
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP4O10/TfOH mediated domino condensation–cyclization of amines with diacids: a route to indolizidine alkaloids under catalyst- and solvent-free conditions†
Ketan S. Mandrekar and
Santosh G. Tilve *
*
School of Chemical Sciences, Goa University, Taleigao, Goa 403206, India. E-mail: stilve@unigoa.ac.in
First published on 15th June 2022
Abstract
A domino condensation–cyclization method is developed to synthesize indolizidine alkaloids using a P4O10/TfOH reagent system without the employment of either a catalyst or solvent. The use of a few aliphatic and aromatic dicarboxylic acids is shown along with various primary amines. This method is suitable for synthesizing pyrrolo[2,1-a]isoquinolines, pyrido[2,1-a]isoquinolines, and isoindolo[1,2-a]isoquinolinones in excellent yields. When phthalic acid is used, a workup with either NaBH4 or a saturated NaHCO3 solution provided 12b-H or 12b-OH isoindolo[1,2-a]isoquinolinones, respectively.
Indolizidine alkaloids are structurally important molecules that belong to the broader class of natural products.1 These indolizidine alkaloids are isolated as lysine-derived metabolites from various animal and plant sources, like higher plants, invertebrates, vertebrates, fungi, and bacteria.2 With their core structure of a fused six- and five-membered ring system with a bridgehead nitrogen atom, indolizidine compounds constitute 25–30% of all alkaloids.3 Benzoindolizidine alkaloids that contain benzene rings fused to one of the indolizidine rings are also known in nature. A few selected examples are shown in Fig. 1. For instance, crispine contains a benzene ring attached to the indolizidine piperidine ring. Jamtine and erythraline, in addition to the benzene ring, have a six membered cyclohexene ring attached to indolizidine pyrrolidine. In elaeocarpaceae, a phenanthrene ring is attached to the indolizidine piperidene ring. In nuevamine, a benzene ring is attached to both the indolizidine piperidine and pyrrolidine rings. Alkaloids like lamellarin, which are well studied for their wide range of biological activity, contain an indolizidine ring fused to a benzene and a coumarin ring, making them unique in the indolizidine family.
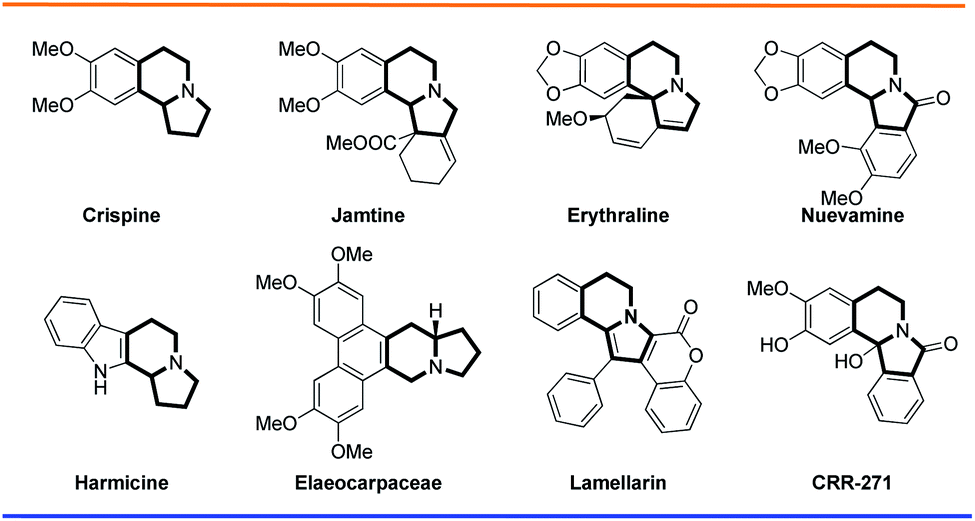 | ||
| Fig. 1 Selected bioactive molecules containing indolizidine rings. | ||
Among current pharmaceuticals, ∼40% are derived from or inspired by natural products.4 Scientists have extensively worked on the construction of indolizidine core structures from various synthons.5 The indolizidine compound class shows a wide range of promising biological activities such as cytotoxicity,6 antibacterial activity, antiviral activity,7 antioxidant activity,8 antileishmanial activity,9 antinociceptive activity,10 etc. Manufactured compounds like CRR-271 act as poly(ADP-ribose) polymerase-1 inhibitors, while other derivatives have been prepared and tested for antioxidant activity for lipid peroxidation at a cellular level and for protection against genomic DNA damage at a nuclear level.11
