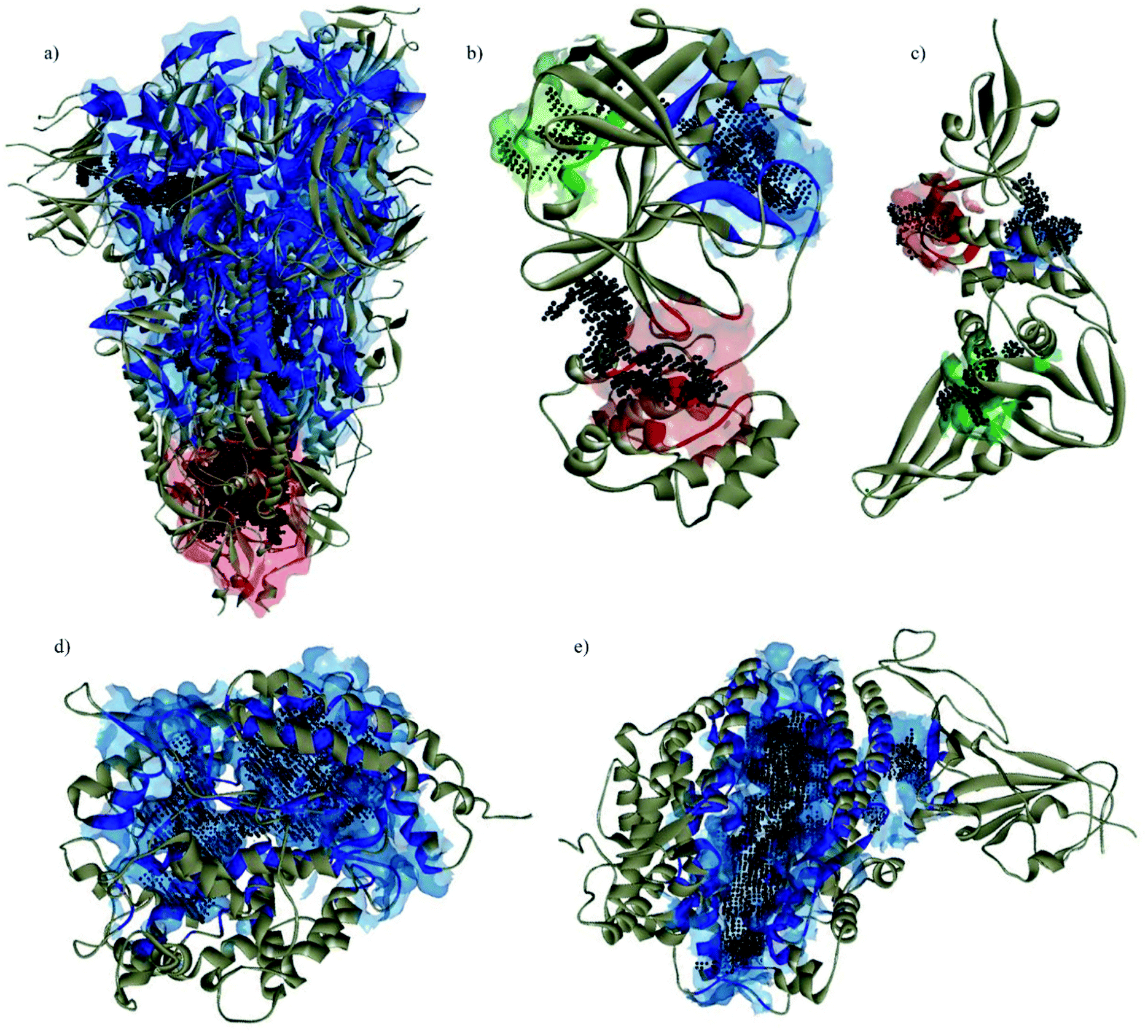 | ||
| Fig. 3 Active site pockets predicted by CastP server (blue, red, and green surfaces) and CHARMM-GUI (black dots) of the investigated proteins: (a) spike; (b) Mpro; (c) PLpro; (d) ACE2; (e) spike RBD-ACE2 complex. | ||
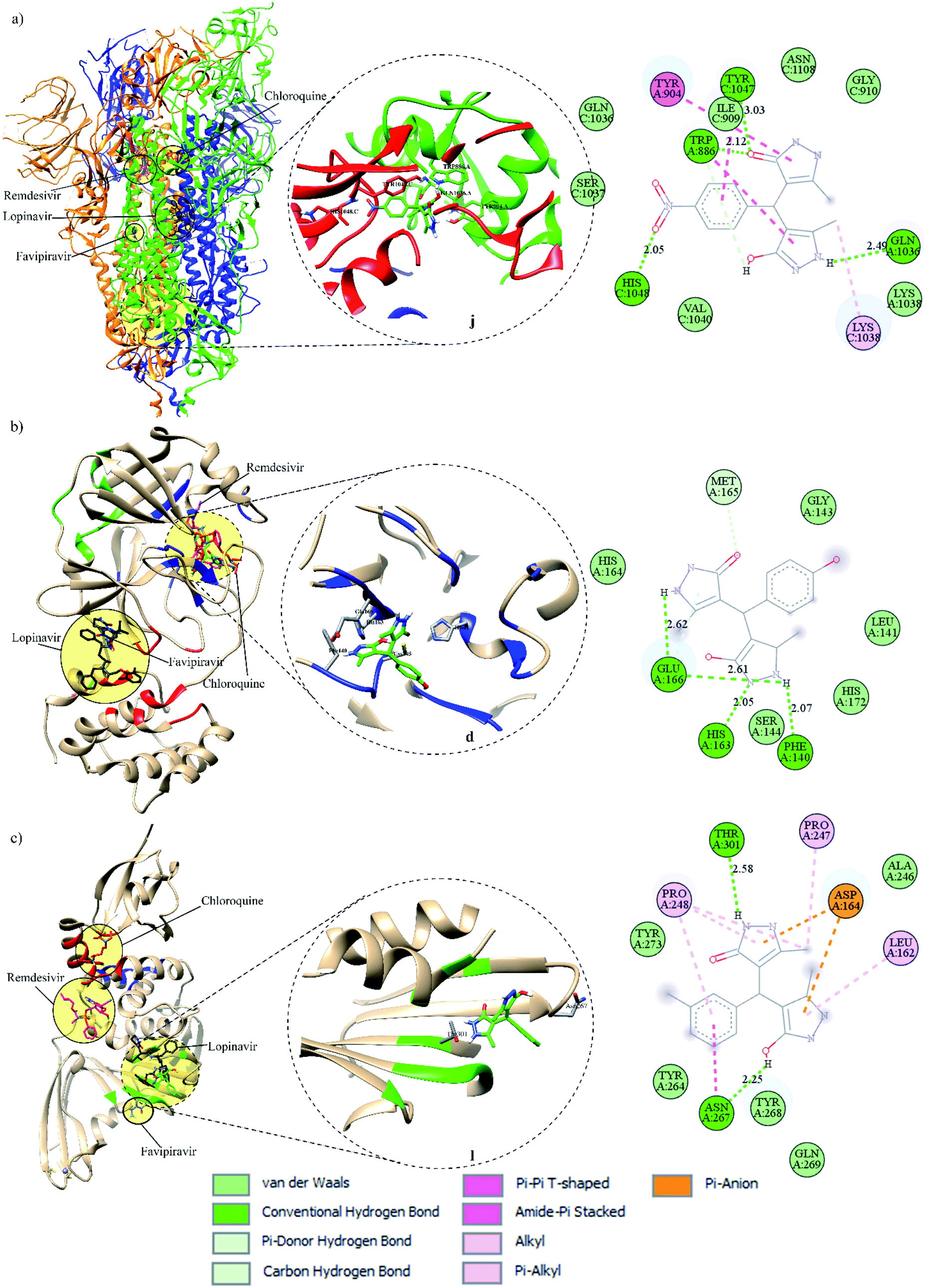 | ||
| Fig. 4 Binding modes of FDA-approved drugs and pyrazolones (left), insight into the binding of the best-screened pyrazolones (middle), and 2D interaction plot of the best-screened compounds (right) to the: (a) spike, (b) Mpro, and (c) PLpro. | ||
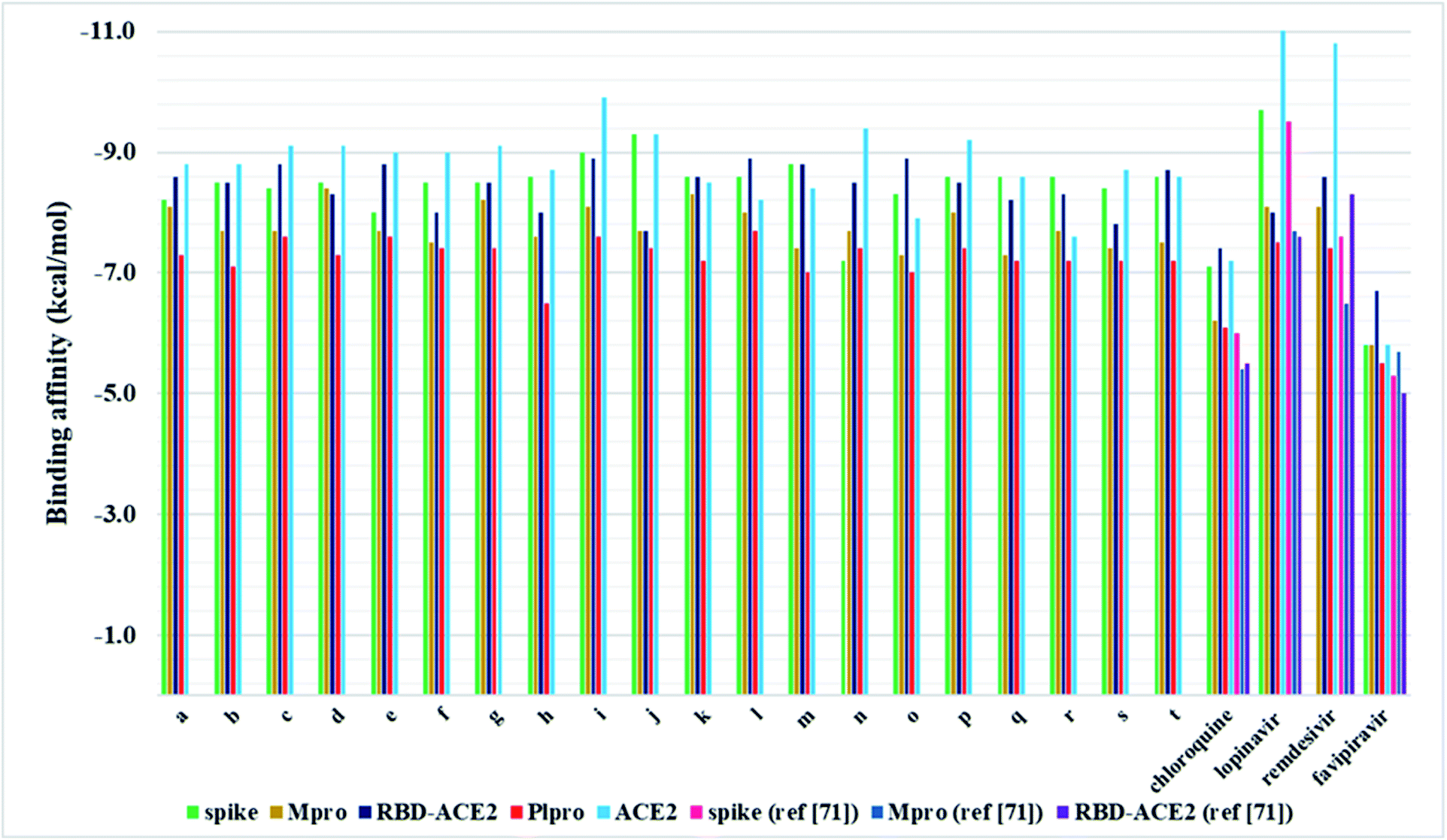 | ||
| Fig. 5 Graphical interpretation of binding affinities for compounds a–t and FDA-approved drugs towards selected proteins. | ||
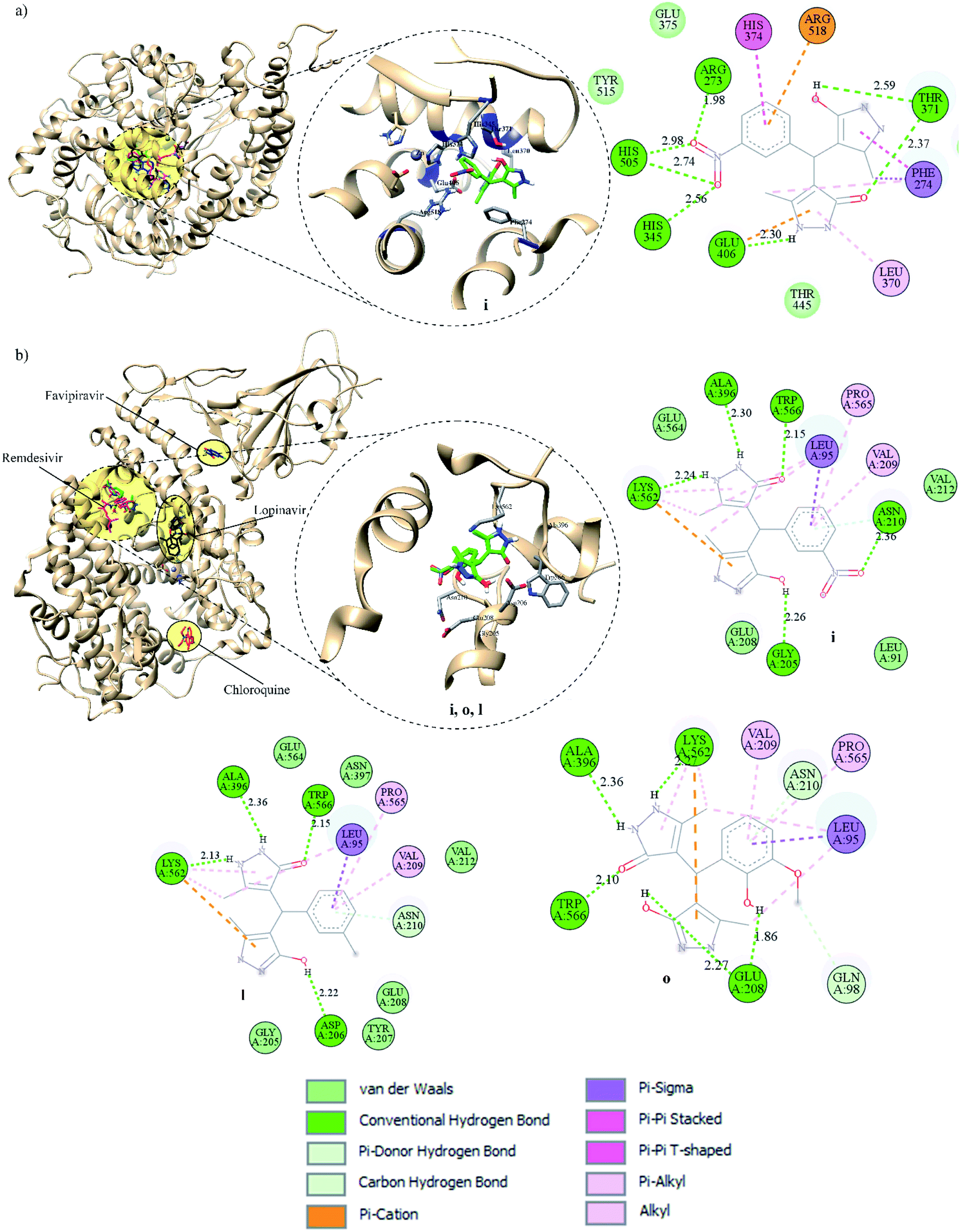 | ||
| Fig. 6 Binding modes of FDA-approved drugs and pyrazolones, insight into the binding of the best-screened pyrazolones, and 2D interaction plot of the best-screened compounds to the (a) ACE2 and (b) Spike RBD-ACE2 complex. | ||
In silico ADME/T profile
Po/w) less than five.72 LogPo/w parameter describes the compound's lipophilicity, which is an important feature for drug absorption. In addition to these rules, Ghose and Veber's criteria are extended to molecular refractivity (40–130 range), number of atoms (from 20–70), number of rotatable bonds (ten or fewer), and polar surface area (no greater than 140 Å2).73,74 The results obtained by SwissADME revealed that basically, all pyrazolones meet these criteria, with some minor violations (Tables S4–S8†). Additionally, most compounds fulfil the Egan and Muegge requirements. Also, the SwissADME oral bioavailability radar, which illustrates the optimal range of each property, showed that pyrazolones generally fulfil the requirements, with some slight saturation and polarity deviations (Fig. S39–S42†).The values of LogPo/w were calculated by different methods (iLOGP, XLOGP3, WLOGP, MLOGP, and Silicos-IT LogP). According to the obtained results, all compounds meet the lipophilicity criteria (Tables S4–S8†). Also, water solubility is an important element for absorption. Three methods (ESOL LogS, Ali LogS, and Silicos-IT LogSw) were used for the calculation of water solubility descriptors. Depending on the calculation method, all pyrazolone analogues were predicted to be soluble or moderately soluble in water (Tables S4–S8†).
The SwissADME Brain Or IntestinaL EstimateD permeation method (BOILED-Egg) predicted high gastrointestinal absorption for almost all compounds (Tables S4–S8†). Similarly, pkCSM prediction of the human intestine absorption percentage for all pyrazolones was in the range of 61.5–76.5% (Tables S9–S28†). Finally, it is important to point out that pkCSM categorised all pyrazolones as P-glycoprotein substrates (Tables S9–S28†). P-glycoprotein (P-gp) is an ATP binding cassette transporter involved in drug uptake and efflux, influencing their plasma and tissue concentrations.75 Here, the substrates of P-gp can act as inhibitors or inducers, where the inhibition of P-gp increases drug bioavailability, while induction reduces it.75 The results obtained by SwissADME differ, i.e., most pyrazolones were not identified as P-gp substrates (Tables S4–S8†).
The blood–brain barrier (BBB) represents a structure that protects the brain from exogenous factors.77 The ability of drugs to permeate BBB is an important parameter for side effects/toxicity considerations.77 Based on the obtained results, both SwissADME and pkCSM predicted that pyrazolones are not able to permeate BBB (Tables S4–S28†). Also, the CNS permeability was predicted according to the log PS values (blood–brain permeability-surface area product). Here, compounds with log PS higher than −2.0 penetrate CNS, whereas those below −3.0 are designated as unable. The pkCSM obtained results for log PS were in the −3.0 to −2.0 range (Tables S9–S28†).