We recently reported our work on constructing lamellarin scaffolds via a tandem Bischler–Napieralski reaction–Michael reaction–oxidation sequence wherein we fabricated fused isoquinoline and pyrrole ring structures around a coumarin ring.12 However, the direct condensation of primary amines and dicarboxylic acids for the construction of the multi-ring structures has been less studied. Moreover, practicing chemists usually require two or more steps to construct the indolizidine ring core from different substrates under various conditions.13 Ramanathan et al. elaborated a precise methodological syntheses of isoindoloisoquinolinones, pyrroloisoquinolinones, and benzo[a]quinolizinones using TfOH. They have also prepared numerous annulated tetrahydroisoquinolines. Imide phenylethylamine surrogates were cyclized using excess TfOH with either succinic anhydrides, phthalic anhydrides, or glutaric anhydride before being reduced with sodium borohydride or water to get the corresponding pyrroloisoquinolinones, isoindoloisoquinolinones, benzo[a]quinolizinones, or hydroxyl derivatives. They synthesized a library of such compounds and even demonstrated the syntheses of many naturally occurring compounds in their racemic form. They have synthesized (±)-crispine A, (±)-trolline/oleracein E, (±)-erythrabine, and (±)-mescalotam from their respective imides in excellent yields. Later, they applied an iminium ion cyclization strategy to unsymmetrical succinimides to synthesize 1- or 2-alkylsubstituted pyrroloisoquinolinones and indolizinoindolones.14
The present study describes a one-pot protocol for constructing an indolizidine ring system from 2-arylethanamines and dicarboxylic acids.
Results and discussion
We examined the reagents required for a reaction between primary amines and dicarboxylic acids and settled on the use of phosphorus pentaoxide (P4O10) to carry out the reaction. Many reports have focused on the cyclization of 1-phenethylpyrrolidine-2,5-dione via an acyliminium ion. Thus, our methodology also depends on a one-pot formation of an acyliminium ion followed by its cyclization and reduction.Initially, a model reaction between 1 and succinic acid 2a was carried out in the presence of 2.5 equivalents of P4O10 in refluxing toluene (Table 1, entry 1). It did not cyclize to form tricyclic compound 3 but instead formed imide 4. Further refluxing in dry xylene, to test whether the formation of 3 was temperature-dependent, also failed to give the desired product (Table 1, entry 2). The use of Eaton's reagent (7.5 wt% P4O10 in MsOH) has been reported for dehydration and cyclization reactions in many cases.15 So, in freshly prepared Eaton's reagent at room temperature another reaction was set between 1 and 2a (Table 1, entry 3). Tricyclic product 3 was formed, after reduction, in a 28% yield and no imide 4 was isolated. The acidity of the reagents or the temperature are factors that could also affect the cyclization. Thus, with the same 7.5 wt% proportion of P4O10, we prepared a mixture of P4O10 in TfOH and carried out the reaction at room temperature (Table 1, entry 4). This reaction produced compound 3 in a 45% yield after the reduction process. With the same mixture of P4O10 and TfOH at 60 °C, a 20% increase in yield was found in comparison to the previous attempt (Table 1, entry 5). When the wt% of the P4O10 in TfOH mixture was increased to 8.3 wt% and the temperature was increased to 100 °C, we could obtain the cyclized compound, after reduction using sodium borohydride, in a 79% yield in just 2 h (Table 1, entry 6).
|
||||
|---|---|---|---|---|
| Entry | Reaction conditions (1)a | Time | Yield | |
| 3 | 4 | |||
| a The reaction was performed with homoveratrylamine 1 (1.5 mmol) and succinic acid 2a (1 mmol) for the time periods indicated; this was followed by reduction with NaBH4 (1 mmol) in MeOH at rt.b n.d. – not detected. | ||||
| 1 | P4O10, dry toluene, Δ | 12 h | n.d.b | 63% |
| 2 | P4O10, dry xylene, Δ | 12 h | n.d. | 65% |
| 3 | Eaton's reagent, rt | 8 h | 28% | n.d. |
| 4 | 7.5 wt% P4O10/TfOH, rt | 6 h | 45% | n.d. |
| 5 | 7.5 wt% P4O10/TfOH, 60 °C | 6 h | 65% | n.d. |
| 6 | 8.3 wt% P4O10/TfOH, 100 °C | 2 h | 79% | n.d. |
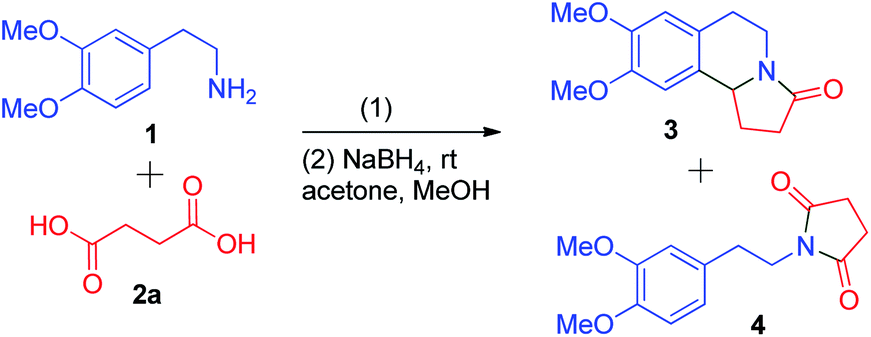
After successfully making 3a, the reactions of other primary amines with succinic acid and glutaric acid were conducted to synthesize tricyclic structures (Scheme 1). The reaction of mono and trimethoxy phenethylamines with succinic acid proceeded smoothly to furnish compounds 3b and 3c in 74% and 75% yields, respectively. Compound 3c is a naturally occurring compound, mescalotam, for which no biological activity study has been reported to date. The indole-containing heterocyclic amine tryptamine, a precursor for naturally occurring compound harmicine, was reacted with succinic acid to afford 3d in an 81% yield. Instead of succinic acid 2a, the five carbon dicarboxylic acid, glutaric acid 2b, was incorporated into our methodology to assess its reactivity. The reaction of 2b with homoveratrylamine and tryptamine gave quinolizidine alkaloids 3e and 3f in 74% and 79% yields, respectively.
 | ||
| Scheme 1 Syntheses of indolizidine and quinolizidine alkaloids. | ||
Furthermore, these compounds were, using reduction and demethylation reactions, utilized to synthesize naturally occurring compounds such as (±)-crispine A, (±)-trolline/oleracein E, and (±)-harmicine. The amide group in compound 3a was reduced using lithium aluminium hydride to get (±)-crispine A 5 in an overall yield of 66% (Scheme 2). Likewise, 3a was demethylated to its dihydroxy form using boron tribromide in dry dichloromethane to afford (±)-trolline/oleracein E 6 in an overall yield of 68% (Scheme 3).