ProTox-II toxicity assessment was related to the prediction of oral toxicity (LD50 and toxicity class), organ toxicity (hepatotoxicity), toxicity endpoints (carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity), as well as Tox21-nuclear receptor signalling and stress response pathways. All compounds were categorised in toxicity class 4 with LD50 values of 800 mg kg−1. Furthermore, all Tox21-nuclear receptor signalling and stress response pathways were observed as inactive in all cases. On the other hand, hepatotoxicity was predicted for all compounds within the class average values, whereas potential carcinogenicity was noted for half of them. Immunotoxicity and cytotoxicity predictions categorised all compounds as inactive, while mutagenicity potential was observed only in a few cases.
Bioactivity predictions
The bioactivity predictions were acquired using SwissTargetPrediction software. Generally, the SwissTargetPrediction charts revealed kinases, proteases, oxidoreductases, and G-protein coupled receptors as the main target classes. Despite the structural similarity, the results differed from compound to compound depending on the present substituents. Bioactivity prediction charts for all compounds are provided in the ESI (Fig. S43–S47†).Conclusions
In the present work, a series of twenty pyrazolone-type compounds a–t were synthesised and characterised by NMR, IR, and UV-Vis spectra. Five of them (m, o, q, r, and t) are newly synthesised compounds and they were additionally characterised by elemental microanalysis and melting points. All spectra were simulated using density functional theory and good agreement was achieved with experimental ones. Additionally, the crystal structure of compound p was determined by single-crystal X-ray diffraction analysis. It turned out that in the analysed pyrazolone, in one ring there is an enol- and in the other ring a keto-form. The compound forms a very strong and directional intramolecular O2–H⋯O1 hydrogen bond resulting from the tautomerisation of pyrazolone rings. The crystal packing is based on strong O–H⋯N and N–H⋯O hydrogen bonds.In silico investigations performed on the spike, Mpro, and PLpro proteins of SARS-CoV-2 revealed higher binding affinity in comparison to selected FDA-approved drugs. Particularly, analogues j and i were identified as the most potent binders of spike protein in comparison to all investigated compounds. Among selected drugs, only lopinavir exerted a slightly higher binding affinity towards spike protein. On the other hand, derivative d expressed the highest in silico inhibitory activity against Mpro in comparison to all other analogues and selected drugs. Pyrazolone l exhibited the highest binding affinity towards PLpro compared to all evaluated compounds and selected drugs. These findings highlighted R2- and R3-substituted pyrazolone analogues as the most potent inhibitors of the SARS-CoV-2 key viral proteins, particularly those bearing –NO2, –OH, and –CH3 groups. Furthermore, the investigations performed on spike RBD-ACE2 complex revealed the highest binding affinity of compound i, whereas for ACE2 the best results were obtained for i, o, and l. These results indicate that pyrazolones could act as potential multitarget antiviral agents, blocking both reproduction and binding of SARS-CoV-2 to human cells.
The obtained in silico ADME/T results revealed good drug-like features, i.e., water-solubility, lipophilicity, and gastrointestinal absorption. Based on the distribution and metabolism predictions, pyrazolones a–t were not categorised as CNS and BBB permeants, nor as inhibitors of crucial cytochrome isoenzymes, which are important features for avoiding drug side effects. Furthermore, toxicity predictions provided generally good results for almost all toxicity parameters.
The obtained results indicate the promising multitarget antiviral potential of the synthesised pyrazolone compounds against SARS-CoV-2 and represent good background for further in vitro experiments.
Experimental section
All chemicals were obtained either from Sigma-Aldrich Co. or Merck & Co. Pyrazolone (5-methyl-2,4-dihydro-3H-pyrazol-3-one) was synthesised from methyl acetoacetate and hydrazine monohydrate employing a common synthetic method. The 1H NMR and 13C NMR spectra were recorded on a Varian Gemini spectrometer (200 MHz for 1H and 50 MHz for 13C) in DMSO-d6 as solvent. The IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer using the KBr plates. The UV-Vis spectra were measured within the 200–600 nm range on the Agilent Technologies, Cary 300 Series UV-Vis Spectrophotometer. Melting points of the newly synthesised compounds were determined on the Melt-Temp capillary melting points apparatus, model 1001. Elemental (C, H, N) microanalysis of new compounds was performed at the University of Belgrade, Faculty of Chemistry.Synthetic procedure
5-Methyl-2,4-dihydro-3H-pyrazol-3-one (1 mmol) was added to the ethanolic solution of the corresponding aromatic aldehyde (0.5 mmol) with a catalytic amount of diethanolamine (20 mol%). The reaction mixture was heated to 80 °C for 3 h. After the completion of the reaction (which was monitored by thin-layer chromatography), the reaction mixture was cooled and the solid product was filtered and washed with minimal amounts of ethanol, water, and ether, respectively. In some cases, the solid products were obtained after cooling the reaction mixture. In this way, most products were obtained in the pure state. For some products, additional purification was achieved by precipitation of the products from ethanol or a mixture of ethanol and water (1:4). Derivative p crystallised from the mixture of ethanol and water (1:2). All products were characterised by 1H NMR, 13C NMR, UV-Vis, and FT-IR spectra, while derivative p was also characterised by X-ray crystallography. Additionally, for the new compounds, elemental microanalysis and melting points were determined. Here, spectral data for new compounds are presented, while for other derivatives are provided in the ESI.†