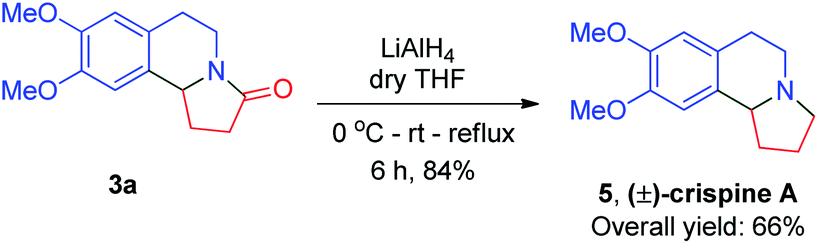 | ||
| Scheme 2 Total synthesis of (±)-crispine A. | ||
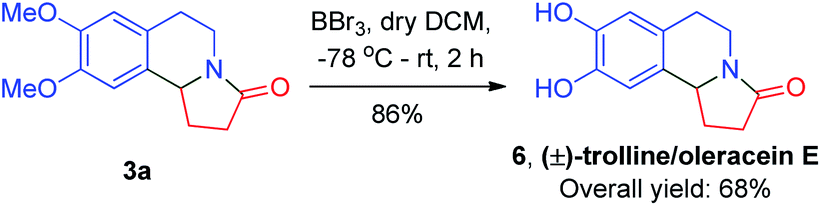 | ||
| Scheme 3 Total synthesis of (±)-trolline/oleracein E. | ||
As past reports do not suggest that mescalotam 3c has any biological activity, we envisaged synthesizing and then studying the bioactivity of its trihydroxy derivative. We successfully synthesized 7 from compound 3c in an 89% yield by complete demethylation using the same method employed to synthesize (±)-trolline/oleracein E (Scheme 4). Using the method employed to synthesize (±)-crispine A, we obtained (±)-harmicine 8 from its amide precursor 3d in an overall yield of 74% starting from tryptamine (Scheme 5).
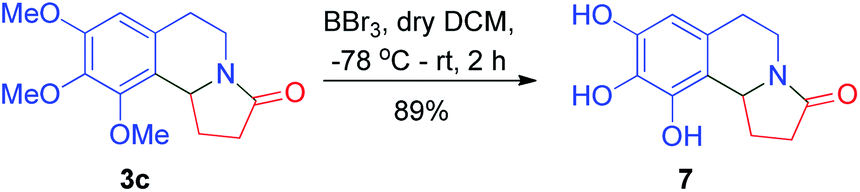 | ||
| Scheme 4 Synthesis of trihydroxy derivative of mescalotam. | ||
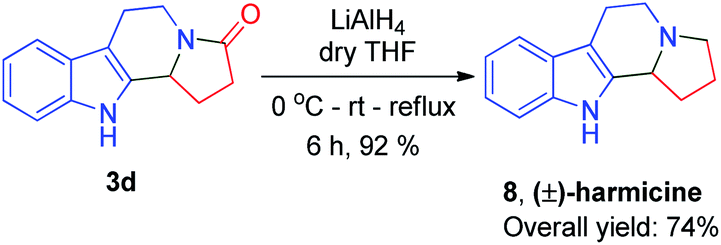 | ||
| Scheme 5 Total synthesis of (±)-harmicine. | ||
In addition, aromatic 1,2-dicarboxylic acids were also reacted with primary amines using the 8.3 wt% P4O10/TfOH reagent system. Four aryl/heteroaryl ethylamines were treated with phthalic acid 2c and heated in an 8.3 wt% P4O10/TfOH mixture at 100 °C followed by reduction of the subsequent iminium ion to produce the final adducts (Scheme 6). Compound 9a was prepared from homoveratrylamine 1 and phthalic acid 2c in an 89% yield. Whereas the monomethoxy compound 9b and the unsubstituted analog 9c were prepared successfully from 3-methoxyphenethylamine and phenylethylamine in 85% and 84% yields, respectively. In addition to simple aryl compounds, a heteroaryl derivative with a pentacyclic ring structure 9d was obtained from tryptamine and phthalic acid 2c in an 87% yield. Successful incorporation of a hydride ion at the 12b-position furnished all the desired isoindoloisoquinolinone compounds successfully.