Single-crystal X-ray diffraction
Single-crystal X-ray diffraction data for compound p were collected on a Bruker APEX-II CCD diffractometer with monochromatised MoKα radiation (λ = 0.71073 Å). Data reduction and empirical absorption correction were performed with the CrysAlisPRO program package.80 The structure was solved by direct methods using SHELXS and refined on F2 by full-matrix least-squares using SHELXL.81 The compound was refined as a two-component inversion twin. H atoms bonded to C atoms were placed in geometrically calculated positions and refined using the riding model with Uiso values constrained to 1.2Ueq or 1.5Ueq of the parent C atoms. The H atoms bonded to O and N atoms were freely refined. Structural analysis was carried out in PLATON82 and Mercury83 which was also used for molecular graphics. Crystallographic details are summarised in Table S2.† X-ray diffraction data of the compound p have been deposited at the Cambridge Crystallographic Data Centre and assigned CCDC 2132092† deposition number.DFT calculations
The Gaussian 09 program package was used to perform all necessary calculations.84 The equilibrium geometries of all pyrazolone analogues were calculated using B3LYP functional in the conjunction with the 6-311+g(d,p) basis set.85–87 To confirm the local minima of the investigated compounds, vibrational analysis was performed (no imaginary frequencies were found). The optimised geometries in the gas phase were used for the simulation of IR spectra (Fig. S21–S23†). IR bands were scaled using the scaling factor obtained using the least-squares method and amount of 0.97. The SMD solvation model implemented in the Gaussian 09 package was utilised to perform calculations in methanol, as well as for the TD-DFT simulation of UV-Vis spectra since it was experimentally used as a solvent.88Molecular docking
The crystal structures of all proteins were acquired from the RSC protein data bank: SARS-CoV-2 spike glycoprotein with an RBD (S) (PDB ID: 6VSB),89 the Mpro in complex with an inhibitor N3 (PDB-ID: 6LU7),10 PLpro (PDB-ID: 6WZU),90 inhibitor bound ACE2 (PDB-ID: 1R4L),91 spike RBD-ACE2 complex (PDB-ID: 6LZG).92 For the preparation of the proteins, as well as for analysis of ligand–macromolecule interactions UCSF Chimera v1.16 software was used, while Discovery Studio Visualizer was used for 2D interpretations.93–95CastP server (https://sts.bioe.uic.edu/castp/)67 and CHARMM-GUI (https://www.charmm-gui.org)68 were used for the prediction of active site pockets and respective amino acid residues. All compounds were subjected to conformational analysis using VeraChem's Vconf 2.0 (VeraChem LLC, Germantown, MA, USA) where the conformations within the threshold of 5 kcal mol−1 were selected for molecular docking. Selected conformations of each compound were subjected to blind docking with the target proteins using AutoDock Vina.96 The grids for the target proteins were set to ensure the whole macromolecules are placed in the corresponding grid box, with a spacing of 1.000 Å. Additionally, selected FDA-approved drugs lopinavir, remdesivir, chloroquine, and favipiravir (PubChem CIDs: 92727, 121304016, 2719, and 492405, respectively) were subjected to docking using the same methodology as described for pyrazolones. The selection was made based on their application in the COVID-19 treatment.97
In silico ADME/T profiling and bioactivity prediction
The insight into physicochemical, pharmacological, toxicological, and drug-like features of pyrazolone derivatives was achieved using SwissADME (https://www.SwissADME.ch/) and pkCSM (https://biosig.unimelb.edu.au/pkCSM/prediction) web tools.77,98 These services were chosen since they provide relevant pharmacokinetic data for small molecule drug candidates (i.e., absorption, distribution, metabolism, excretion, and toxicity).Also, the ProTox-II web service (https://toxnew.charite.de/protox_II/index.php?site=compound_input) was employed for additional evaluation of compounds toxicity.99 The prediction of bioactivity was performed using SwissTargetPrediction. The structures of pyrazolone compounds a–t were converted to simplified molecular-input line entry specification (SMILES) nomenclature and as such imported into selected web services.
Author contributions
Conceptualization, V. P.; methodology, V. P., D. S., and. Z. P.; validation, J. B., and V. M.; formal analysis, V. P, G. B., S. N., and J.B.; investigation, J. B., V. M., D. S., G. B., S. N., and S. T.; resources, V. P., and Z. P.; data curation, J. B. and V. P.; writing—original draft preparation J. B., V. P., and S. N.; writing—review and editing, V. P., Z. P., D. S., and G. B.; visualization, V. P., J. B., and G. B.; supervision, V. P.; project administration, V. P. and Z. P.; funding acquisition, V. P. and Z. P. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Serbian Ministry of Education, Science and Technological Development (Agreement No. 451-03-68/2022-14/200122, 451-03-68/2022-14/200378, 451-03-68/2022-14/200088, and 451-03-68/2022-14/200168).References
- A. Sheikh, J. McMenamin, B. Taylor and C. Robertson, Lancet, 2021, 397, 2461–2462 CrossRef CAS.
- N. Ferguson, A. Ghani, A. Cori, A. Hogan, W. Hinsley and E. Volz, Imp. Coll. COVID-19 Response Team, 2021, pp. 1–10 Search PubMed.
- J. R. C. Pulliam, C. van Schalkwyk, N. Govender, A. von Gottberg, C. Cohen, M. J. Groome, J. Dushoff, K. Mlisana and H. Moultrie, Science, 2022, 376(6593), eabn4947 CrossRef PubMed.
- B. Meng, A. Abdullahi, I. A. T. M. Ferreira, N. Goonawardane, A. Saito, I. Kimura, D. Yamasoba, P. P. Gerber, S. Fatihi, S. Rathore, S. K. Zepeda, G. Papa, S. A. Kemp, T. Ikeda, M. Toyoda, T. S. Tan, J. Kuramochi, S. Mitsunaga, T. Ueno, K. Shirakawa, A. Takaori-Kondo, T. Brevini, D. L. Mallery, O. J. Charles, S. Baker, G. Dougan, C. Hess, N. Kingston, P. J. Lehner, P. A. Lyons, N. J. Matheson, W. H. Ouwehand, C. Saunders, C. Summers, J. E. D. Thaventhiran, M. Toshner, M. P. Weekes, P. Maxwell, A. Shaw, A. Bucke, J. Calder, L. Canna, J. Domingo, A. Elmer, S. Fuller, J. Harris, S. Hewitt, J. Kennet, S. Jose, J. Kourampa, A. Meadows, C. O'Brien, J. Price, C. Publico, R. Rastall, C. Ribeiro, J. Rowlands, V. Ruffolo, H. Tordesillas, B. Bullman, B. J. Dunmore, S. Gräf, J. Hodgson, C. Huang, K. Hunter, E. Jones, E. Legchenko, C. Matara, J. Martin, F. Mescia, C. O'Donnell, L. Pointon, J. Shih, R. Sutcliffe, T. Tilly, C. Treacy, Z. Tong, J. Wood, M. Wylot, A. Betancourt, G. Bower, C. Cossetti, A. De Sa, M. Epping, S. Fawke, N. Gleadall, R. Grenfell, A. Hinch, S. Jackson, I. Jarvis, B. Krishna, F. Nice, O. Omarjee, M. Perera, M. Potts, N. Richoz, V. Romashova, L. Stefanucci, M. Strezlecki, L. Turner, E. M. D. D. De Bie, K. Bunclark, M. Josipovic, M. Mackay, H. Butcher, D. Caputo, M. Chandler, P. Chinnery, D. Clapham-Riley, E. Dewhurst, C. Fernandez, A. Furlong, B. Graves, J. Gray, S. Hein, T. Ivers, E. Le Gresley, R. Linger, M. Kasanicki, R. King, N. Kingston, S. Meloy, A. Moulton, F. Muldoon, N. Ovington, S. Papadia, C. J. Penkett, I. Phelan, V. Ranganath, R. Paraschiv, A. Sage, J. Sambrook, I. Scholtes, K. Schon, H. Stark, K. E. Stirrups, P. Townsend, N. Walker, J. Webster, E. P. Butlertanaka, Y. L. Tanaka, J. Ito, K. Uriu, Y. Kosugi, M. Suganami, A. Oide, M. Yokoyama, M. Chiba, C. Motozono, H. Nasser, R. Shimizu, K. Kitazato, H. Hasebe, T. Irie, S. Nakagawa, J. Wu, M. Takahashi, T. Fukuhara, K. Shimizu, K. Tsushima, H. Kubo, Y. Kazuma, R. Nomura, Y. Horisawa, K. Nagata, Y. Kawai, Y. Yanagida, Y. Tashiro, K. Tokunaga, S. Ozono, R. Kawabata, N. Morizako, K. Sadamasu, H. Asakura, M. Nagashima, K. Yoshimura, P. Cárdenas, E. Muñoz, V. Barragan, S. Márquez, B. Prado-Vivar, M. Becerra-Wong, M. Caravajal, G. Trueba, P. Rojas-Silva, M. Grunauer, B. Gutierrez, J. J. Guadalupe, J. C. Fernández-Cadena, D. Andrade-Molina, M. Baldeon, A. Pinos, J. E. Bowen, A. Joshi, A. C. Walls, L. Jackson, D. Martin, K. G. C. Smith, J. Bradley, J. A. G. Briggs, J. Choi, E. Madissoon, K. B. Meyer, P. Mlcochova, L. Ceron-Gutierrez, R. Doffinger, S. A. Teichmann, A. J. Fisher, M. S. Pizzuto, A. de Marco, D. Corti, M. Hosmillo, J. H. Lee, L. C. James, L. Thukral, D. Veesler, A. Sigal, F. Sampaziotis, I. G. Goodfellow, N. J. Matheson, K. Sato and R. K. Gupta, Nature, 2022, 603, 706–714 CrossRef CAS PubMed.
- Y. Cao, J. Wang, F. Jian, T. Xiao, W. Song, A. Yisimayi, W. Huang, Q. Li, P. Wang, R. An, J. Wang, Y. Wang, X. Niu, S. Yang, H. Liang, H. Sun, T. Li, Y. Yu, Q. Cui, S. Liu, X. Yang, S. Du, Z. Zhang, X. Hao, F. Shao, R. Jin, X. Wang, J. Xiao, Y. Wang and X. S. Xie, Nature, 2022, 602, 657–663 CrossRef CAS PubMed.
- L. Liu, S. Iketani, Y. Guo, J. F. W. Chan, M. Wang, L. Liu, Y. Luo, H. Chu, Y. Huang, M. S. Nair, J. Yu, K. K. H. Chik, T. T.-T. Yuen, C. Yoon, K. K. W. To, H. Chen, M. T. Yin, M. E. Sobieszczyk, Y. Huang, H. H. Wang, Z. Sheng, K.-Y. Yuen and D. D. Ho, Nature, 2022, 602, 676–681 CrossRef CAS PubMed.
- S. Cele, L. Jackson, D. S. Khoury, K. Khan, T. Moyo-Gwete, H. Tegally, J. E. San, D. Cromer, C. Scheepers, D. G. Amoako, F. Karim, M. Bernstein, G. Lustig, D. Archary, M. Smith, Y. Ganga, Z. Jule, K. Reedoy, S.-H. Hwa, J. Giandhari, J. M. Blackburn, B. I. Gosnell, S. S. Abdool Karim, W. Hanekom, M.-A. Davies, M. Hsiao, D. Martin, K. Mlisana, C. K. Wibmer, C. Williamson, D. York, R. Harrichandparsad, K. Herbst, P. Jeena, T. Khoza, H. Kløverpris, A. Leslie, R. Madansein, N. Magula, N. Manickchund, M. Marakalala, M. Mazibuko, M. Moshabela, N. Mthabela, K. Naidoo, Z. Ndhlovu, T. Ndung’u, N. Ngcobo, K. Nyamande, V. Patel, T. Smit, A. Steyn, E. Wong, A. von Gottberg, J. N. Bhiman, R. J. Lessells, M.-Y. S. Moosa, M. P. Davenport, T. de Oliveira, P. L. Moore and A. Sigal, Nature, 2022, 602, 654–656 CrossRef CAS PubMed.
- U. Rutwick Surya and N. Praveen, VirusDisease, 2021, 32, 46–54 CrossRef CAS PubMed.
- M. Hosseini, W. Chen, D. Xiao and C. Wang, Precision Clinical Medicine, 2021, 4, 1–16 CrossRef PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- J. Qiao, Y.-S. Li, R. Zeng, F.-L. Liu, H.-R. Luo, C. Huang, Y.-F. Wang, J. Zhang, B. Quan, C. Shen, X. Mao, X. Liu, W. Sun, W. Yang, X. Ni, K. Wang, L. Xu, Z.-L. Duan, Q.-C. Zou and S. Yang, Science, 2021, 371, eabf1611 CrossRef PubMed.
- M. E. Sobhia, K. Ghosh, S. Sivangula, S. Kumar and H. Singh, J. Biomol. Struct. Dyn., 2021, 1–11 CrossRef PubMed.
- D. W. Kneller, G. Phillips, H. M. O'Neill, R. Jedrzejczak, L. Stols, P. Langan, A. Joachimiak, L. Coates and A. Kovalevsky, Nat. Commun., 2020, 11, 7–12 CrossRef PubMed.
- L. Zhang, D. Lin, Y. Kusov, Y. Nian, Q. Ma, J. Wang, A. Von Brunn, P. Leyssen, K. Lanko, J. Neyts, A. De Wilde, E. J. Snijder, H. Liu and R. Hilgenfeld, J. Med. Chem., 2020, 63, 4562–4578 CrossRef CAS PubMed.
- M. D. Sacco, C. Ma, P. Lagarias, A. Gao, J. A. Townsend, X. Meng, P. Dube, X. Zhang, Y. Hu, N. Kitamura, B. Hurst, B. Tarbet, M. T. Marty, A. Kolocouris, Y. Xiang, Y. Chen and J. Wang, Sci. Adv., 2020, 6, eabe0751 CrossRef CAS PubMed.