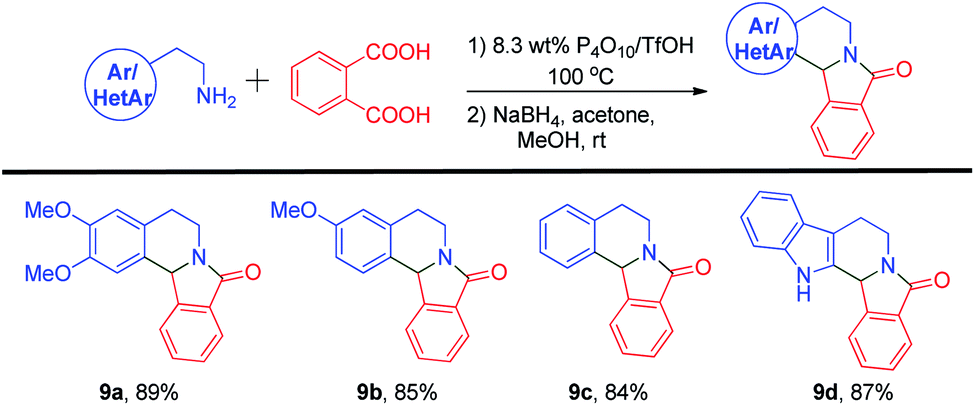 | ||
| Scheme 6 Syntheses of isoindolo[2,1-a]isoquinolinone alkaloids. | ||
After accomplishing the target molecules, we synthesized their 12b-hydroxy analogs via a saturated sodium bicarbonate workup (Scheme 7). The respective aryl/heteroaryl ethylamines 11a, 11b, 11c, and 11d were condensed with two different phthalic acid derivatives. When condensed with simple phthalic acid 2c, four 12b-hydroxy isoindoloisoquinolinone derivatives, 10a, 10b, 10c, and 10d, were prepared from their corresponding amines using just a simple basic workup. Novel analogs like 10e, 10f, and 10g were prepared using 4-nitrophthalic acid 2d. When 2d was fused with homoveratrylamine, 10e was obtained in a 91% yield. The reaction of 3-methoxyphenethyl amine went smoothly to form 10f in an 89% yield. Also, tryptamine-derived 12b-hydroxy analog 10g was synthesized in a slightly higher yield than the other amines.
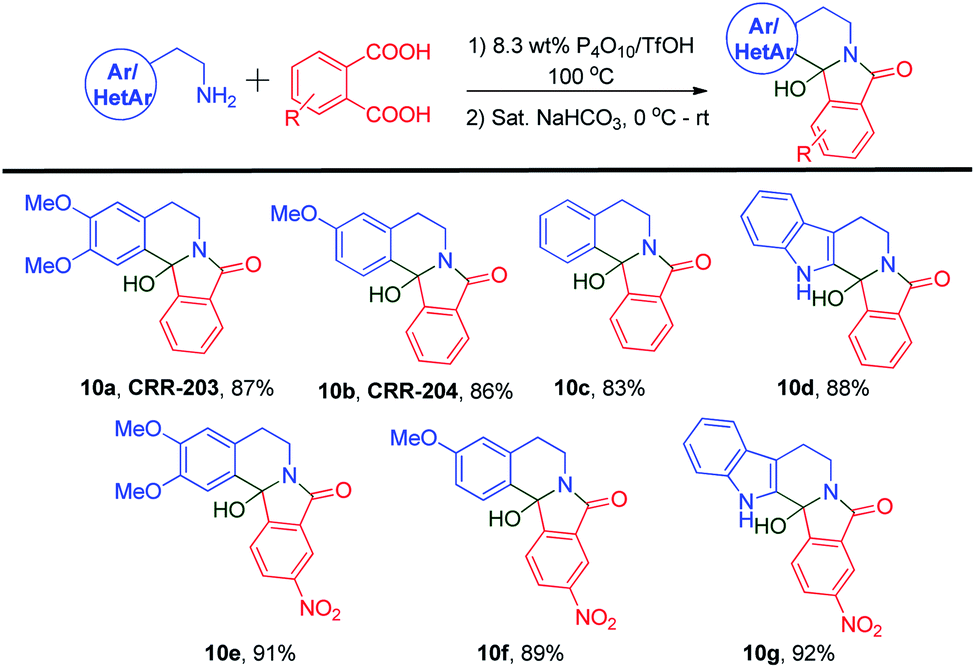 | ||
| Scheme 7 Syntheses of 12b-hydroxyisoindolo[2,1-a]isoquinolinone alkaloids. | ||
A speculative mechanism is proposed in Scheme 8. The combination of P4O10/TfOH produces triflic anhydride which induces the activation of the diacid. The resulting mixed anhydride is attacked by the amine to form the amide acid. The amide acid is further activated by triflic anhydride to form the imide via a mixed anhydride. The imide undergoes a Bischler–Napieralski reaction mediated by triflic anhydride. Reduction of the resulting salt delivers the final product.