- Z. Fu, B. Huang, J. Tang, S. Liu, M. Liu, Y. Ye, Z. Liu, Y. Xiong, W. Zhu, D. Cao, J. Li, X. Niu, H. Zhou, Y. J. Zhao, G. Zhang and H. Huang, Nat. Commun., 2021, 12, 488 CrossRef CAS PubMed.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K. P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Nature, 2020, 587, 657–662 CrossRef CAS PubMed.
- K. Al Adem, A. Shanti, C. Stefanini and S. Lee, Pharmaceuticals, 2020, 13, 1–34 CrossRef PubMed.
- M. Gheblawi, K. Wang, A. Viveiros, Q. Nguyen, J.-C. Zhong, A. J. Turner, M. K. Raizada, M. B. Grant and G. Y. Oudit, Circ. Res., 2020, 126, 1456–1474 CrossRef CAS PubMed.
- J. Yang, S. J. L. Petitjean, M. Koehler, Q. Zhang, A. C. Dumitru, W. Chen, S. Derclaye, S. P. Vincent, P. Soumillion and D. Alsteens, Nat. Commun., 2020, 11, 4541 CrossRef CAS PubMed.
- S. Choudhary, Y. S. Malik and S. Tomar, Front. Immunol., 2020, 11, 1664 CrossRef CAS PubMed.
- S. Bharadwaj, E. I. Azhar, M. A. Kamal, L. H. Bajrai, A. Dubey, K. Jha, U. Yadava, S. G. Kang and V. D. Dwivedi, J. Biomol. Struct. Dyn., 2020, 1–16 CAS.
- Y. Han and P. Král, ACS Nano, 2020, 14, 5143–5147 CrossRef CAS PubMed.
- G. Jaiswal and V. Kumar, PLoS One, 2020, 15, 1–15 CrossRef PubMed.
- F. X. Domínguez-Villa, N. A. Durán-Iturbide and J. G. Ávila-Zárraga, Bioorg. Chem., 2021, 106, 104497 CrossRef PubMed.
- R. Nandini Asha, B. Ravindran Durai Nayagam and N. Bhuvanesh, Bioorg. Chem., 2021, 112, 104967 CrossRef CAS PubMed.
- A. Ali, N. Sepay, M. Afzal, N. Sepay, A. Alarifi, M. Shahid and M. Ahmad, Bioorg. Chem., 2021, 110, 104772 CrossRef CAS PubMed.
- N. Atatreh, S. Hasan, B. R. Ali and M. A. Ghattas, Acta Pharm., 2021, 71, 325–333 CrossRef CAS.
- V. A. Obakachi, N. D. Kushwaha, B. Kushwaha, M. C. Mahlalela, S. R. Shinde, I. Kehinde and R. Karpoormath, J. Mol. Struct., 2021, 1241, 130665 CrossRef CAS PubMed.
- R. Ramajayam, K. P. Tan, H. G. Liu and P. H. Liang, Bioorg. Med. Chem., 2010, 18, 7849–7854 CrossRef CAS PubMed.
- V. Kumar, K. P. Tan, Y. M. Wang, S. W. Lin and P. H. Liang, Bioorg. Med. Chem., 2016, 24, 3035–3042 CrossRef CAS PubMed.
- J. Sahoo, C. R. Sahoo, P. K. Nandini Sarangi, S. K. Prusty, R. N. Padhy and S. K. Paidesetty, Eur. J. Med. Chem., 2020, 186, 111911 CrossRef CAS PubMed.
- X. Fan, X. Zhang, L. Zhou, K. A. Keith, E. R. Kern and P. F. Torrence, Bioorg. Med. Chem. Lett., 2006, 16, 3224–3228 CrossRef CAS PubMed.
- H. E. Gaffer, S. Abdel-Fattah, H. A. Etman and E. Abdel-Latif, J. Heterocycl. Chem., 2017, 54, 331–340 CrossRef CAS.
- H. M. Aly, N. M. Saleh and H. A. Elhady, Eur. J. Med. Chem., 2011, 46, 4566–4572 CrossRef CAS PubMed.
- N. A. Khalil, E. M. Ahmed, K. O. Mohamed, Y. M. Nissan and S. A. B. Zaitone, Bioorg. Med. Chem., 2014, 22, 2080–2089 CrossRef CAS PubMed.
- M. T. El Sayed, M. A. M. S. El-Sharief, E. S. Zarie, N. M. Morsy, A. R. Elsheakh, A. Voronkov, V. Berishvili and G. S. Hassan, Bioorg. Med. Chem. Lett., 2018, 28, 952–957 CrossRef CAS PubMed.
- M. H. Norman, L. Liu, M. Lee, N. Xi, I. Fellows, N. D. D'Angelo, C. Dominguez, K. Rex, S. F. Bellon, T. S. Kim and I. Dussault, J. Med. Chem., 2012, 55, 1858–1867 CrossRef CAS PubMed.
- M. A. Abdelgawad, M. B. Labib, W. A. M. Ali, G. Kamel, A. A. Azouz and E. S. EL-Nahass, Bioorg. Chem., 2018, 78, 103–114 CrossRef CAS PubMed.
- K. Ochiai, S. Takita, A. Kojima, T. Eiraku, N. Ando, K. Iwase, T. Kishi, A. Ohinata, Y. Yageta, T. Yasue, D. R. Adams and Y. Kohno, Bioorg. Med. Chem. Lett., 2012, 22, 5833–5838 CrossRef CAS PubMed.
- X. K. Qian, J. Zhang, P. F. Song, Y. S. Zhao, H. Y. Ma, Q. Jin, D. D. Wang, X. Q. Guan, S. Y. Li, X. Z. Bao and L. W. Zou, Bioorg. Med. Chem., 2021, 40, 116187 CrossRef CAS PubMed.
- T. M. A. Eldebss, A. M. Farag, M. M. Abdalla and A. A. Khedr, J. Heterocycl. Chem., 2019, 56, 765–780 CrossRef CAS.
- Z. T. Muhseen, S. Kadhim, Y. I. Yahiya, E. A. Alatawi, F. F. Aba Alkhayl and A. Almatroudi, Biology, 2021, 10, 1310 CrossRef CAS PubMed.
- H. You, X. Su and G. Su, Arch. Pharm., 2020, 353, 1–10 CrossRef PubMed.