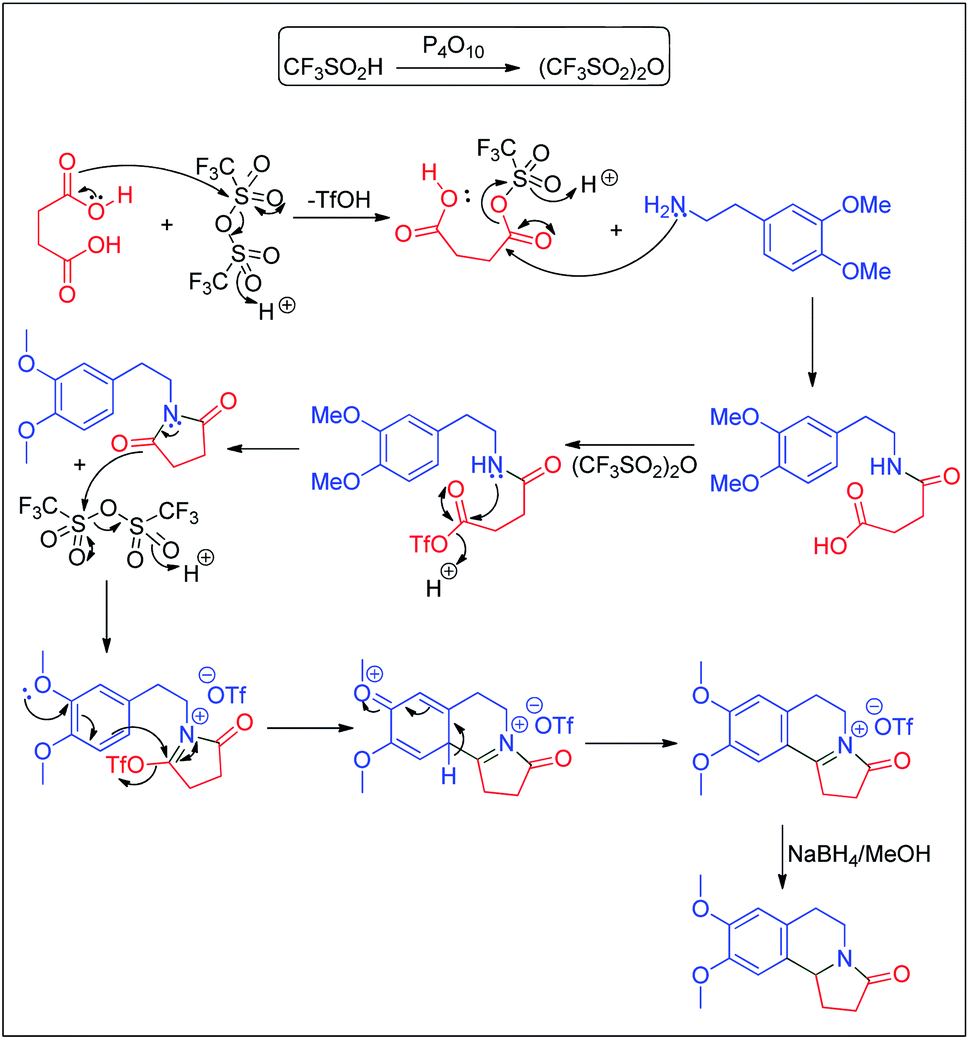 | ||
| Scheme 8 Plausible mechanism for the formation of pyrrolo[2,1-a]isoquinolinone. | ||
Conclusions
The present methodology allows the construction of indolizidine and quinolizidine rings adjacent to benzene rings. The reactions proceed via single pot domino reactions between phenethylamine derivatives and aliphatic and aromatic dicarboxylic acids in the presence of an 8.3 wt% mixture of P4O10/TfOH. In total, seventeen 12b-H and 12b-OH derivatives of pyrrolo[2,1-a]isoquinoline and isoindolo[1,2-a]isoquinoline alkaloids are synthesized in very good yields. The utility of this methodology was demonstrated by synthesizing naturally occurring compounds such as, (±)-crispine A, (±)-trolline/oleracein E, (±)-mescalotam and (±)-harmicine from commercially available synthons. A novel trihydroxy derivative of mescalotam was also synthesized successfully. Compounds such as CRR-203, CRR-204 were also synthesized in excellent yields.Author contributions
SGT was involved in overall supervision, while KSM was involved in project planning, execution, experiments, and manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KSM acknowledges UGC for a NFOBC fellowship and SGT for a UGC-BSR faculty fellowship.Notes and references
- I. Chakraborty and S. Jana, Synthesis, 2013, 45, 3325 CrossRef CAS.
- J. P. Michael, Simple Indolizidine and Quinolizidine Alkaloids, Alkaloids: Chem. Biol., 2016, 75, 1–498 CAS.
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed; (b) D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed; (c) J. P. Michael, Beilstein J. Org. Chem., 2007, 3, 27 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139–165 RSC.
- H. El-Subbagh, T. Wittig, M. Decker, S. Elz, M. Nieger and J. Lehmann, Arch. Pharm., 2002, 335, 443448 CrossRef.
- (a) D. L. J. Clive, Z. Li and M. Yu, J. Org. Chem., 2007, 72, 5608–5617 CrossRef CAS PubMed; (b) K. A. Tehrani, M. D'hooghe and N. D. Kimpe, Tetrahedron, 2003, 59, 3099 CrossRef CAS; (c) W. H. Fearson and K.-C. Lin, Tetrahedron Lett., 2003, 131, 7571 Search PubMed; (d) N. Zill, A. Schoenfelder, N. Girard, M. Taddei and A. Mann, J. Org. Chem., 2012, 77, 2246 CrossRef CAS PubMed; (e) D. Berthold, A. G. A. Geissler, S. Giofre and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 9994 CrossRef CAS PubMed; (f) S. V. Kauloorkar, V. Jha, G. Jogdand and P. Kumar, Org. Biomol. Chem., 2014, 12, 4454 RSC.
- R. F. Wang, X. W. Yang, C. M. Ma, S. Q. Cai, J. N. Li and Y. Shoyama, Heterocycles, 2004, 63, 1443 CrossRef CAS.
- C. Narajji, M. D. Karvekar and A. K. Das, S. Afr. J. Chem., 2008, 61, 53 CAS.
- Z. Yang, C. Liu, L. Xiang and Y. Zheng, Phytother. Res., 2009, 23, 10321035 Search PubMed.
- H. M. Spindola, D. B. Vendramini-Costa, M. T. Rodrigues Jr, M. A. Foglio, R. A. Pilli and J. E. Carvalho, Pharmacol., Biochem. Behav., 2012, 102, 133 CrossRef CAS PubMed.
- (a) S. H. Cheon, J. S. Park, J. Y. Lee, Y. N. Lee, B. H. Chung, B. G. Choi, W. J. Cho, S. U. Choi and C. O. Lee, Arch. Pharmacal Res., 2001, 24, 276 CrossRef CAS PubMed; (b) T. Radovits, L. Seres, D. Gero, I. Berger, C. Szabo, M. Karck and G. Szabo, Exp. Gerontol., 2007, 42, 676 CrossRef CAS PubMed; (c) R. Pellicciari, E. Camaioni, G. Costantino, L. Formentini, P. Sabbatini, F. Venturoni, G. Eren, D. Bellocchi, A. Chiaruqi and F. Moroni, ChemMedChem, 2008, 3, 914 CrossRef CAS PubMed.
- A. Suyavaran, C. Ramamurthy, R. Mareeswaran, Y. V. Shanthi, J. Selvakumar, S. Mangalaraj, M. S. Kumar, C. R. Ramanathan and C. Thirunavukkarasu, Bioorg. Med. Chem., 2015, 23, 488 CrossRef CAS PubMed.
- K. S. Mandrekar, H. K. Kadam and S. G. Tilve, Eur. J. Org. Chem., 2018, 2018, 6665 CrossRef CAS.