- Z. Zhou and Y. Zhang, J. Chil. Chem. Soc., 2015, 60, 2992–2996 CrossRef CAS.
- A. Vafaee, A. Davoodnia and M. Pordel, Res. Chem. Intermed., 2015, 41, 8343–8354 CrossRef CAS.
- J. Safaei-Ghomi, B. Khojastehbakht-Koopaei and S. Zahedi, Chem. Heterocycl. Compd., 2015, 51, 34–38 CrossRef CAS.
- R. Ramesh, N. Nagasundaram, D. Meignanasundar, P. Vadivel and A. Lalitha, Res. Chem. Intermed., 2017, 43, 1767–1782 CrossRef CAS.
- M. Zarghani and B. Akhlaghinia, RSC Adv., 2015, 5, 87769–87780 RSC.
- J. Safaei-Ghomi, B. Khojastehbakht-Koopaei and H. Shahbazi-Alavi, RSC Adv., 2014, 4, 46106–46113 RSC.
- R. Khalifeh, R. Shahimoridi and M. Rajabzadeh, Catal. Lett., 2019, 149, 2864–2872 CrossRef CAS.
- Z. Zhou and Y. Zhang, Green Chem. Lett. Rev., 2014, 7, 18–23 CrossRef.
- Z.-Z. Sheng, M.-M. Huang, T. Xue, F. Xia and H.-H. Wu, RSC Adv., 2020, 10, 34910–34915 RSC.
- J. E. Gannon, M. C. Adams and E. O. Bennett, Microbios, 1978, 23, 7–18 CAS.
- R. J. West and S. J. Gonsior, Environ. Toxicol. Chem., 1996, 15, 472–480 CrossRef CAS.
- F. Mancia, P. Vasileios, O. B. Clarke and J. P. Vendome, Rational drug design targeting resistant Gram-negative bacterial infections to polymyxin-class antibiotics, WIPO Pat., WO2017083859A1, 2017 Search PubMed.
- A. M. Zonouz, M. Beiranvand, R. Mohammad-Rezaei and S. Naderi, Lett. Org. Chem., 2020, 17, 548–554 CrossRef CAS.
- W. Wang, S.-X. Wang, X.-Y. Qin and J.-T. Li, Synth. Commun., 2005, 35, 1263–1269 CrossRef CAS.
- R. Ramesh, N. Nagasundaram, D. Meignanasundar, P. Vadivel and A. Lalitha, Res. Chem. Intermed., 2017, 43, 1767–1782 CrossRef CAS.
- E. Soleimani, S. Ghorbani, M. Taran and A. Sarvary, C. R. Chim., 2012, 15, 955–961 CrossRef CAS.
- H.-K. Fun, R. Kia, K. S. Girish and B. Kalluraya, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o66–o67 CrossRef CAS PubMed.
- B. Kumar, H. Mahajan, S. Paul, R. Kant and V. K. Gupta, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o805–o806 CrossRef CAS PubMed.
- H.-K. Fun, S. R. Jebas, K. S. Girish and B. Kalluraya, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o2301–o2302 CrossRef CAS PubMed.
- C. Pettinari, F. Marchetti, R. Pettinari, D. Martini, A. Drozdov and S. Troyanov, J. Chem. Soc., Dalton Trans., 2001, 1790–1797 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- G. A. Bogdanović, B. D. Ostojić and S. B. Novaković, Cryst. Growth Des., 2018, 18, 1303–1314 CrossRef.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, Nucleic Acids Res., 2018, 46, W363–W367 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- P. K. Panda, M. N. Arul, P. Patel, S. K. Verma, W. Luo, H.-G. Rubahn, Y. K. Mishra, M. Suar and R. Ahuja, Sci. Adv., 2020, 6, 1–15 Search PubMed.
- H. Yang, W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei, J. Ziebuhr, R. Hilgenfeld, K. Y. Yuen, L. Wong, G. Gao, S. Chen, Z. Chen, D. Ma, M. Bartlam and Z. Rao, PLoS Biol., 2005, 3, e324 CrossRef PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- A. Finch and P. Pillans, Aust. Prescr., 2014, 37, 137–139 CrossRef.
- A. Mansoor and N. Mahabadi, Volume of Distribution, Treasure Island, FL, 2022 Search PubMed.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- P. Manikandan and S. Nagini, Curr. Drug Targets, 2018, 19, 38–54 CAS.
- K. Mortelmans and E. Zeiger, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2000, 455, 29–60 CrossRef CAS PubMed.
- Rigaku Oxford Diffraction, CrysAlisPro Softw. Syst, Rigaku Corp., Oxford, UK, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2009, 65, 148–155 CAS.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. a. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- J. Osipiuk, S.-A. Azizi, S. Dvorkin, M. Endres, R. Jedrzejczak, K. A. Jones, S. Kang, R. S. Kathayat, Y. Kim, V. G. Lisnyak, S. L. Maki, V. Nicolaescu, C. A. Taylor, C. Tesar, Y.-A. Zhang, Z. Zhou, G. Randall, K. Michalska, S. A. Snyder, B. C. Dickinson and A. Joachimiak, Nat. Commun., 2021, 12, 743 CrossRef CAS PubMed.
- P. Towler, B. Staker, S. G. Prasad, S. Menon, J. Tang, T. Parsons, D. Ryan, M. Fisher, D. Williams, N. A. Dales, M. A. Patane and M. W. Pantoliano, J. Biol. Chem., 2004, 279, 17996–18007 CrossRef CAS PubMed.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu, K.-Y. Yuen, Q. Wang, H. Zhou, J. Yan and J. Qi, Cell, 2020, 181, 894–904.e9 CrossRef CAS PubMed.
- D. Simijonović, E. E. Vlachou, Z. D. Petrović, D. J. Hadjipavlou-Litina, K. E. Litinas, N. Stanković, N. Mihović and M. P. Mladenović, Bioorg. Chem., 2018, 80, 741–752 CrossRef PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- S. D. D. S. BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, v 21.1.0.20298 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- Faheem, B. K. Kumar, K. V. G. C. Sekhar, S. Kunjiappan, J. Jamalis, R. Balaña-Fouce, B. L. Tekwani and M. Sankaranarayanan, Bioorg. Chem., 2020, 104, 104269 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, Nucleic Acids Res., 2018, 46, W257–W263 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2132092. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra02542f |
| This journal is © The Royal Society of Chemistry 2022 |