- (a) S. Agarwal, O. Kataeva, U. Schmidt and H. J. Knölker, RSC Adv., 2013, 3, 1089 RSC; (b) S. Dhanasekaran, V. Bisai, R. A. Unhale, A. Suneja and V. K. Singh, Org. Lett., 2014, 16, 6068 CrossRef CAS PubMed; (c) J. C. Orejarena Pacheco, A. Lipp, A. M. Nauth, F. Acke, J. P. Dietz and T. Opatz, Chem.–Eur. J., 2016, 22, 5409 CrossRef CAS PubMed; (d) C. Yan, Y. Liu and Q. Wang, Org. Lett., 2015, 17, 5714 CrossRef CAS PubMed; (e) K. Murai, K. Matsuura, H. Aoyama and H. Fujioka, Org. Lett., 2016, 18, 1314 CrossRef CAS PubMed; (f) R. A. Talk, A. Duperray, X. Li and I. Coldham, Org. Biomol. Chem., 2016, 14, 4908 RSC; (g) F. Souquet, W. Drici, S. A. Fayssal, I. Lazouni, S. Thueillon and J. A. Pérard-Viret, Synthesis, 2020, 52, 2970 CAS; (h) B. V. M. Teodoro, J. T. M. Correia and F. Coelho, J. Org. Chem., 2015, 80, 2529 CrossRef CAS PubMed; (i) M. J. Albaladejo, M. J. González-Soria and F. Alonso, J. Org. Chem., 2016, 81, 9707 CrossRef CAS PubMed.
- (a) J. Selvakumar, R. S. Rao, V. Srinivasapriyan, S. Marutheeswaran and C. R. Ramanathan, Eur. J. Org. Chem., 2015, 2015, 2175 CrossRef CAS; (b) J. Selvakumar, S. Mangalaraj, K. M. M. Achari, K. Mukund and C. R. Ramanathan, Synthesis, 2017, 49, 1053 CAS.
- (a) P. E. Eaton, G. R. Carlson and J. T. Lee, J. Org. Chem., 1973, 38, 4071 CrossRef CAS; (b) K. B. Hansen, J. Balsells, S. Dreher, Y. Hsiao, M. Kubryk, M. Palucki, N. Rivera, D. Steinhuebel, J. D. Armstrong, D. Askin and E. J. Grabowski, Org. Process Res. Dev., 2005, 9, 634 CrossRef CAS; (c) D. Zewge, C. Y. Chen, C. Deer, P. G. Dormer and D. L. Hughes, J. Org. Chem., 2007, 72, 4276 CrossRef CAS PubMed; (d) D. J. Skalitzky, J. T. Marakovits, K. A. Maegley, A. Ekker, X. H. Yu, Z. Hostomsky, S. E. Webber, B. W. Eastman, R. Almassy and J. Li, J. Med. Chem., 2003, 46, 210 CrossRef CAS PubMed; (e) R. L. Dorow, P. M. Herrinton, R. A. Hohler, M. T. Maloney, M. A. Mauragis, W. E. McGhee, J. A. Moeslein, J. W. Strohbach and M. F. Veley, Org. Process Res. Dev., 2006, 10, 493 CrossRef CAS; (f) C. R. Pandit, R. P. Polniaszek and J. K. Thottathil, Synth. Commun., 2002, 32, 2427 CrossRef CAS; (g) D. Zhao, D. L. Hughes, D. R. Bender, A. M. DeMarco and P. J. Reider, J. Org. Chem., 1991, 56, 3001 CrossRef CAS; (h) S. Kano, T. Yokomatsu, Y. Yuasa and S. Shibuya, Chem. Pharm. Bull., 1985, 33, 340 CrossRef CAS; (i) D. Gala, V. H. Dahanukar, J. M. Eckert, B. S. Lucas, D. P. Schumacher and I. A. Zavialov, Org. Process Res. Dev., 2004, 8, 754 CrossRef CAS; (j) J. Chae and S. L. Buchwald, J. Org. Chem., 2004, 69, 3336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound data. See https://doi.org/10.1039/d2ra02534e |
| This journal is © The Royal Society of Chemistry 2022 |